DOI:
10.1039/C7QO00786H
(Review Article)
Org. Chem. Front., 2018,
5, 273-287
Total syntheses of pyrroloazocine indole alkaloids: challenges and reaction discovery
Received
31st August 2017
, Accepted 23rd October 2017
First published on 24th October 2017
Abstract
Lapidilectines, grandilodines, lundurines and tenuisines are indole alkaloids, isolated from plants of Kopsia genus. They feature a common indole-fused pyrroloazocine core, whose construction poses a significant synthetic challenge. In this review, we discuss the reported strategies for the total synthesis of this family of alkaloids with a focus on the different methods used for the construction of spiro[cyclohexane-2-indoline] and indole-pyrroloazocine intermediates, introduction of indole-fused cyclopropane as well as other new methodologies uncovered in the course of the total syntheses. In closing, the existing hypothesis of the biosynthetic origin and relationships of the pyrroloazocine indole alkaloids are presented.
 Mariia S. Kirillova | Mariia S. Kirillova was born in Bryansk (Russia) in 1988 and graduated in chemistry from the Gubkin Russian State University of Oil and Gas in Moscow (Russia) in 2011. She was awarded a pre-doctoral fellowship from the Government of Catalonia to work on the synthesis of complex natural products at the Institute of Chemical Research of Catalonia (ICIQ) with Prof. Antonio M. Echavarren and obtained her Ph.D. in 2016. She continued as a post-doctoral associate until the summer of 2017. In 2015, she worked with the group of Prof. Corey R. J. Stephenson (University of Michigan) as a summer exchange student. Recently, she joined the University of Zurich as a post-doctoral researcher in the group of Prof. Cristina Nevado. |
 Fedor M. Miloserdov | Fedor Miloserdov was born in Moscow in 1990 and started to work in the research laboratory while at high school (Moscow Chemical Lyceum). He graduated from the Mendeleev University of Chemical Technology of Russia (Moscow), in 2012. During his university study, he made a series of short stays with the group of Prof. Vladimir Grushin (Institute of Chemical Research of Catalonia, ICIQ, Tarragona, Spain). He continued his education with Vladimir Grushin at ICIQ, and obtained his Ph.D. degree in 2015. After that he became a Postdoctoral Fellow in the group of Prof. Antonio Echavarren (ICIQ), where he is currently working in the areas of total synthesis and gold catalysis. |
 Antonio M. Echavarren | Antonio M. Echavarren received his PhD from the Universidad Autónoma de Madrid (UAM, 1982) with Prof. Francisco Fariña. After a postdoctoral stay in Boston College with Prof. T. Ross Kelly, he joined the UAM as an Assistant Professor. Following a two year period as a NATO-fellow with Prof. John K. Stille in Fort Collins (Colorado State University), he joined the Institute of Organic Chemistry of the CSIC in Madrid. In 1992, he returned to the UAM as a Professor of Organic Chemistry and in 2004, he moved to Tarragona as a Group Leader at the Institute of Chemical Research of Catalonia (ICIQ). He has been a Liebig Lecturer (Organic Division, German Chemical Society, 2006), an Abbot Lecturer in Organic Chemistry (University of Illinois at Urbana-Campaign, 2009), a Schulich Visiting Professor (Technion, Haifa, 2011), Sir Robert Robinson Distinguished Lecturer (University of Liverpool, 2011), a Novartis Lecturer in Organic Chemistry (Massachusetts Institute of Technology, 2015) and a Kurt Alder Lecturer 2017 (University of Cologne). In 2012, he got a European Research Council Advanced Grant and in 2014, he became the president of the 49th EUCHEM Conference on Stereochemistry (Bürgenstock conference). Prof. Echavarren is a member of the International Advisory Board of Organic & Biomolecular Chemistry, Chemical Society Reviews, Advanced Synthesis and Catalysis, and Organic Letters, a member of the Editorial Board of Chemistry European Journal, and an Associate Editor of Chemical Communications. He is a Fellow of the Royal Society of Chemistry. He received the 2004 Janssen-Cylag Award in Organic Chemistry and the 2010 Gold Medal of the Royal Spanish Chemical Society and an Arthur C. Cope Scholar Award from the ACS. |
1. Introduction
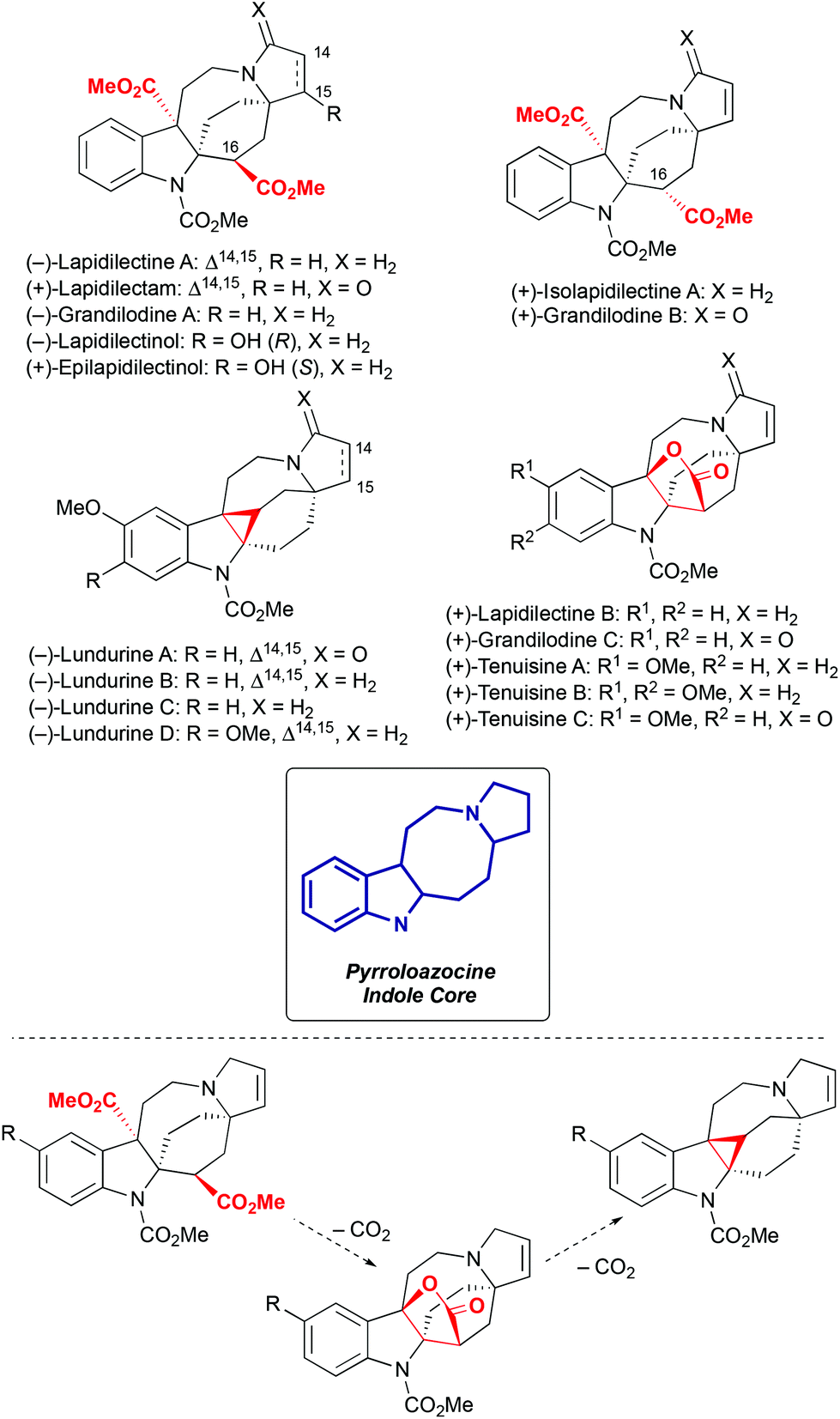
Pyrroloazocine indole alkaloids is a family comprising sixteen natural compounds isolated from two closely related plant species, Kopsia grandifolia1–3 and Kopsia tenuis4–6 (Scheme 1). Kopsia grandifolia as a species was described in 2004 in the D. J. Middleton classification,7 and was previously known as Kopsia lapidilecta.1,2 In the modern classification,7Kopsia lapidilecta describes another plant that grows on Natuna islands in Indonesia, while Kopsia grandifolia is distributed over peninsular Malaysia, and Kopsia tenuis is endemic to the Sarawak region on the North of the Borneo island.
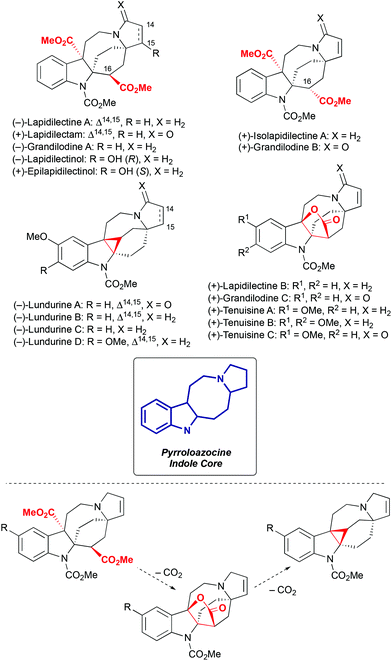 |
| | Scheme 1 Pyrroloazocine indole alkaloids and their biosynthetic relationship. | |
All sixteen pyrroloazocine indole alkaloids have a common pyrroloazocine core and they are biosynthetically related by a series of decarboxylation events8 (Scheme 1). These natural compounds can be further divided into three subcategories based on their key structural features: diester, lactone or cyclopropane. Diester natural compounds were isolated from the Kopsia grandifolia plant as both epimers at the C16 position2,3 (here and further in the manuscript we will use the numbering defined in the isolation reports).1–6 Cyclopropane-containing lundurines were obtained from Kopsia tenuis,4,5 and lactones were isolated from both Kopsia tenuis and Kopsia grandifolia.1,3,4,6 Initially, the structure of tenuisines was wrongly assigned to be dimeric,6 and later corrected4 to monomeric structures with a lactone moiety. Interestingly, all members of this alkaloid family isolated from the Kopsia tenua plant feature methoxylation at the indole ring.
Only preliminary studies of biological activities of pyrroloazocine indole alkaloids have been reported. Lundurines B and D showed cytotoxicity toward B15 melanoma cells,4 and lundurines B, D, grandilodines A, C and lapidilectine B were capable of reversing multidrug resistance in vincristine-resistant KB (VJ300) cells.3,4
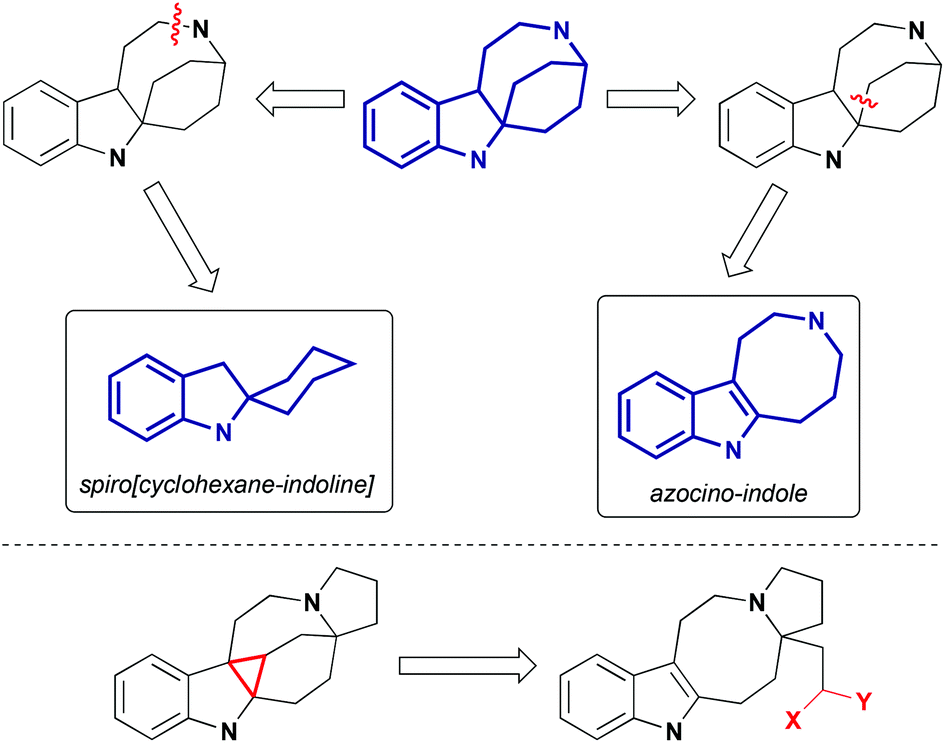
In recent years, pyrroloazocine indole alkaloids attracted considerable attention from the synthetic community, and the total syntheses of lundurines A–C, grandilodines B, C and lapidilectine B have been reported. For lundurines A–C, grandilodine C and lapidilectine B, asymmetric approaches were developed as well. Depending on how the rigid indole-fused azabicyclo[4,2,2] skeleton is constructed, syntheses of pyrroloazocine indole alkaloids fall into two categories: via spiro[cyclohexane-2-indoline] and via azocine-containing intermediates (Scheme 2). The use of the latter pathway for the synthesis of lundurines requires another difficult step – the cyclopropanation of an indole moiety.
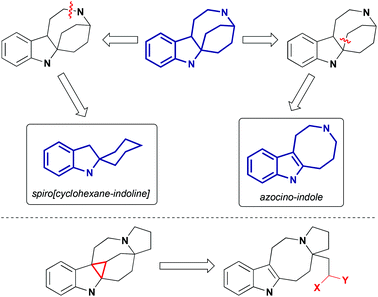 |
| | Scheme 2 Retrosynthetic approaches to the indole-fused 2-azabicyclo[4,2,2]-skeleton. Cyclopropanation of indole for the synthesis of lundurines. | |
In this review, we discuss the reported methods to achieve the synthesis of the key spiro[cyclohexane-2-indoline] and azocino-indole intermediates, as well as other important transformations that were necessary to complete the synthesis of the natural products. In the context of lundurine syntheses,9 the indole cyclopropanation, and some key transformations of the cyclopropane moiety will be discussed in more detail. Finally, the current biosynthetic proposal for the origin of pyrroloazocine indole alkaloids will be presented.
2. Synthesis via spiro[cyclohexane-2-indoline] intermediates
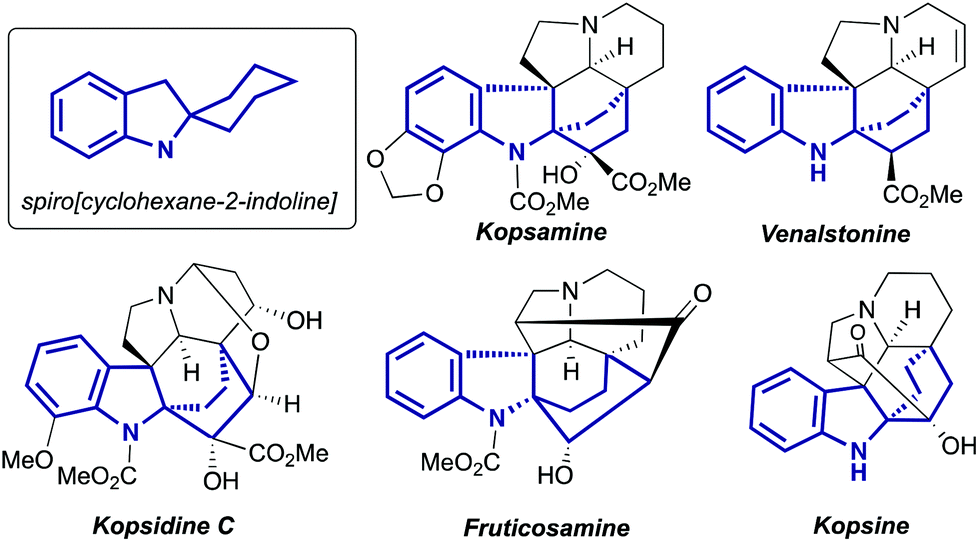
Indolines containing a 2-spiro fused cyclohexane ring are well known compounds. This moiety is presented in numerous indole alkaloids isolated from plants of genus Kopsia8 (Fig. 1), including kopsamine, isolated for the first time by Gorter in 1920.10 To the best of our knowledge, the first spiro[cyclohexane-2-indoline] was synthesized nine decades ago, by thermal alkali-mediated cyclization of an amino acid obtained by means of Strecker synthesis (Scheme 3).11 Nowadays there are several well-established routes to spiro[cyclohexane-2-indolines], and some of them were successfully utilized in the synthesis of pyrroloazocine indole alkaloids.
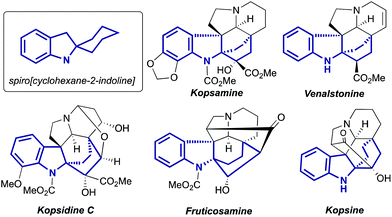 |
| | Fig. 1 Examples of natural compounds with the highlighted spiro[cyclohexane-2-indoline] part. | |
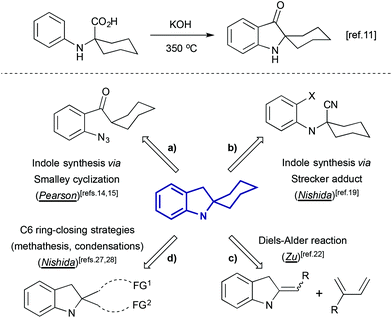 |
| | Scheme 3 The first synthesis of spiro[cyclohexane-2-indolines],11 and retrosynthetic approaches to indole spirofused cyclohexanes applied to the total synthesis of pyrroloazocine indole alkaloids. | |
One of these methods is Smalley cyclization (Scheme 3a). Enolizable aryl ketones bearing an ortho-azido group can be involved in base-induced cyclization that constructs the indoline ring, and in case of a cyclohexyl substituent, this leads to the formation of spiro[cyclohexane-2-indolines].12,13 This transformation was employed in the total synthesis of lapidilectine B, developed by Pearson et al.14,15 A similar reaction was later applied to enamine-bearing substrates,16 and recently a practical Cu-catalyzed cascade of ortho-bromoarene azidation/Smalley cyclization has been reported.17
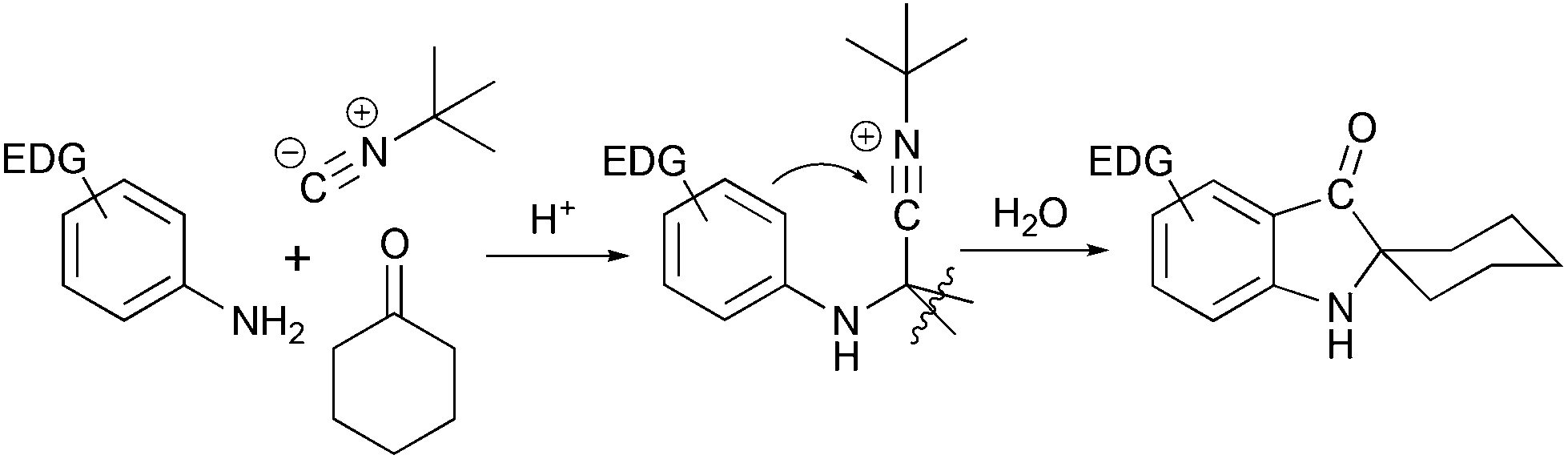
Another method of assembly of spiro[cyclohexane-2-indoline] compounds relays on the construction of a C–C bond between an ortho-bromo aryl moiety and a cyano-group in Strecker type intermediates (Scheme 3b). This transformation was initially developed as a radical cyclization method.18 However such conditions were not suitable for the application in the total synthesis, and an alternative protocol utilizing i-PrMgCl was developed.19 A similar transformation from a retrosynthetic perspective is the acid-catalyzed Ugi condensation/Hoesh reaction cascade between electron-rich anilines, cyclohexanone and t-BuNC20 (Scheme 4).
