
Ir-Catalyzed reactions in natural product synthesis
Pengquan
Chen
,
Yuecheng
Wu
,
Shifa
Zhu
 ,
Huanfeng
Jiang
and
Zhiqiang
Ma
*
,
Huanfeng
Jiang
and
Zhiqiang
Ma
*
Key Lab of Functional Molecular Engineering of Guangdong Province, School of Chemistry & Chemical Engineering, South China University of Technology, Wushan Road-381, Guangzhou 510641, People's Republic of China. E-mail: cezqma@scut.edu.cn
First published on 28th September 2017
Abstract
This review highlights the recent applications of Ir-catalyzed reactions in the total synthesis of natural products. On the basis of the type of Ir-catalyzed reaction, the applications will be grouped into four main sections, including transfer hydrogenation reactions, hydrogenation reactions, allylic substitution reactions and C–H borylation reactions. The examples presented demonstrate the high efficiency of the entitled reactions in the formation of chemical bonds and the construction of naturally occurring skeletons.
 Pengquan Chen | Pengquan Chen was born in Shandong Province, China, in 1990. He received his B.S. degree in Chemistry from Qingdao University in 2014. He then started his graduate research in South China University of Technology as a M.S. candidate under the supervision of Prof. Huanfeng Jiang. He is currently a Ph.D. candidate in the laboratory of Prof. Zhiqiang Ma. His research interests include methodology development and natural product synthesis. |
 Yuecheng Wu | Yuecheng Wu received his B.S. degree in Chemistry from China Three Gorges University in 2015. He then joined South China University of Technology as a graduate student under the supervision of Prof. Zhiqiang Ma. He is currently involved in the total synthesis of natural products. |
 Shifa Zhu | Shifa Zhu obtained his B.S. degree in Chemistry from Hunan University in 2001. He did Ph.D. studies at Shanghai Institute of Organic Chemistry (SIOC), Chinese Academy of Science, under the supervision of Prof. Shizheng Zhu. Then he moved to Syracuse University to undertake postdoctoral research in Prof. Yan-Yeung Luk's group in 2006. In 2007, he joined Prof. X. Peter Zhang's group in the University of South Florida. In 2009, he got his first academic position in South China University of Technology as Associate Professor. In 2013, he was promoted as Professor. Research interests of Prof. Zhu's group include synthetic methodology, transition-metal catalysis, alkyne chemistry, and carbene chemistry. |
 Huanfeng Jiang | Huanfeng Jiang received his Ph.D. from Shanghai Institute of Organic Chemistry (SIOC) with Professor Xiyan Lu in 1993. And then he joined Guangzhou Institute of Chemistry as a research fellow. In 2003, he moved to the South China University of Technology (SCUT) as the Leading Professor of Chemistry. He received Chinese Chemical Society-BASF Young Investigator's Award in 2002 and National Natural Science Funds for Distinguished Young Scholar in 2006. His research interests focus on synthetic methodology, green and sustainable chemistry. |
 Zhiqiang Ma | Zhiqiang Ma obtained his B.S. degree from Lanzhou University in 2001, and his Ph.D. degree from Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, in 2007, working under the supervision of Prof. Hongbin Zhai. He joined Prof. Chuo Chen's lab at UT Southwestern Medical Center as a Postdoctoral Fellow to work on the total synthesis of dimeric pyrrole–imidazole alkaloids. He is currently a faculty member of South China University of Technology. His research interests include natural product synthesis, target-oriented methodology development, and medicinal chemistry. |
1. Introduction
Transition-metal-catalyzed organic chemical reactions,1–4 as versatile tools in the field of synthetic organic chemistry, have received wide attention and experienced tremendous development. In particular, research on Ir-catalyzed reactions in diverse aspects has witnessed significant progress. Taking advantage of these well-established methods, a vast range of structurally challenging frameworks have been rapidly synthesized from readily available precursors, allowing access to the corresponding natural products. Additionally, the combination of Ir-catalyzed reactions with other reactions has the ability to furnish a structurally complex, naturally occurring skeleton in a tandem fashion, further demonstrating the high efficiency and great utility of Ir-complexes in organic synthesis.Many excellent reviews regarding Ir-catalyzed reactions, with particular emphasis on methodological development, have been reported.5–10 Due to the rapid growth of the field and the appearance of a number of instructive total syntheses involving Ir-catalyzed reactions as key steps in recent years, we will introduce the applications of the entitled reactions in natural product synthesis (Scheme 1). The review is divided into four main sections based on the type of Ir-catalyzed reaction, including transfer hydrogenation reactions, hydrogenation reactions, allylic substitution reactions and C–H borylation reactions.
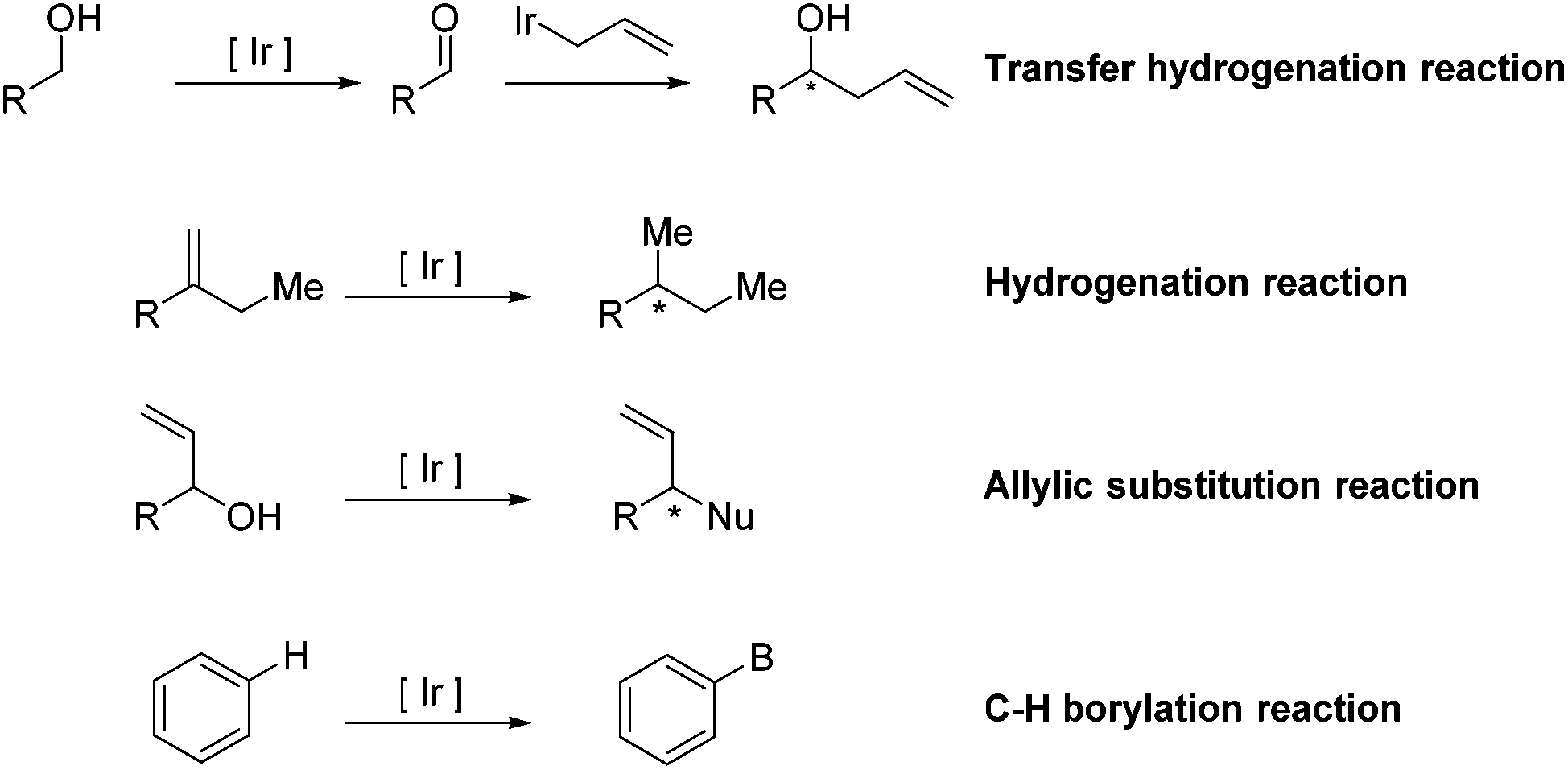 | ||
| Scheme 1 General Ir-catalyzed reactions used in the natural product synthesis. | ||
2. Ir-Catalyzed transfer hydrogenation reactions
Transfer hydrogenation reactions are widely applied in organic synthesis.6 In the 1960s, Henbest and co-workers11 discovered that cyclohexanol and dihydrochalcone could be obtained from cyclohexanone or chalcone through transfer hydrogenation, where IrCl3-(DMSO)3 or HIrCl2(DMSO)3 was used as the catalyst. After decades, Ir-catalyzed transfer hydrogenation reactions have gained extensive development6,12 and found applications in the natural product synthesis. This section will be divided into two parts, (1) Ir-catalyzed carbonyl allylation via transfer hydrogenation; (2) Ir-catalyzed oxidation.2.1 Ir-Catalyzed carbonyl allylation via transfer hydrogenation
In 2009, Krische and co-workers disclosed enantioselective carbonyl allylations, crotylations and tert-prenylations promoted by an Ir-complex. Compared to conventional methods, where stoichiometric metallic nucleophiles are often required, the current strategy instead uses a substoichiometric Ir-complex.13 The authors pointed out that primary alcohols function as both hydrogen donors and aldehyde precursors. First, dehydrogenation of the primary alcohol enabled by an Ir-complex through transfer hydrogenation generates an aldehyde, which is attacked by allyiridium nucleophile A2 formed in situ from allyl acetate, delivering the product and completing the asymmetric carbonyl allylation and crotylation process (Scheme 2). As a result, C–C bond formation via Ir-mediated primary alcohol C–H functionalization can be achieved.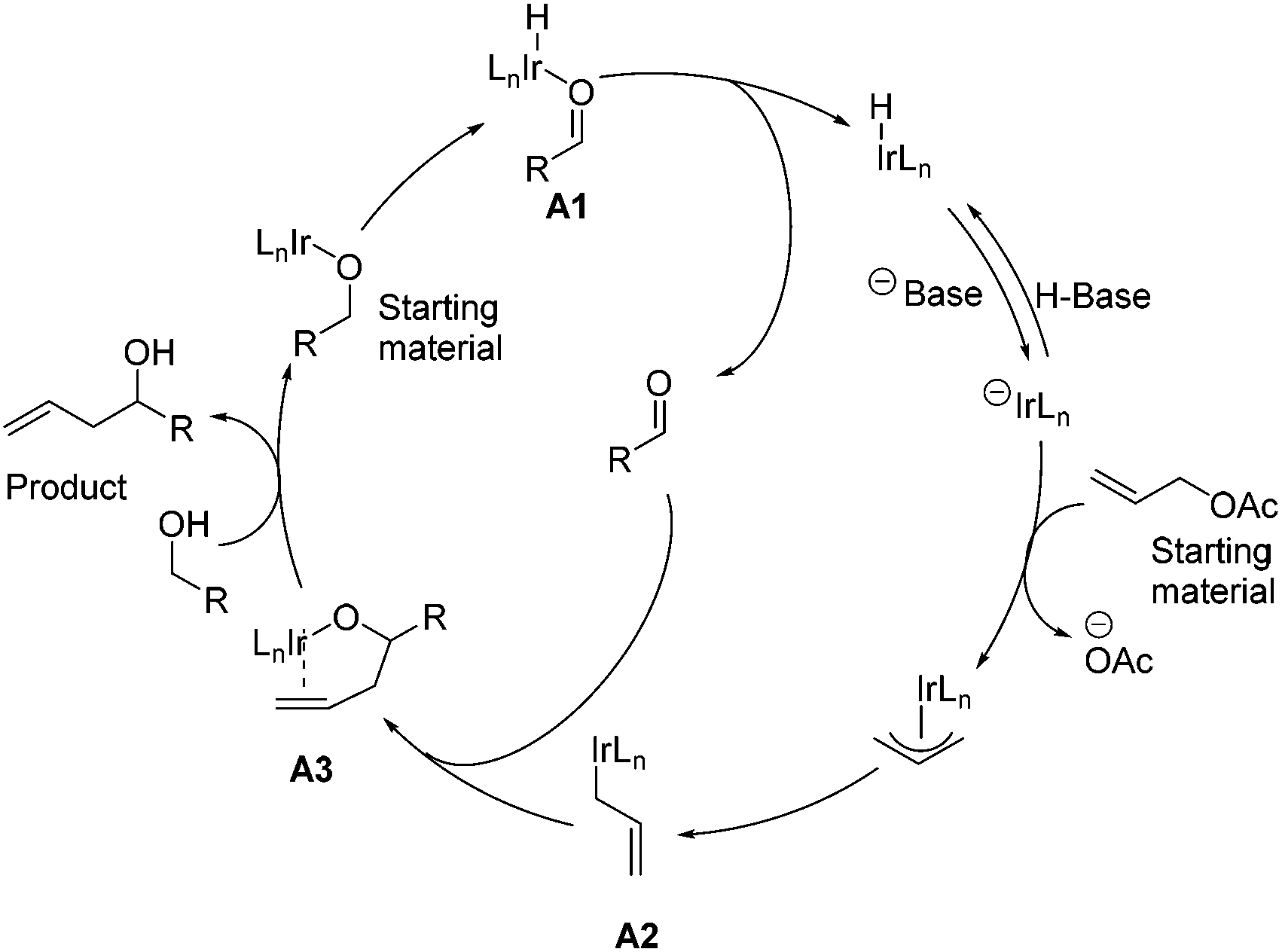 | ||
| Scheme 2 Postulated mechanism of Ir-catalyzed carbonyl allylation via transfer hydrogenation. | ||
The aforementioned asymmetric carbonyl allylation was further utilized by the Krische group to achieve the synthesis of a polyketide natural product (+)-roxation (Scheme 3).14 In the course of the authors’ study, seven C–C bonds were formed through asymmetric carbonyl allylation. Double C-allylation of 1,3-propanediol using ortho-cyclometalated iridium C,O-benzoate formed in situ from [Ir(cod)Cl]2, (R)-Cl,MeO-BIPHEP, 4-chloro-3-nitrobenzoic acid, and allyl acetate led to 1 in 70% yield with 99% ee, two additional steps furnished 2, thus completing the “first iteration”. Subsequent two double allylation reactions and synthetic transformations gave 3 as a single diastereomer, completing the “second and third iterations”. Elongation of diol 3 over three steps provided intermediate 4, poised for the pivotal crotylation reaction. Exposure of 4 to iridium C,O-benzoate complex (S)-cat2 gave rise to adduct 5 in 85% yield. Ultimately, the preparation of (+)-roxation was finalized in seven additional steps.
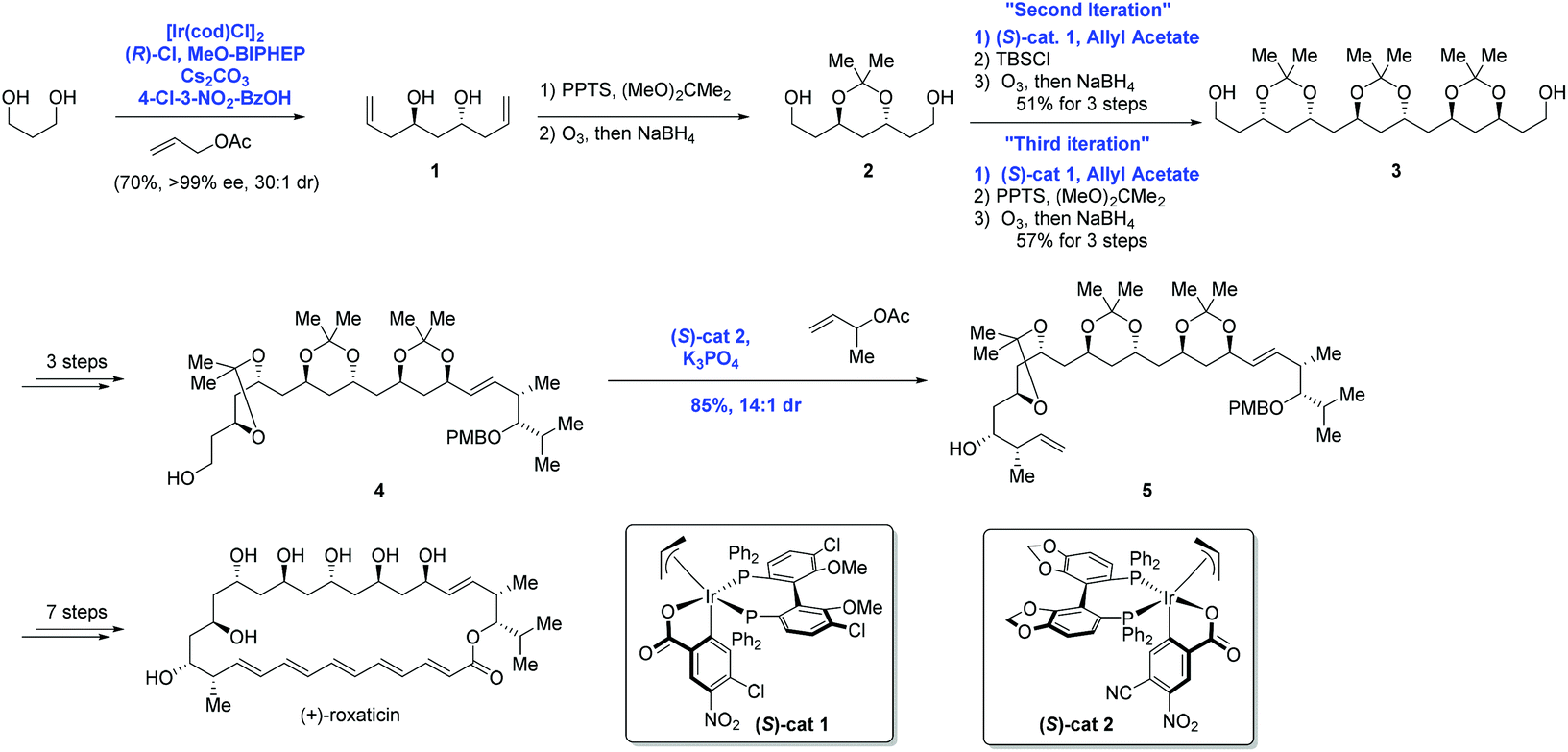 | ||
| Scheme 3 Synthesis of (+)-roxaticin. | ||
In the Krische group's subsequent study, asymmetric carbonyl allylation was used to achieve the synthesis of (+)-zincophorin methyl ester in 13 steps (longest linear sequence) (Scheme 4).15 (+)-Zincophorin, an iconic antibiotic, possesses potent in vivo activity against Gram-positive bacteria, and has attracted a number of research groups to pursue its total synthesis. Among them, Krische and co-workers envisioned making (+)-zincophorin methyl ester by the union of fragment A and fragment B. To obtain fragment A, anti-crotylation of allylic alcohol 6 in the presence of (R)-cat2 gave the homoallylic alcohol 7 with good diastereoselectivity (dr = 5.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Alcohol 7 was further elongated to deliver fragment A over two steps. To obtain fragment B, diol 8 experienced two-directional double anti-crotylation catalyzed by (S)-cat2 followed by iodoetherification to generate iodide 9. Eight additional steps then gave fragment B.
:1). Alcohol 7 was further elongated to deliver fragment A over two steps. To obtain fragment B, diol 8 experienced two-directional double anti-crotylation catalyzed by (S)-cat2 followed by iodoetherification to generate iodide 9. Eight additional steps then gave fragment B.
