
Photoinduced hydroxylperfluoroalkylation of styrenes†
Tongbi
Chen
a,
Yong
Guo
 *b,
Ke
Sun
c,
Li-Zhu
Wu
d,
Wen-Qiang
Liu
d,
Chao
Liu
b,
Yangen
Huang
*a and
Qing-Yun
Chen
*b
*b,
Ke
Sun
c,
Li-Zhu
Wu
d,
Wen-Qiang
Liu
d,
Chao
Liu
b,
Yangen
Huang
*a and
Qing-Yun
Chen
*b
aCollege of Chemistry, Chemical Engineering and Biotechnology, Donghua University, 2999 North Renmin Road, Shanghai 201620, P. R. of China. E-mail: hyg@dhu.edu.cn
bKey Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, P. R. of China. E-mail: yguo@sioc.ac.cn; chenqy@sioc.ac.cn
cState Key Laboratory of the Discovery and Development of Novel Pesticide, Shenyang Sinochem Agrochemicals R&D Ltd., Shenyang 110021, P. R. of China
dKey Laboratory of Photochemical Conversion and Optoelectronic Materials, Technical Institute of Physics and Chemistry and University of Chinese Academy of Sciences, Chinese Academy of Sciences, Beijing 100190, P. R. China
First published on 8th January 2018
Abstract
A transition metal- and organophotocatalyst-free hydroxylperfluoroalkylation of styrenes under visible-light irradiation was developed. In this protocol, an electron donor–acceptor complex (perfluoroalkyl iodide and tertiary amine) was employed for the introduction of a perfluorinated chain and molecular oxygen was used as a green oxidant for the generation of hydroxyl groups. Various styrenes successfully undergo the reaction affording the corresponding fluorine-containing alcohols in up to 96% yields.
Introduction
Over the past several years, impressive developments in the field of photoredox catalysis using visible light have occurred.1 However, studies on the photochemistry of electron donor–acceptor (EDA) complexes are rare.2 In some reactions, catalyst-free systems only lead to a slightly better result than that with a photocatalyst.3 In the 1990s, we published several reports on the generation of fluoroalkyl and perfluoroaryl radicals from EDA complexes by UV irradiation.4 UV light has a high energy, making it hard to control the chemical selectivity. On the other hand, visible-light is a viable alternative for designing a reaction with mild conditions and good functional group tolerance. Visible-light promoted fluoroalkylations without the use of a photocatalyst were reported by Yu in 2016 and later by Chen in 2017 for the synthesis of 2-fluoroalkylated quinoxalines, perfluoroalkyl-substituted phenanthridines, β-iodo-perfluoroalkylated alkenes/alkynes, and perfluoroalkylated arenes.5 These pioneering reactions should encourage the further design of catalytic visible-light promoted reactions.Difunctionalization of carbon–carbon double bonds is a useful strategy for organic synthesis, and there are numerous such reactions involving fluoroalkylation.6 However, the hydroxylperfluoroalkylation of alkenes is challenging. Previous successful examples required expensive electrophilic trifluoromethylating reagents, toxic tin reagents, stoichiometric amounts of copper, or showed a lack of selectivity.7 In 2017, Tsui reported the hydroxytrifluoromethylation of alkenes in the presence of bis(pinacolato)diboron (B2Pin2) using CuCF3 derived from fluoroform in air.7c Recently, we disclosed a visible light-induced hydroxyltrifluoroethylation of styrenes with CF3CH2I and O2 using Ir(ppy)3 as a photocatalyst.8 As part of our continued interest in the difunctionalization and fluoroalkylation of alkenes,9 we applied perfluoroalkyl iodides as electron acceptors as well as fluoroalkylating reagents in photoredox reactions. We observed the “No Catalyst is Better” phenomenon which was mentioned several years ago by MacMillan,3a and has received more attention by Aggarwal and Wu very recently.10 Although metal-mediated electron transfers and sulfinatodehalogenations are commonly used for the generation of perfluoroalkyl radicals, visible light-promoted reactions can occur under oxygen or air, thus facilitating the introduction of a hydroxy group, which is difficult to realize under other reaction conditions.11,12 Herein, we report the hydroxylperfluoroalkylation of styrenes under visible light without the use of any photocatalyst or other additives besides amines under air.