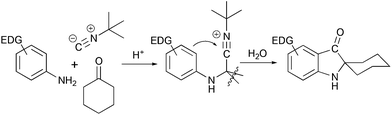 |
| | Scheme 4 Ugi condensation/Hoesh reaction cascade in the synthesis of spiro[cyclohexane-2-indolines].20 | |
The Diels–Alder cycloaddition reaction is one of the most useful synthetic tools for the construction of cyclohexane rings, including the one presented in spiro[cyclohexane-2-indolines]. In such transformations, the indoline-containing part can play the role of a diene or dienophile. For the latter, several examples of cycloaddition reactions involving indole-ylidenes and dimethyl butadiene were reported,21 and, recently, this strategy was applied for the total synthesis of racemic grandilodine B22 (Scheme 3c). The Diels–Alder reaction between an indoline-containing diene and a dienophile has also been widely used in the synthesis of indole alkaloids23 (Scheme 5).
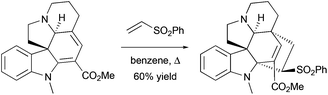 |
| | Scheme 5 [4 + 2]-cycloaddition reaction between indoline-containing diene and vinyl sulfone in the total synthesis of indole alkaloids.23a | |
An alternative approach to spiro[indoline-cyclohexanes] is based on C6 ring-closing transformations (Scheme 3d), such as aldol-type reactions,24,25 olefin metathesis,26 and Dieckmann condensation.27,28 The last two approaches were developed by the group of Nishida in the course of the total synthesis of lundurines.
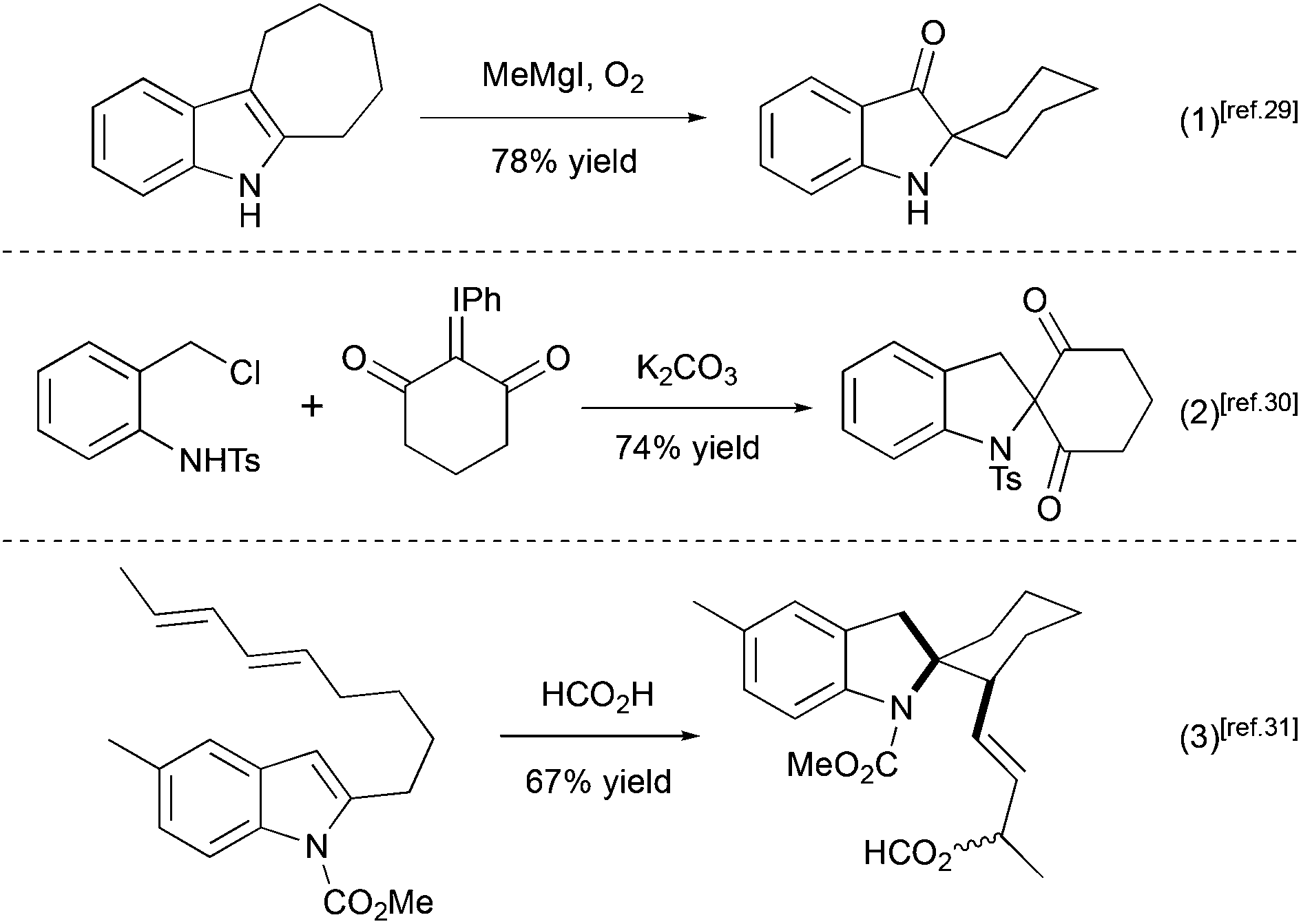
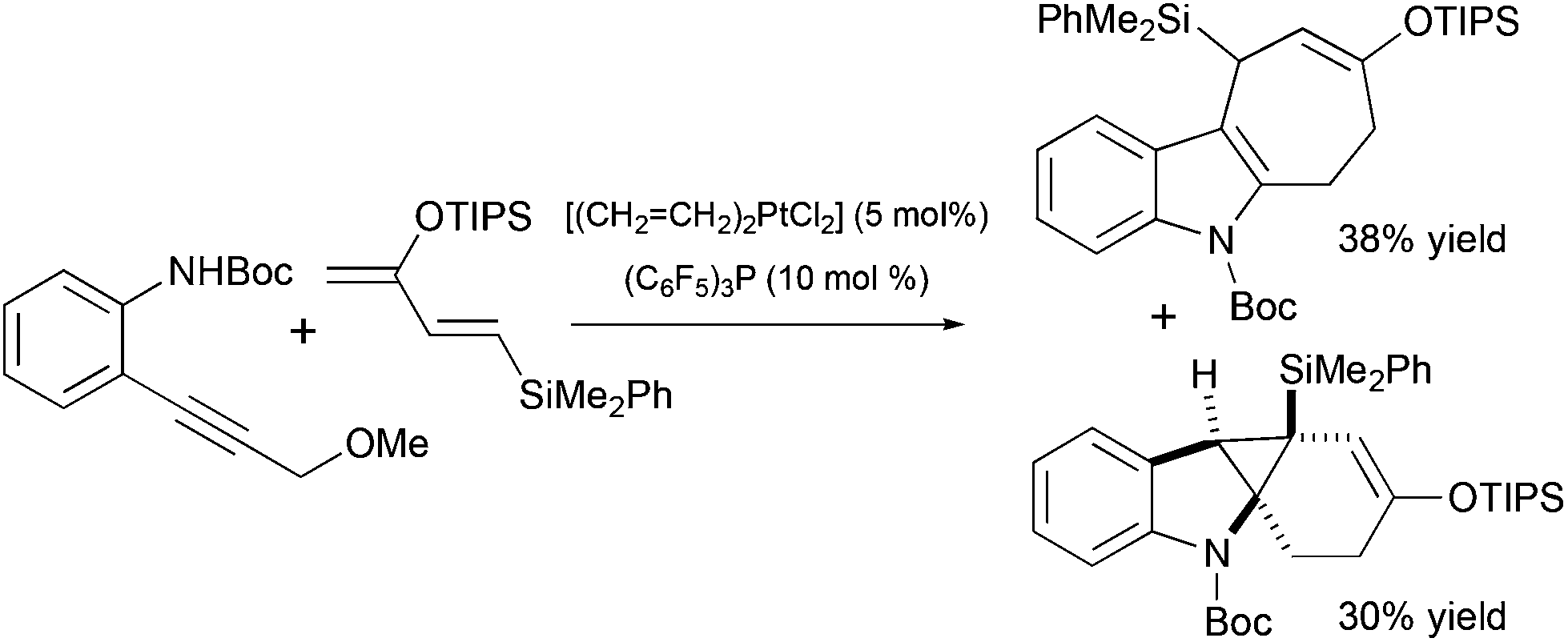
Several other methods for the synthesis of spiro[cyclohexane-2-indoline] were reported, including an alkylation/oxidative rearrangement of cyclohepta[b]indoles29 (Scheme 6, eqn (1)), addition of iodonium ylides to ortho-chloromethyl aryl amides30 (Scheme 6, eqn (2)), and spirocyclization of conjugated dienes with protonated indoles31 (Scheme 6, eqn (3)). The platinum-catalyzed formal [4 + 3] cycloaddition can also give rise to cyclohexane-2-indoline spirocyclic products containing an additional cyclopropyl moiety32 (Scheme 7). Although these products resemble the lundurines, the [4 + 3] cycloaddition reaction has not yet been applied to the synthesis of these natural compounds.
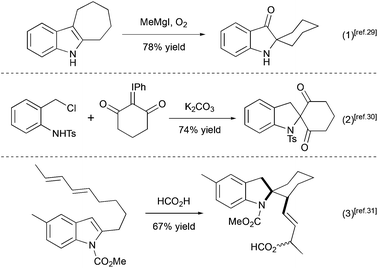 |
| | Scheme 6 Examples of spiro[cyclohexane-2-indoline] synthesis.29–31 | |
 |
| | Scheme 7 Cyclopropane-containing spiro[cyclohexane-2-indoline] as a product in Pt-catalyzed formal [4 + 3] cycloaddition.32 | |
2.1 Total synthesis of racemic lapidilectine B by Pearson et al.
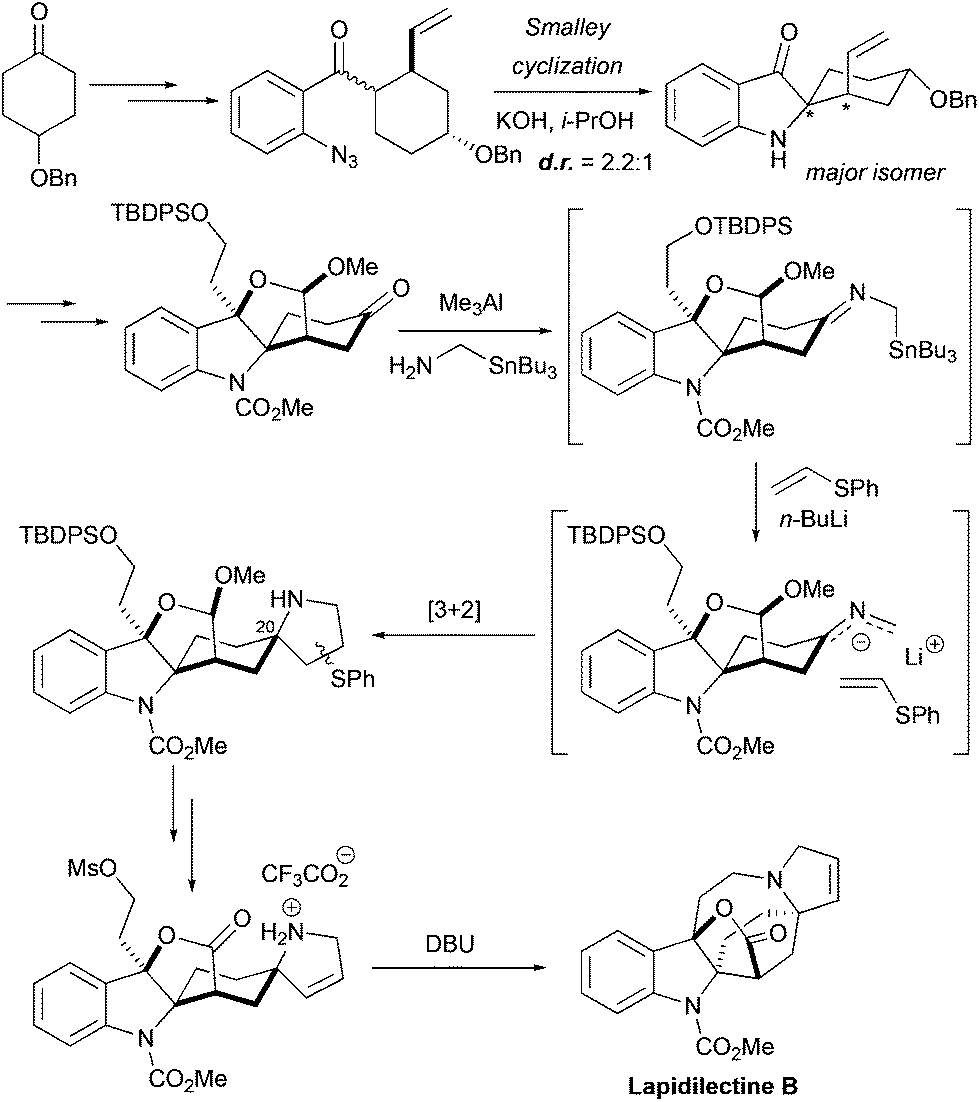
The very first total synthesis of pyrroloazocine indole alkaloid, racemic lapidilectine B, was accomplished by the group of Pearson in 200114,15 (Scheme 8). The spiro[cyclohexane-2-indoline] fragment was constructed by Smalley cyclization of the substituted cyclohexyl ketone. Because of the unsymmetrical nature of the cyclohexyl substituent, the product was obtained as a 2.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of two diastereomers, and the major one was further used in the synthesis. The functionalization of 3-oxiindole and the cleavage of a double bond led to the lactol ether intermediate bearing a keto group. This ketone became the source of 2-azaallyllithium species that were subsequently introduced into the [3 + 2] cycloaddition reaction with phenyl vinyl sulfide, providing the pyrrolidine core of the molecule with the correct relative configuration at C20. The elimination of the sulfoxide moiety provided the required unsaturation in the pyrroline. The lactole ether fragment was oxidized to the lactone, and finally an intramolecular alkylation of the pyrroline nitrogen with alkyl mesylate closed the azocine ring, leading to lapidilectine B. The approach developed by Pearson et al. required ca. 25 steps with ca. 0.5% overall yield,14 which was further improved to ca. 1%.15
:1 mixture of two diastereomers, and the major one was further used in the synthesis. The functionalization of 3-oxiindole and the cleavage of a double bond led to the lactol ether intermediate bearing a keto group. This ketone became the source of 2-azaallyllithium species that were subsequently introduced into the [3 + 2] cycloaddition reaction with phenyl vinyl sulfide, providing the pyrrolidine core of the molecule with the correct relative configuration at C20. The elimination of the sulfoxide moiety provided the required unsaturation in the pyrroline. The lactole ether fragment was oxidized to the lactone, and finally an intramolecular alkylation of the pyrroline nitrogen with alkyl mesylate closed the azocine ring, leading to lapidilectine B. The approach developed by Pearson et al. required ca. 25 steps with ca. 0.5% overall yield,14 which was further improved to ca. 1%.15
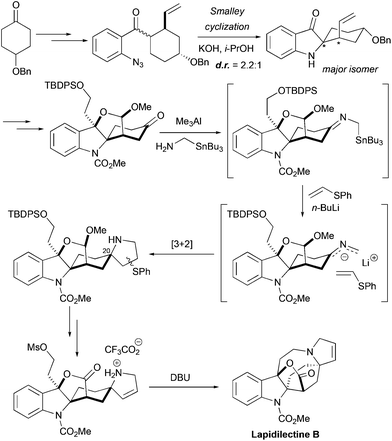 |
| | Scheme 8 Total synthesis of racemic lapidilectine B by Pearson et al.14,15 | |
2.2 Total synthesis of lundurines by Nishida et al., 1st generation
The first total synthesis of lundurines was developed by the group of Nishida, and was reported in 2014.26 First the synthesis of racemic lundurine B was accomplished, and shortly after the initial publication, the same group published another approach to racemic lundurines A and B,27 that will be discussed separately, as a 2nd generation synthesis. The primary synthetic approach featured an early construction of a cyclopropane motif (Scheme 9). The indole ring was assembled at one side of cyclopropane by a copper-mediated aromatic amination, and the lactone ring at the other side of a cyclopropane was opened. The side chains were elongated by means of a Wittig olefination providing a precursor for ring-closing metathesis. The ring closing metathesis was performed under standard conditions producing the spiro[cyclohexane-2-indoline]. After several steps, an amino-ethanol moiety was attached to a side chain of a molecule, and an N,O-acetal of cyclohexanone was generated, building the azocine ring. The pyrroline ring was constructed by another ring closing metathesis step, and finally the Boc protecting group was converted into the methyl carbamate, completing the synthesis of racemic lundurine B, in 29 steps and ca. 1% overall yield.