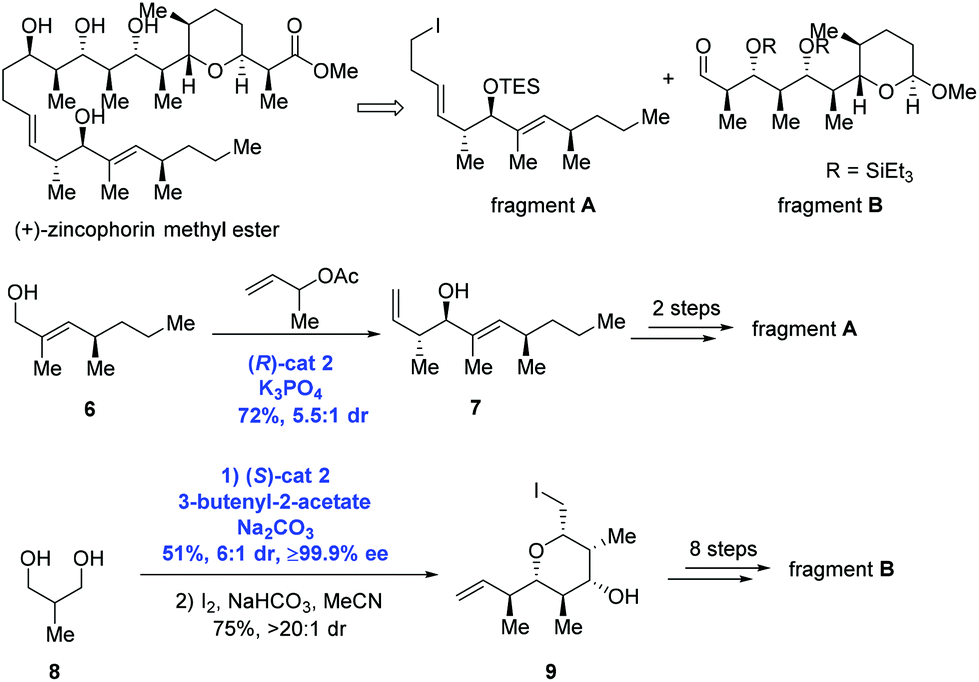 | ||
| Scheme 4 Synthesis of (+)-zincophorin methyl ester. | ||
Having the success in hand, Krische and co-workers expanded the methodology to enable the formation of an all-carbon quaternary stereocenter, allowing efficient and modular syntheses of terpenoid natural products oridamycin A, triptoquinones B and C, and isoiresin (Scheme 5).16 Of note is that the class of terpenoids was conventionally prepared through biomimetic polyene cascade reactions. To this end, the key intermediate 12 containing the all-carbon quaternary stereocenter was accessed from the reaction of alcohol 10 and isoprene oxide 11 catalyzed by (R)-cat3 derived from 4-CN-3-NO2-benzoic acid and (R)-Tol-BINAP, in high yield (90%) and with excellent diastereoselectivity (dr = 34:1) and enantioselectivity (98% ee). The common intermediate 13 prepared from 12 in a sequence of three steps underwent different reaction modes, featuring Suzuki/Friedel–Crafts and Diels–Alder cycloaddition to deliver the natural products oridamycin A, triptoquinones B and C, and isoiresin, respectively.
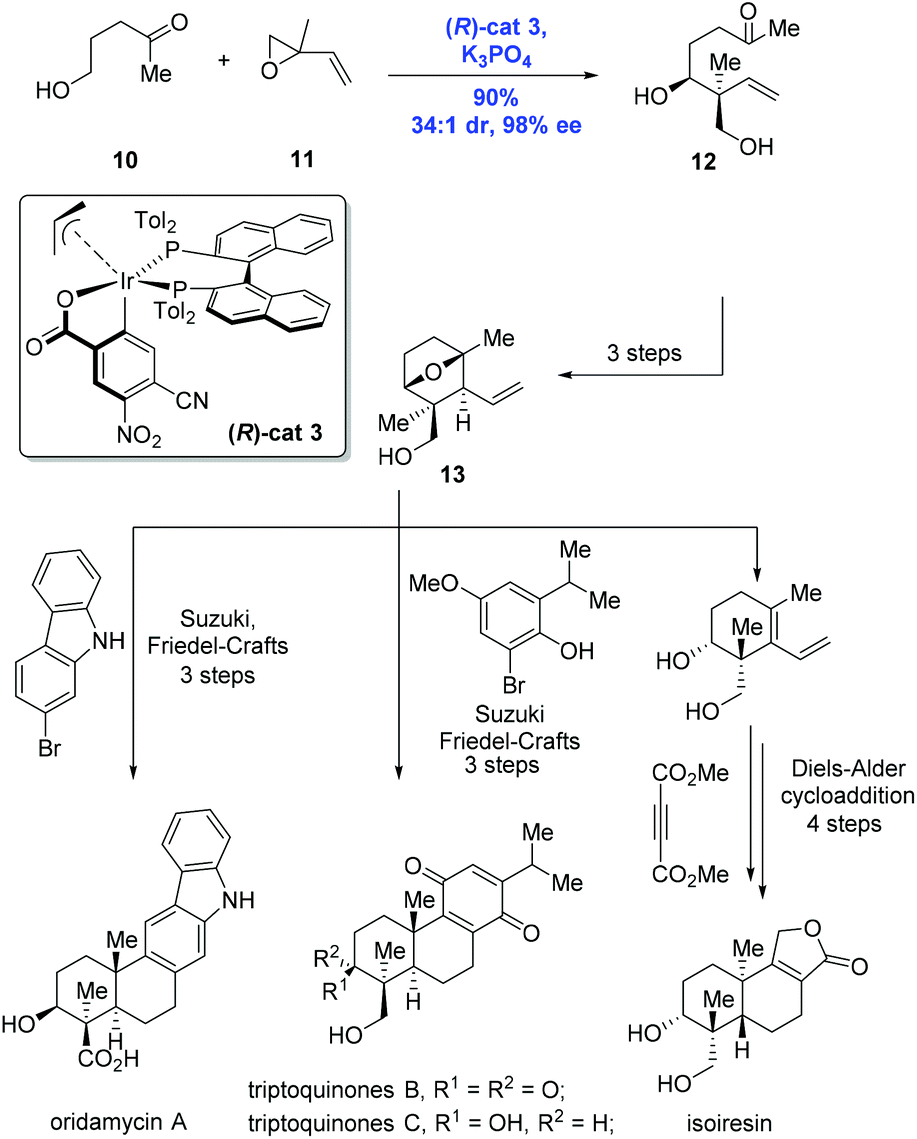 | ||
| Scheme 5 Syntheses of oridamycin A, triptoquinones B and C, and isoiresin. | ||
In addition to the representative polyketides discussed in Schemes 3–5, other polyketides completed by the Krische group,6b,17a like bryostatin 7,17b cyanolide A,17c 6-deoxyerythronolide B,17d swinholide A17e and cryptocaryol A,17f are collected in Fig. 1.
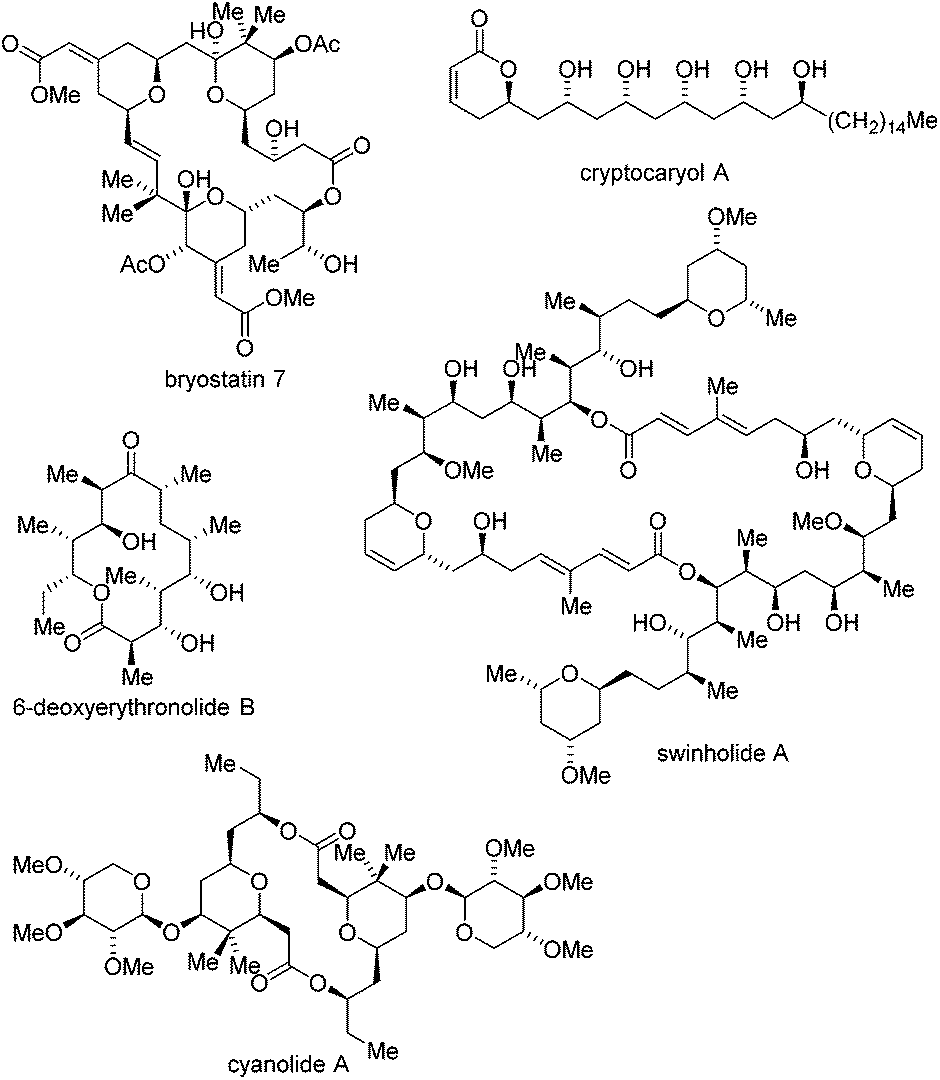 | ||
| Fig. 1 Other polyketides synthesized by the Krische group. | ||
By using Ir-catalyzed enantioselective carbonyl allylation developed by Krische, O'Doherty and co-workers achieved the asymmetric syntheses of (+)-goniothalamin, (+)-goniothalamin oxide and 7,8-bis-epi-goniothalamin oxide in 2009 (Scheme 6).18 Under Krische's catalysis system, trans-cinnamyl alcohol 14 reacted with allyl acetate to provide the homoallylic alcohol 15 in 68% yield with high enantioselectivity (90% ee). A few additional steps were performed to deliver the corresponding natural products, respectively.
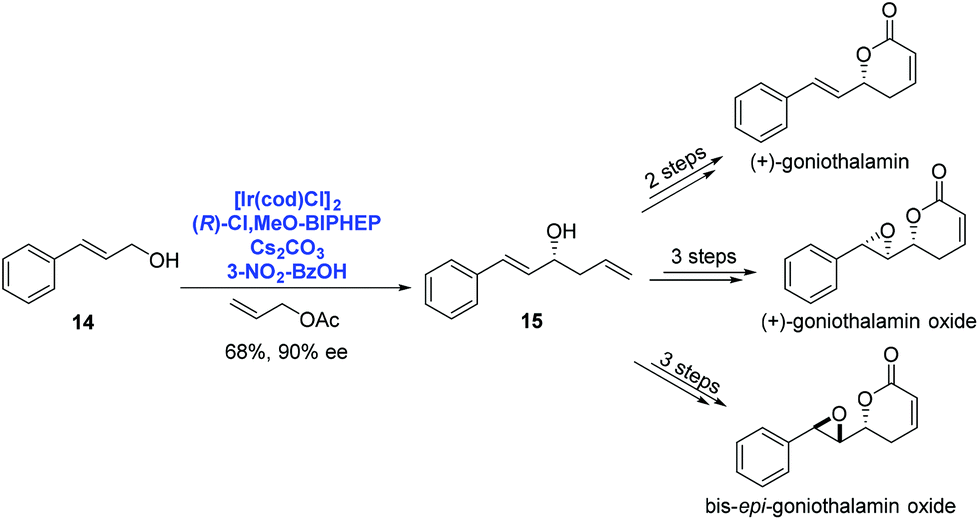 | ||
| Scheme 6 Syntheses of (+)-goniothalamin, (+)-goniothalamin oxide and 7,8-bis-epi-goniothalamin oxide. | ||
2.2 Ir-Catalyzed oxidation
Ir-Catalyzed oxidation reactions, through transfer hydrogenation, have found applications in the natural product synthesis as well. This sub-section will present two syntheses involving transfer hydrogenation itself as the key step. In 2008, Galano and co-workers reported an Ir-catalyzed transfer hydrogenation to achieve the total synthesis of 15-F2t-IsoP 1 (Scheme 7).19 In the process of synthesis, they found that oxidation of diol 16 with PDC or IBX only delivered 20% of the inseparable mixture of 17a and 17b in a 1:1 ratio and a separable lactols 18a and 18b in a 1:1 ratio in good yields. Remarkably, regioselective oxidation could be achieved upon the treatment of 16 with Cp*Ir[OCH2C(C6H5)2NH], affording the lactones 17 in 91% yield with a high ratio (98:2) favoring the desired lactone 17a. 15-F2t-IsoP 1 was then obtained in eight steps from 17a.
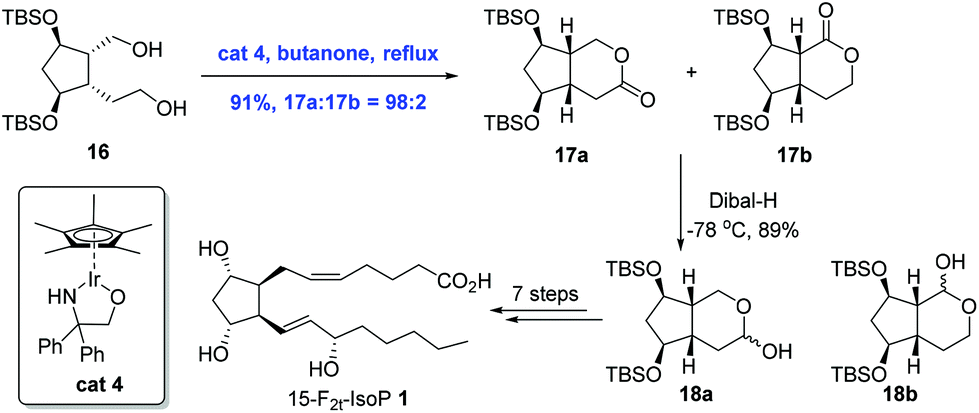 | ||
| Scheme 7 Synthesis of 15-F2t-IsoP 1. | ||
Catalytic asymmetric synthesis is an effective strategy for the synthesis of chiral compounds. Specifically, the efficient asymmetric synthesis of chiral hydroxy ketones is a key technology in the natural product synthesis. Because overreduction remains a problem in the asymmetric reduction of diketones, a common method for the preparation of chiral hydroxy ketones, oxidative desymmetrization can serve as an alternative.20 In 2009, Suzuki and co-workers20,21 reported an innovative strategy for the catalytic oxidative desymmetrization of meso-diols using a chiral iridium catalyst through transfer hydrogenation to accomplish the syntheses of ottelione and scyphostatin (Scheme 8). In practice, the key common intermediate 20 was prepared from the meso-diol 19via oxidative desymmetrization in the presence of chiral iridium catalyst (R,R)-cat5 and t-BuOK in moderate yield (60%) and with high ee (>99%).
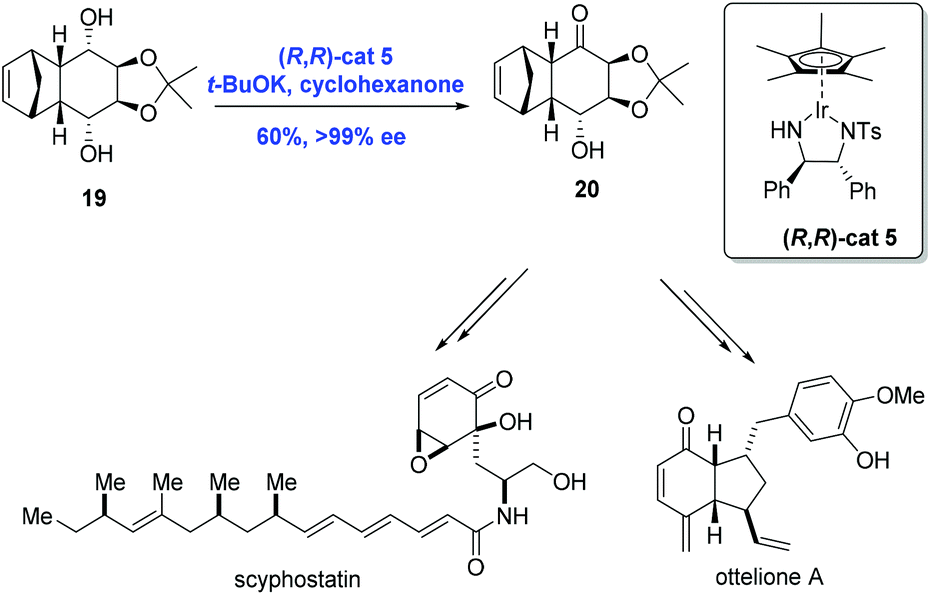 | ||
| Scheme 8 Syntheses of scyphostatin and ottelione A. | ||
3. Ir-Catalyzed hydrogenation reactions
The asymmetric hydrogenation of olefins, ketones, and imines is one of the most powerful methods in asymmetric catalysis. Particularly, asymmetric hydrogenation of olefins has achieved outstanding results in the preparation of chiral compounds.22 Ruthenium and rhodium are effective catalysts in the asymmetric hydrogenation of functionalized olefins, in which coordinating groups are required in the substrates to ensure high stereoselectivity.22 General asymmetric hydrogenation of unfunctionalized olefins remained an unmet challenge until the discovery of Ir-complexes.7,23 With respect to the mechanism of Ir-catalyzed hydrogenation of olefins, two possible pathways, namely the Ir(I)/Ir(III) cycle and the Ir(III)/Ir(V) cycle, were proposed as depicted in Scheme 9.7 Both cycles employ B1 as the key intermediate. In the Ir(I)/Ir(III) cycle, intermediate B1 gives the hydrogenation product after reductive elimination. Alternatively, in the Ir(III)/Ir(V) cycle, the oxidative addition of intermediate B1 with hydrogen occurs, delivering the key intermediate B6, which undergoes reductive elimination to afford the hydrogenation product.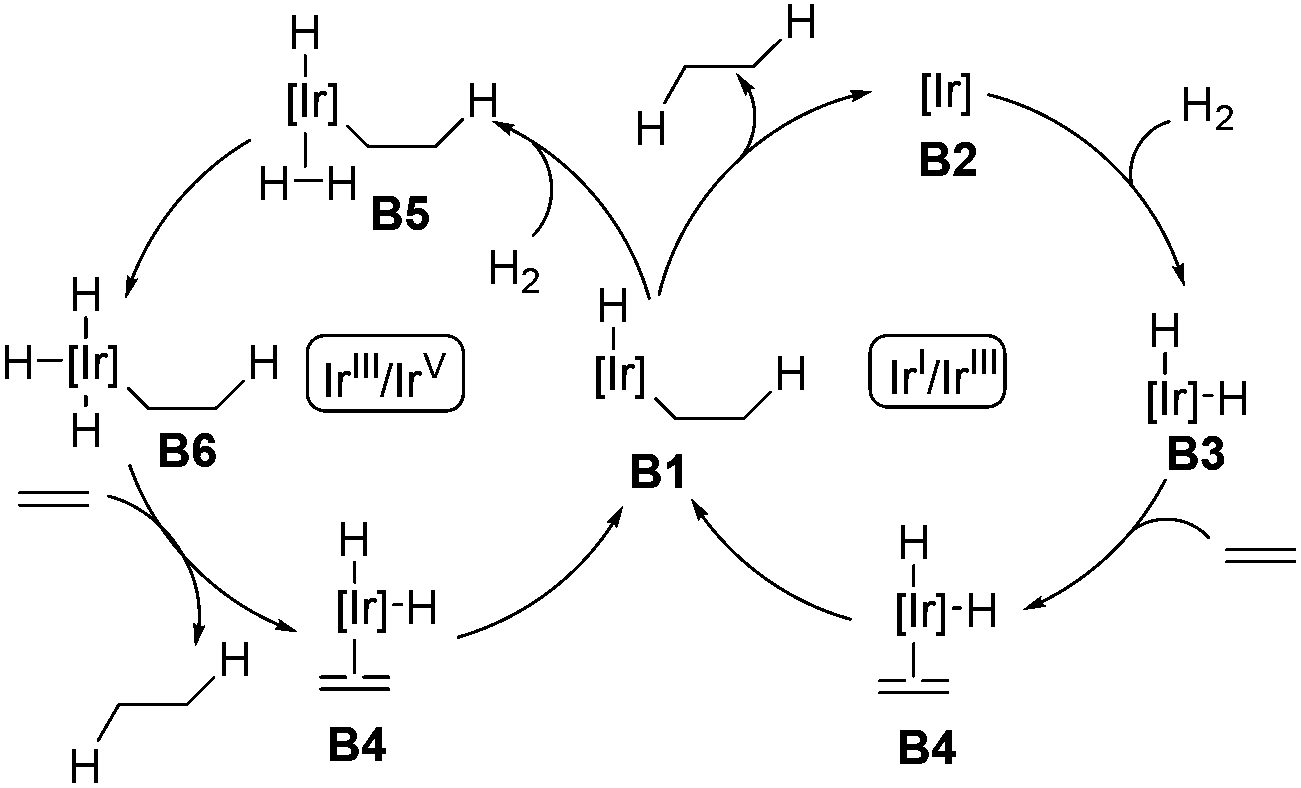 | ||
| Scheme 9 Two proposed mechanisms of the Ir-catalyzed hydrogenation of olefins. | ||
The first homogeneous hydrogenation achiral iridium catalyst which could reduce a series of unfunctionalized olefins, including multisubstituted olefins, was reported by Crabtree and co-workers.24 After that, Crabtree's catalyst and its chiral analogues were employed in the natural product synthesis. In 1996, Hart and co-workers introduced the selective hydrogenation reaction using Crabtree's catalyst system ([Ir(cod)py(PCy3)PF6]/CH2Cl2) to achieve the syntheses of (±)-axamide-1 and (±)-axisonitrile-1 (Scheme 10).25 Selective hydrogenation of the isopropenyl group of the diene 21 enabled by the iridium catalyst under hydrogen gave (±)-axamide-1 in 39% yield. Subsequent treatment of (±)-axamide-1 with p-toluenesulfonyl chloride delivered (±)-axisonitrile-1 in 88% yield.