Results and discussion
The protocol was initially evaluated with styrene (1a) as a model substrate; it was reacted with 2 equivalents of n-C4F9I (2) and 2 equivalents of iPr2NEt in various solvents under visible light irradiation with a 24 W fluorescent lamp under air (Table 1). Among the common solvents tested including acetonitrile, DMF, THF, and DMSO, all of them promoted the reaction and DMSO was the best, giving product 3a in 75% yield (Table 1, entries 1–4; detected by 19F NMR spectroscopy). A blue light (15 W × 2) was applied, and gave the product in 81% yield, while irradiation with green light (15 W × 2) provided product 3a in a low yield (13%) (Table 1, entries 5 and 6). To our delight, employing N,N,N′,N′-tetramethylethane-1,2-diamine (TMEDA) as a base under irradiation with blue light provided 3a in 91% yield and increasing the concentration from 0.1 M to 0.2 M enhanced the yield to 99% (Table 1, entries 7 and 8). Notably, 3a could not be obtained in the dark, in the absence of the base, or under a nitrogen atmosphere (Table 1, entries 9–11).|
|
||||
|---|---|---|---|---|
| Entrya | Light | Solvent | Base | Yieldb (%) |
| a Reaction conditions: 1a (0.2 mmol), 2 (0.4 mmol), base (0.4 mmol), anhydrous solvent (2 mL), visible light, 24 h, under air. b Yields were determined by 19F NMR spectroscopy using benzotrifluoride as the internal standard. c DMSO (1 mL). d The reaction was carried out under a nitrogen atmosphere. | ||||
| 1 | White light | MeCN | iPr2NEt | 48 |
| 2 | White light | DMF | iPr2NEt | 60 |
| 3 | White light | THF | iPr2NEt | 37 |
| 4 | White light | DMSO | iPr2NEt | 75 |
| 5 | Blue light | DMSO | iPr2NEt | 81 |
| 6 | Green light | DMSO | iPr2NEt | 13 |
| 7 | Blue light | DMSO | TMEDA | 91 |
| 8c | Blue light | DMSO | TMEDA | 99 |
| 9 | Dark | DMSO | TMEDA | 0 |
| 10 | Blue light | DMSO | — | 0 |
| 11d | Blue light | DMSO | TMEDA | 0 |
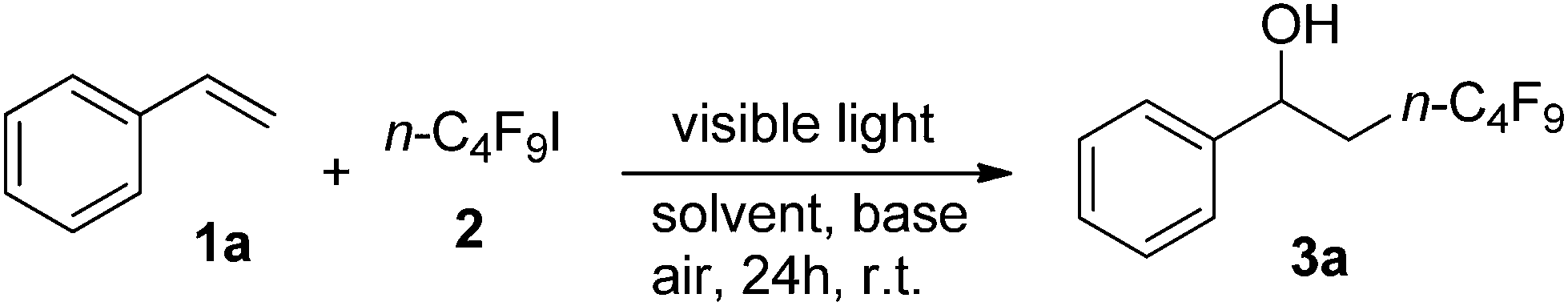
With the optimized conditions in hand (Table 1, entry 8), we began to explore the substrate scope of the hydroxylperfluoroalkylation. Inspiringly, a wide array of aromatic alkenes were subjected to the protocol and numerous functional groups were tolerated (Table 1). Styrenes bearing different halogens produced the corresponding products (3b, 3c, 3d, 3n, 3o, and 3q) in good yields (from 85 to 92%). Styrenes with electron-donating substituents (3e, 3f, 3g, 3h, 3p, 3r, and 3s) afforded the corresponding products in moderate to excellent yields (from 70 to 96%). Some disubstituted alkenes underwent the desired reaction rendering the corresponding products (3j, 3k, 3l, and 3m) effectively. Remarkably, styrenes with electron-withdrawing groups efficiently gave the corresponding products (3i) in 70% yield. In addition, this reaction could be applied to naphthalene and heterocyclic aryl alkenes such as vinylnaphthalenes (3t and 3u), a benzothiophene derivative (3v), a thiazole derivative (3w), 2-vinylpyridine (3x), 2-vinylpyrazine (3y), and a steroid derivative (3z) to give the corresponding products in moderate to good yields (from 63 to 87%). A short or perfluorinated chain (C2F5 or n-C6F13) was successfully introduced in products 3aa and 3ab. The reaction could also be scaled up to 5 mmol to give 1.41 g (84%) of product 3a in 72 h (Table 2).