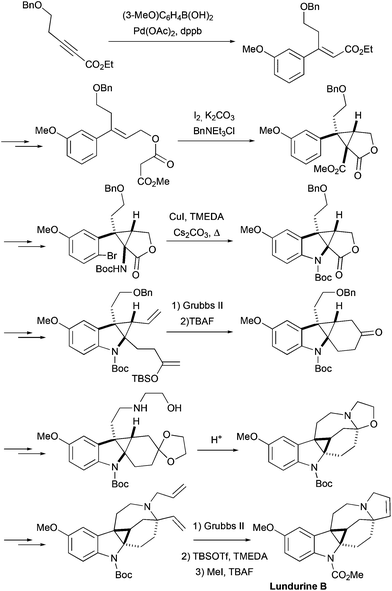 |
| | Scheme 9 Total synthesis of racemic lundurine B by Nishida et al., 1st generation.26 | |
2.3 Total synthesis of lundurines by Nishida et al., 2nd generation
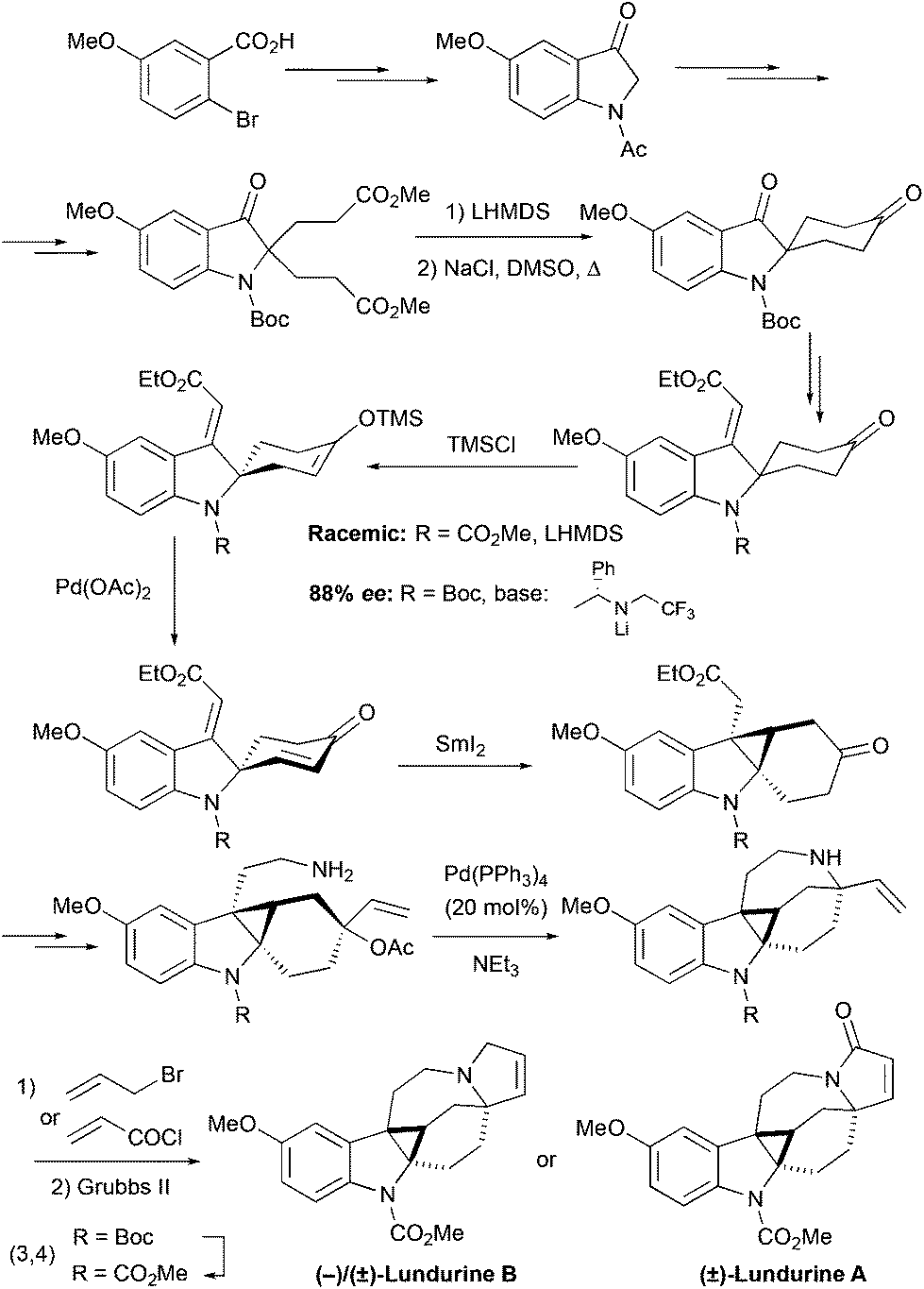
The second generation synthesis of the lundurine by the group of Nishida27,28 retrosynthetically resembles the synthesis of lapidilectine B by Pearson et al.14,15 First, the spiro[cyclohexane-2-indoline] was constructed by Dieckmann condensation (Scheme 10). The unsaturated ethyl ester was introduced by the ethoxyacetylene addition/Meyer–Schuster rearrangement sequence, and the silyl enol ether precursor for the Saegusa-Ito oxidation was generated, desymmetrizing the molecule. It was previously reported that 4-t-Bu-cyclohexanone can be desymmetrized upon treatment with chiral lithium amides.33 The application of this methodology to the Boc-protected spiro[cyclohexane-2′-indoline]-4-one provided the corresponding chiral product with 88% ee, that was employed for the asymmetric synthesis of (−)-lundurine B.28 The obtained 1,4-diene was further treated with SmI2, that triggered radical cyclization thereby producing the cyclopropane-containing product.34,35 The modification of functional groups resulted in the formation of allylic acetate, containing an amino-group, which was cyclized in the presence of a Pd-catalyst producing an azocine ring. Like in their 1st generation of synthesis, Nishida and coworkers constructed a pyrroline ring at the last stage by ring-closing metathesis, providing racemic lundurines A and B in 31 steps and ca. 1.5% overall yield.27 An asymmetric synthesis of (−)-lundurine B was accomplished following a similar synthetic strategy, where the Boc-protecting group was converted into methyl carbamate in the last steps (total 30 steps, ca. 1% overall yield, 88% ee).28
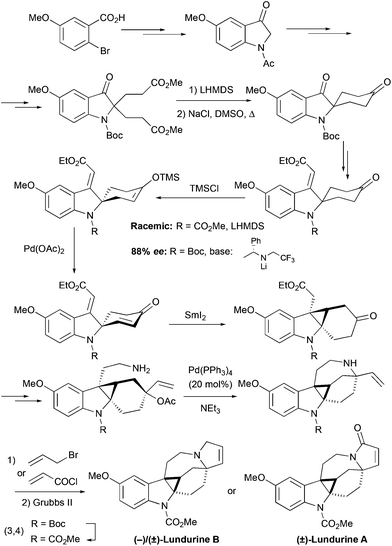 |
| | Scheme 10 2nd generation total synthesis of lundurines by Nishida et al.27,28 | |
2.4 Total synthesis of (+)-grandilodine C and (+)-lapidilectine B by Nishida et al.
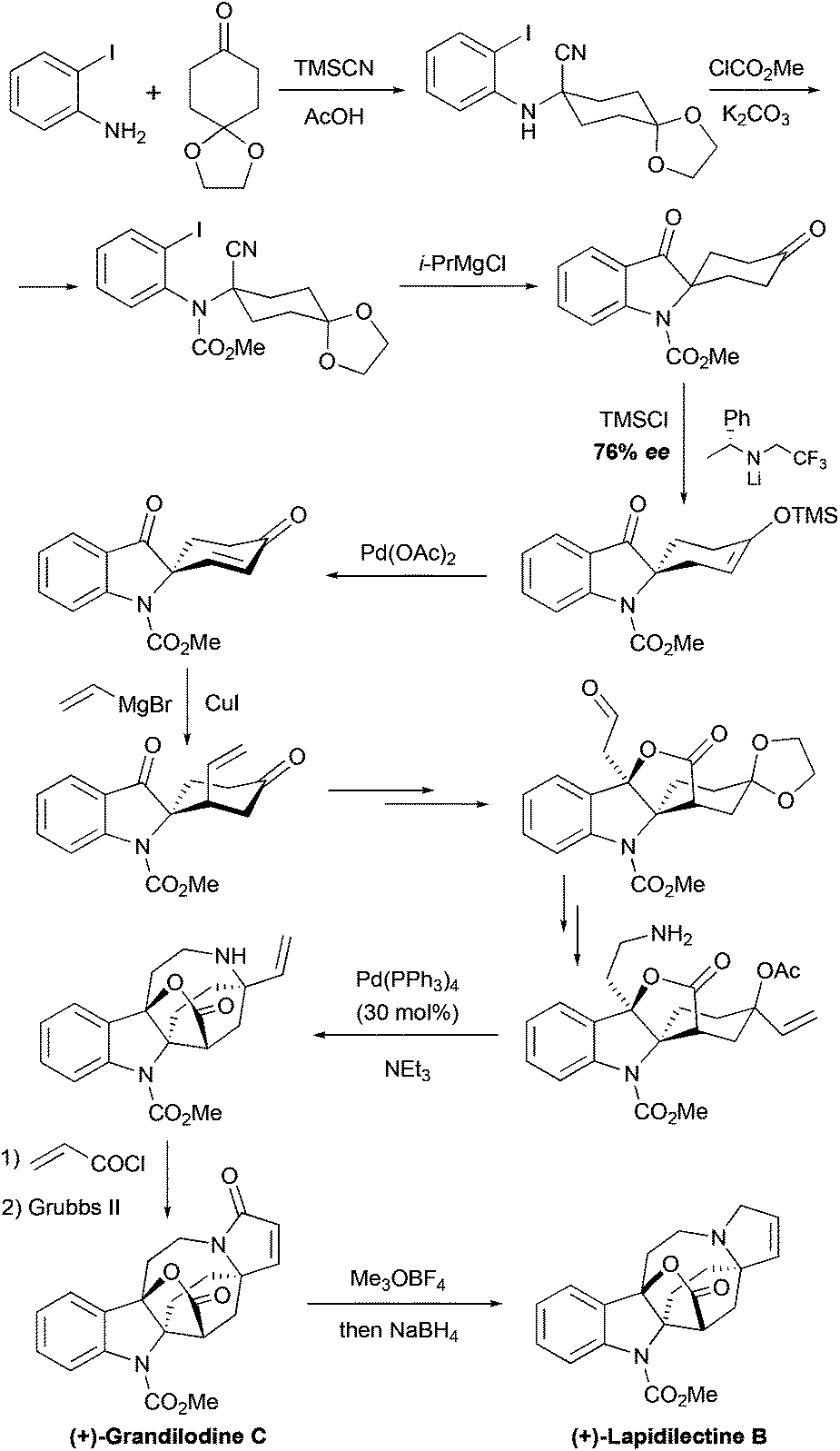
The group of Nishida further elaborated their approach to pyrroloazocine indole alkaloids in the course of the asymmetric total synthesis of lactone-containing grandilodine C and lapidilectine B.19 Similar to their 2nd generation synthesis of the lundurines, the desymmetrization of spiro[cyclohexane-2′-indoline]-4-one was the first key step. The synthetic sequence to this starting material was shortened from 11 steps27 to only 3, and the indole ring was constructed by i-PrMgCl-mediated cyclization in the Strecker adduct (Scheme 11). In this study, the desymmetrization reaction could be performed directly on the diketone substrate, containing additionally the methyl carbamate protecting group on the indole nitrogen, instead of Boc (see above), with good 76% ee, which could be increased to 91% ee by crystallization. The next copper-mediated conjugated vinylation occurred with high diastereoselectivity, providing an unsaturated side-chain, that after ozonolysis and oxidation, it became a part of a lactone ring. The modification of functional groups resulted in the formation of allylic acetate, containing an amino-group. An analogous intermediate was involved in the total synthesis of lundurines.27 Similarly the Pd-catalyzed allylic substitution was employed to assemble an azocine moiety, and the metathesis reaction was used for the formation of unsaturated pyrrolidone in (+)-grandilodine C. A further reduction of amide gave (+)-lapidilectine B. The described synthesis of (+)-grandilodine A (76% ee)36 has 18 steps and ca. 8.5% overall yield.19
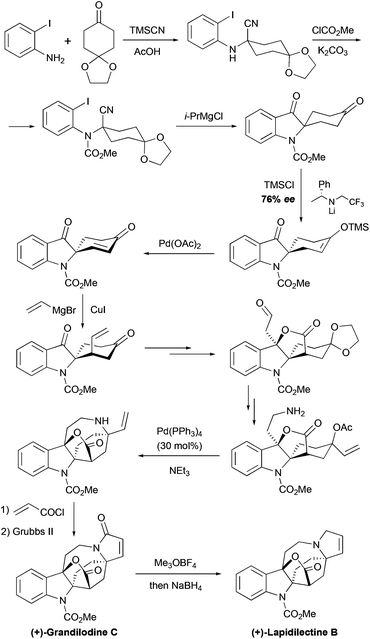 |
| | Scheme 11 Total synthesis of (+)-grandilodine C and (+)-lapidilectine B by Nishida et al.19 | |