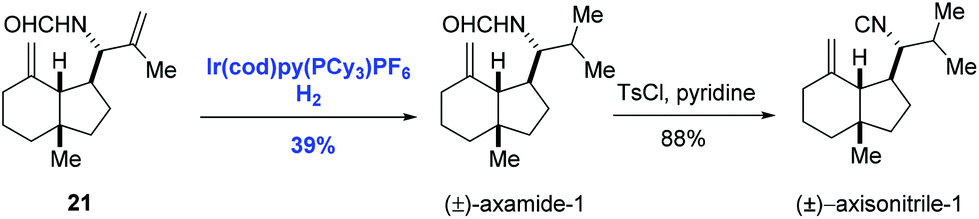 | ||
| Scheme 10 Syntheses of (±)-axamide-1 and (±)-axisonitrile-1. | ||
Besides disubstituted olefins, asymmetric hydrogenation of tri- and tetra-substituted olefins is also important in organic synthesis. Based on the perfect performance of Crabtree's catalyst, Burgess and co-workers developed new iridium complexes of the N-heterocyclic carbene oxazoline ligands to accomplish the synthesis of (−)-spongidepsin (Scheme 11).26 They found that hydrogenation with this Ir-complex was able to reduce di- and tri-substituted allylic alcohols with high stereoselectivity and low catalyst loading. In the event, allylic alcohol 22 was converted into 23 in 93% yield (dr = 34:1) enabled by iridium catalyst (R)-cat6. In parallel, hydrogenation of 24 with iridium catalyst (S)-cat6 gave 25 in 97% yield and with high diastereoselectivity (dr = 61:1) after removal of the protection group. Finally, the natural product (−)-spongidepsin was furnished through the union of fragment C and fragment D which were prepared from 23 and 25, respectively.
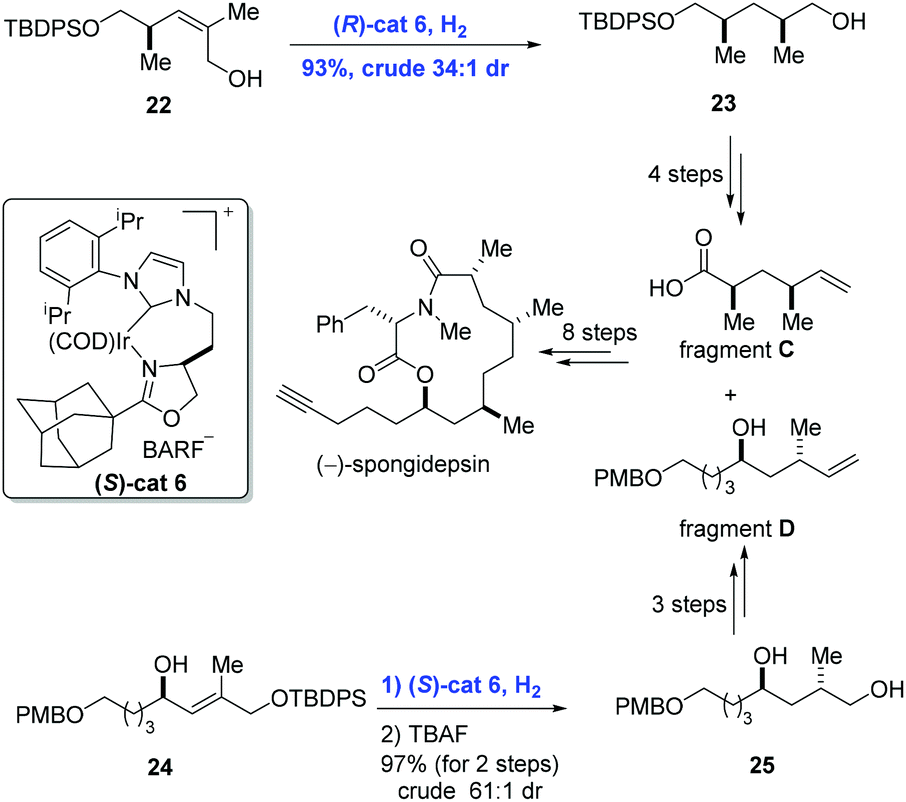 | ||
| Scheme 11 Synthesis of (−)-spongidepsin. | ||
In 2009, the first asymmetric syntheses of (+)-mutisianthol and its enantiomer were achieved by Silva and co-workers, in which the pivotal asymmetric hydrogenation of nonfunctional olefins was mediated by the chiral Ir-complex (Scheme 12).27 While exposure of olefin 26 to hydrogen in the presence of Ir-complex cat7 gave 27, the hydrogenation of 26 under similar conditions with Ir-complex cat8 delivered the enantiomer of 27. Subsequent synthetic transformations from 27 and its enantiomer featuring a ring-contraction reaction led to the natural products (+)-mutisianthol and (−)-mutisianthol, respectively.
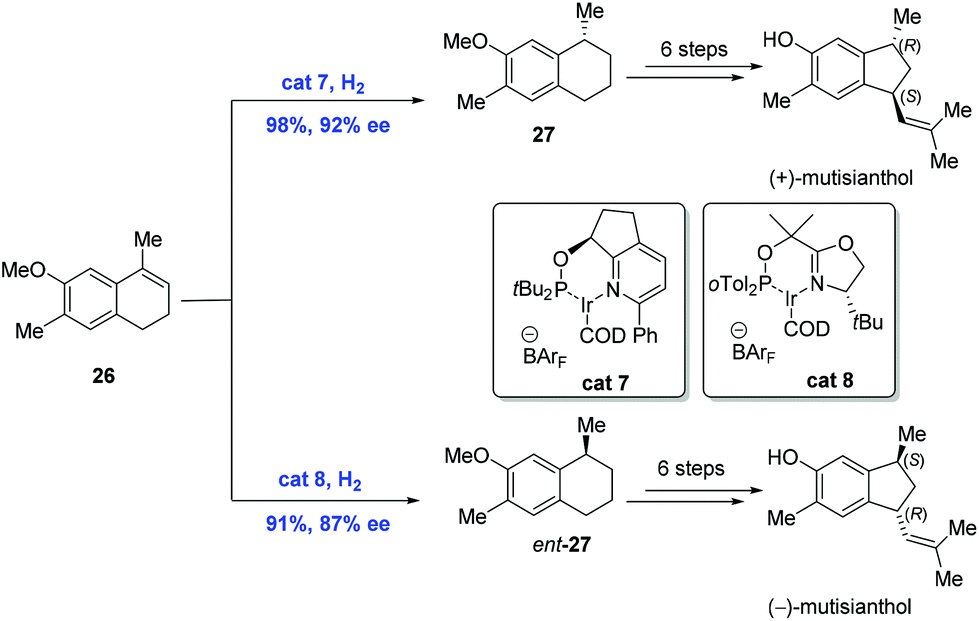 | ||
| Scheme 12 Syntheses of (+)-mutisianthol and its enantiomer. | ||
4-Alkyl-4-aryl butanoic acids are important synthons for the synthesis of a broad range of natural products. The preparation of 4-alkyl-4-aryl butanoic acids or their derivatives has been a subject of research for a long time. Many interesting methods have been invented to access the synthons, allowing the syntheses of diverse natural products,28,29 like pseudopterosins,29a kendomycin,29b (−)-sesquichamaenol29c and (−)-heritol,29d just to name a few (Scheme 13).
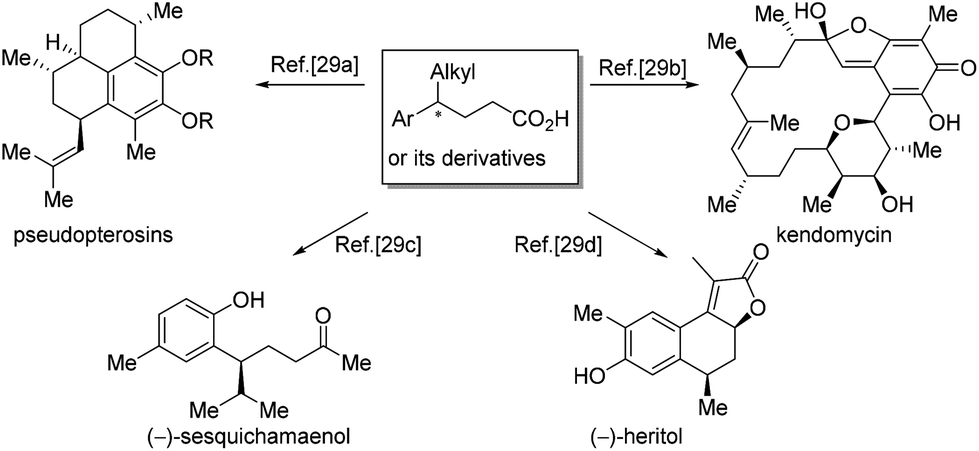 | ||
| Scheme 13 Representative synthesis from chiral 4-alkyl-4-aryl butanoic acids or their derivatives. | ||
In 2012, Zhou and co-workers described a highly enantioselective hydrogenation of β,γ-unsaturated acids using an iridium catalyst with the chiral spiro phosphine-oxazoline ligand (Scheme 14).28 A series of 4-alkyl-4-aryl butanoic acids were synthesized under these conditions. For example, the hydrogenation of 4-alkyl-4-aryl-3-butenoic acids with a low loading of Ir-complex (Sa,S)-cat9 gave rise to chiral acids 28 and 29 in high yield and with good enantioselectivity, respectively, each of which was advanced to the corresponding natural products (R)-curcumene, (R)-xanthorrhizol and (R)-aristelegone-A, featuring chain elongation and Friedel–Crafts acylation.
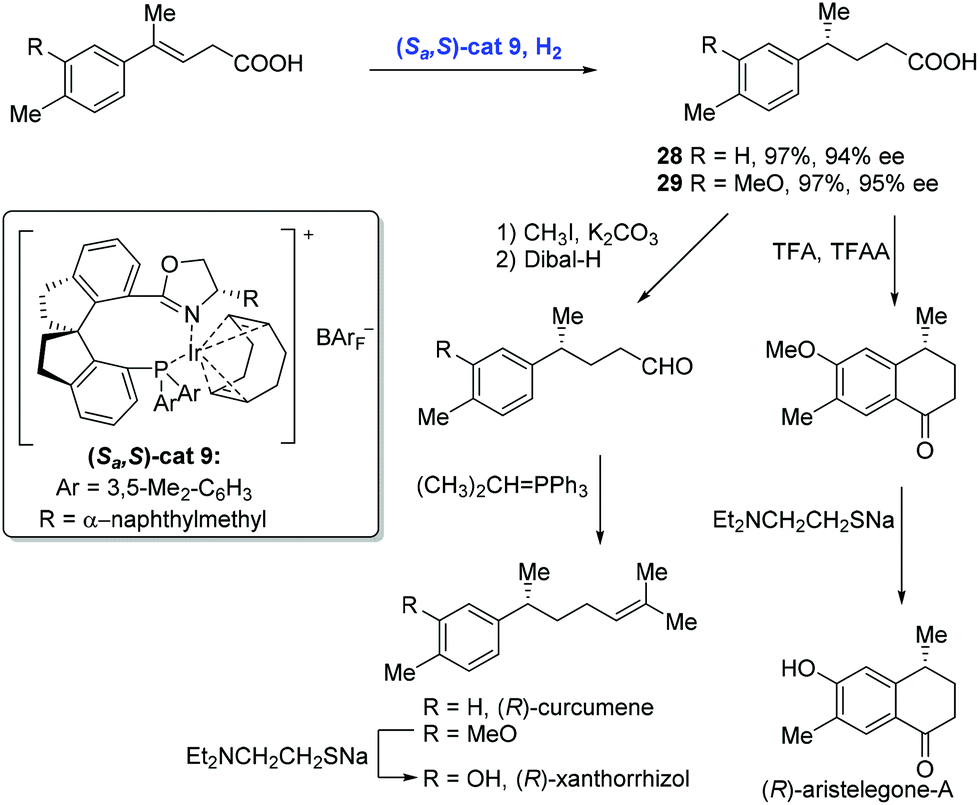 | ||
| Scheme 14 Syntheses of (R)-curcumene, (R)-xanthorrhizol and (R)-aristelegone-A. | ||
In the subsequent research, Zhou and co-workers extended the Ir-catalyzed enantioselective hydrogenation to unsaturated heterocyclic carboxylic acids, such as N-heterocyclic acids and O-heterocyclic acids, in an analogous catalytic system (Scheme 15).30 (R)-Nipecotic acid, a potent γ-aminobutyric acid reuptake inhibitor previously produced over ten steps, was obtained directly from unsaturated carboxylic acid 30 under the present neutral conditions in 99% yield with 96% ee by the Zhou group. At the same time, the hydrogenation of unsaturated N-heterocyclic acid 31 under similar conditions prior to treatment with hydrochloric acid gave rise to (R)-tiagabine. It is remarkable that the hydrogenation showed excellent regioselectivity since the double bond conjugated to the thiophene groups remained intact.
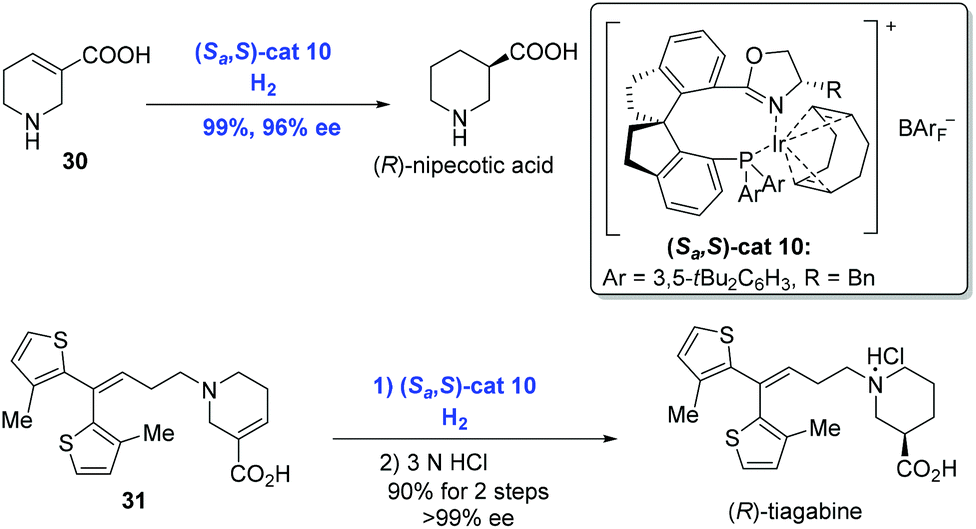 | ||
| Scheme 15 Syntheses of (R)-nipecotic acid and (R)-tiagabine. | ||
In addition to olefins, Ir-catalyzed asymmetric hydrogenation is capable of reducing imines. In general, transition-metal-catalyzed asymmetric hydrogenation of cyclic imines has become an efficient strategy to construct N-heterocycles,31 although the hydrogenation of pyridyl-containing cyclic imines still remains a challenge due to the deactivation of transition-metal catalysts by the pyridine moiety. In 2014, Zhou and co-workers developed a strategy, by placing a substituent at the position adjacent to the pyridine N atom, to achieve the enantioselective hydrogenation of pyridyl-containing cyclic imines using an Ir-complex (Scheme 16).32 Under the conditions of (Sa,S)-cat11, iodine and hydrogen, cyclic imine 32 underwent hydrogenation to afford the product 33 or 34 in high yield with excellent enantioselectivity. Subsequent transformations delivered four pyrrolidine and piperidine alkaloids, respectively.