Next, we evaluated the effect of a photocatalyst (Table 3). We chose two reaction conditions for comparison and compared the results with or without the photocatalyst. Condition A represents the optimized conditions (1 equivalent of 1a, 2 equivalents of 2, 2 equivalents of TMEDA, anhydrous DMSO, blue light (15 W × 2), 48 h, and under an air atmosphere). Under condition A, we found that the yield was more than 90% on 0.1 mmol, 0.5 mmol, and 1 mmol scales without the use of a photocatalyst (Table 3, entries 1–3). However, if the photocatalyst Ir(ppy)3 was added to the reaction, interesting results were observed (Table 3, entries 4–6). On the 0.1 mmol scale, the yield did not change much (Table 3, entry 4 vs. entry 1). However, the yield decreased dramatically when the scale was 0.5 mmol (44% yield), and almost no product was detected in the 1 mmol scale reaction (Table 3, entries 5 and 6). In these experiments, we concluded that the photocatalyst had a negative effect on the visible light-induced hydroxylperfluoroalkylation reaction of styrenes, especially when the reaction was conducted on a large scale (1 mmol).
|
|
||||
|---|---|---|---|---|
| Entry | Condition | Scale | Catalyst | Yielda |
| a Isolated yield. b Yields were determined by 19F NMR spectroscopy using benzotrifluoride as the internal standard. | ||||
| 1 | Condition A: blue light TMEDA DMSO (c = 0.2 M) | 0.1 mmol | — | 99%b |
| 2 | 0.5 mmol | — | 90% | |
| 3 | 1 mmol | — | 91% | |
| 4 | 0.1 mmol | Ir(ppy)3 (1%) | 94%b | |
| 5 | 0.5 mmol | Ir(ppy)3 (1%) | 41% | |
| 6 | 1 mmol | Ir(ppy)3 (1%) | Traceb | |
| 7 | Condition B: white light iPr2NEt DMSO (c = 0.1 M) | 0.1 mmol | — | 95%b |
| 8 | 0.5 mmol | — | 84% | |
| 9 | 1 mmol | — | 83% | |
| 10 | 0.1 mmol | Ir(ppy)3 (1%) | 97%b | |
| 11 | 0.5 mmol | Ir(ppy)3 (1%) | 55% | |
| 12 | 1 mmol | Ir(ppy)3 (1%) | 43% | |
In order to further confirm the above conclusion, we carried out the reaction under condition B. Condition B was the optimal conditions in the presence of a photocatalyst (for the condition screening for reactions with a photocatalyst, see ESI Tables S3 and S4†). 2 equivalents of perfluorobutyl iodide 2 (to styrene 1a) were used and 2 equivalents of Hünig's base were used. The reaction was carried out in DMSO under irradiation with a 24 W white fluorescent lamp under air (Table 3, entries 7–12). Very interestingly, when the reaction was carried out on the 0.1 mmol scale, both conditions (with or without catalyst) gave excellent yields of the products (95% in entry 7 vs. 97% in entry 10). Surprisingly, the yield decreased when the reaction was conducted on a larger scale (55% yield at 0.5 mmol scale in entry 11 or 43% yield at 1.0 mmol scale in entry 12) when the reaction was carried out in the presence of a photocatalyst. In contrast, the yields of the reaction did not fluctuate largely when the reaction was conducted on a larger scale without a catalyst (entries 8 and 9). Therefore, we explored the substrate scope using condition B on the 0.5 mmol scale (see ESI Table S5†). Most of the substrates gave the products in 40 to 85% yields. However, pure 3h and 3i could not be obtained under condition B. Therefore, when the reaction was conducted without a photocatalyst, it was more efficient and had a wider substrate scope as compared to that with a photocatalyst.