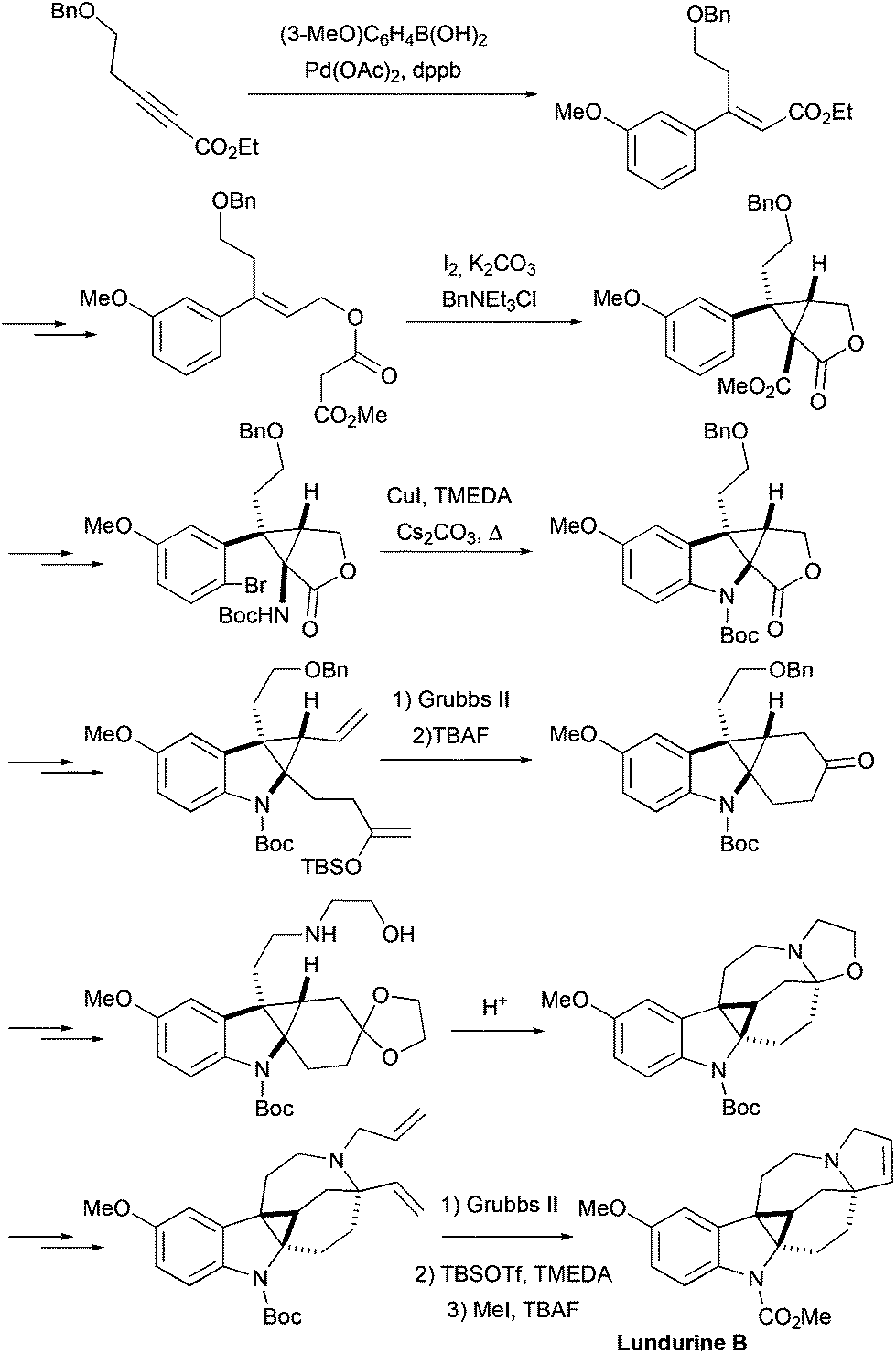
2.5 Total synthesis of racemic grandilodine B by Zu et al.
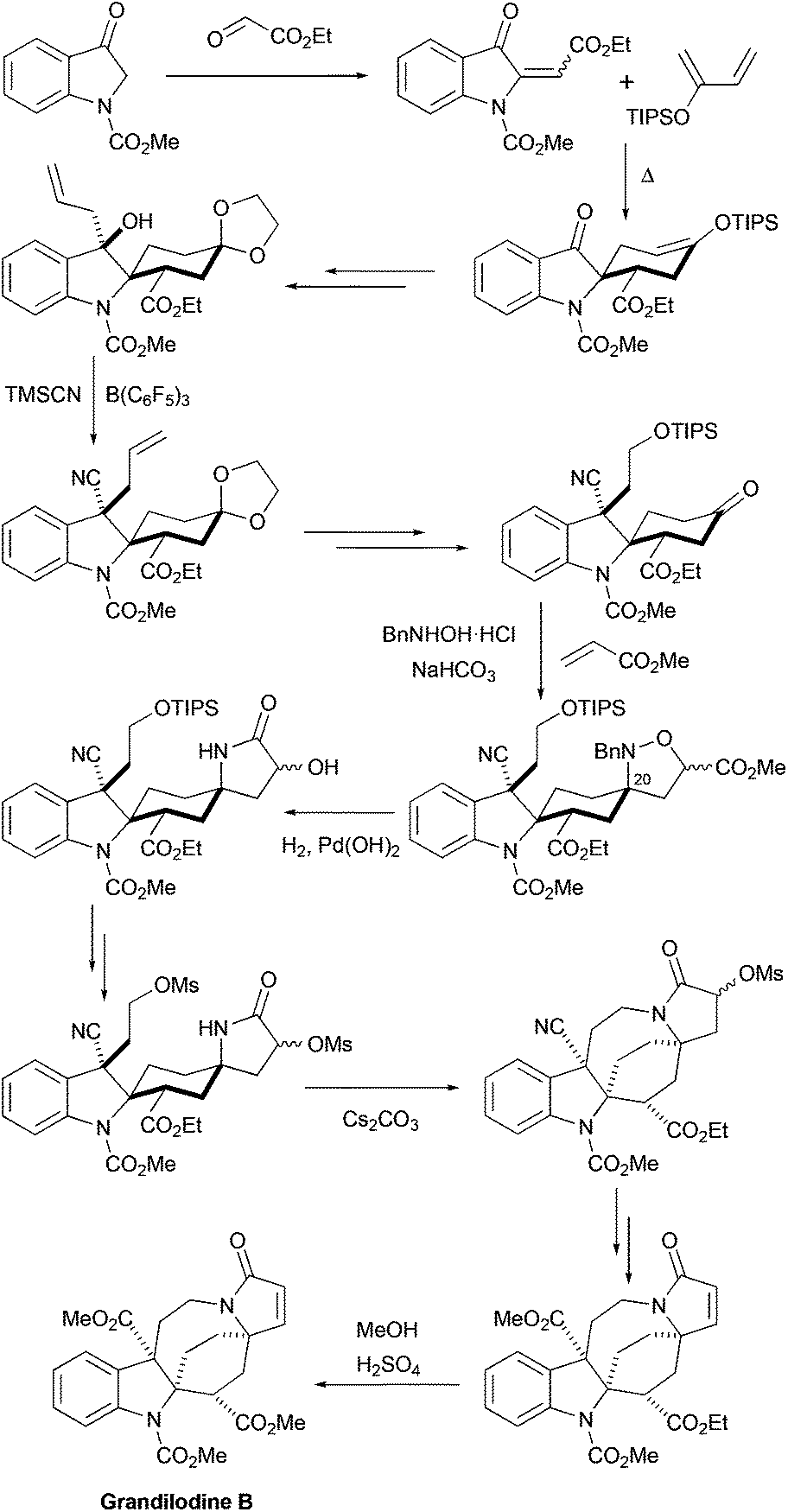
More recently, the first total synthesis of a diester-type pyrroloazocine indole alkaloid, grandilodine B, has been developed by Zu and coworkers (Scheme 12).22 Their approach has similarities with the above-mentioned synthesis of lapidilectine B by Pearson et al.14,15 The spiro[cyclohexane-2-indoline] tricycle was efficiently constructed in only two steps taking advantage of the Diels–Alder reaction that proceeded with an excellent yield and a good level of diastereoselectivity (4:1 α/β ratio). The major diastereomer was further functionalized, the 3-oxyindole moiety was allylated, and the benzylic hydroxyl group was substituted with cyanide, which was later transformed into a methyl ester. Similar to Pearson et al., the pyrroline ring was introduced by [3 + 2] cycloaddition, and the azocine ring was closed by N-alkylation reactions. The 4-cyclohexanone presented in the molecule was transformed into a nitrone, which reacted with methyl acrylate thereby providing an isoxazolidine.37 The N–O bond and the benzyl group were cleaved in the course of hydrogenation leading to the formation of a lactam ring.38 The intramolecular N-alkylation of the lactam led to the azocine ring. Finally, cyano- and ethyl ester-groups were transformed into methyl esters, thereby completing the synthesis of racemic grandilodine B with a total of 19 steps and ca. 2.5% overall yield.
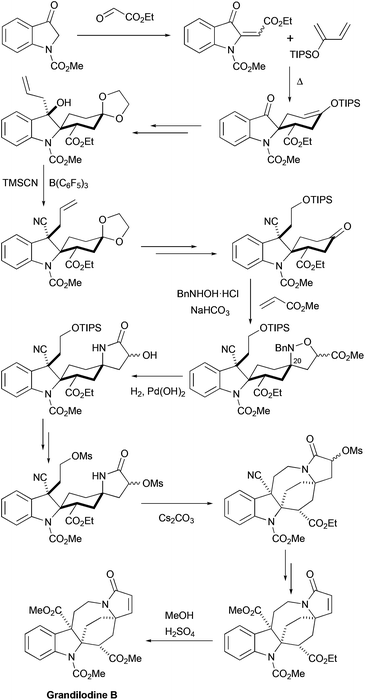 |
| | Scheme 12 Total synthesis of racemic grandilodine B by Zu et al.22 | |
3. Synthesis via fused pyrroloazocine-indole intermediates
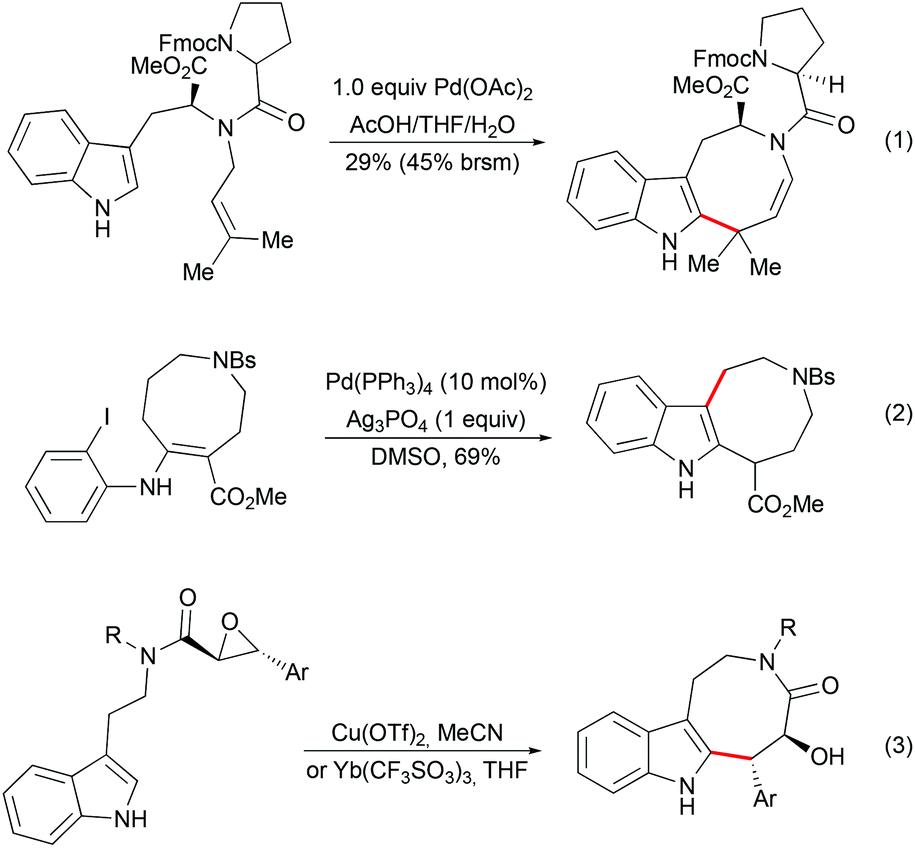
As an alternative to the spiro-fused indoline strategy, the construction of indole-containing an 8-membered ring followed by late-stage indole cyclopropanation has been explored by several groups. Diverse methodologies have been developed for the synthesis of indoloazocine tricyclic scaffolds. Palladium-mediated dihydroindoloazocine cyclization of N-prenyl-(S)-tryptophan derivatives has been used as a key step in the synthesis of (+)-austamide and related alkaloids by Corey (Scheme 13, eqn (1)).39 Nishida and co-workers reported the formation of the azocino-indole subunit from o-halophenylenaminoester by deconjugation of a double bond followed by intramolecular Heck reactions (Scheme 13, eqn (2)).40 Lewis acid-mediated epoxide opening of tryptamine-derived trans-epoxyamides followed by 8-exo-tet cyclization have been applied as a key step in the syntheses of (+)-balasubramide41 and its derivatives (Scheme 13, eqn (3)).42
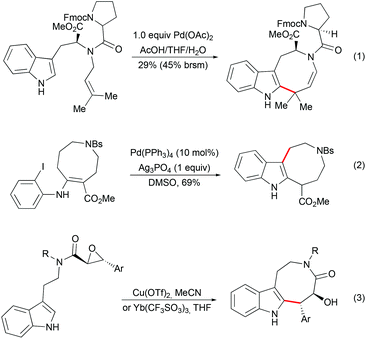 |
| | Scheme 13 Examples of indoloazocine synthesis. | |
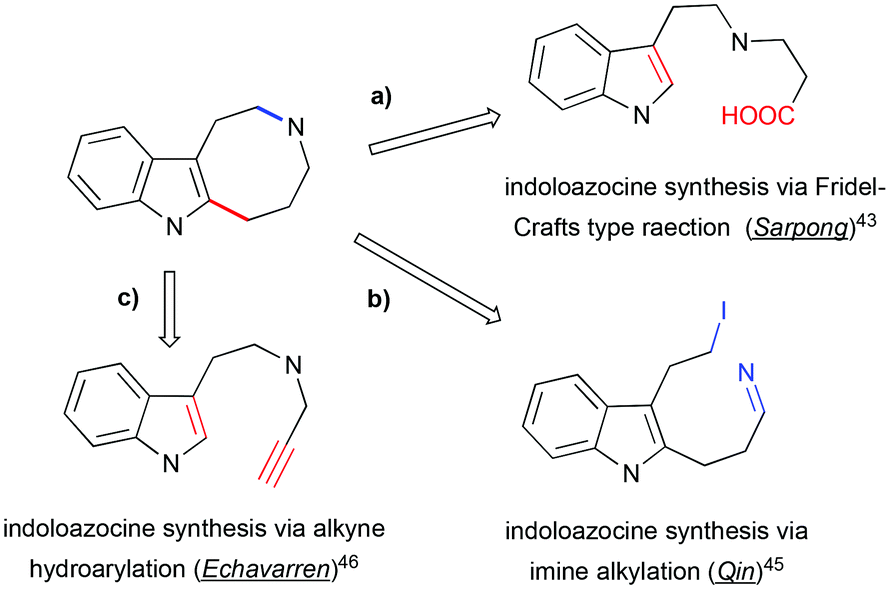
A number of methods have been developed for the assembly of indoloazocines to build up the rigid polycyclic skeleton of Kopsia alkaloid. One of the first syntheses of the lapidilectine tetracyclic core developed by Sarpong et al. relies on Friedel–Crafts (or Friedel–Crafts-type) reactions (Scheme 14, path a).43 Similar indole acylation was reported in the synthesis of substituted indoloazocines from tryptamine-derived (sulfonamido)methyl acrylic acid.44
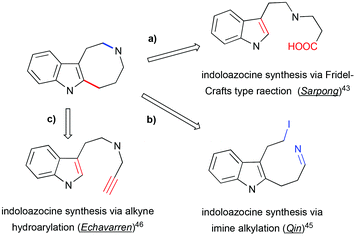 |
| | Scheme 14 Retrosynthetic approaches to indoloazocine applied in the total synthesis of pyrroloazocine indole alkaloids. | |
In the synthesis of lundurine A by the group of Qin, the azocinoindole ring system was assembled by intramolecular alkylation of imine by primary iodide (Scheme 14, path b).45 Intramolecular hydroarylation of alkynes with electron-rich heteroarenes, such as indoles, provide a fast access to the azocinoindole skeleton. Due to the remarkable affinity of gold(I) towards alkynes, gold salts demonstrate high efficiency in catalyzing these processes.46 Mercury salts are also competent in catalyzing hydroarylation reactions.47
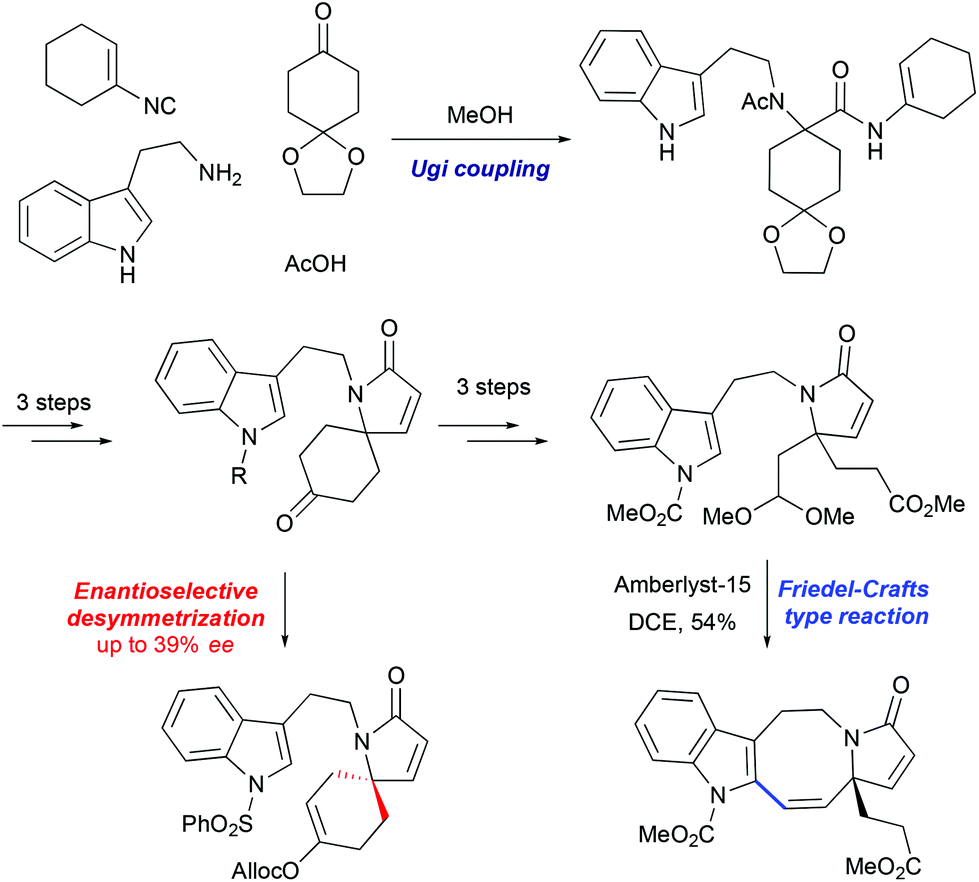
3.1 Synthesis of the lapidilectine tetracyclic core by Sarpong et al., 8-membered ring via Friedel–Crafts acylation
An 11-step synthesis of the tetracyclic core of Kopsia indole alkaloid was developed by Sarpong based on the Friedel–Crafts acylation of indole to form the 8-membered ring (Scheme 15).43 The synthesis commenced with a four-component Ugi reaction between tryptamine, isocyanocyclohexene, a substituted cyclohexanone, and acetic acid to form the coupling adduct that was converted into the key spiro-fused lactam. This intermediate was further opened in 3 steps: formation of TBS enol ether, its oxidative cleavage and subsequent formation of dimethyl acetal, furnishing the precursor for the Friedel–Crafts-type transformation. The cyclization was promoted by Amberlyst-15, as a source of acid, to afford indoloazocine in 54% yield. Preliminary studies of enantioselective deprotonation of spiro-fused ketone with chiral amine bases demonstrated the possibility of stereoselective desymmetrization, although only moderate enantiomeric excess (up to 39%) was achieved.
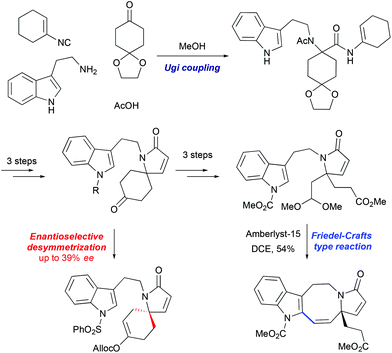 |
| | Scheme 15 Synthesis of lapidilectine tetracyclic core by Sarpong et al.43 | |
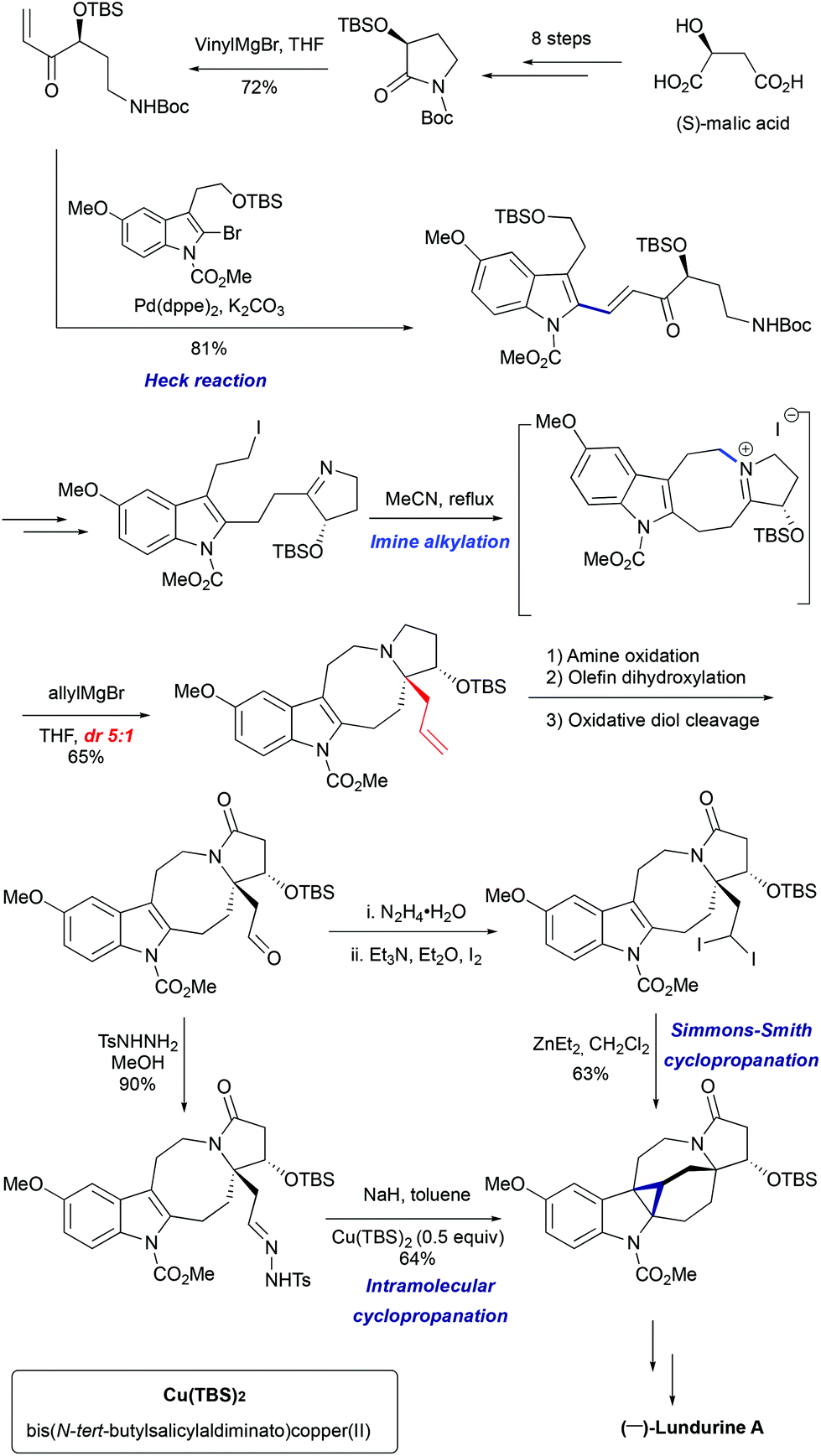
3.2 Total synthesis of (−)-lundurine A by Qin et al., 8-membered ring via imine alkylation
In 2015, Qin's group reported the asymmetric synthesis of (−)-lundurine A and determined its absolute configuration (Scheme 16).45a The primary iodide intermediate, required to construct the polyhydropyrroloazocine scaffold, was prepared from N-protected pyrrolidinone in 7 steps. The pyrrolidinone fragment was allowed to react with vinylmagnesium bromide, followed by palladium-catalyzed Heck coupling of resulting enone with 2-bromoindole to give the 2-alkenyl substituted indole. The manipulation of the functional group yielded the desired cyclic imine, the precursor for the indoloazocine synthesis. Intramolecular N-alkylation led to the 8-membered ring and formation of iminium cation that sets the stage for the diastereoselective generation of tetrasubstituted carbon stereocenter C20. The addition of allylmagnesium bromide from the less hindered face of the iminium cation allowed the generation of stereocenter C20, providing the product as a 5:1 mixture of diastereomers. The hexacyclic core of lundurine A was efficiently constructed by employing a late stage Simmons–Smith cyclopropanation. Simple functional group modification provided the natural product (−)-lundurine A in approximately 2% overall yield.