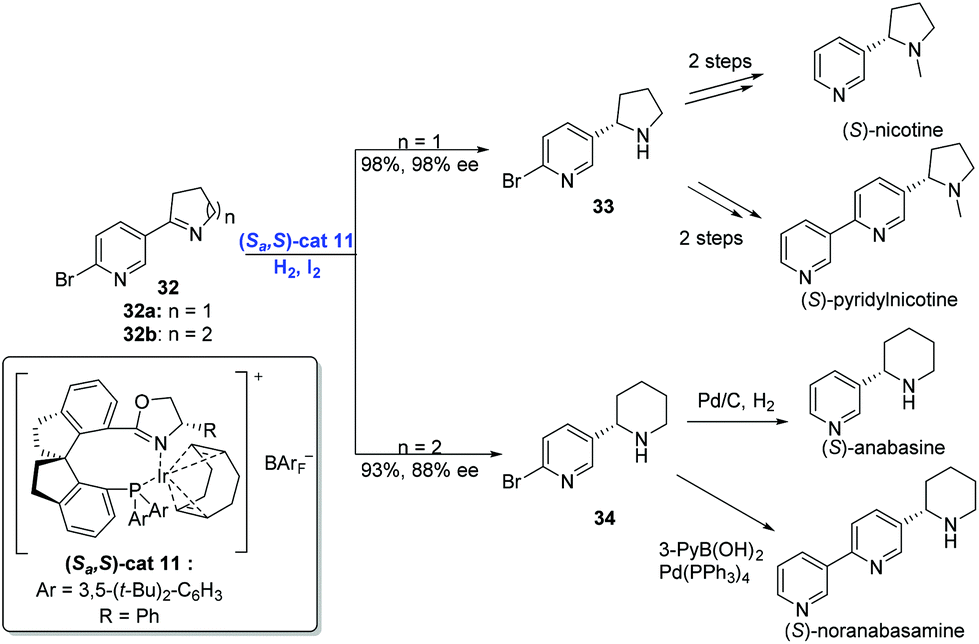 | ||
| Scheme 16 Syntheses of chiral pyrrolidine and piperidine alkaloids. | ||
With the development of Ir-catalyzed enantioselective hydrogenation of olefins and imines, Zhou and co-workers further expanded the methodology to the asymmetric reduction of the carbonyl group to achieve the syntheses of (−)-mesembrine, hamigerans and crinine-type alkaloids (Scheme 17).
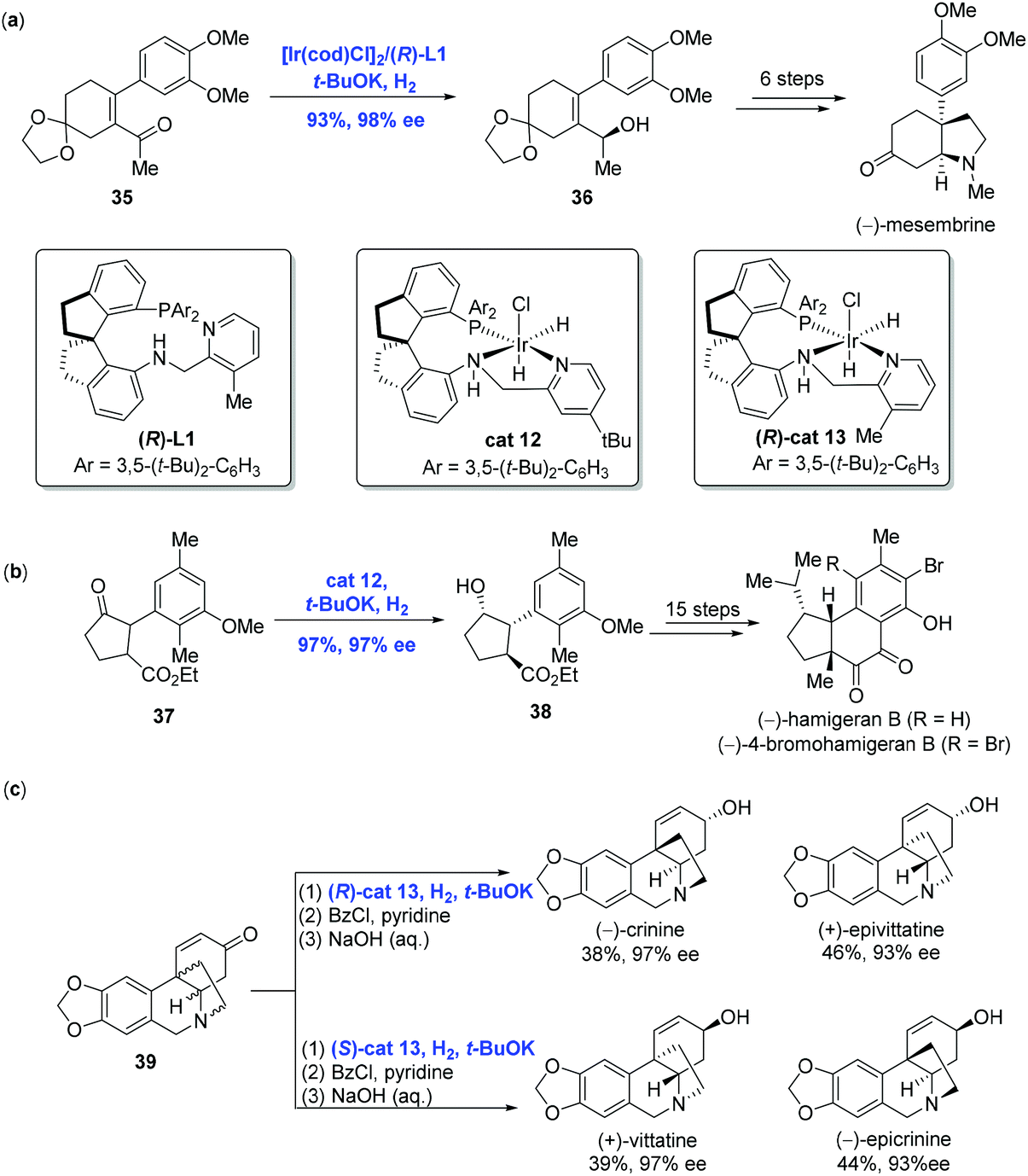 | ||
| Scheme 17 Syntheses of (−)-mesembrine, (−)-hamigeran B and crinine-type alkaloids. | ||
(−)-Mesembrine bearing a chiral arylated quaternary carbon center is a potent serotonin reuptake inhibitor. In Zhou and co-workers’ synthetic effort toward (−)-mesembrine, the asymmetric hydrogenation of α-substituted α,β-unsaturated acyclic ketone 35 in the presence of [Ir(cod)Cl]2 and spiro pyridine-aminophosphine ligand (R)-L1 led to the corresponding chiral allylic alcohol 36 in 93% yield with 98% ee (Scheme 17a).33 Additional synthetic transformations, including a Johnson–Claisen rearrangement used to reach the quaternary carbon center, were conducted to complete the synthesis of (−)-mesembrine.
Hamigerans, a family of natural products showing strong in vitro activity against herpes and polio viruses, have received great attention from the synthetic community to pursue their synthesis. Recently, Zhou and co-workers developed a new strategy for the asymmetric hydrogenation of ketones to achieve (−)-hamigeran B and (−)-4-bromohamigeran B (Scheme 17b).34 To accomplish this goal, trans-ketone 37 was hydrogenated to give cyclopentanol 38 in 97% yield with 97% ee and significantly high diastereomeric ratio (trans/cis > 99/1) via dynamic kinetic resolution enabled by Ir-complex cat12. In comparison, the same reaction using the Ru-complex gave 38 in only moderate yield (30–46%) and enantioselectivity (76–82% ee). In addition, the hydrogenation could be conducted on a gram scale with comparable yield and selectivity, indicating the practicality of the current Ir-catalyzed hydrogenation. Starting from 38, a sequence of fifteen steps was performed to complete the synthesis of (−)-hamigeran B.
Crinine-type alkaloids belong to the Amaryllidaceae alkaloid family featuring an antipodal chiral 5,10b-ethanophenanthridine core skeleton. Taking advantage of the Ir-catalyzed asymmetric hydrogenation of racemic cycloenones developed by themselves in 2017, Zhou and co-workers synthesized twenty-four crinine-type alkaloids and eight analogues successfully (Scheme 17c).35 For example, both (−)-crinine and (+)-epivittatine were produced directly from racemic cycloenone 39 using chiral spiro iridium catalyst (R)-cat13. On the other hand, (S)-cat13 resulted in (+)-vittatine and (−)-epicrinine. Of note is that the stereochemistry of the hydrogenation was dictated by the configuration of the catalyst.
4. Ir-Catalyzed allylic substitution reactions
Transition-metal-catalyzed asymmetric allylic substitution reaction is considered as an unique method for the construction of C–C and C–X bonds.36,37 The Ir-catalyzed asymmetric allylic substitution reaction has the ability to form branched allylic stereogenic centers, a character that distinguishes Ir catalysts from the commonly used Pd catalysts. As shown in Scheme 18, Ir-catalyzed asymmetric allylic substitution proceeds with the binding of iridium to the olefin at first, the resulting intermediate C1 then ionizes to give complex C2, which is attacked by the nucleophile to provide the product and regenerate iridium after the disassociation of the metal from intermediate C3.8,36b,e This section will be grouped into two parts according to the type of bond formed, including C–C and C–N bonds.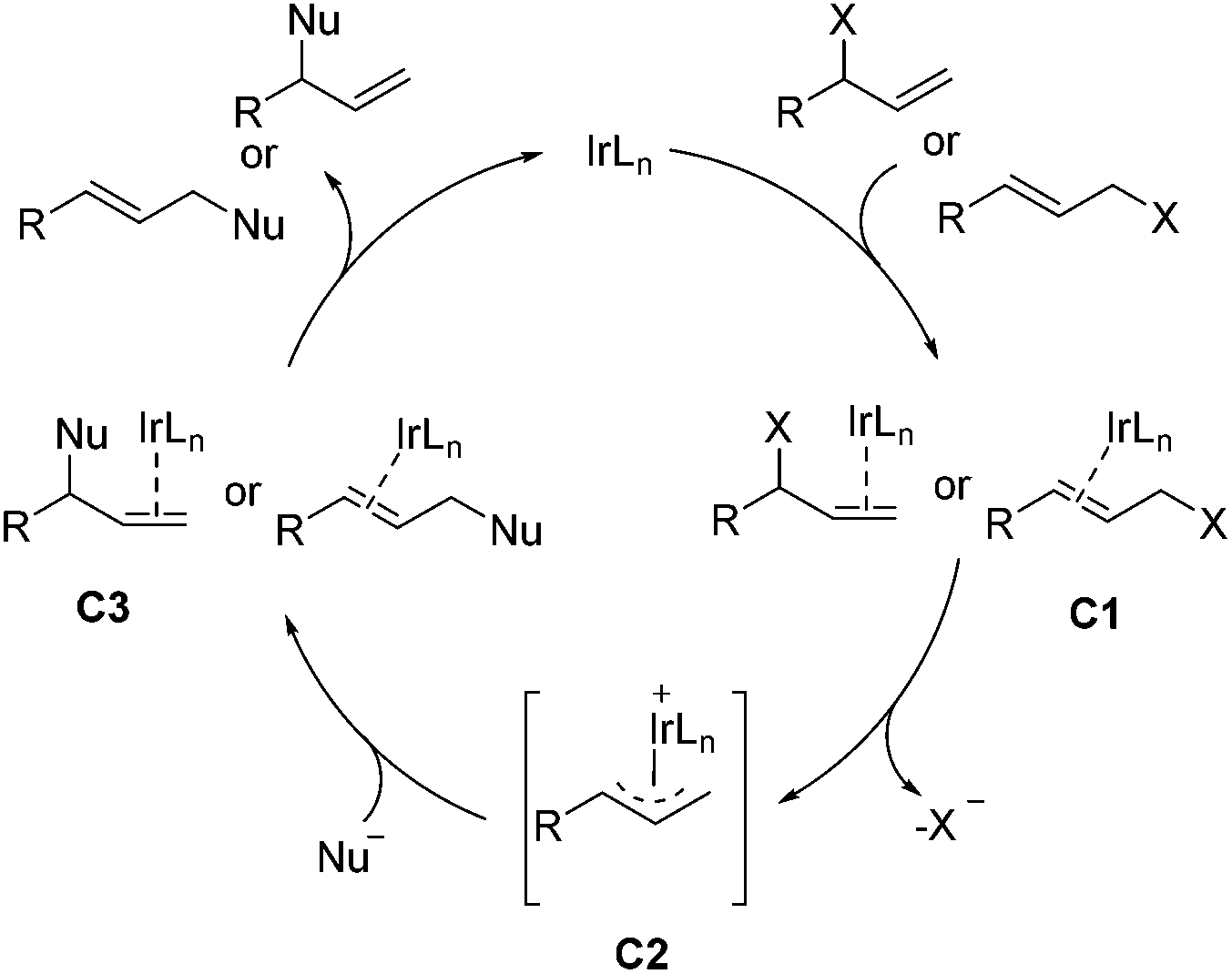 | ||
| Scheme 18 Mechanism of Ir-catalyzed allylic substitution reactions. | ||
4.1 Ir-Catalyzed allylic substitution reactions for formation of C–C bond
Catalytic enantioselective cyclizations of polyenes could be conventionally achieved in the presence of Brønsted/Lewis acid catalysts or organocatalysts to construct polycyclic frameworks present in natural products.38 Distinct from the traditional strategy, in 2012, Carreira and co-workers39 first reported an innovative enantioselective polyene cyclization cascade reaction using branched racemic allyl alcohols as initiators enabled by the transition metal iridium. As depicted in Scheme 19a, under the conditions of the catalyst [Ir(cod)Cl]2, ligand (R)-L2, promoter Zn(OTf)2 and solvent DCE, polyene 40 was converted into polycyclic 41via the transition state 42. The authors found that a broad range of carbo- and heterocyclic ring systems, including alkoxy-substituted aromatics, pyrroles, indoles, and furan heterocycles all could undergo a cyclization reaction in high yield and with excellent enantioselectivity. Meanwhile, they found that the polyene 43 could be elaborated to tetracyclic product 44 (43% yield, 99.5% ee), together with byproducts 45 (30% yield) presumably as a mixture of mono- and bis-cyclization products, which converged into tetracyclic product 44 upon treating with trifluoroacetic acid (Scheme 19b).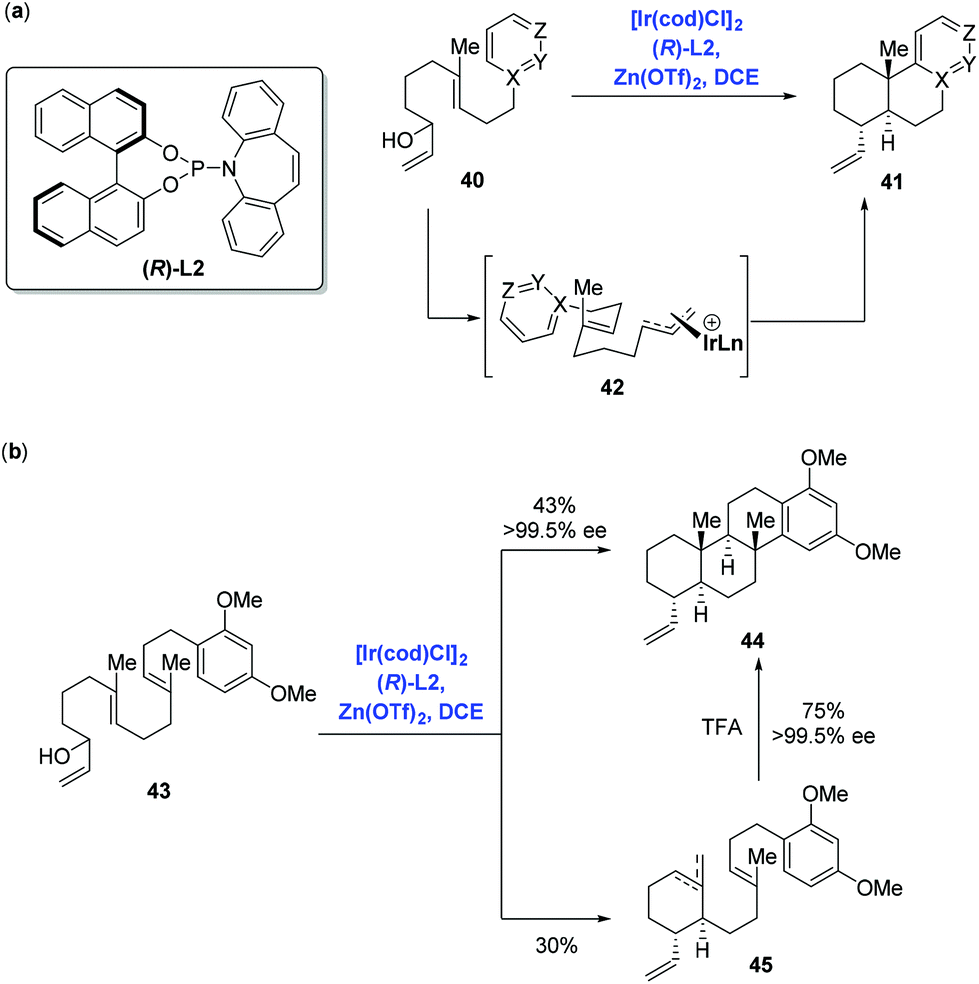 | ||
| Scheme 19 Ir-Catalyzed enantioselective polyene cyclization. | ||
Having this methodology established, Carreira and co-workers sought to expand it in order to complete the synthesis of asperolide C (Scheme 20a),40 a tetranorlabdane diterpenoid isolated from Aspergillus wentii EN-48.41 Using the standard conditions aforementioned, polyene 46 proceeded without complication through the cyclization cascade reaction to give the desired decalin 47 in 73% yield with excellent stereoselectivity (dr = 9:1, er = 98:2), in which allyl silane instead of arenes functioned as the terminating group. Sequential synthetic transformations allowed the authors to complete the first total synthesis of asperolide C. Furthermore, Carreira and co-workers applied the catalysis system into the asymmetric α-allylation of aldehydes (Scheme 20b).42 In the event, under the conditions of the catalyst [Ir(cod)Cl]2, promoter Zn(OTf)2, ligand (R)-L2 or (S)-L2, Jørgensen amine (R)-L3 or (S)-L3, α-allylation of aldehyde 49 with allylic alcohol 48 gave rise to all possible stereoisomers of 50 in a stereodivergent fashion. Subsequent transformations allowed them to access all stereoismers of Δ9-tetrahydrocannabinol.