Next we concentrated on the reaction mechanism (Scheme 1). In the 19F NMR spectra of pure perfluorobutyl iodide and its mixtures with TMEDA in DMSO, a 1.7 ppm downfield shift in the signal at around −70 ppm, which assigned for the terminal –CF2I group of n-C4F9I, was observed. Consequently, we proposed that there was an interaction between the nitrogen of TMEDA and the iodine atom of n-C4F9I. Analogous chemical shift changes have been reported in other cases between fluoroalkyl iodides and nitrogen-containing molecules.4,13 Notably, aromatic amines did not promote the reaction, but most tertiary aliphatic amines worked (see the ESI, Table S1†). On the other hand, the use of hydroquinone and 1,4-dinitrobenzene partially or completely inhibited the reaction (Scheme 1a). Based on the aforementioned phenomena, a mechanism is proposed in Scheme 1b. A perfluoroalkyl iodide (n-CF3(CF2)nI n = 1, 3 or 5) and TMEDA would produce electron donor–acceptor complex A where the C–I bond is activated. A reacts with styrene subsequently under visible light irradiation generating benzyl radical B. The capture of O2 from air by radical intermediate B gives peroxide radical C, which provides the final product 3. To shed further light on the mechanism, we measured the quantum yields (Φ) of the reaction over 2 h. Quantum yields of 0.20% and 0.23% were observed, which suggested that a chain propagation pathway was unlikely.14 A color change of the reaction was not observed when the reactants were mixed, which was also confirmed by UV-visible light analysis.
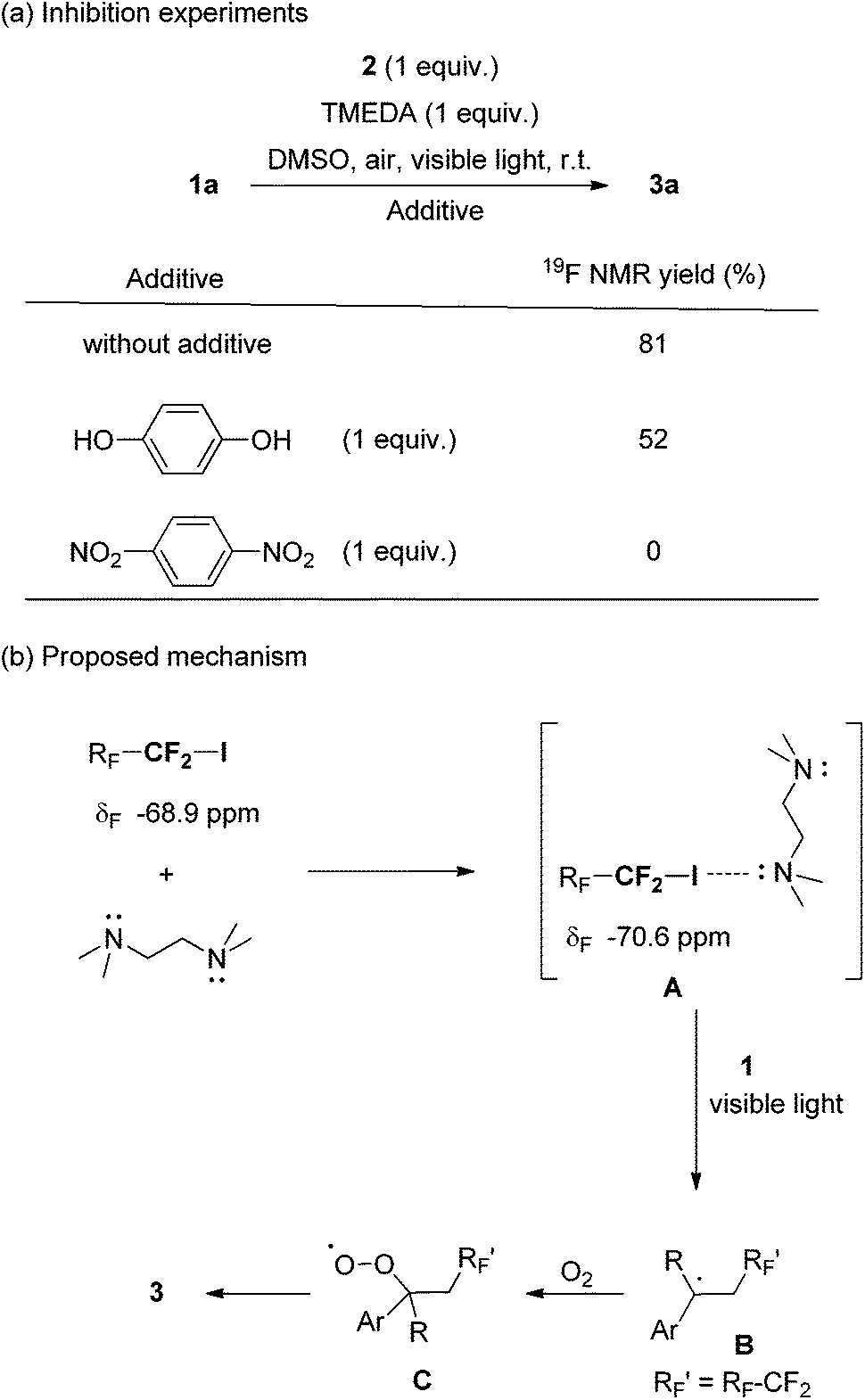 | ||
| Scheme 1 Mechanistic study of the visible light-induced hydroxylperfluoroalkylation of styrenes under air. | ||