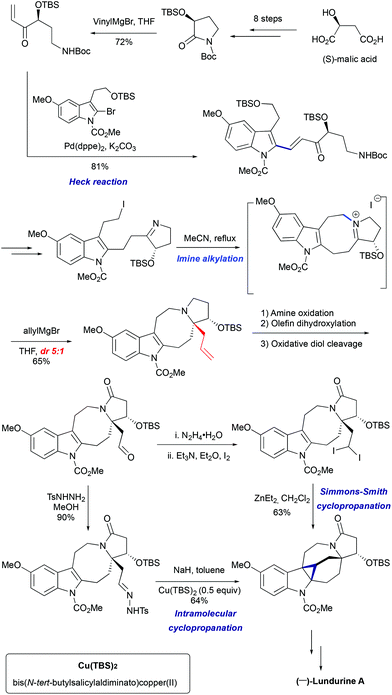 |
| | Scheme 16 Total synthesis of (−)-lundurine A by Qin et al.45 | |
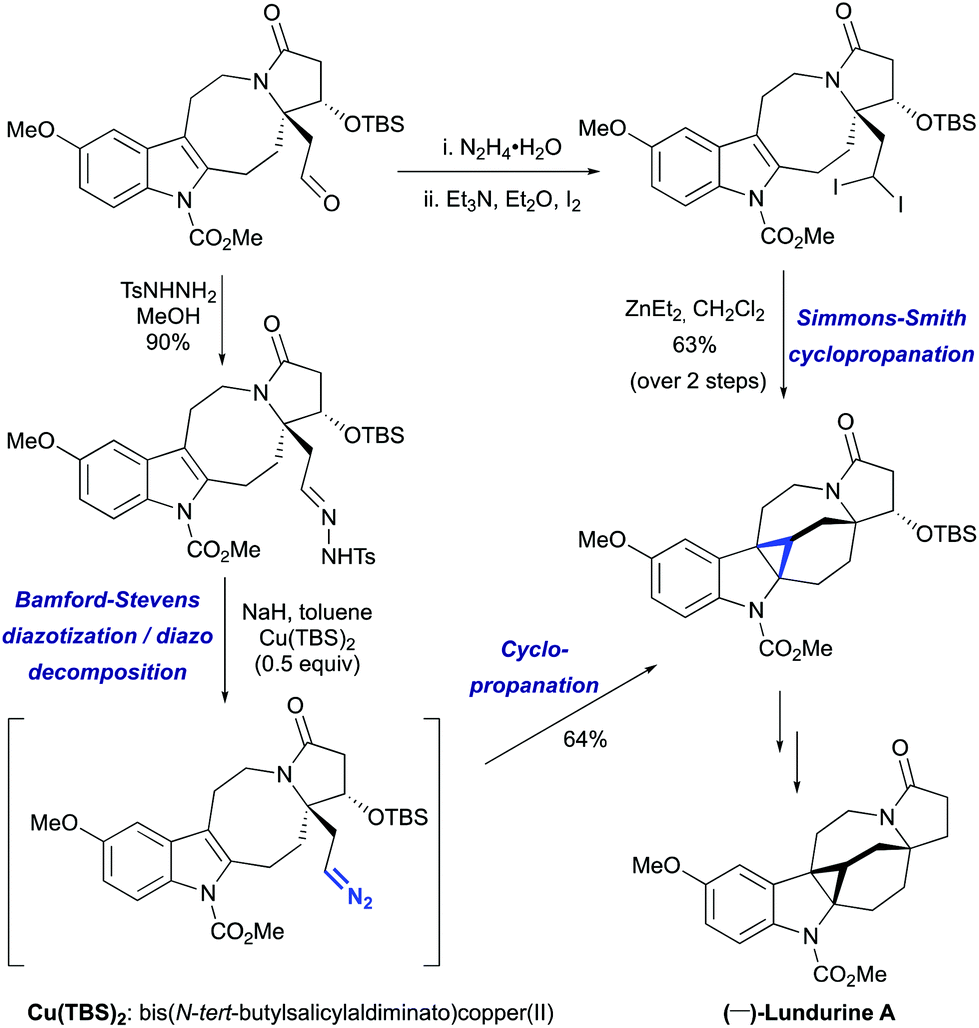
However, the lengthy preparation of pyrrolidinone was a significant issue in this first approach.45a The second asymmetric synthesis of (−)-lundurine A by Qin et al. relied on a diazo cyclopropanation strategy (Scheme 16).45b The intermediate aldehyde was converted into tosyl hydrazone, that underwent a one-pot Bamford-Stevens diazotization/diazo decomposition/cyclopropanation cascade promoted by Cu(TBS)2 to yield the corresponding hexacycle.
3.3 Construction of the 8-membered ring via alkyne hydroarylation
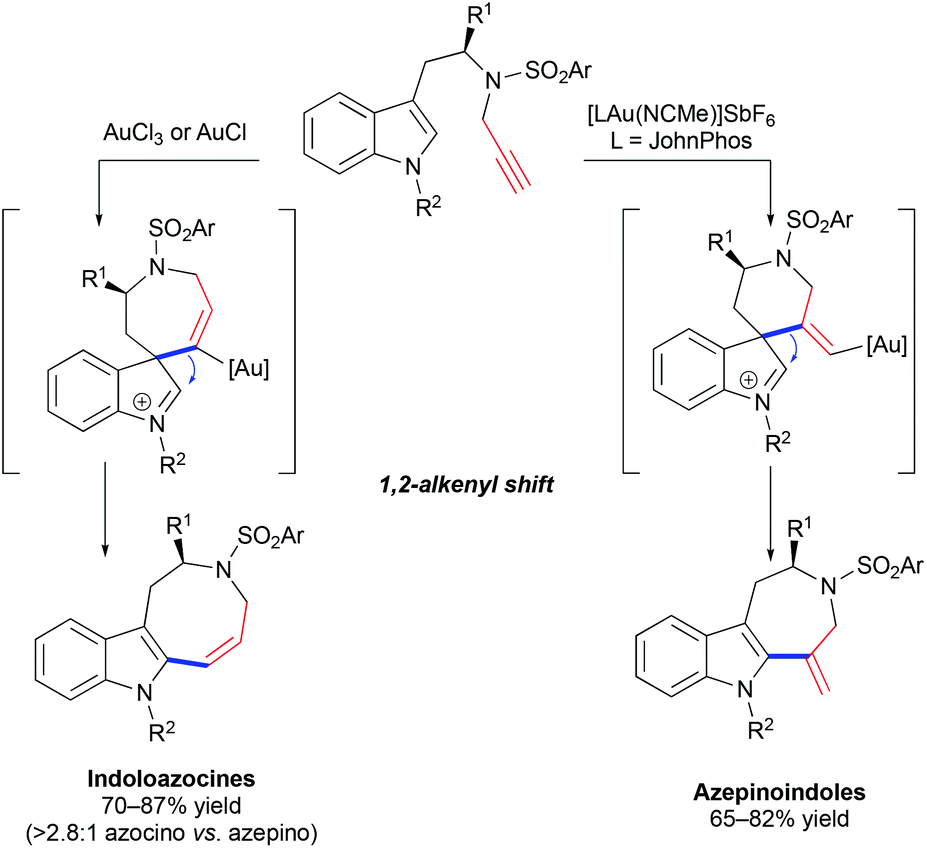
The hydroarylation of alkynes with indoles is a facile and convenient method for the synthesis of alkenyl heteroarenes and polycyclic skeletons from easily accessible precursors.48 The group of Echavarren demonstrated that alkenylation of indole with terminal alkynes results in the formation of indoloazocines in the presence of a catalytic amount of AuCl or AuCl3 by 7-endo-dig cyclization and a subsequent 1,2-alkene shift (Scheme 17).46 In contrast, the use of an electrophilic [JohnPhosAu(NCMe)]SbF6 catalyst leads to 7-membered azepinoindoles presumably through an initial 6-exo-pathway. The cycloisomerization of propargylated 2-substituted tryptamine derivatives results in the formation of isolable spirocyclic 2-methyleneindoline, which supported the initial hypothesis that the reaction proceeds through an initial attack by the most nucleophilic 3-position of indole onto alkyne.
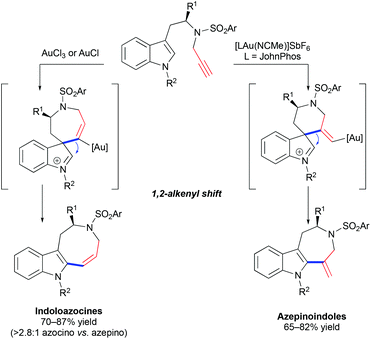 |
| | Scheme 17 Cycloisomerization of propargylated tryptamine derivatives by spirocyclic indolenine intermediates. | |
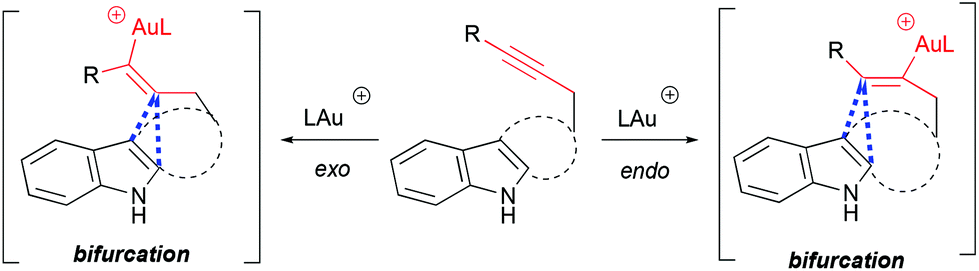
The mechanism and selectivity of gold-catalyzed cyclization of indole-ynes yielding indoloazocines have been studied by DFT calculations and experimentally.49 These studies suggest that reaction pathways bifurcate after the formation of the alkyne–gold complex, leading to α- or β-alkenylation routes (Scheme 18).
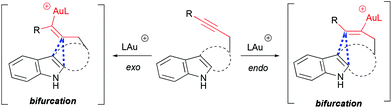 |
| | Scheme 18 Bifurcation point of cycloisomerization pathways. | |
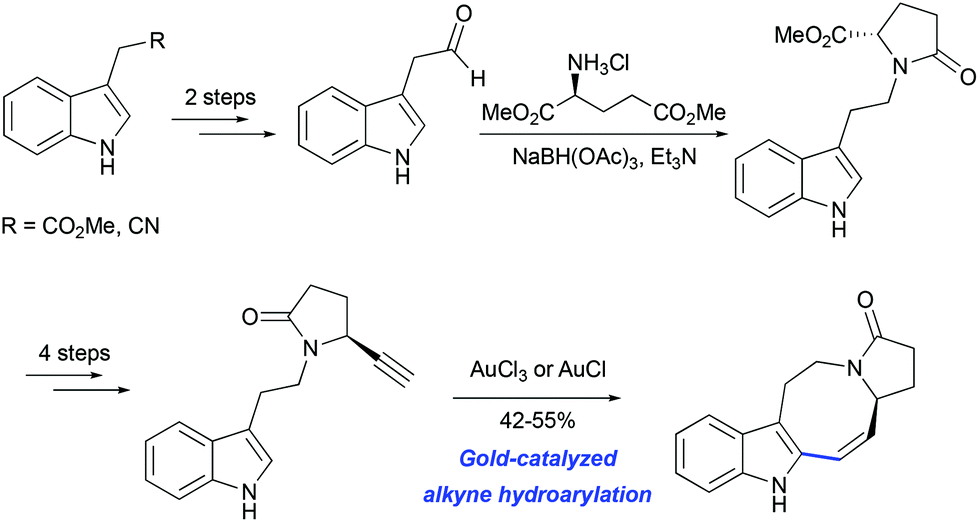
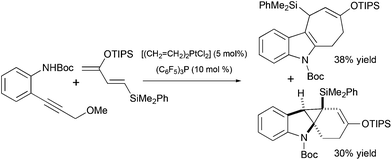
The potential utility of gold-catalyzed cyclization of propargylated indole derivatives has been tested in model studies for the synthesis of pyrroloazocine Kopsia alkaloids by Echavarren and co-workers. An enantiopure precursor for the hydroarylation step was obtained from methyl 2-(1H-indol-3-yl) acetate in 7 steps (Scheme 19).50 The formation of the tetracycle was achieved in 55% yield using AuCl3 as the catalyst. A similar outcome was obtained with AuCl suggesting that gold(III) might be reduced to gold(I) under the reaction conditions.
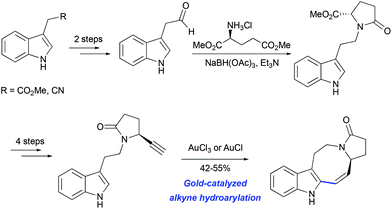 |
| | Scheme 19 Synthesis of lapidilectine tetracyclic core by Echavarren et al.50 | |