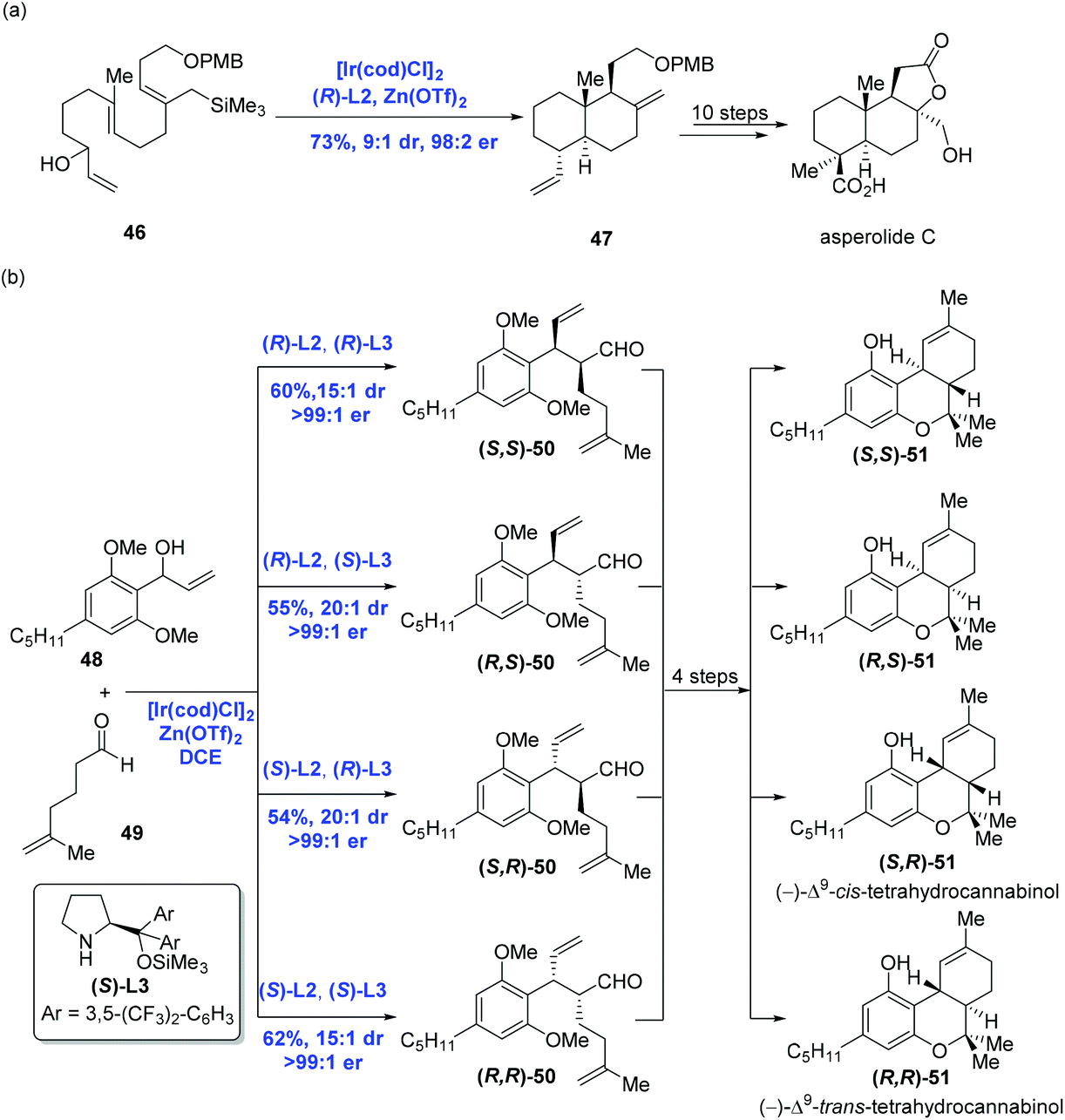 | ||
| Scheme 20 Syntheses of asperolide C and all stereoismers of Δ9-tetrahydrocannabinol. | ||
Inspired by Carreira's Ir-catalyzed enantioselective polyene cyclization, Li and co-workers accomplished the first total syntheses of taiwaniadducts B, C, and D, in which both important diterpenoid intermediates (54 and 57) were prepared using the cascade cyclization (Scheme 21).43 To this end, treating allylic alcohol 52 with Carreira's catalyst system gave rise to tricycle 53 in 69% yield and with over 99% ee. A following 11-step sequence led to 54 from 53. In parallel, subjecting allylic alcohol 55 to the standard conditions followed by BF3·OEt2-mediated cyclization led to 56, where the pendant primary hydroxyl group of 55 acted as the terminating nucleophile. An 11-step sequence gave 57 from 56. With both intermediates 54 and 57 being formed, the authors then intended to effect the planned intermolecular Diels–Alder reaction. After screening numerous conditions, the authors found that Er(fod)3 was able to enable cycloaddition to provide taiwaniadduct B core in 52% yield, along with taiwaniadduct C core in 21% yield. Both adducts were converted to the corresponding natural products in steps. To achieve taiwaniadduct D, additional three steps featuring a carbonyl–ene cyclization were performed from taiwaniadduct B core.
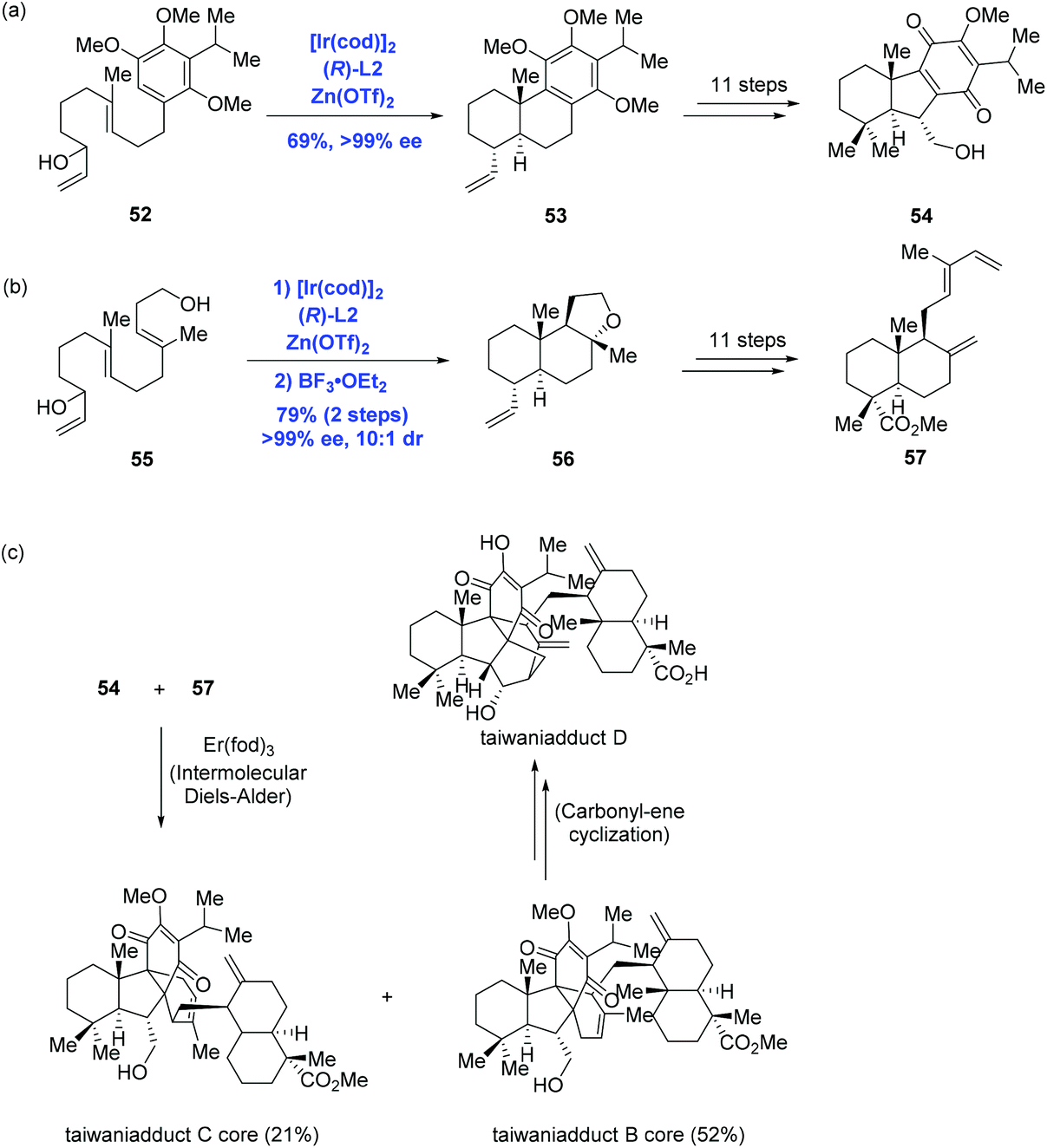 | ||
| Scheme 21 Syntheses of taiwaniadducts B, C and D. | ||
In their subsequent research, Li and co-workers extended the Ir-catalyzed polyene cyclization to indolosesquiterpenoids and accomplished the first total synthesis of mycoleptodiscin A (Scheme 22).44 In the event, the authors conducted the Ir-catalyzed asymmetric polyene cyclization under standard conditions to forge the tetracyclic core structure mycoleptodiscin A, giving 59 in 21% yield, along with a mixture of presumed cascade interruption products 61 and 62. The mixture could be funnelled to tetracyclic 59 in 87% yield upon treating with BF3·OEt2. Distinct from previous reports, where Ir-catalyzed polyene cyclizations were used more often to assemble bi- and tricyclic ring systems, the case here served as an elegant example for the preparation of tetracyclic architectures. To complete the synthesis, sequential synthetic transformations mainly used for assembling the indole system were performed.
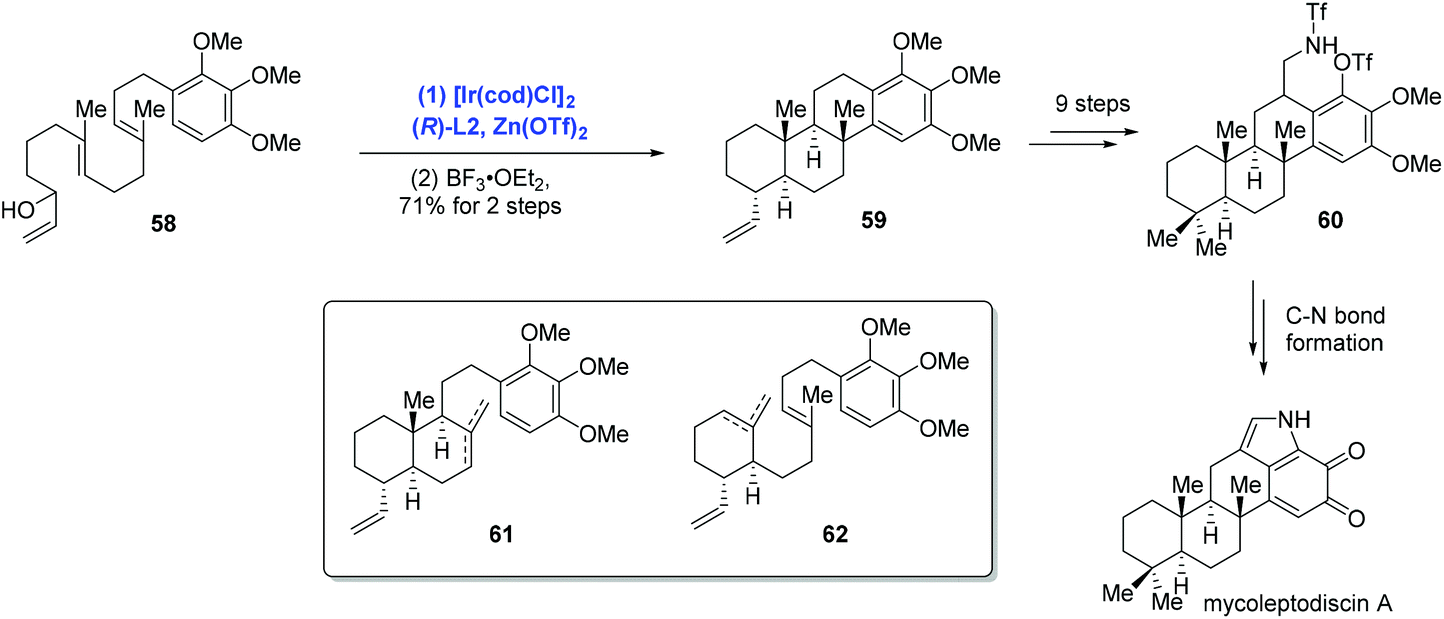 | ||
| Scheme 22 Synthesis of mycoleptodiscin A. | ||
Recently, asymmetric dearomatization reactions of indoles and pyridines have made great progress.45 In 2014, inspired by Carreira's catalysis system, You and co-workers achieved an Ir-catalyzed intermolecular asymmetric dearomatization of substituted indoles with allylic alcohols (Scheme 23a).46 In the presence of [Ir(cod)Cl]2, (S)-L2 and Fe(OTf)2 as the Lewis acid, the asymmetric dearomatization of indole 63 with allylic alcohol 64 proceeded to construct the pyrroloindoline unit with an all-carbon quaternary stereogenic center. It is noteworthy that the current allylic substitution gave a linear product instead of a branched product, presumably due to the steric bulkiness of the nucleophile. To complete the synthesis, two transformations were employed. In the same year, Carreira and co-workers reported an Ir-catalyzed selective reverse prenylation of 3-substituted-1H-indoles to achieve the synthesis of (−)-brevicompanine (Scheme 23b).47 In the event, a reaction of tryptophan amino ester 66 and (Boc)-carbonate 67 in the presence of [Ir(cod)Cl]2, KHMDS and 9-BBN-n-C6H13 resulted in product (−)-exo-68 in 58% yield with excellent site selectivity (b/l > 20:1) and diastereoselectivity (exo/endo > 20:1). Carreira's reaction is complementary to that of You's since high branched to linear selectivity was observed in Carreira's case. Starting from (−)-exo-68, (−)-brevicompanine was then concluded with two more steps.
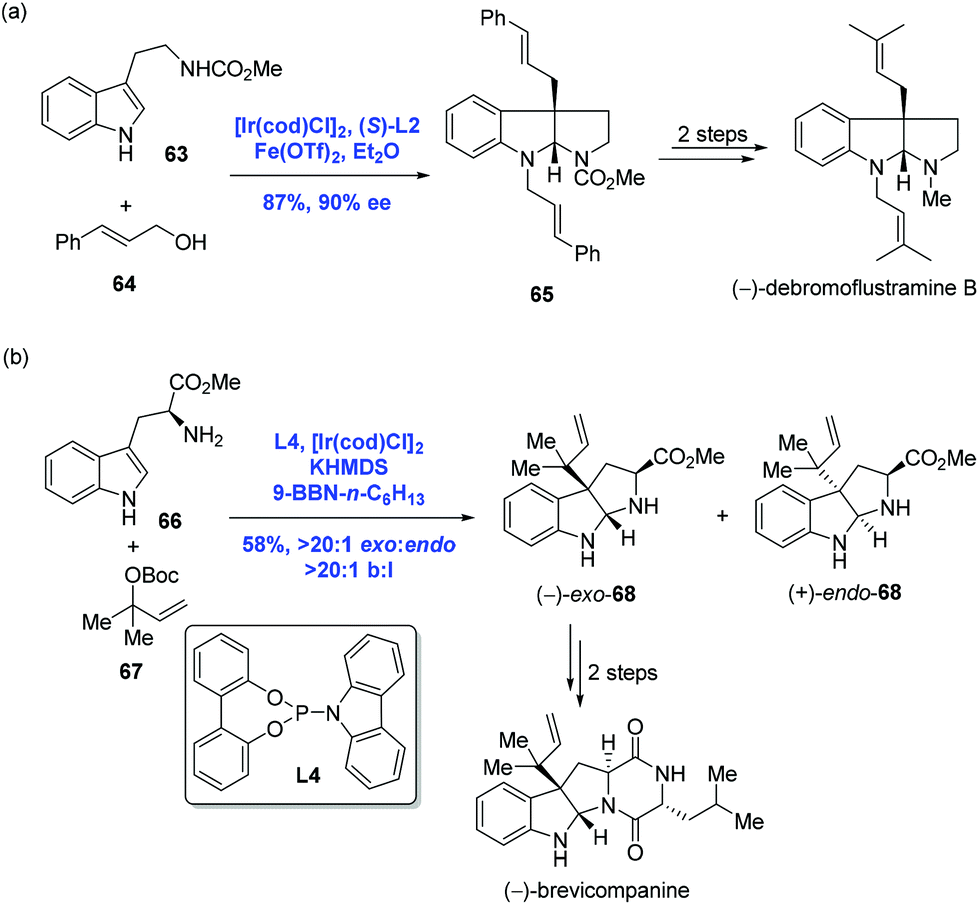 | ||
| Scheme 23 Syntheses of (−)-debromoflustramine B and (−)-brevicompanine. | ||
Inspired by the pioneering studies from the groups of Trost48a and Walsh,48b in which palladium was employed to effect an asymmetric allylic alkylation with 2-methylpyridine derivatives, You and co-workers developed an Ir-catalyzed asymmetric allylic alkylation of 2-methylpyridines to accomplish the synthesis of (−)-lycopladine A (Scheme 24).48c Allylic alkylation of 2-methylpyridine 69 with carbonate 70 gave the desired product 71 in 67% yield and 90% ee. A sequence of eleven steps was then performed to complete the synthesis of (−)-lycopladine A.
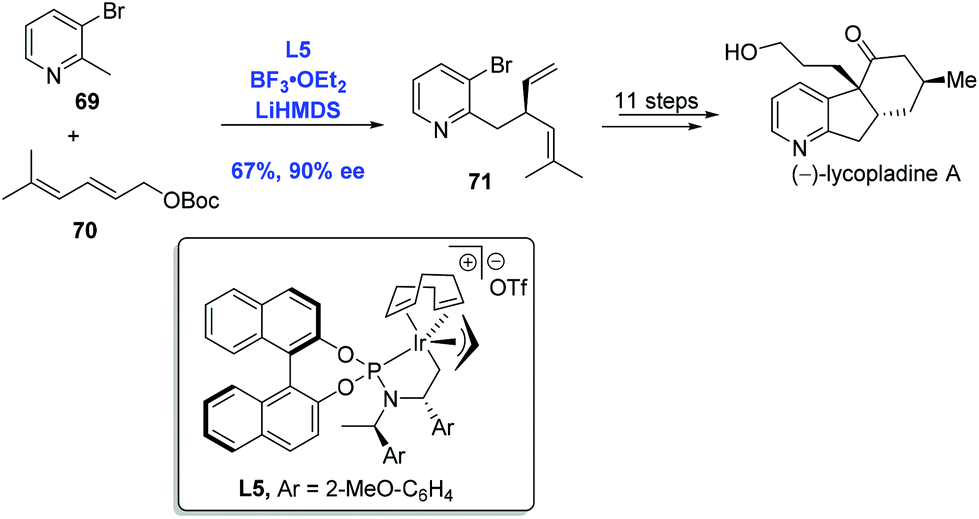 | ||
| Scheme 24 Synthesis of (−)-lycopladine A. | ||
The akuammiline family of alkaloids comprising a class of monoterpene indole alkaloids displays a broad range of biological activities spanning from anticancer to antimalarial. Due to the diverse bioactivities and unique intricate architectures, akuammilines have been targeted by many laboratories through distinctly different synthetic strategies.49 Taking advantage of the pioneering studies from two research groups, Ir-catalyzed enantioselective polyene cyclization by Carreira39,40,43,44 and Ir-catalyzed intramolecular allylic dearomatization of indoles by You,46,50 Yang and co-workers have developed an Ir-catalyzed intramolecular allylic alkylation through the dearomatization of the functionalized indole to achieve the synthesis of the monoterpene indole alkaloid (−)-aspidophylline A (Scheme 25).51 Enantioselective dearomatization of 2,3-disubstituted indole 72 under the Carreira catalysis system [Ir(cod)Cl]2 and (R)-L2 gave the tetracyclic indole system 73 and 74 in a 5:1 ratio with excellent ee values for both isomers. Remarkably, the tetracyclic core of the akuammilines bearing two contiguous quaternary stereogenic centers was assembled in a cascade fashion. Starting from isomer 73, (−)-aspidophylline A was prepared in ten steps featuring C3–H azidation and Ni-catalyzed intramolecular cyclization.