In summary, we developed a transition metal- and organophotocatalyst-free hydroxylperfluoroalkylation reaction that occurs under visible light irradiation. In this reaction, an electron donor–acceptor complex (RFI-amine) acts as an inexpensive fluoroalkylating reagent and oxygen serves as an environmentally friendly oxidant. Various styrenes undergo the reaction to afford the corresponding products in up to 96% yields.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support of our work by the National Natural Science Foundation of China (No. 21737004, 21672239, 21421002, and 21302207), and the Innovation Program of Shanghai Municipal Education Commission (No. 13ZZ047).References
- (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (b) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed; (c) N. A. Romero, D. A. Nicewicz, D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850 CrossRef PubMed; (d) Y. Guo, M.-W. Huang, X.-L. Fu, C. Liu, Q.-Y. Chen, Z.-G. Zhao, B.-Z. Zeng and J. Chen, Chin. Chem. Lett., 2017, 28, 719 CrossRef CAS; (e) J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2016, 45, 2044 RSC; (f) Q. Liu and L.-Z. Wu, Natl. Sci. Rev., 2017, 4, 359 CrossRef.
- (a) L. Wozniak, J. J. Murphy and P. Melchiorre, J. Am. Chem. Soc., 2015, 137, 5678 CrossRef CAS PubMed; (b) C. G. S. Lima, T. M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389 CrossRef CAS.
- (a) P. V. Pham, D. A. Nagib and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2011, 50, 6119 CrossRef CAS PubMed; (b) L. Li, Q.-Y. Chen and Y. Guo, J. Fluorine Chem., 2014, 167, 79 CrossRef CAS.
- (a) Q.-Y. Chen and Z.-T. Li, J. Chem. Soc., Perkin Trans. 1, 1992, 1443 RSC; (b) Q.-Y. Chen, Z.-T. Li and C.-M. Zhou, J. Chem. Soc., Perkin Trans. 1, 1993, 2457 RSC; (c) Q.-Y. Chen and Z.-T. Li, J. Chem. Soc., Perkin Trans. 1, 1993, 645 RSC; (d) Q.-Y. Chen and Z.-T. Li, J. Chem. Soc., Perkin Trans. 1, 1993, 1705 RSC; (e) P. Cao, Z.-Y. Long and Q.-Y. Chen, Molecules, 1997, 2, 11 CrossRef CAS.