3.4 Total synthesis of (−)-lundurine A–C by Echavarren et al.
A unified approach towards racemic and enantioselective total syntheses of the natural products lundurines A–C has been developed, based on a previously elaborated method for the construction of the 8-membered ring by gold-catalyzed hydroheteroarylation (Scheme 20).51
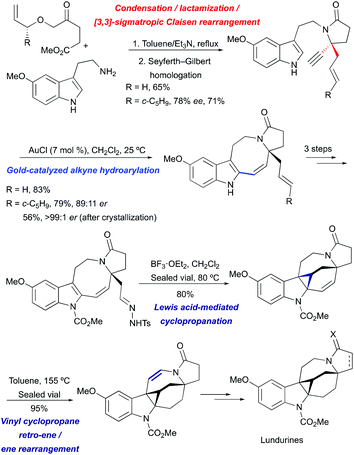 |
| | Scheme 20 Total synthesis of lundurines A–C by Echavarren et al.51 | |
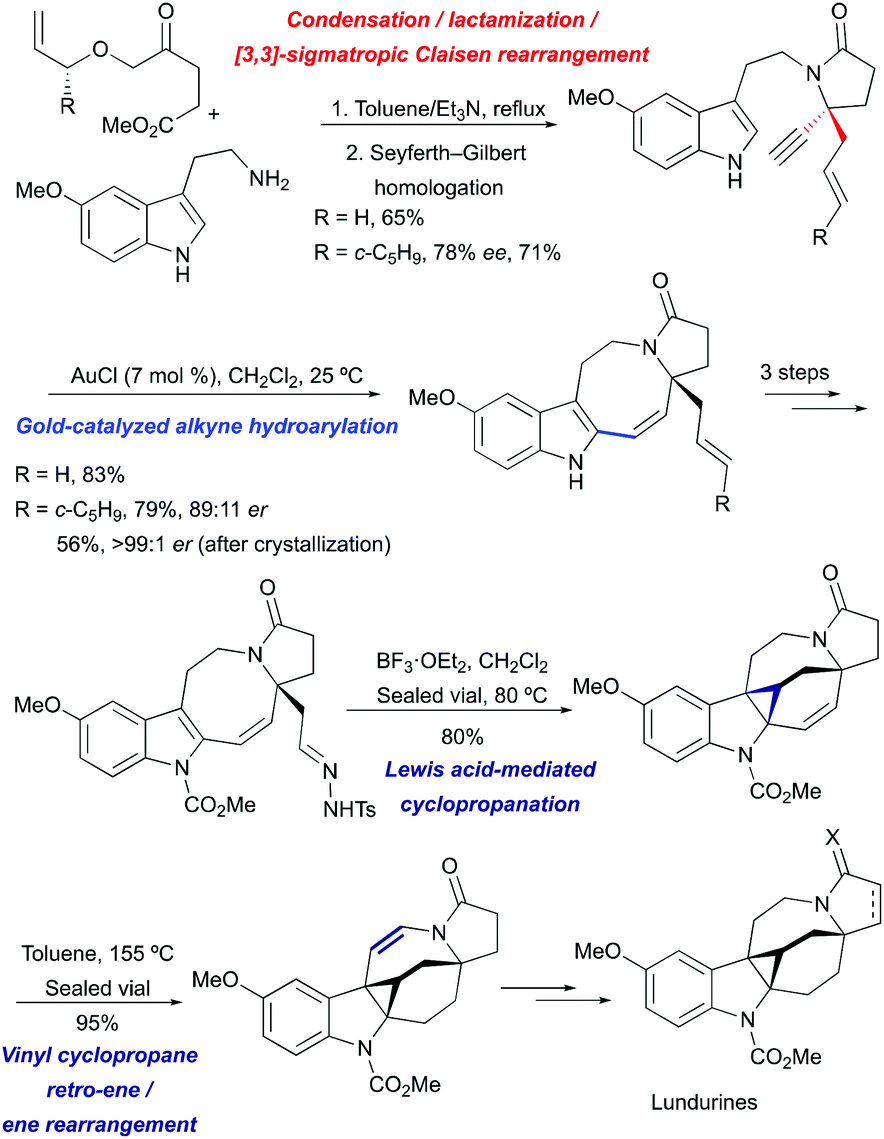
The indole pyrroloazocine ring system of lundurines was constructed in 3 steps starting from commercially available 5-methoxytryptamine and an allyl oxoester by a tandem condensation/lactamization/[3,3]-sigmatropic Claisen rearrangement, followed by Seyferth–Gilbert homologation and gold(I)-mediated cycloisomerization of the resulting indole-yne precursor. The latter transformation proceeded with perfect 8-endo selectivity and high yields, enabling the fastest and most efficient synthesis of the tetracyclic core of the pyrroloazocine indole alkaloids. The protection of indole nitrogen with methyl carbamate, followed by the oxidative cleavage of an exocyclic double bond and condensation with tosyl hydrazide yielded a diazo precursor for a Lewis acid-mediated intramolecular indole cyclopropanation. The desired hexacycle was obtained in 80% yield when tosyl hydrazone was treated with BF3·OEt2 at 80 °C. The migration of the double bond to the opposite side of the hexahydroazocine ring was achieved by simple heating through vinyl cyclopropane retro-ene/ene rearrangement providing enamide. Hydride reductions of the olefin under acidic conditions led to an advanced cyclopropyl-fused indoline intermediate. This common intermediate was ultimately converted into lundurines A–C within a few additional steps.51
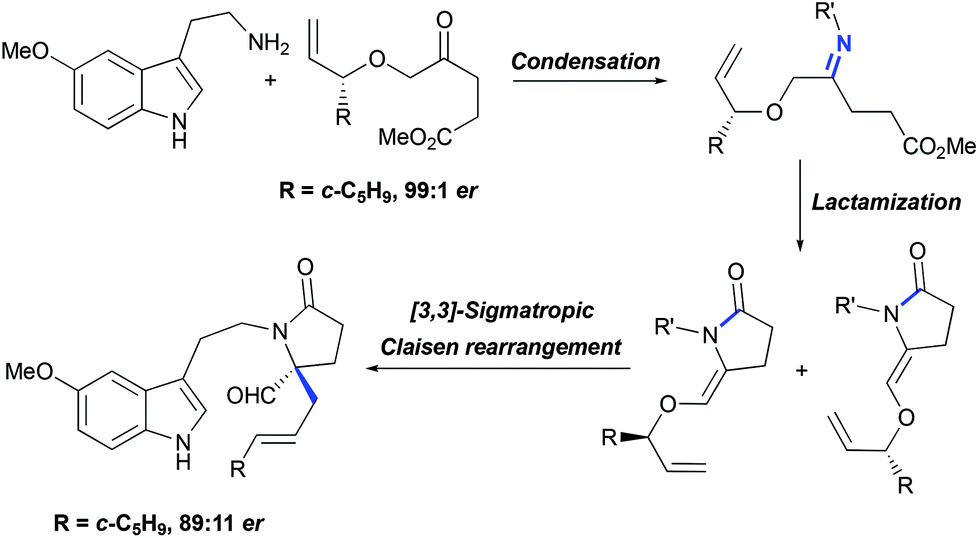
For the asymmetric synthesis of lundurines A–C, the generation of the stereocenter C20 was achieved by the implementation of a practical chirality transfer from the chiral auxiliary on the allyl fragment (Scheme 21). The enantiopure oxoester was condensed with 5-methoxytryptamine under basic conditions to prevent Pictet-Spendler type reactions. The resulting imine undergoes lactamization to form a mixture of E- and Z-enamides, following the Claisen rearrangement of allylvinyl ether thereby leading to the desired aldehyde in an 89:11 enantiomeric ratio.51
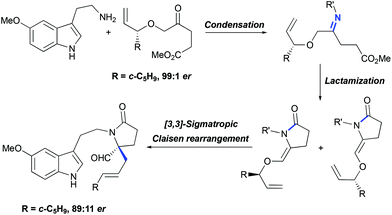 |
| | Scheme 21 Generation of tetrasubstituted carbon stereocenter C20 in the total synthesis of lundurines A–C by Echavarren et al.51 | |
4. Synthesis and reactivity of indoline-fused cyclopropanes
The construction of the cyclopropyl-fused indoline core of the lundurines poses a significant synthetic challenge. A number of methods have been developed for both inter- and intra-molecular cyclopropanation by transition metal-catalyzed decomposition of diazo compounds, typically employing rhodium or coper salts.52 For some systems, enantioselective versions have also been developed.53
4.1 Undesired reactivity in indoles cyclopropanation step in the course of lundurines total syntheses
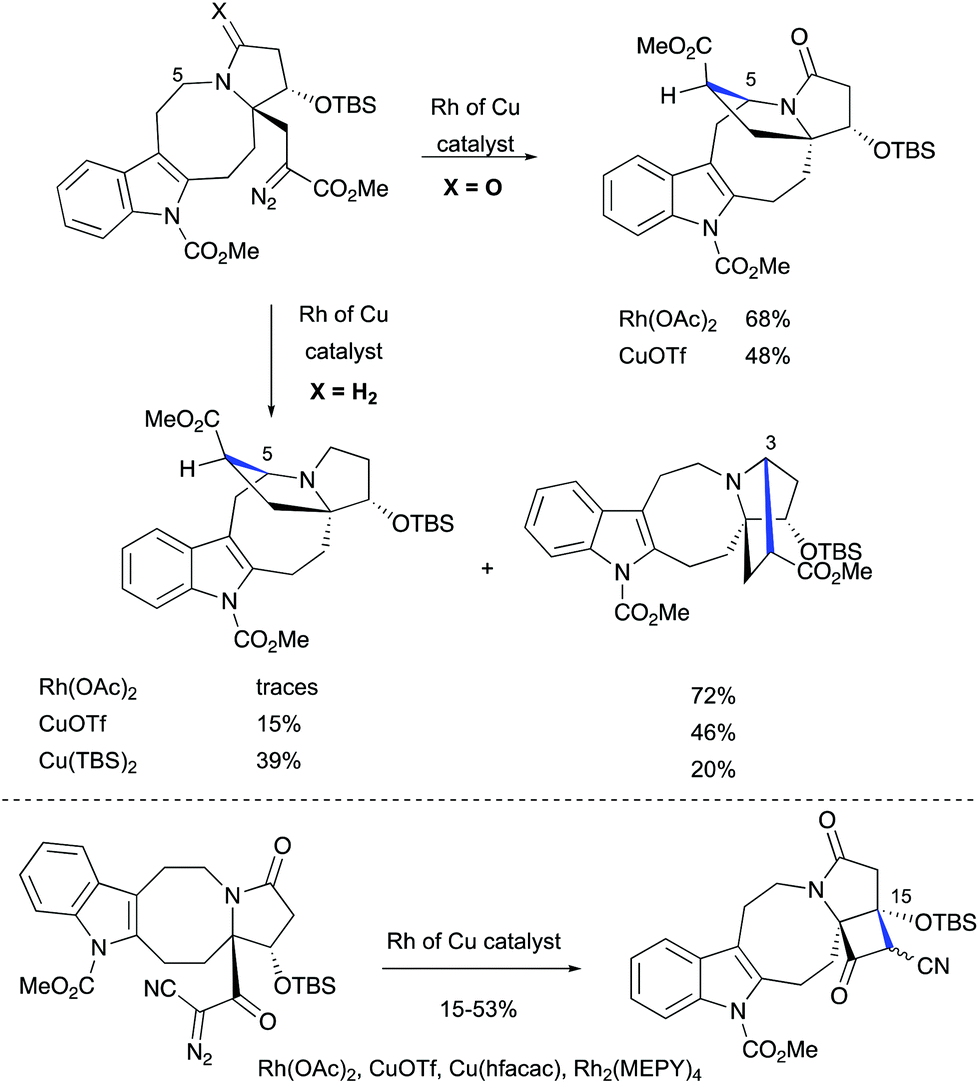
The challenge of constructing a hexacyclic lundurine core with a cyclopropyl moiety fused to an indoline by using a diazo cyclopropanation strategy is well illustrated from the work of Qin's group.45b An extensive investigation of the transition metal-catalyzed intramolecular cyclopropanation of indole pyrroloazocine ring systems closely related to the lundurines revealed an unexpected reactivity of metalo-carbene species. In the presence of copper or rhodium salts, the decomposition of α-diazocarboxylates resulted in the formation of C–H insertion products (at C3 and C5 positions) instead of the anticipated reaction of the carbene with an indole double bond (Scheme 22). In case of α-diazocyanide, the insertion at C15–H was predominant leading to a cyclobutanone product. The steric hindrance, conformational features of the 8-membered ring, as well as the presence of electron-withdrawing groups stabilizing the diazo compound could possibly explain the preferential formation of the insertion products. Ultimately, the successful application of the diazocyclopropanation strategy was achieved using a less stable alkyl diazo compound generated in situ from the corresponding tosyl hydrazone. Cascade Bamford–Stevens diazotization/diazo decomposition/cyclopropanation mediated by Cu(TBS)2 occurred with a similar efficiency in comparison with the previously used Simmons–Smith reaction and afforded cyclopropyl-fused indoline in a 64% yield (Scheme 23).
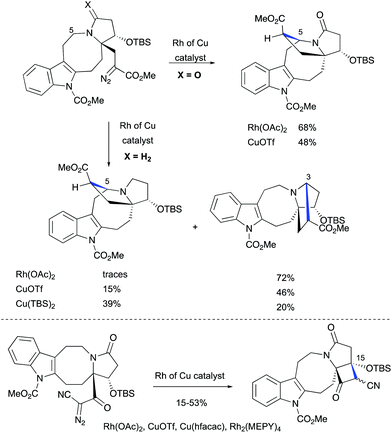 |
| | Scheme 22 Transition metal-catalyzed intramolecular C–H functionalizations of indole pyrroloazocine ring systems by Qin et al.45 | |
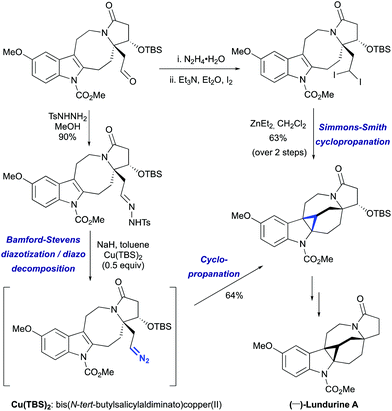 |
| | Scheme 23 The construction of the hexacyclic lundurine core by cyclopropanation by Qin et al.45 | |
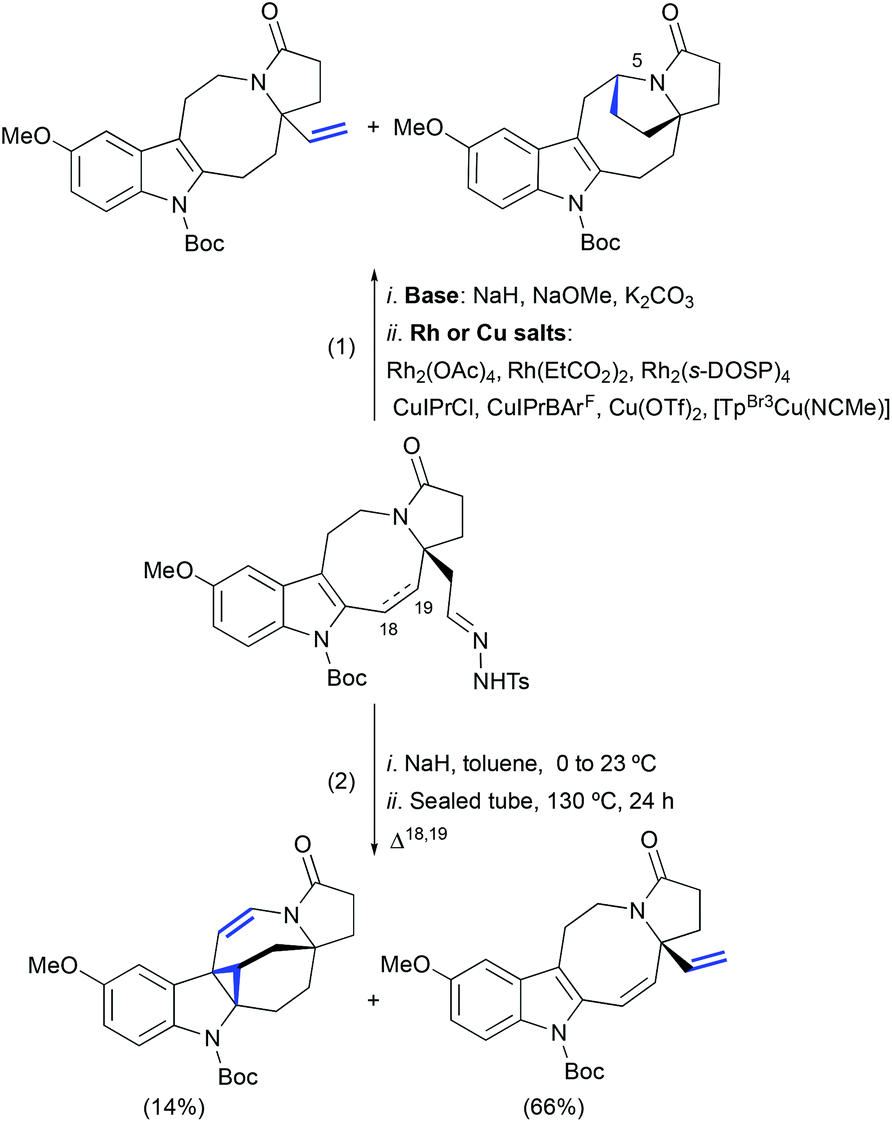
In our model studies towards the synthesis of lundurines, we have considered the use of Bamford–Stevens diazotization/a transition metal-catalyzed diazo decomposition/indole cyclopropanation strategy (Scheme 24, eqn (1)).54 Despite our efforts in the systematic screening of bases, catalysts, and temperatures, an undesired vinyl-substituted tetracycle was obtained as the major product in all cases. An unexpected formal C–H insertion at the C5 position was occurring in the presence of transition metal salts. However, in the absence of a metal catalyst, the vinyl derivative was obtained as the only product. Interestingly, when transition metal-free conditions were used for the system featuring a partially unsaturated hexahydroazocine ring system, the cyclopropane-containing hexacycle was obtained in low yield with a concomitant migration of the double bond to the opposite side of the hexahydroazocine ring (Scheme 24, eqn (2)).
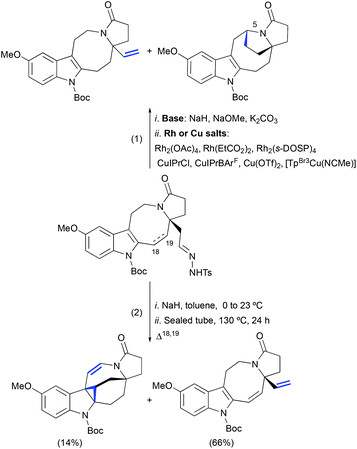 |
| | Scheme 24 Transition metal-catalyzed intramolecular cyclopropanation of indole pyrroloazocine ring systems vs. C–H insertion or elimination.51,54 | |
4.2 Lewis acid-mediated [3 + 2] cycloaddition of tosyl hydrazones to alkenes as a cyclopropanation strategy
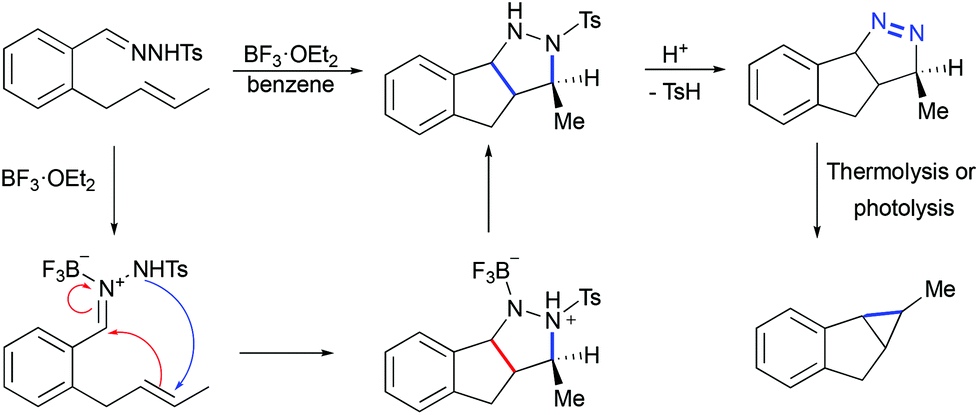
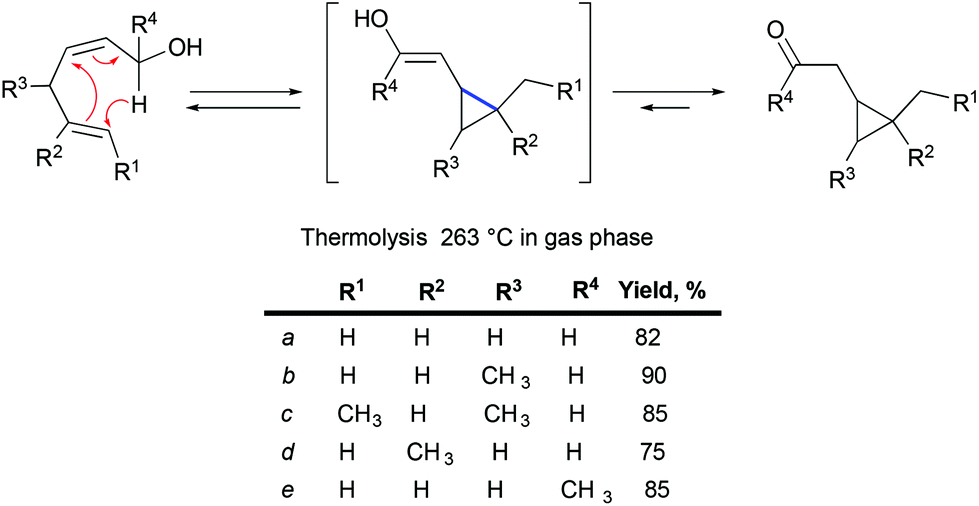
Less known Lewis acid-mediated 1,3-dipolar cycloaddition of tosyl hydrazones to olefins yielding corresponding pyrazolines followed by a loss of dinitrogen is a very interesting alternative to diazotization/transition metal catalyzed cyclopropanation. Thus, when o-(trans-2-butenyl)benzaldehyde tosylhydrazone was treated with BF3·OEt2, indenopyrazole was obtained as the only product (Scheme 25).55 This transformation was proposed to begin by the coordination of boron trifluoride to the imine nitrogen atom of the hydrazone, followed by a stepwise formal 1,3-dipolar cycloaddition. The chromatography purification of indenopyrazole resulted in the loss of p-toluenesulphenic acid, thereby yielding the corresponding indenopyrazoline. The latter, upon thermolysis or photolysis, is converted into a cyclopropane with the loss of dinitrogen.