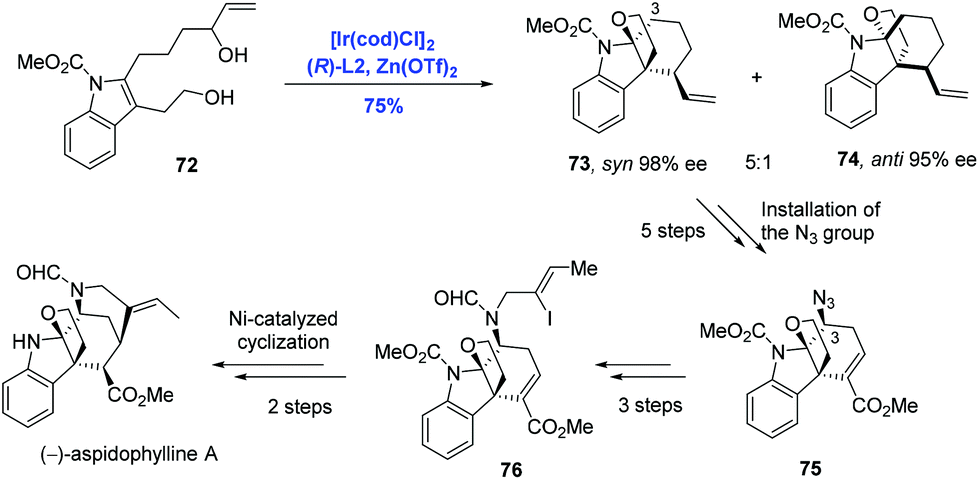 | ||
| Scheme 25 Synthesis of (−)-aspidophylline A. | ||
The further application of Ir-catalyzed enantioselective allylic alkylation by Yang in the indole system was illustrated in the total synthesis of (−)-alstoscholarisine A, the most active member of the indole alkaloid family that is responsible for promoting adult NSC proliferation at low concentrations (Scheme 26).52 In their synthetic approach, the tricycle 78 was synthesized via an intramolecular allylic alkylation of the functionalized indole 77 under the conditions of [Ir(cod)Cl]2, (R)-L2 and Sc(OTf)3 in 75% yield and 99% ee. Compared to the authors’ previous methodology as shown in Scheme 25, where C3 of the indole system served as the nucleophile, C2 of the indole system was envisioned as the nucleophile in the current case, demonstrating that diverse complex chiral polycyclic indole systems could be readily accessed just by judicious decoration of indole precursors. Moreover, it is worth noting that the newly formed stereogenic center (C16) in the key step was used to efficiently dictate the stereochemical outcome of the subsequent 1,4-addition/Aldol tandem reactions. (−)-Alstoscholarisine A was then concluded from 80 in five more steps.
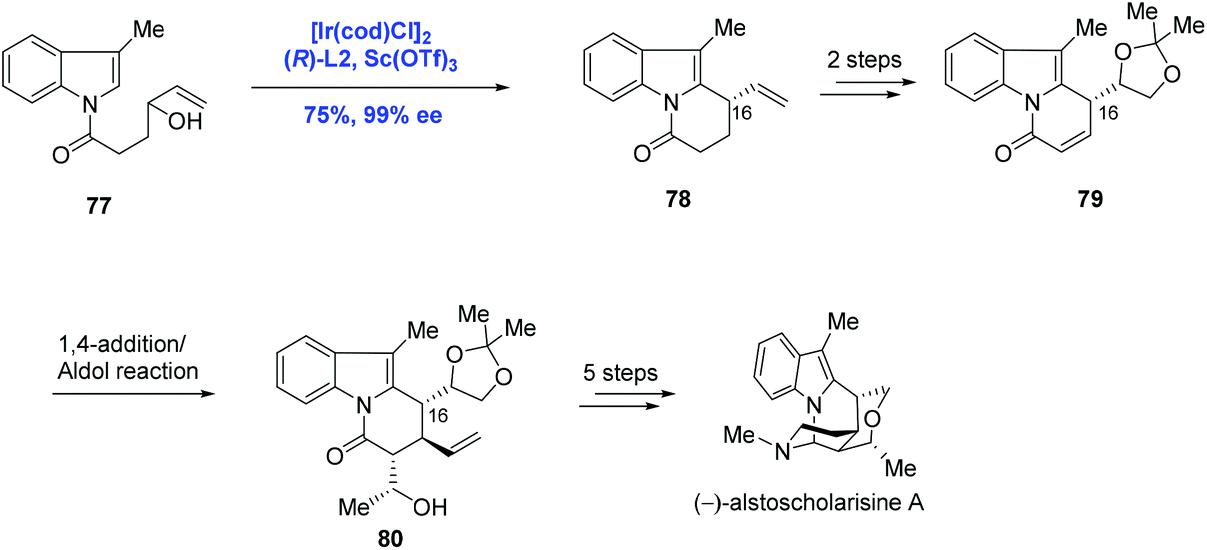 | ||
| Scheme 26 Synthesis of (−)-alstoscholarisine A. | ||
In their subsequent research, Yang and co-workers strategically used an Ir-catalyzed asymmetric intermolecular cascade cyclization to construct the CDEF tetracyclic core of communesins in one step (Scheme 27).53 Ir-Catalyzed tandem allylation/cyclization between indole 81 and t-butyl carbonate 82 proceeded to provide the desired product 83 (83:84, >10:1) in 56% yield and 93% ee. A sequence of sixteen steps was then operated to give rise to the natural product (−)-communesin F.
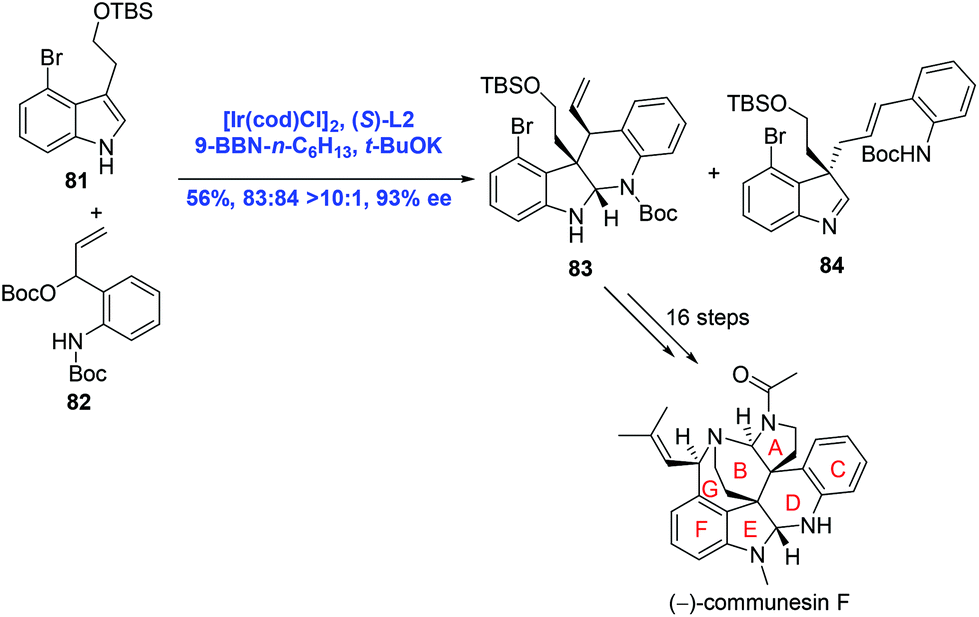 | ||
| Scheme 27 Synthesis of (−)-communesin F. | ||
4.2 Ir-Catalyzed allylic substitution reactions for formation of C–N bond
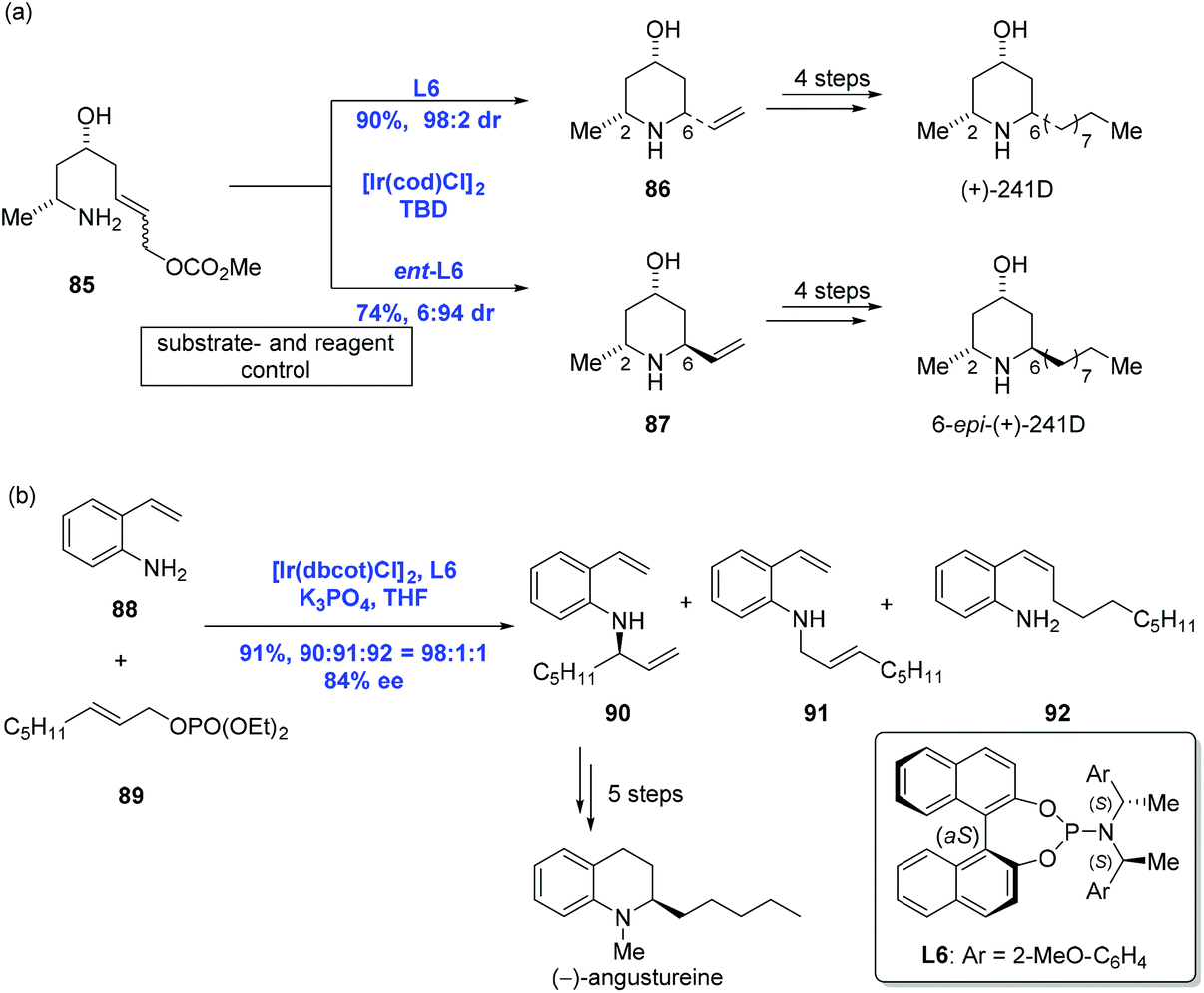
Ir-Catalyzed asymmetric allylic amination is a convenient and reliable method for the construction of nitrogen heterocycles, which has been explored in-depth.54,55 In 2009, Helmchen and co-workers communicated a stereoselective intramolecular allylic amination to accomplish the syntheses of (+)-241D and its C6 epimer. While the diastereoselective synthesis of piperidine via substrate control could only access one of a pair of diastereomers, the authors pointed out that Ir-catalyzed allylic amination was able to selectively achieve either diastereomeric cyclization products by reagent control (Scheme 28a).56,57 For example, to complete the syntheses of (+)-241D and its C6 epimer, amine 85 (E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :Z = 9:1) underwent an Ir-catalyzed reaction under [Ir(cod)Cl]2 and ligand L6 to give 2,6-cis-piperidine 86 in high yield with perfect diastereoselectivity (dr = 98:2). Complementarily, 2,6-trans-piperidine 87 was obtained as the major product under similar conditions with ligand ent-L6. Additional four steps led to (+)-241D and its C6 epimer, respectively. Aside from the success, a series of other piperidine alkaloids like (+)-prosophylline, (+)-prosopinine, and (+)-prosopine were synthesized using the same strategy.
:Z = 9:1) underwent an Ir-catalyzed reaction under [Ir(cod)Cl]2 and ligand L6 to give 2,6-cis-piperidine 86 in high yield with perfect diastereoselectivity (dr = 98:2). Complementarily, 2,6-trans-piperidine 87 was obtained as the major product under similar conditions with ligand ent-L6. Additional four steps led to (+)-241D and its C6 epimer, respectively. Aside from the success, a series of other piperidine alkaloids like (+)-prosophylline, (+)-prosopinine, and (+)-prosopine were synthesized using the same strategy.
 | ||
| Scheme 28 Syntheses of (+)-241D, its C6 epimer and (−)-angustureine. | ||
In 2011, You and Helmchen and co-workers realized an Ir-catalyzed intermolecular asymmetric allylic amination with an allylic phosphate under similar conditions used in the above intramolecular version except for K3PO4 (Scheme 28b).58 A reaction between 2-vinylaniline 88 and allylic phosphate 89 led to the desired amination product 90 in 91% yield. Five more synthetic transformations were then conducted to obtain (−)-angustureine.
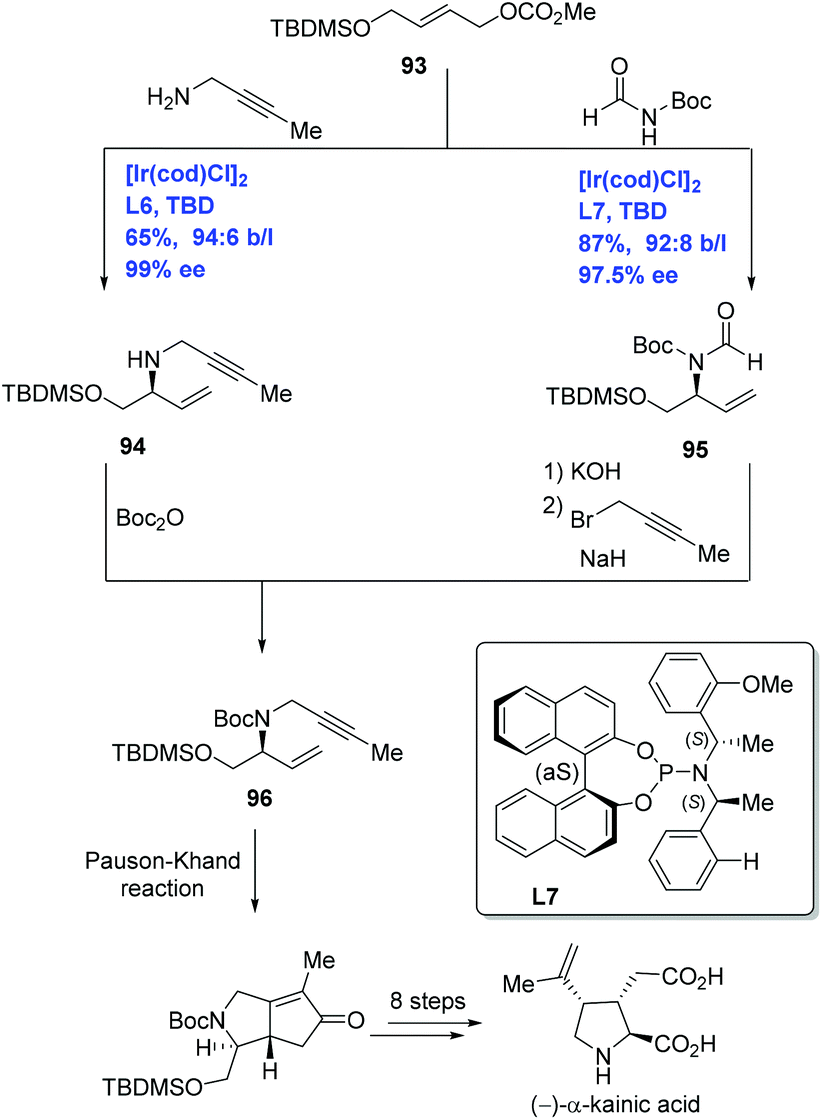
(−)-α-Kainic acid, the parent member of the kainoid family,59 is used as a neuroexcitatory agent relevant to Alzheimer's disease and Huntington's chorea. Given kainic acid's significant bioactivities and high cost, it has captivated synthetic chemists. Over sixty publications devoted to its synthesis have been disclosed.60 Helmchen and co-workers have developed Ir-catalyzed allylic amination to achieve the enantioselective total synthesis of (−)-α-kainic acid (Scheme 29).61 Treating carbonate 93 with but-2-ynylamine or N,N-diacylamine in the presence of [Ir(cod)Cl]2 and different P,N-ligands delivered allylic amination products 94 and 95 with high ee and excellent ratio of the branched to linear substitution product, respectively. Allylic amines 94 and 95 were each advanced to the key intermediate 1,6-enyne 96 by two complementary approaches. The synthesis of (−)-α-kainic acid was completed in nine additional steps from 96 featuring an intramolecular Pauson–Khand reaction.
 | ||
| Scheme 29 Synthesis of (−)-α-kainic acid. | ||
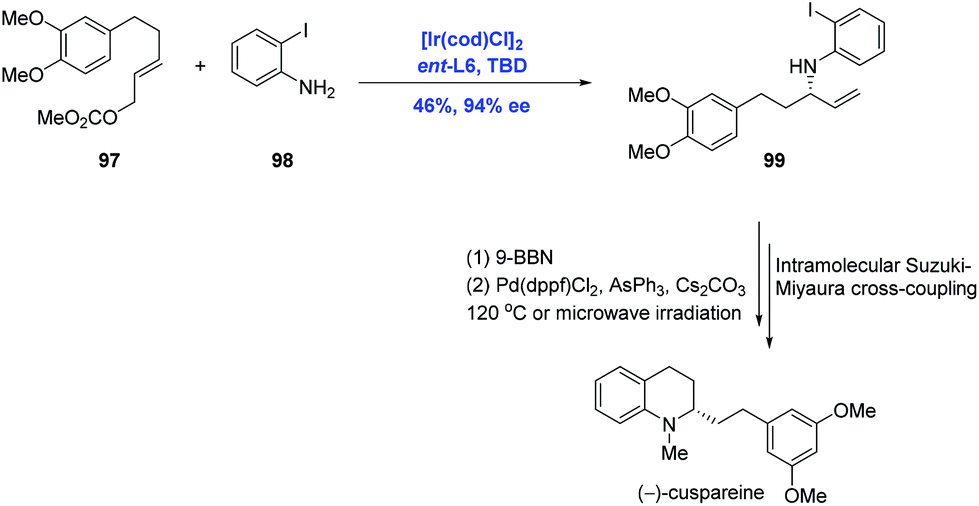
Tetrahydroquinoline alkaloids, such as cusparine, galipine and cuspareine, exhibit pronounced biological activities.62 The asymmetric hydrogenation of quinoline is the most used reaction to assemble the tetrahydroquinoline fragment.63 In their continuing application of Ir-catalyzed allylic amination, Helmchen and co-workers finished the syntheses of tetrahydroquinoline alkaloids (−)-cuspareine and (+)-angustureine (Scheme 30).64 In practice, the Ir-catalyzed allylic amination of carbonate 97 and 2-haloanilines 98 under standard conditions gave rise to allylic amine 99 with 94% ee and excellent regioselectivity, albeit in low (46%) yield. Subsequent hydroboration and Suzuki–Miyaura coupling were characterized to access (−)-cuspareine.