- (a) X. Sun, W. Wang, Y. Li, J. Ma and S. Yu, Org. Lett., 2016, 18, 4638 CrossRef CAS PubMed; (b) Y. Wang, J. Wang, G.-X. Li, G. He and G. Chen, Org. Lett., 2017, 19, 1442 CrossRef CAS PubMed.
- (a) A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950 CrossRef CAS PubMed; (b) E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598 RSC; (c) H. Egami and M. Sodeoka, Angew. Chem., Int. Ed., 2014, 53, 8294 CrossRef CAS PubMed; (d) B.-H. Zhang, J.-J. Kong, Y. Huang, Y.-G. Lou, X.-F. Lia and C.-Y. He, Chin. Chem. Lett., 2017, 28, 1751 CrossRef CAS; (e) Y. Xu, Z. Wu, J. Jiang, Z. Ke and C. Zhu, Angew. Chem., Int. Ed., 2017, 56, 4545 CrossRef CAS PubMed; (f) Z. Wu, D. Wang, Y. Liu, L. Huan and C. Zhu, J. Am. Chem. Soc., 2017, 139, 1388 CrossRef CAS PubMed; (g) X. Tang and A. Studer, Chem. Sci., 2017, 8, 6888 RSC; (h) X.-Y. Jiang and F.-L. Qing, Angew. Chem., Int. Ed., 2013, 52, 14177 CrossRef CAS PubMed.
- (a) M. Yoshida, M. Ohkoshi, N. Aoki, Y. Ohnuma and M. Iyoda, Tetrahedron Lett., 1999, 40, 5731 CrossRef CAS; (b) Y. Yasu, T. Koike and M. Akita, Angew. Chem., Int. Ed., 2012, 51, 9567 CrossRef CAS PubMed; (c) X. Yang, L. He and G. C. Tsui, Org. Lett., 2017, 19, 2446 CrossRef CAS PubMed; (d) Y. Yang, Y. Liu, Y. Jiang and D. A. Vicic, J. Org. Chem., 2015, 80, 6639 CrossRef CAS PubMed.
- L. Li, M. Huang, C. Liu, J.-C. Xiao, Q.-Y. Chen and Y. Guo, Org. Lett., 2015, 17, 4714 CrossRef CAS PubMed.
- F. Huan, Q.-Y. Chen and Y. Guo, J. Org. Chem., 2016, 81, 7051 CrossRef CAS PubMed.
- (a) A. Fawcett, J. Pradeilled, Y. Wang, T. Wutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283 CrossRef CAS PubMed; (b) Q.-Y. Meng, X.-W. Gao, T. Lei, Z. Liu, F. Zhan, Z.-J. Li, J.-J. Zhong, H. Xiao, K. Feng, B. Chen, Y. Tao, C.-H. Tung and L.-Z. Wu, Sci. Adv., 2017, 3, e1700666 CrossRef PubMed.
- Q.-Y. Chen and Z.-Y. Yang, J. Fluorine Chem., 1985, 28, 399 CrossRef CAS.
- (a) C.-P. Zhang, Q.-Y. Chen, Y. Guo, J.-C. Xiao and Y.-C. Gu, Chem. Soc. Rev., 2012, 41, 4536 RSC; (b) Z.-Y. Long and Q.-Y. Chen, J. Org. Chem., 1999, 64, 4775 CrossRef CAS PubMed.
- (a) A. Bruckmann, M. A. Pena and C. Bolm, Synlett, 2008, 900 CAS; (b) S. Dordonne, B. Crousse, D. B. Delpon and J. Legros, Chem. Commun., 2011, 47, 5855 RSC.
- X.-D. Xia, L.-Q. Lu, W.-Q. Liu, D.-Z. Chen, Y.-H. Zheng, L.-Z. Wu and W.-J. Xiao, Chem. – Eur. J., 2016, 22, 8432 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qo00946a |
| This journal is © the Partner Organisations 2018 |