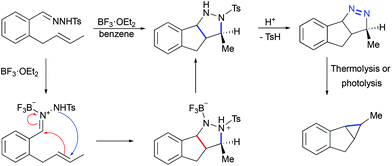 |
| | Scheme 25 Proposed mechanism of indenopyrazoline formation by Lewis acid-mediated [3 + 2]-cycloaddition of hydrazones to olefins.55 | |
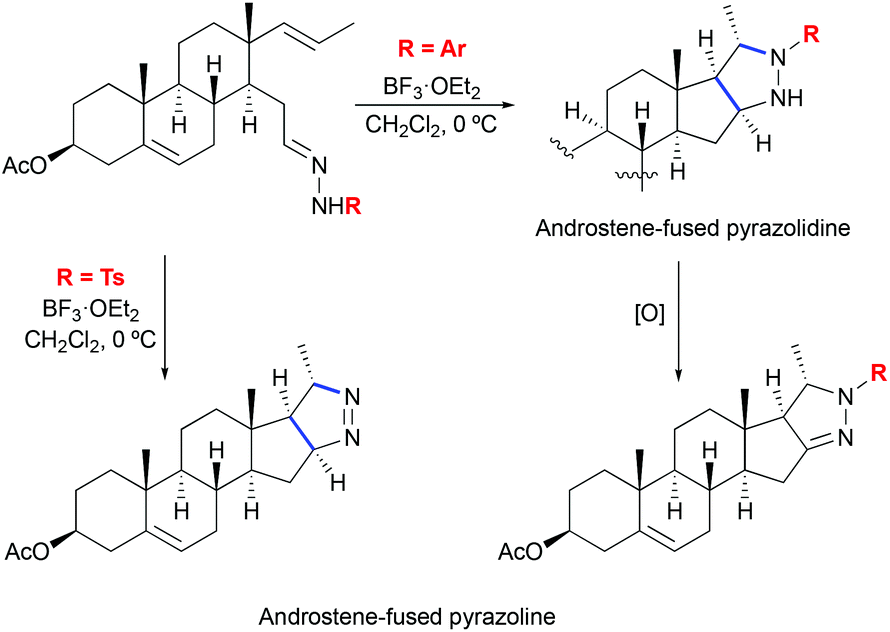
The formation of pyrazolines by 1,3-dipolar cycloaddition between hydrazones and olefins was also reported in the context of steroid systems (Scheme 26).56 Various aryl substituted hydrazones were shown to successfully undergo stereoselective cycloaddition reactions providing androstene-fused pyrazoline derivatives. In the case of o,p-dinitrophenyl hydrazones, pyrazolidines were obtained instead. This finding shows that the reaction rate is dependent on the electronic characteristic of the substituent and supports the proposed stereochemical model of cycloaddition. Additionally, computational studies on the mechanism of the pyrazoline formation have been disclosed.56
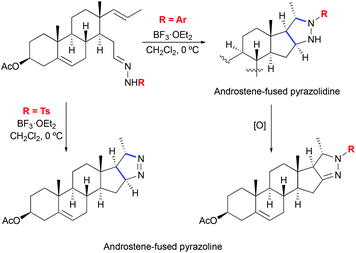 |
| | Scheme 26 Formation of androstene-fused pyrazolines by Lewis acid-mediated [3 + 2] cycloaddition of hydrazones to olefins.56 | |
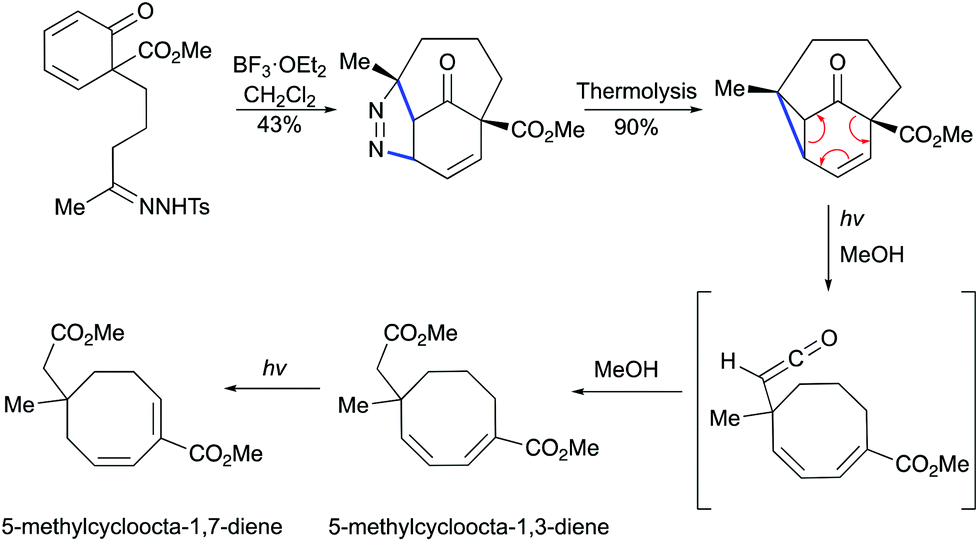
Moreover, in 1986, the group of Schultz demonstrated that a vinylcyclopropane can be generated by BF3-mediated [3 + 2] cycloaddition/thermal pyrazoline decomposition (Scheme 27).57 Thermally induced dinitrogen extrusion resulted in a tricyclic product. The irradiation of the vinylcyclopropane with Pyrex-filtered light in methanol provided a 1:2:1 mixture of starting materials and a mixture of isomeric l,3- and 1,7-cyclooctadienes. Longer irradiation of the mixture resulted in the formation of 1,7-cyclooctadiene in an almost quantitative yield.
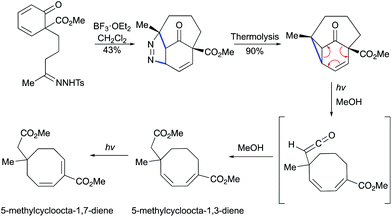 |
| | Scheme 27 Formation of cyclooctadienes by Lewis acid-mediated [3 + 2] cycloaddition and photochemical cleavage.57 | |
4.3 [3 + 2] cycloaddition of tosyl hydrazones to alkenes in total synthesis of lundurines. Unexpected isomerization of vinyl cyclopropane and similar literature precedents
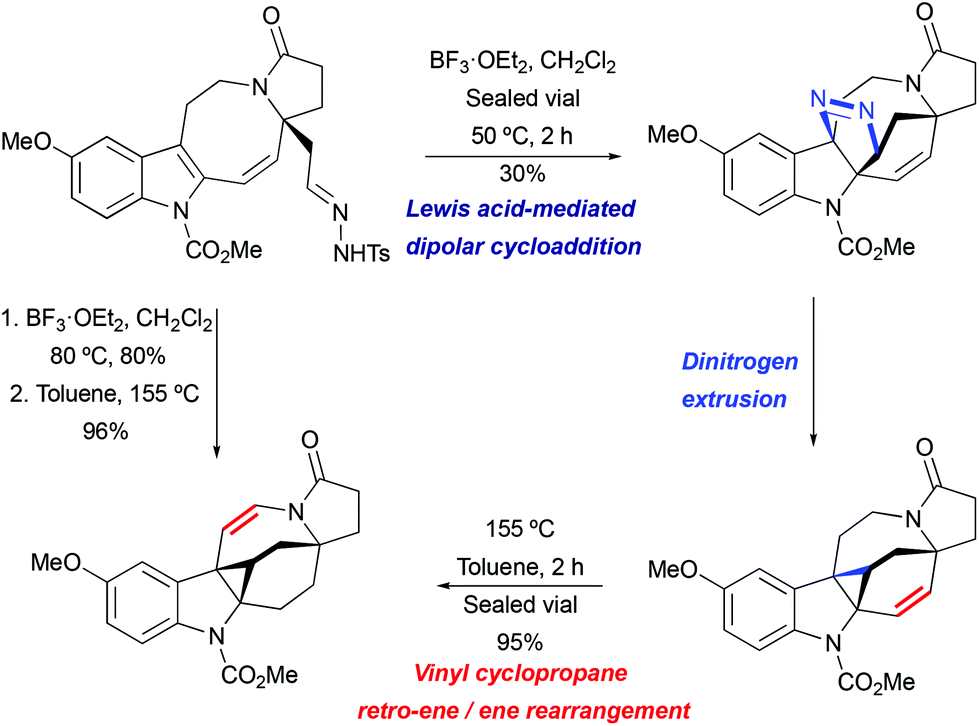
Lewis acid-mediated [3 + 2] cycloaddition/thermal pyrazoline decomposition was found to be the most efficient strategy for the key indole cyclopropanation step in the course of the lundurine synthesis by Echavarren et al. (Scheme 28).51 Under similar to the above mentioned acidic conditions (BF3·OEt2 in CH2Cl2, at 80 °C), the desired hexacycle was obtained in 80% yield. The isolated pyrazoline intermediate was converted to indoline-fused cyclopropane upon heating or slowly while being stored.
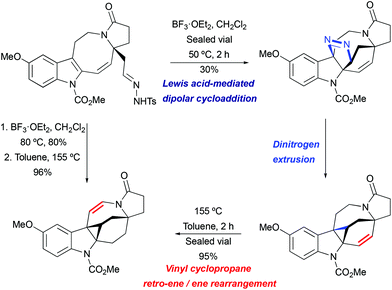 |
| | Scheme 28 The construction of the hexacyclic lundurine core by cyclopropanation by Echavarren et al.51 | |
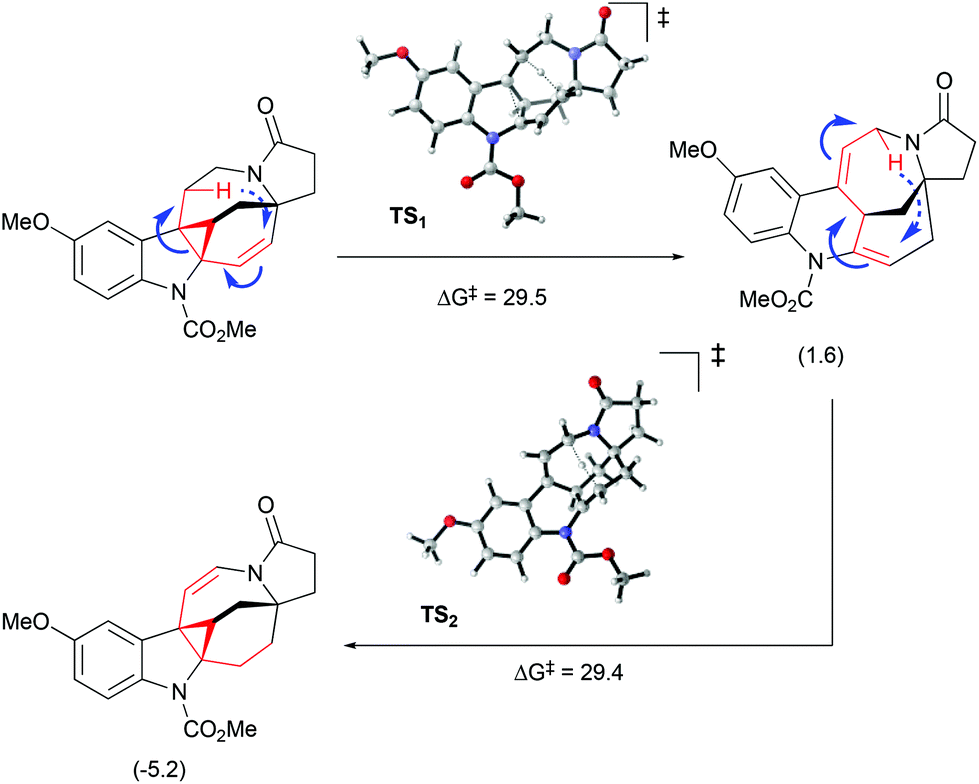
Interestingly, the product of direct cyclopropanation undergoes a puzzling thermal isomerization upon which the double bond migrates to the opposite side of the hexahydroazocine ring (Scheme 28). This transformation was proposed to proceed by a homodienyl retro-ene rearrangement followed by the reverse process. DFT calculations (Scheme 29), demonstrated that a 1,5-hydrogen shift leads to the formation of an 1,4-diene intermediate, which undergoes intramolecular ene reactions to form a new vinyl cyclopropane moiety. The formation of a more stable conjugated N-acylenamine is a driving force of this transformation. Both transition states for 1,5-hydrogen shifts were found to be similar in energy (ca. 29.5 kcal mol−1). The computed barrier for an alternative mechanism proceeding by direct dyotropic rearrangement was found to be unaccessibly high (>80 kcal mol−1). Additionally, a similar ene-reaction was observed separately in a pyrroloazocine diene substrate (Scheme 30).58
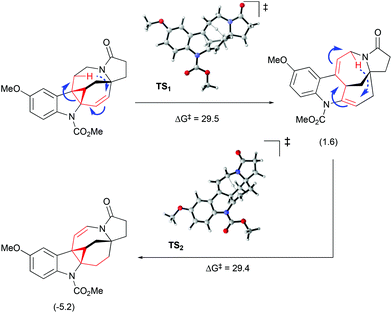 |
| | Scheme 29 Olefin migration by homodienyl retro-ene rearrangement/ene-reaction.51 | |
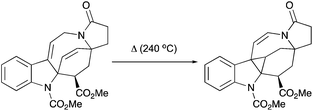 |
| | Scheme 30 Thermal isomerization of 1,4-diene into vinyl cyclopropane by an ene-reaction. | |
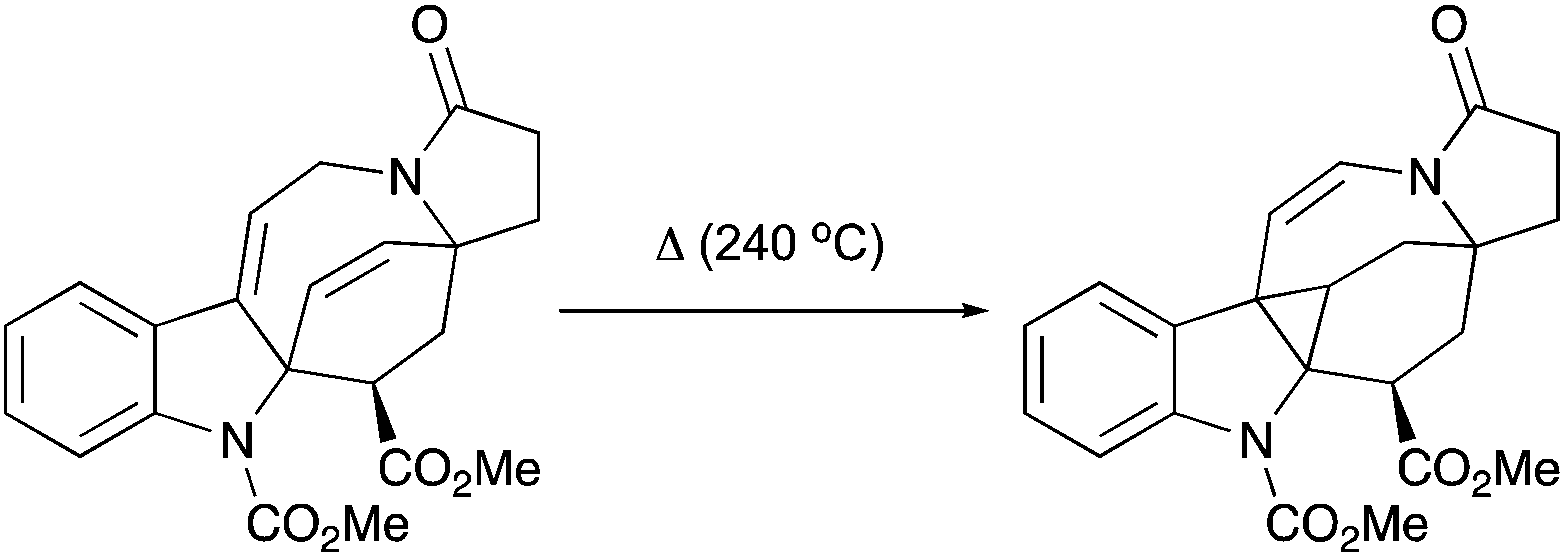
An irreversible formation of skipped dienes from vinylcyclopropane has been previously reported in the context of cyclic and bicyclic systems.59 A mechanistic study of intramolecular dienyl and homodienyl 1,5-hydrogen shifts in the context of cyclic (5- to 9-membered rings) and open chain systems has been performed.60 The isomerization of bicyclo[6.1.0]octen-2-ene through homodienyl retro-ene rearrangement takes place at 150–170 °C, with an activation energy of ca. 33 kcal mol−1. Only one precedent for the reverse process, the rearrangement of skipped dienes into vinyl cyclopropanes under thermal conditions, has been reported for the oxy-homodienyl substrate (Scheme 31), and requires temperatures of around 260 °C (activation energies of 41–43.5 kcal mol−1).61
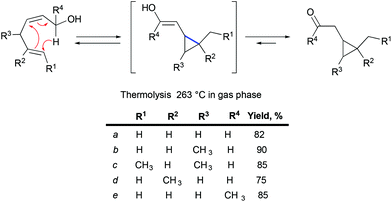 |
| | Scheme 31 The rearrangement of skipped dienes into vinyl cyclopropanes under thermal conditions. | |
5. Biosynthesis of pyrroloazocine indole alkaloids
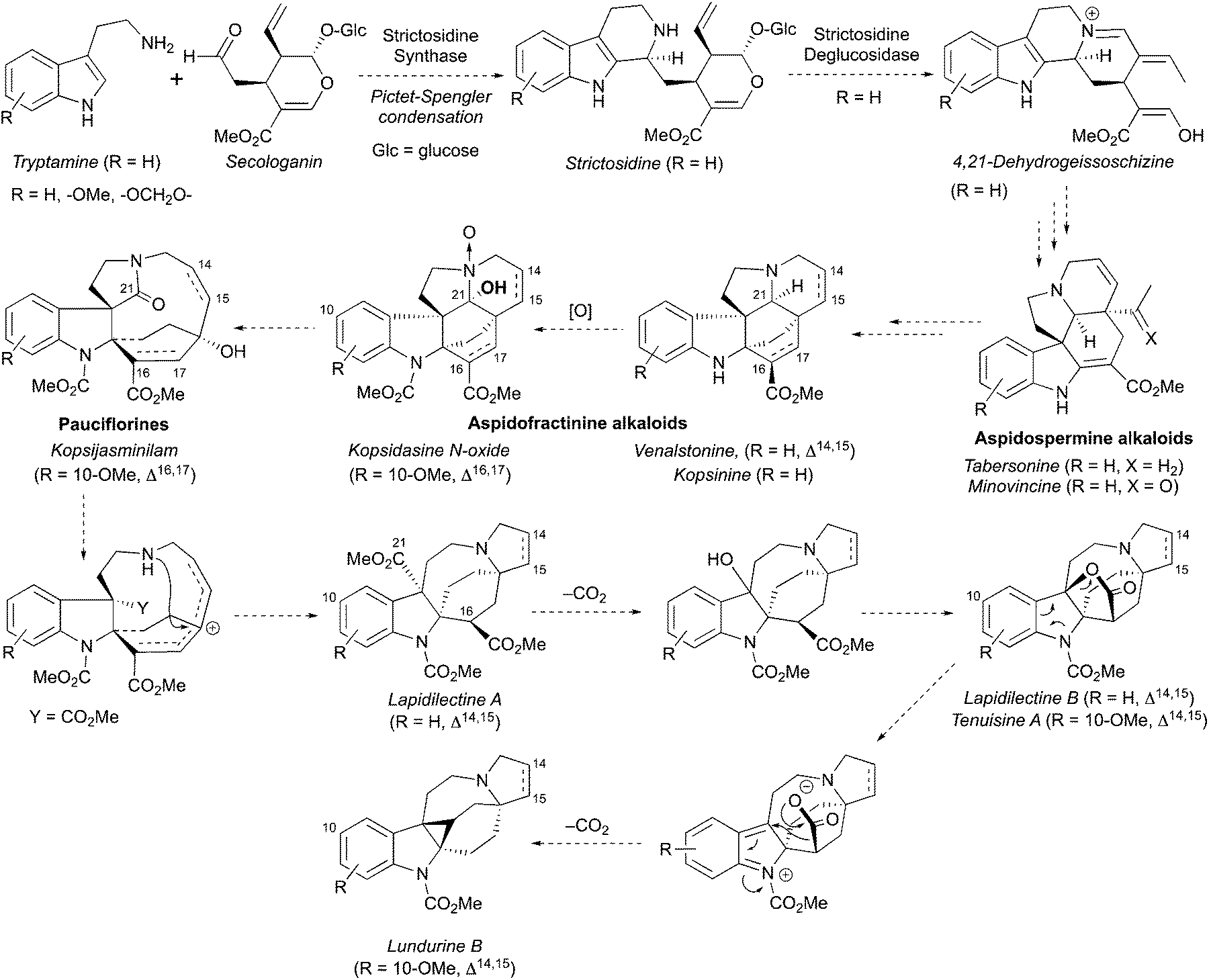
A hypothesis for the origin of pyrroloazocine indole alkaloids and the biosynthetic relationships within this family has been reported.8 The pyrroloazocine indole alkaloids belong to a broader class of monoterpene indole alkaloids, which are derivatives of iridoid terpene secologanin. Secologanin is first condensed with tryptamine in a Pictet–Spengler reaction providing strictosidine (Scheme 32).62 Deglycosylation of strictosidine then provides 4,21-dehydrogeissoschizine, which after a series of rearrangements gives aspidosperma type alkaloids, such as tabersonine or minovincine.62 The latter was proposed63 to be a precursor for aspidofractinines, alkaloids that are predominant in Kopsia species.8 For example, grandilodines and lapidilectines were isolated from Kopsia grandifolia together with aspidofractinine alkaloids venalstonine2 and kopsinine.3 Further oxidation at C21 provides alkaloids like kopsidasine, and its N-oxide, that were proposed to be a source of pauciflorine type alkaloids, such as kopsijasminilam.8 Pauciflorines were considered as precursors for diester-type pyrroloazocine indole alkaloids, such as lapidilectine A.8 subsequent oxidative decarboxylation of benzylic methyl ester and lactonization would lead to alkaloids like lapidilectine B and tenuisine A. Next, the decarboxylation of the lactone would result in the formation of cyclopropane-containing lundurines, which was proposed to proceed by a heterolytic Krapcho-type mechanism. It is worth noting that both decarboxylations of quaternary methyl ester and a lactone are rather unusual, and no experimental support for these transformations has been disclosed yet.