 | ||
| Scheme 30 Synthesis of (−)-cuspareine. | ||
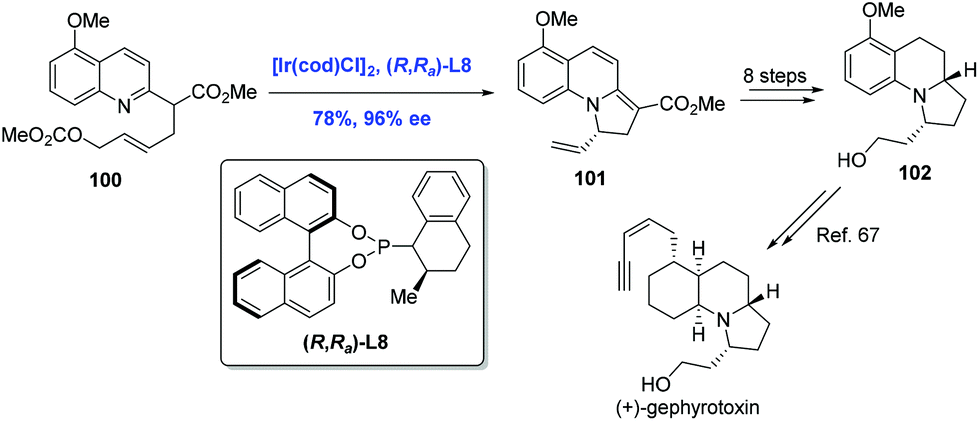
Gephyrotoxin, which was isolated from the skin extracts of Columbian frog,65 features a tricyclic perhydropyrrolo[1,2-a]quinoline structure. In 2015, You and co-workers communicated the first Ir-catalyzed intramolecular asymmetric allylic dearomatization reaction of pyridines, pyrazines, quinolines, and isoquinolines (Scheme 31).66 Taking advantage of this newly developed method, the authors completed a formal synthesis of (+)-gephyrotoxin. The key intermediate 101 was prepared from pyridine derivative 100 in 78% yield. Tricycle 101 was then elaborated to 102, an advanced intermediate in Ito's synthesis of gephyrotoxin.67
 | ||
| Scheme 31 Formal synthesis of (+)-gephyrotoxin. | ||
5. Ir-Catalyzed C–H borylation reactions
Organoboron compounds are versatile synthetic intermediates in organic synthesis. On one hand, organoborons serve as precursors for a broad range of functional groups, like amines, alcohols, and aldehydes. On the other hand, the Miyaura–Suzuki reaction, one of the most general methods for the formation of C–C bonds, adopts boronic acids as one of the coupling substances. In recent years, the transition-metal-catalyzed borylation of hydrocarbons has been extensively studied.68 In 2002, Miyaura and Hartwig and co-workers achieved the direct borylation of arenes under mild conditions of [Ir(cod)Cl]2/bpy, B2pin2 without solvent, providing a simple means for the synthesis of arylboronates.69 Moreover, they expanded this methodology to the borylation of heteroarenes.70 The Ir-catalyzed C–H borylation may occur by means of an IrIII/IrV cycle (Scheme 32).68f Oxidative addition of arene species with the intermediate D1 IrIII, which is generated from Ir(I) catalyst and pin2B2, results in D2 Ir(V). Reductive elimination of D2 produces the borylated product and intermediate D3, which could undergo the oxidative addition of pin2B2 or pinBH, followed by reductive elimination to regenerate D1.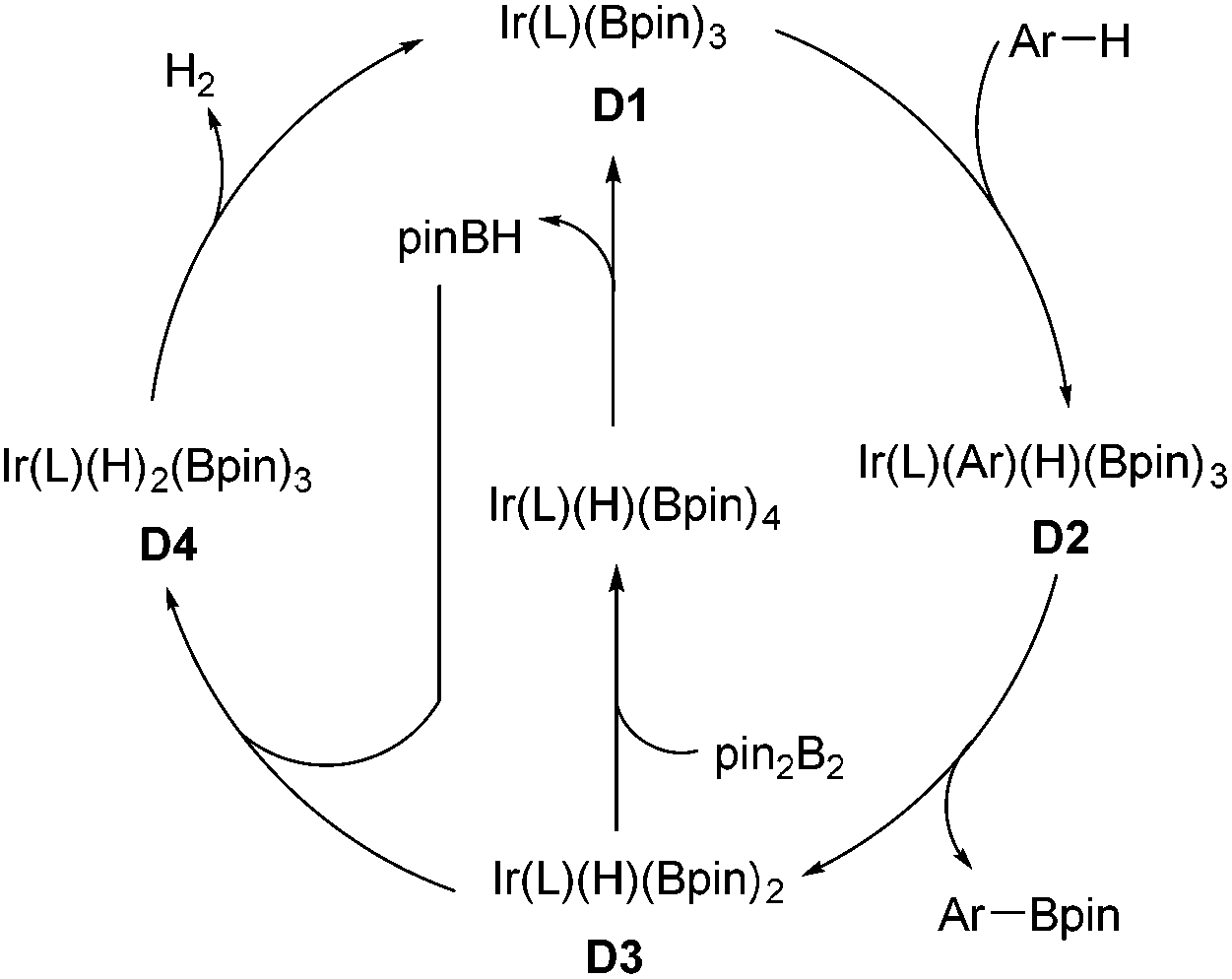 | ||
| Scheme 32 The possible mechanism of Ir-catalyzed C–H borylation reactions. | ||
Encouraged by the above direct borylation reactions, Sarpong and co-workers applied it to the synthesis of complanadine A in 2010.71 Structurally, complanadine A is an unsymmetrical dimer of lycodine (C2–C3′ linkage, Scheme 33), a character posing the synthetic challenge. In the event, convergent coupling of the enamide 103 and amine 104 followed by functional group operations gave the triflate 105. Pd-catalyzed reduction of 105 removed the triflate group, delivering 106 in 90% yield and setting the stage for the key reaction. Under the conditions of [Ir(cod)(OMe)]2, di-tert-butylbipyridine (dtpy), and diboronpinacolato ester, the C3′ position of the pyridine part of 106 was borylated to give product 107 in 75% yield, the site selectivity was attributed to the steric hindrance. Finally, the synthesis of complanadine A was achieved using Pd-catalyzed Suzuki coupling prior to deprotection of the Boc group.
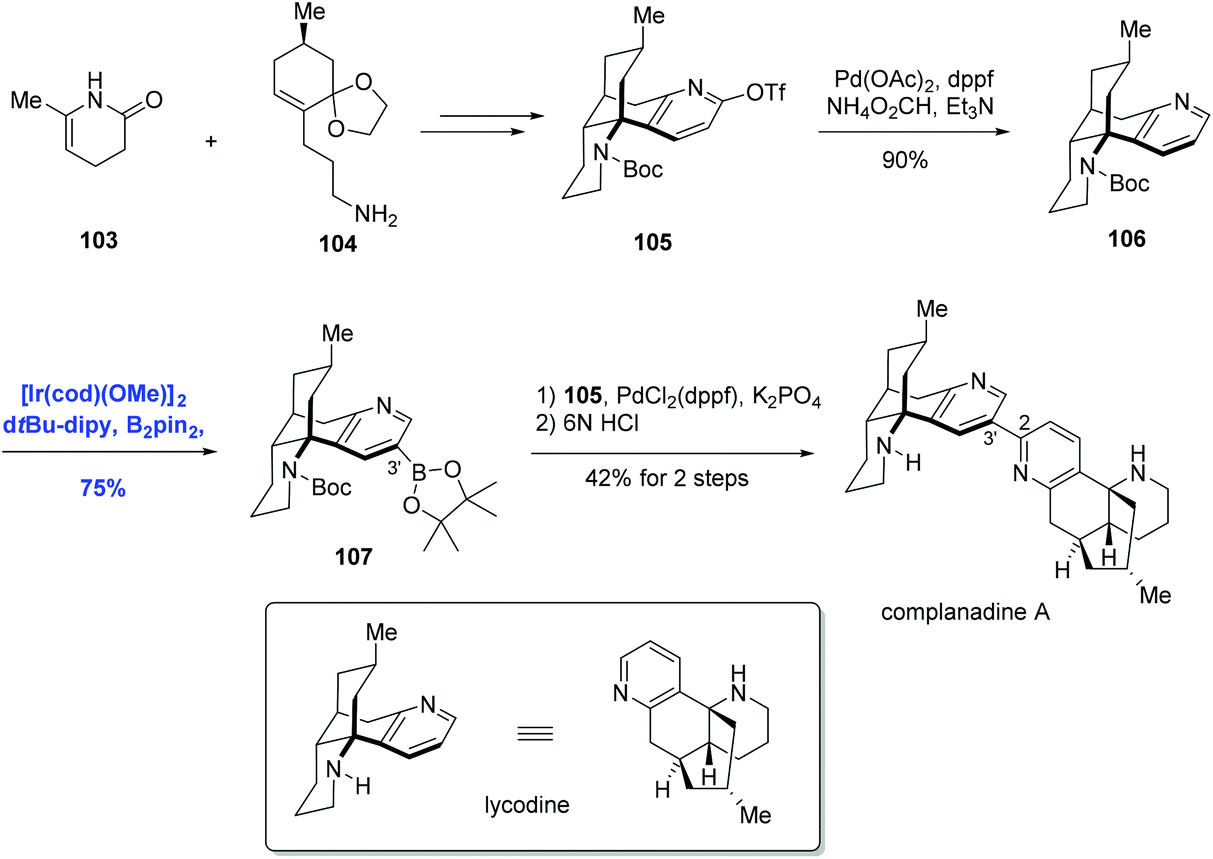 | ||
| Scheme 33 Synthesis of complanadine A. | ||
Aryl halides are traditionally prepared by the halogenation of arenes. Most halogenation reactions occur at the ortho or para position; methods to synthesize meta-substituted haloarenes are limited. Recently, Hartwig reported the meta halogenation of 1,3-disubstituted arenes through sequential Ir-catalyzed arene borylations and ipso halogenations to accomplish the syntheses of (−)-taiwaniaquinone H and (−)-taiwaniaquinol B (Scheme 34).72,73 To reach this goal, sterically controlled Ir-catalyzed borylation of arene 108 was conducted to produce the aryl boronate ester 109, and the bromination of 109 with copper(II) bromide produced 110. In stark contrast, bromination of 108 under traditional conditions occurred at the ortho positions of the two methoxy groups. With 110 in hand, an enantioselective α-arylation of a 2-methylcyclohexanone derivative proceeded smoothly to generate ketone 111. Additional seven steps finalized the synthesis of (−)-taiwaniaquinone H. Meanwhile, (−)-taiwaniaquinol B was obtained from 111 in nine steps.
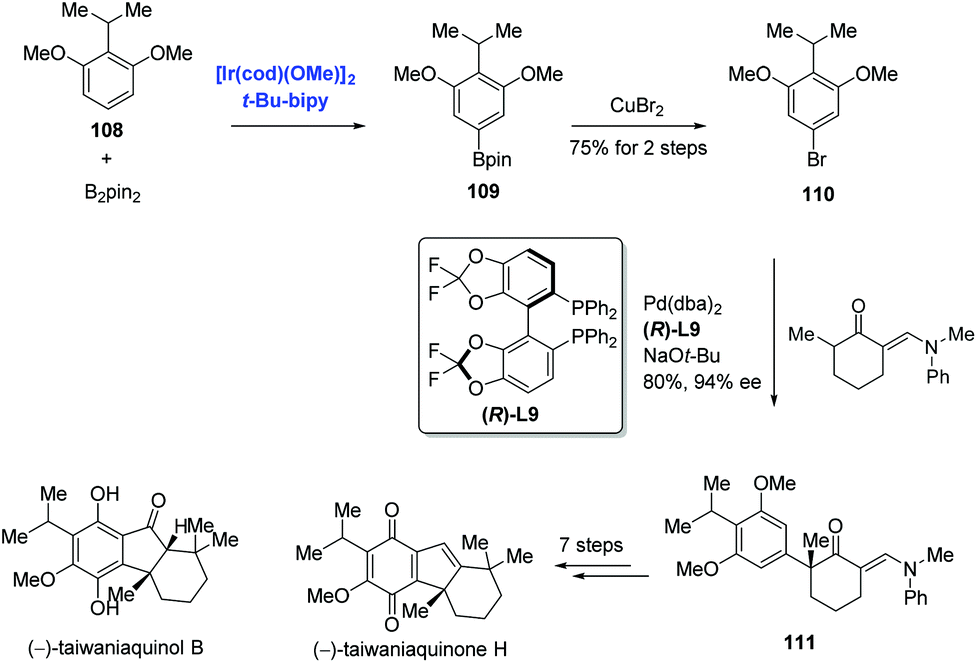 | ||
| Scheme 34 Enantioselective total synthesis of (−)-taiwaniaquinone H and (−)-taiwaniaquinol B. | ||
The indole frameworks are ubiquitous in natural products. C–H functionalization of indole and its derivatives has become the research focus of many groups. In addition to pyridine and benzene systems as exemplified in Schemes 33 and 34, Ir-catalyzed C–H borylation was extended to the indole substrate by Miyaura and Hartwig.70 Indole natural product (−)-goniomitine belonging to the Aspidosperma family was isolated by Husson in 1987. Among the numerous synthetic approaches towards (−)-goniomitine, Jia and co-workers’ strategy was characterized by an Ir-catalyzed C–H borylation of indole and Suzuki–Miyaura coupling (Scheme 35).74 Under the conditions of pinBH, [Ir(cod)(OMe)]2, and 4,4′-di-tert-butyl-2,2′-bipyridine (dtbpy), indole 112 was activated at the C2 position to provide 113 in 86% yield. Subsequent Suzuki–Miyaura coupling of 113 and vinyl iodide 114 with concomitant amidation afforded 115. (−)-Goniomitine was then accessed with additional six steps.
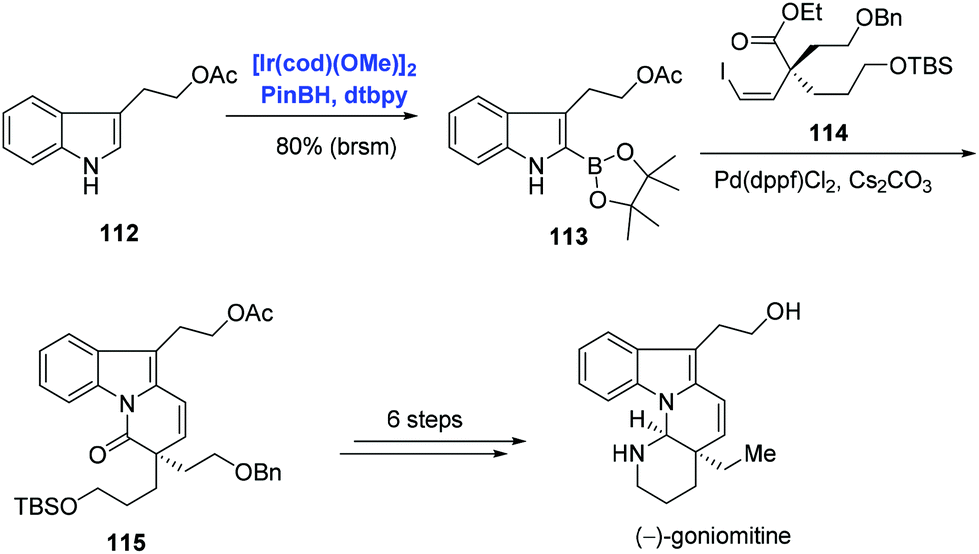 | ||
| Scheme 35 Synthesis of (−)-goniomitine. | ||
Aside from the above-mentioned C2–H borylation of indole, Baran and co-workers reported Ir-catalyzed C6–H borylation of a tryptophan derivative and applied it to the syntheses of verruculogen and fumitremorgin A (Scheme 36).75 The tryptophan derivative 116, which contains a bulky TIPS group on the indole nitrogen to suppress the reaction at C2 and C7 positions, was borylated at the C6 position at will. Subsequent copper-catalyzed Chan–Lam coupling with methanol afforded the methoxy 117 in 65% yield and with 8:1 (C6:C5) selectivity. Then verruculogen and fumitremorgin A were obtained from 118 featuring an endoperoxide-forming cyclization with the aldehyde.
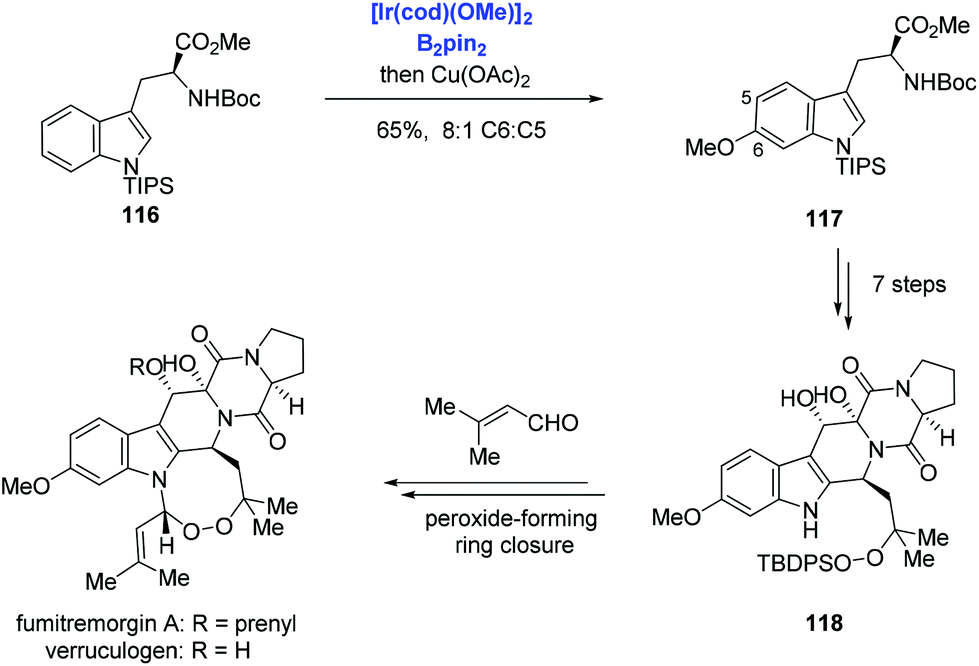 | ||
| Scheme 36 Syntheses of verruculogen and fumitremorgin A. | ||
6. Conclusions
Iridium is considered as a powerful and versatile catalyst in asymmetric synthesis. Diverse reaction types promoted by Ir-complexes have been developed, such as transfer hydrogenation (oxidation), hydrogenation, allylic substitution, C–H functionalization (C–H borylation), isomerization reactions and a few others. More importantly, Ir-catalyzed reactions have found applications in the synthesis of diverse natural products. The examples presented in the review demonstrate the high efficiency of the entitled reaction in the formation of chemical bonds and the construction of naturally occurring skeletons. Although significant progress has been made in the field, in-depth applications of Ir-catalyzed reactions in a more complex structural setting are highly encouraged. On one hand, problems encountered during the study may offer organic chemists chances to develop new iridium catalyst systems, thus broadening the current reaction types and providing new tools for synthetic organic chemists. On the other hand, successful implementations of such studies will further facilitate access to biologically active natural products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Program on Key Research Project (2016YFA0602900), the Science and Technology Program of Guangzhou (201707010073), the “1000 Youth Talents Plan”, the National Natural Science Foundation of China (21672073), and the Fundamental Research Funds for the Central Universities, SCUT, for the financial support.References
- For reviews on Au-catalyzed reactions in natural product synthesis, see: (a) M. Rudolph and A. S. K. Hashmi, Chem. Soc. Rev., 2012, 41, 2448–2462 RSC; (b) D. Pflästerer and A. S. K. Hashmi, Chem. Soc. Rev., 2016, 45, 1331–1367 RSC.