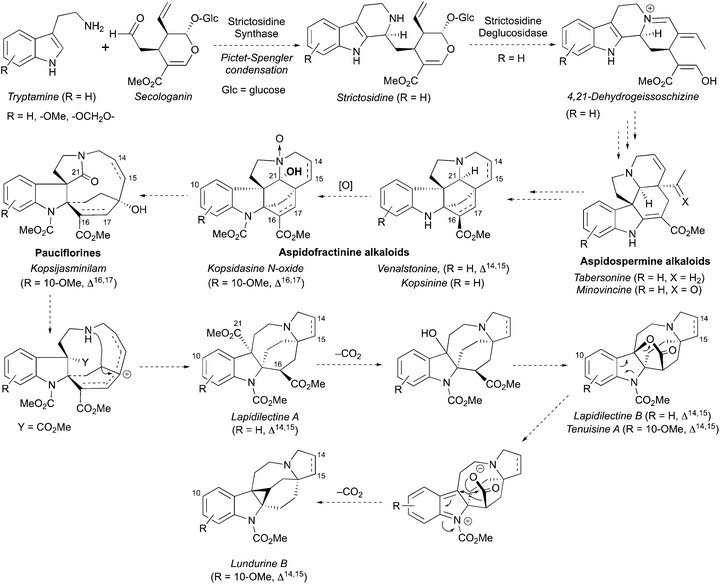 |
| | Scheme 32 Proposed biosynthesis of pyrroloazocine indole alkaloids. | |
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge the funding from MINECO/FEDER, UE (CTQ2016-75960-P), MINECO-Severo Ochoa Excellence Acreditation 2014–2018, (SEV-2013-0319), the European Research Council (Advanced Grant No. 321066), the AGAUR (2014 SGR 818 and FI fellowship to MSK), and CERCA Programme/Generalitat de Catalunya.
References
- K. Awang, T. Sévenet, A. H. A. Hadi, B. David and M. Païs, Tetrahedron Lett., 1992, 33, 2493–2496 CrossRef CAS.
- K. Awang, T. Sévenet, M. Païs and A. H. A. Hadi, J. Nat. Prod., 1993, 56, 1134–1139 CrossRef CAS.
- W.-S. Yap, C.-Y. Gan, Y.-Y. Low, Y.-M. Choo, T. Etoh, M. Hayahi, K. Komiyama and T.-S. Kam, J. Nat. Prod., 2011, 74, 1309–1312 CrossRef CAS PubMed.
- T.-S. Kam, K.-H. Lim, K. Yoganathan, M. Hayashi and K. Komiyama, Tetrahedron, 2004, 60, 10739–10745 CrossRef CAS.
- T.-S. Kam, K. Yoganathan and C.-H. Chuah, Tetrahedron Lett., 1995, 36, 759–762 CrossRef CAS.
- T.-S. Kam, K. Yoganathan, H.-Y. Li and N. Harada, Tetrahedron, 1997, 53, 12661–12670 CrossRef CAS.
- D. J. Middleton, Harvard Pap. Bot., 2004, 9, 89–142 Search PubMed.
-
T.-S. Kam and K.-H. Lim, in The Alkaloids: Chemistry and Biology, ed. G. A. Cordell, Academic Press, USA, 2008, vol. 66, pp. 1–111 Search PubMed.
- For the recent review about lundurines see:
S. Arai, M. Nakajima and A. Nishida, in The Alkaloids: Chemistry and Biology, ed. H.-J. Knölker, 2017, vol. 78, p. 167 Search PubMed.
- T. Sévenet, L. Allorge, B. David, K. Awang, A. H. A. Hadi, C. Kan-Fan, J.-C. Quirion, F. Remy, H. Schaller and L. E. Teo, J. Ethnopharmacol., 1994, 41, 147–183 CrossRef.
- R. L. Betts, R. Muspratt and S. G. P. Plant, J. Chem. Soc., 1927, 1310–1314 RSC.
- M. A. Ardakani and R. K. Smalley, Tetrahedron Lett., 1979, 49, 4769–4772 CrossRef.
- M. Azadi-Arkadani, M. A. Alkhader, J. H. Lippiatt, D. I. Patel, R. K. Smalley and S. Higson, J. Chem. Soc., Perkin Trans. 1, 1986, 1107–1111 RSC.
- W. H. Pearson, Y. Mi, I. Y. Lee and P. Stoy, J. Am. Chem. Soc., 2001, 123, 6724–6725 CrossRef CAS PubMed.
- W. H. Pearson, I. Y. Lee, Y. Mi and P. Stoy, J. Org. Chem., 2004, 69, 9109–9122 CrossRef CAS PubMed.
- Y.-w. Ren, X. Wang, W. Wang, B. Li, Z.-j. Shi and W. Zhang, Tetrahedron Lett., 2011, 52, 192–195 CrossRef CAS.
- Y. Goriya and C. V. Ramana, Chem. Commun., 2013, 49, 6376–6378 RSC.
- R. Sulsky, J. Z. Gougoutas, J. DiMarco and S. A. Biller, J. Org. Chem., 1999, 64, 5504–5510 CrossRef CAS PubMed.
- M. Nakajima, S. Arai and A. Nishida, Angew. Chem., Int. Ed., 2016, 55, 3473–3476 CrossRef CAS PubMed.
- J. S. Schneekloth Jr., J. Kim and E. J. Sorensen, Tetrahedron, 2009, 65, 3096–3101 CrossRef PubMed.
- J.-Y. Mérour, L. Chichereau, E. Desarbre and P. Gadonneix, Synthesis, 1996, 519–524 CrossRef.
- C. Wang, Z. Wang, X. Xie, X. Yao, G. Li and L. Zu, Org. Lett., 2017, 19, 1828–1830 CrossRef CAS PubMed.
- See, for example:
(a) M. E. Kuehne and P. J. Seaton, J. Org. Chem., 1985, 50, 4790–4796 CrossRef CAS;
(b) D. Schinzer and M. Kalesse, Synlett, 1989, 34–35 CAS;
(c) P. Le Ménez, J. Sápi and N. Kunesch, J. Org. Chem., 1989, 54, 3216–3218 CrossRef;
(d) E. Wenkert and S. Liu, J. Org. Chem., 1994, 59, 7677–7682 CrossRef CAS;
(e) P. Magnus, L. Gazzard, L. Hobson, A. H. Payne, T. J. Rainey, N. Westlund and V. Lynch, Tetrahedron, 2002, 58, 3423–3443 CrossRef CAS;
(f) D. Gagnon and C. Spino, J. Org. Chem., 2009, 74, 6035–6041 CrossRef CAS PubMed;
(g) S. B. Jones, B. Simmons, A. Mastracchio and D. W. C. MacMillan, Nature, 2011, 475, 183–188 CrossRef CAS PubMed;
(h) S. Harada, T. Sakai, K. Takasu, K.-i. Yamada, Y. Yamamoto and K. Tomioka, Chem. – Asian J., 2012, 7, 2196–2198 CrossRef CAS PubMed;
(i) X. Wu, J. Huang, B. Guo, L. Zhao, Y. Liu, J. Chen and W. Cao, Adv. Synth. Catal., 2014, 356, 3377–3382 CrossRef CAS.
- M. Dufour, J.-C. Gramain, H.-P. Husson, M.-E. Sinibaldi and Y. Troin, Tetrahedron Lett., 1989, 30, 3429–3432 CrossRef CAS.
- C. J. Whipp and F. G.-L. de Turiso, Tetrahedron Lett., 2008, 49, 5508–5510 CrossRef CAS.
- M. Hoshi, O. Kaneko, M. Nakajima, S. Arai and A. Nishida, Org. Lett., 2014, 16, 768–771 CrossRef CAS PubMed.
-
(a) S. Arai, M. Nakajima and A. Nishida, Angew. Chem., Int. Ed., 2014, 53, 5569–5572 CrossRef CAS PubMed;
(b) Corrigendum: S. Arai, M. Nakajima and A. Nishida, Angew. Chem., Int. Ed., 2014, 53, 14295 CrossRef CAS.
- M. Nakajima, S. Arai and A. Nishida, Chem. – Asian J., 2015, 10, 1065–1070 CrossRef CAS PubMed.
- J.-G. Rodriguez and A. S. Andrès, J. Heterocycl. Chem., 1991, 28, 1293–1299 CrossRef.
- H. Huang, Y. Yang, X. Zhang, W. Zeng and Y. Liang, Tetrahedron Lett., 2013, 54, 6049–6052 CrossRef CAS.
- H. Abe, N. Miyagawa, S. Hasegawa, T. Kobayashi, S. Aoyagi, C. Kibayashi, T. Katoh and H. Ito, Tetrahedron Lett., 2015, 56, 921–924 CrossRef CAS.
- H. Kusama, H. Sogo, K. Saito, T. Suga and N. Iwasawa, Synlett, 2013, 1364–1370 CAS.
- K. Aoki and K. Koga, Tetrahedron Lett., 1997, 38, 2505–2506 CrossRef CAS.
- H. Y. Harb and D. J. Procter, Synlett, 2012, 6–20 CAS.
- J. Inanaga, Y. Handa, T. Tabuchi and K. Otsubo, Tetrahedron Lett., 1991, 32, 6557–6558 CrossRef CAS.
- We assumed the 76% ee, because the procedure for crystallization to 91% ee was not described, and its yield was not included into the final yield calculations by Nishida et al.19 Additionally, the second step of the synthesis has 100% brsm yield, while the conversion of the starting material was only 33%.
- L. L. Anderson, Asian J. Org. Chem., 2016, 5, 9–30 CrossRef CAS.
- B. B. Snider and H. Lin, J. Am. Chem. Soc., 1999, 121, 7778–7786 CrossRef CAS.
-
(a) P. S. Baran and E. J. Corey, J. Am. Chem. Soc., 2002, 124, 7904–7905 CrossRef CAS PubMed;
(b) P. S. Baran, C. A. Guerrero and E. J. Corey, J. Am. Chem. Soc., 2003, 125, 5628–5629 CrossRef CAS PubMed.
-
(a) T. Watanabe, S. Arai and A. Nishida, Synlett, 2004, 907–909 CAS;
(b) T. Watanabe, S. Arai and A. Nishida, Tetrahedron, 2009, 65, 1327–1335 CrossRef.
-
(a) L. Yang, G. Deng, D.-X. Wang, Z.-T. Huang, J.-P. Zhu and M.-X. Wang, Org. Lett., 2007, 9, 1387–1390 CrossRef CAS PubMed;
(b) M. B. Johansen, A. B. Leduc and M. A. Kerr, Synlett, 2007, 2593–2595 CAS.
-
(a) C. Zheng, Y. Li, Y. Yang, H. Wang, H. Cui, J. Zhang and G. Zhao, Adv. Synth. Catal., 2009, 351, 1685–1691 CrossRef CAS;
(b) M. Juárez-Calderón, D. M. Aparicio, D. Gnecco, J. R. Juárez, L. Orea, A. Mendoza, F. Sartillo-Piscil, E. del Olmo and J. L. Terán, Tetrahedron Lett., 2013, 54, 2729–2732 CrossRef;
(c) L. Fuentes, M. Hernández-Juarez, J. L. Terán, L. Quintero and F. Sartillo-Piscil, Synlett, 2013, 878–882 CAS;
(d) J. Li, J. Li, Y. Xu, Y. Wang, L. Zhang, L. Ding, Y. Xuan, T. Pang and H. Lin, Nat. Prod. Res., 2016, 30, 800–805 CrossRef CAS PubMed.
- E. E. Schultz, B. G. Pujanauski and R. Sarpong, Org. Lett., 2012, 14, 648–651 CrossRef CAS PubMed.
- G. Pandey, R. Kant and S. Batra, Tetrahedron Lett., 2015, 56, 930–933 CrossRef CAS.
-
(a) S. Jin, J. Gong and Y. Qin, Angew. Chem., Int. Ed., 2015, 54, 2228–2231 CrossRef CAS PubMed;
(b) H.-X. Huang, S.-J. Jin, J. Gong, D. Zhang, H. Song and Y. Qin, Chem. – Eur. J., 2015, 21, 13284–13290 CrossRef CAS PubMed.
-
(a) C. Ferrer and A. M. Echavarren, Angew. Chem., Int. Ed., 2006, 45, 1105–1109 CrossRef CAS PubMed;
(b) C. Ferrer, H. M. Amijs and A. M. Echavarren, Chem. – Eur. J., 2007, 13, 1358–1373 CrossRef CAS PubMed.
-
(a) V. A. Peshkov, O. P. Pereshivko and E. V. Van der Eycken, Adv. Synth. Catal., 2012, 354, 2841–2848 CrossRef CAS;
(b) P. A. Donets, K. Van Hecke, L. Van Meervelt and E. V. Van der Eycken, Org. Lett., 2009, 11, 3618–3621 CrossRef CAS PubMed.
-
(a)
M. E. Muratore and A. M. Echavarren, Gold-catalyzed hydroarylation of alkynes, in The Chemistry of Organogold Compounds, Part 2, PATAI's Chemistry of Functional Group, ed. Z. Rappoport, J. F. Liebman and I. Marek, John Wiley & Sons, Ltd, Chichester, UK, 2014, ch. 16 Search PubMed;
(b)
M. S. Kirillova, F. M. Miloserdov and A. M. Echavarren, Catalytic hydroarylation of carbon-carbon multiple bonds, ed. L. Ackermann, T. B. Gunnoe and L. G. Habgood, Wiley-VCH, 2017, in press Search PubMed.
-
(a) L. Zhang, L. Chang, H. Hu, H. Wang, Z.-J. Yao and S. Wang, Chem. – Eur. J., 2014, 20, 2925–2932 CrossRef CAS PubMed;
(b) L. Zhang, Y. Wang, Z.-J. Yao, S. Wang and Z.-X. Yu, J. Am. Chem. Soc., 2015, 137, 13290–13300 CrossRef CAS PubMed.
- C. Ferrer, A. Escribano-Cuesta and A. M. Echavarren, Tetrahedron, 2009, 65, 9015–9020 CrossRef CAS.
- M. S. Kirillova, M. E. Muratore, R. Dorel and A. M. Echavarren, J. Am. Chem. Soc., 2016, 138, 3671–3674 CrossRef CAS PubMed.
- Review:
(a) H. M. L. Davies and S. J. Hedley, Chem. Soc. Rev., 2007, 36, 1109–1119 RSC. Some selected examples:
(b) B. Zhang and A. G. H. Wee, Chem. Commun., 2008, 4837–4839 RSC;
(c) B. Zhang and A. G. H. Wee, Org. Biomol. Chem., 2012, 10, 4597 RSC;
(d) J. M. Fraile, K. L. Jeune, J. A. Myaoral, N. Ravasio and F. Zaccheria, Org. Biomol. Chem., 2013, 11, 4327–4332 RSC.
-
(a) G. Özüduru, T. Schubach and M. M. K. Boysen, Org. Lett., 2012, 14, 4990–4993 CrossRef PubMed;
(b) H. Xu, Y.-P. Li, Y. Cai, G.-P. Wang, S.-F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2017, 139, 7697–7700 CrossRef CAS PubMed.
-
M. S. Kirillova, PhD Thesis, Rovira i Virgili University, 2016.
- A. Padwa and H. Ku, J. Org. Chem., 1980, 45, 3756–3766 CrossRef CAS.
- É. Frank, Z. Mucsi, I. Zupkó, B. Réthy, G. Falkay, G. Schneider and J. Wölfling, J. Am. Chem. Soc., 2009, 131, 3894–3904 CrossRef PubMed.
- A. G. Schultz and K. K. Eng, Tetrahedron Lett., 1986, 27, 2331–2334 CrossRef CAS.
-
F. M. Miloserdov
, unpublished results.
-
(a) W. von E. Doering and W. R. Roth, Angew. Chem., Int. Ed. Engl., 1963, 2, 115–122 CrossRef;
(b) R. J. Ellis and H. M. Frey, Proc. Chem. Soc., 1964, 221 Search PubMed;
(c) W. Grimme, Chem. Ber., 1965, 756–763 CrossRef CAS;
(d) P. A. Parziale and J. A. Berson, J. Am. Chem. Soc., 1990, 112, 1650–1652 CrossRef CAS;
(e) P. A. Parziale and J. A. Berson, J. Am. Chem. Soc., 1990, 113, 4595–4606 CrossRef;
(f) T. Hudlicky, T. N. Kutchan and S. M. Naqvi, Org. React., 1985, 33, 247–335 CAS.
- D. S. Glass, R. S. Boikess and S. Winstein, Tetrahedron Lett., 1966, 10, 999–1008 CrossRef.
- F.-G. Klärner, W. Rüngeler and W. Maifeld, Angew. Chem., 1981, 93, 613–614 CrossRef.
- S. E. ÓConnor and J. J. Maresh, Nat. Prod. Rep., 2006, 23, 532–547 RSC.
- M. E. Kuehne, Y.-L. Li and C.-Q. Wei, J. Org. Chem., 2000, 65, 6434–6440 CrossRef CAS PubMed.
|
| This journal is © the Partner Organisations 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Fedor M.
Miloserdov
a,
Fedor M.
Miloserdov