- For a review on Ti-catalyzed reactions in natural product synthesis, see: S. P. Morcillo, D. Miguel, A. G. Campaña, L. Álvarez de Cienfuegos, J. Justicia and J. M. Cuerva, Org. Chem. Front., 2014, 1, 15–33 RSC.
- For reviews on Rh-catalyzed reactions in natural product synthesis, see: (a) P. B. Brady and V. Bhat, Eur. J. Org. Chem., 2017, 5179–5190 CrossRef CAS; (b) F. J. Lombard and M. J. Coster, Org. Biomol. Chem., 2015, 13, 6419–6431 RSC; (c) T. Kan, T. Asakawa and M. Inai, Heterocycles, 2016, 92, 31–43 CrossRef CAS.
- For reviews on Pd-catalyzed reactions in natural product synthesis, see: (a) Y. Bai, D. C. Davis and M. J. Dai, J. Org. Chem., 2017, 82, 2319–2328 CrossRef CAS PubMed; (b) C. Hui and J. Xu, Tetrahedron Lett., 2016, 57, 2692–2696 CrossRef CAS; (c) D. N. Garad and S. B. Mhaske, Org. Lett., 2016, 18, 3862–3865 CrossRef CAS PubMed; (d) M. M. Heravi, E. Hashemi and F. Azimian, Tetrahedron, 2014, 70, 7–21 CrossRef CAS; (e) D. Wang and S. H. Gao, Org. Chem. Front., 2014, 1, 556–566 RSC; (f) M. M. Heravi and E. Hashemi, Tetrahedron, 2012, 68, 9145–9178 CrossRef CAS; (g) L. F. Tietze and A. Düfert, Pure Appl. Chem., 2010, 82, 1375–1392 CrossRef CAS; (h) L. F. Tietze and T. Kinzel, Pure Appl. Chem., 2007, 79, 629–650 CrossRef CAS; (i) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS PubMed; (j) T. Graening and H. Schmalz, Angew. Chem., Int. Ed., 2003, 42, 2580–2584 CrossRef CAS PubMed; (k) A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945–2963 CrossRef CAS PubMed.
- For general reviews on Ir-complex and Ir-catalyzed reactions, see: (a) Iridium Catalysis, ed. P. G. Andersson, Springer, Berlin, Heidelberg, Germany, Top. Organomet. Chem., 2011, p. 34 Search PubMed; (b) R. Crabtree, Acc. Chem. Res., 1979, 12, 331–337 CrossRef CAS; (c) P. P. Deutsch and R. Eisenberg, Chem. Rev., 1988, 88, 1147–1161 CrossRef CAS.
- For reviews on Ir-catalyzed transfer hydrogenation reactions, see: (a) T. Suzuki, Chem. Rev., 2011, 111, 1825–1845 CrossRef CAS PubMed; (b) A. R. Dechert-Schmitt, D. C. Schmitt, X. Gao, T. Itohand and M. J. Krische, Nat. Prod. Rep., 2014, 31, 504–513 RSC; (c) Y. Obora and Y. Ishii, Synlett, 2011, 30–51 CrossRef CAS; (d) I. Shin and M. J. Krische, Top. Curr. Chem., 2016, 372, 85–102 CrossRef CAS PubMed.
- For reviews on Ir-catalyzed hydrogenation reactions, see: (a) S. F. Zhu and Q. L. Zhou, Acc. Chem. Res., 2017, 50, 988–1001 CrossRef CAS PubMed; (b) Y. Zhu and K. Burgess, Acc. Chem. Res., 2012, 45, 1623–1636 CrossRef CAS PubMed; (c) S. J. Roseblade and A. Pfaltz, Acc. Chem. Res., 2007, 40, 1402–1411 CrossRef CAS PubMed.
- For reviews on Ir-catalyzed allylic substitution reactions, see: (a) Transition Metal Catalyzed Enantioselective Allylic Substitution in Organic Synthesis, ed. U. Kazmaier, Springer, Berlin, Germany, Top. Organomet. Chem., 2011, vol. 38 Search PubMed; (b) C. X. Zhuo, C. Zheng and S. L. You, Acc. Chem. Res., 2014, 47, 2558–2573 CrossRef CAS PubMed; (c) P. Tosatti, A. Nelson and S. P. Marsden, Org. Biomol. Chem., 2012, 10, 3147–3163 RSC; (d) G. Helmchen, A. Dahnz, P. Dubon, M. Schelwies and R. Weihofen, Chem. Commun., 2007, 675–691 RSC; (e) J. F. Hartwig and L. M. Stanley, Acc. Chem. Res., 2010, 43, 1461–1475 CrossRef CAS PubMed.
- For a review on Ir-catalyzed isomerization reactions, see: H. h. Li and C. Mazet, Acc. Chem. Res., 2016, 49, 1232–1241 CrossRef CAS PubMed.
- For a review on Ir-catalyzed dehydrogenation reactions, see: J. Choi, A. H. Roy MacArthur, M. Brookhart and A. S. Goldman, Chem. Rev., 2011, 111, 1761–1779 CrossRef CAS PubMed.
- (a) Y. M. Y. Haddad, H. B. Henbest, J. Husbands and T. R. B. Mitchell, Proc. Chem. Soc., 1964, 361 CAS; (b) J. Trocha-Grimshaw and H. B. Henbest, Chem. Commun., 1967, 544 RSC.
- (a) A. Camus, G. Mestroni and G. Zassinovich, J. Mol. Catal., 1979, 6, 231–233 CrossRef CAS; (b) A. Nait Ajjou, Tetrahedron Lett., 2001, 42, 13–15 CrossRef CAS; (c) A. Nait Ajjou and J. L. Pinet, Can. J. Chem., 2005, 83, 702–710 CrossRef.
- S. B. Han, I. S. Kimab and M. J. Krische, Chem. Commun., 2009, 7278–7287 RSC.
- S. B. Han, A. Hassan, I. S. Kim and M. J. Krische, J. Am. Chem. Soc., 2010, 132, 15559–15561 CrossRef CAS PubMed.
- Z. A. Kasun, X. Gao, R. M. Lipinski and M. J. Krische, J. Am. Chem. Soc., 2015, 137, 8900–8903 CrossRef CAS PubMed.
- J. Feng, F. Noack and M. J. Krische, J. Am. Chem. Soc., 2016, 138, 12364–12367 CrossRef CAS PubMed.
- (a) J. J. Feng, Z. A. Kasun and M. J. Krische, J. Am. Chem. Soc., 2016, 138, 5467–5478 CrossRef CAS PubMed; (b) Y. Lu, S. K. Woo and M. J. Krische, J. Am. Chem. Soc., 2011, 133, 13876–13879 CrossRef CAS PubMed; (c) A. R. Waldeck and M. J. Krische, Angew. Chem., Int. Ed., 2013, 52, 4470–4473 CrossRef CAS PubMed; (d) X. Gao, S. K. Woo and M. J. Krische, J. Am. Chem. Soc., 2013, 135, 4223–4226 CrossRef CAS PubMed; (e) I. Shin, S. Hong and M. J. Krische, J. Am. Chem. Soc., 2016, 138, 14246–14249 CrossRef CAS PubMed; (f) F. Perez, A. R. Waldeck and M. J. Krische, Angew. Chem., Int. Ed., 2016, 55, 5049–5052 CrossRef CAS PubMed.
- P. Harsh and G. A. O'Doherty, Tetrahedron, 2009, 65, 5051–5055 CrossRef CAS PubMed.
- C. Oger, Y. Brinkmann, S. Bouazzaoui, T. Durand and J. M. Galano, Org. Lett., 2008, 10, 5087–5090 CrossRef CAS PubMed.
- T. Suzuki, K. Ghozati, T. Katoh and H. Sasai, Org. Lett., 2009, 11, 4286–4288 CrossRef CAS PubMed.
- T. Suzuki, K. Ghozati, D. Zhou, T. Katoh and H. Sasai, Tetrahedron, 2010, 66, 7562–7568 CrossRef CAS.
- (a) J. M. Brown, in Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, Berlin, 1999, ch. 5.1, vol. I, pp. 121–182 Search PubMed; (b) W. S. Knowles, Angew. Chem., Int. Ed., 2002, 41, 1998–2007 CrossRef CAS; (c) R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008–2022 CrossRef CAS; (d) K. Kallstrom, I. Munslow and P. G. Andersson, Chem. – Eur. J., 2006, 12, 3194–3200 CrossRef PubMed.
- (a) A. Lightfoot, P. Schnider and A. Pfaltz, Angew. Chem., Int. Ed., 1998, 37, 2897–2899 CrossRef CAS; (b) D. G. Blackmond, A. Lightfoot, A. Pfaltz, T. Rosner, P. Schnider and N. Zimmermann, Chirality, 2000, 12, 442–449 CrossRef CAS PubMed; (c) J. Blankenstein and A. Pfaltz, Angew. Chem., Int. Ed., 2001, 40, 4445–4447 CrossRef CAS; (d) P. G. Cozzi, N. Zimmermann, R. Hilgraf, S. Schaffner and A. Pfaltz, Adv. Synth. Catal., 2001, 343, 450–454 CrossRef CAS; (e) F. Menges and A. Pfaltz, Adv. Synth. Catal., 2002, 344, 40–44 CrossRef CAS; (f) A. Pfaltz, J. Blankenstein, R. Hilgraf, E. Hörmann, S. McIntyre, F. Menges, M. Schönleber, S. P. Smidt, B. Wüstenberg and N. Zimmermann, Adv. Synth. Catal., 2003, 345, 33–43 CrossRef CAS.
- (a) R. H. Crabtree, H. Felkin and G. E. Morris, J. Organomet. Chem., 1977, 141, 205–215 CrossRef CAS; (b) R. H. Crabtree, H. Felkin, T. Fillebeen-Kahn and G. E. Morris, J. Organomet. Chem., 1979, 168, 183–195 CrossRef CAS.
- A. C. Guevel and D. J. Hart, J. Org. Chem., 1996, 61, 473–479 CrossRef CAS PubMed.
- Y. Zhu, A. Loudet and K. Burgess, Org. Lett., 2010, 12, 4392–4395 CrossRef CAS PubMed.
- G. G. Bianco, H. M. C. Ferraz, A. M. Costa, L. V. Costa-Lotufo, C. Pessoa, M. O. de Moraes, M. G. Schrems, A. Pfaltz and L. F. Silva Jr., J. Org. Chem., 2009, 74, 2561–2566 CrossRef CAS PubMed.
- S. Song, S. F. Zhu, S. Yang, S. Li and Q. L. Zhou, Angew. Chem., Int. Ed., 2012, 51, 2708–2711 CrossRef CAS PubMed.
- (a) D. J. Mans, G. A. Cox and T. V. Rajan Babu, J. Am. Chem. Soc., 2011, 133, 5776–5779 CrossRef CAS PubMed; (b) T. Magauer, H. J. Martin and J. Mulzer, Angew. Chem., Int. Ed., 2009, 48, 6032–6036 CrossRef CAS PubMed; (c) Y. Kiyotsuka, Y. Katayama, H. P. Acharya, T. Hyodo and Y. Kobayashi, J. Org. Chem., 2009, 74, 1939–1951 CrossRef CAS PubMed; (d) S. P. Chavan, M. Thakkar and U. R. Kalkote, Tetrahedron Lett., 2007, 48, 643–646 CrossRef CAS.
- S. Song, S. F. Zhu, L. Y. Pu and Q. L. Zhou, Angew. Chem., Int. Ed., 2013, 52, 6072–6075 CrossRef CAS PubMed.
- (a) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029–3069 CrossRef CAS PubMed; (b) T. C. Nugent and M. Ei-Shazly, Adv. Synth. Catal., 2010, 352, 753–819 CrossRef CAS; (c) J. H. Xie, S. F. Zhu and Q. L. Zhou, Chem. Rev., 2011, 111, 1713–1760 CrossRef CAS PubMed; (d) C. Li and J. Xiao, J. Am. Chem. Soc., 2008, 130, 13208–13209 CrossRef CAS PubMed; (e) M. Chang, W. Li, G. Hou and X. Zhang, Adv. Synth. Catal., 2010, 352, 3121–3125 CrossRef CAS; (f) F. Chen, Z. Y. Ding, J. Qin, T. L. Wang, Y. M. He and Q. H. Fan, Org. Lett., 2011, 13, 4348–4351 CrossRef CAS PubMed; (g) M. Chang, W. Li and X. Zhang, Angew. Chem., Int. Ed., 2011, 50, 10679–10681 CrossRef CAS PubMed; (h) Q. A. Chen, M. W. Chen, C. B. Yu, L. Shi, D. S. Wang, Y. Yang and Y. G. Zhou, J. Am. Chem. Soc., 2011, 133, 16432–16435 CrossRef CAS PubMed; (i) Q. A. Chen, K. Gao, Y. Duan, Z. S. Ye, L. Shi, Y. Yang and Y. G. Zhou, J. Am. Chem. Soc., 2012, 134, 2442–2448 CrossRef CAS PubMed; (j) Z. Y. Ding, F. Chen, J. Qin, Y. M. He and Q. H. Fan, Angew. Chem., Int. Ed., 2012, 51, 5706–5710 CrossRef CAS PubMed.
- C. Guo, D. W. Sun, S. Yang, S. J. Mao, X. H. Xu, S. F. Zhu and Q. L. Zhou, J. Am. Chem. Soc., 2015, 137, 90–93 CrossRef CAS PubMed.
- Q. Q. Zhang, J. H. Xie, X. H. Yang, J. B. Xie and Q. L. Zhou, Org. Lett., 2012, 14, 6158–6161 CrossRef CAS PubMed.
- H. Lin, L. J. Xiao, M. J. Zhou, H. M. Yu, J. H. Xie and Q. L. Zhou, Org. Lett., 2016, 18, 1434–1437 CrossRef CAS PubMed.
- X. D. Zuo, S. M. Guo, R. Yang, J. H. Xie and Q. L. Zhou, Chem. Sci., 2017, 8, 6202–6206 RSC.
- (a) B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395–422 CrossRef CAS PubMed; (b) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2944 CrossRef CAS PubMed; (c) Z. Lu and S. Ma, Angew. Chem., Int. Ed., 2008, 47, 258–297 CrossRef CAS PubMed; (d) G. Poli, G. Prestat, F. Liron and C. Kammerer-Pentier, Top. Organomet. Chem., 2012, 38, 1–64 Search PubMed; (e) B. M. Trost, T. Zhang and J. D. Sieber, Chem. Sci., 2010, 1, 427–440 RSC; (f) B. M. Trost, J. Org. Chem., 2004, 69, 5813–5837 CrossRef CAS PubMed; (g) C. Moberg, Top. Organomet. Chem., 2012, 38, 209–234 CrossRef; (h) O. Belda and C. Moberg, Acc. Chem. Res., 2004, 37, 159–167 CrossRef CAS PubMed; (i) J.-M. Begouin, J. E. M. N. Klein, D. Weickmann and B. Plietker, Top. Organomet. Chem., 2012, 38, 269–320 CrossRef; (j) S. R. Harutyunyan, T. den Hartog, K. Geurts, A. J. Minnaard and B. L. Feringa, Chem. Rev., 2008, 108, 2824–2852 CrossRef CAS PubMed; (k) C. A. Falciola and A. Alexakis, Eur. J. Org. Chem., 2008, 3765–3780 CrossRef CAS; (l) A. Alexakis, J. E. Backvall, N. Krause, O. Pamies and M. Dieguez, Chem. Rev., 2008, 108, 2796–2823 CrossRef CAS PubMed.
- (a) B. M. Trost and C. Lee, in Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley-VCH, New York, 2nd edn, 2000, pp. 593–649 Search PubMed; (b) A. Pfaltz and M. Lautens, in Comprehensive Asymmetric Catalysis I-III, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, Berlin, 1999, pp. 833–884 Search PubMed.
- (a) K. Ishihara, S. Nakamura and H. Yamamoto, J. Am. Chem. Soc., 1999, 121, 4906–4907 CrossRef CAS; (b) K. Ishihara, H. Ishibashi and H. Yamamoto, J. Am. Chem. Soc., 2001, 123, 1505–1506 CrossRef CAS; (c) H. Ishibashi, K. Ishihara and H. Yamamoto, J. Am. Chem. Soc., 2004, 126, 11122–11123 CrossRef CAS PubMed; (d) Y. J. Zhao, B. Li, L. J. S. Tan, Z. L. Shen and T. P. Loh, J. Am. Chem. Soc., 2010, 132, 10242–10244 CrossRef CAS PubMed; (e) K. Surendra and E. J. Corey, J. Am. Chem. Soc., 2012, 134, 11992–11994 CrossRef CAS PubMed; (f) A. Sakakura, A. Ukai and K. Ishihara, Nature, 2007, 445, 900–903 CrossRef CAS PubMed; (g) S. Rendeler and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 5027–5029 CrossRef PubMed; (h) R. R. Knowles, S. Lin and E. N. Jacobsen, J. Am. Chem. Soc., 2010, 132, 5030–5032 CrossRef CAS PubMed.
- M. A. Schafroth, D. Sarlah, S. Krautwald and E. M. Carreira, J. Am. Chem. Soc., 2012, 134, 20276–20278 CrossRef CAS PubMed.
- O. F. Jeker, A. G. Kravina and E. M. Carreira, Angew. Chem., Int. Ed., 2013, 52, 12166–12169 CrossRef CAS PubMed.
- H. F. Sun, X. M. Li, L. Meng, C. M. Cui, S. S. Gao, C. S. Li, C. G. Huang and B. G. Wang, J. Nat. Prod., 2012, 75, 148–152 CrossRef CAS PubMed.
- M. A. Schafroth, G. Zuccarello, S. Krautwald, D. Sarlah and E. M. Carreira, Angew. Chem., Int. Ed., 2014, 53, 13898–13901 CrossRef CAS PubMed.
- J. Deng, S. P. Zhou, W. H. Zhang, J. Li, R. F. Li and A. Li, J. Am. Chem. Soc., 2014, 136, 8185–8188 CrossRef CAS PubMed.
- S. P. Zhou, H. Chen, Y. J. Luo, W. H. Zhang and A. Li, Angew. Chem., Int. Ed., 2015, 54, 6878–6882 CrossRef CAS PubMed.
- (a) A. R. Pape, K. P. Kaliappan and E. P. Kündig, Chem. Rev., 2000, 100, 2917–2940 CrossRef CAS PubMed; (b) F. López Ortiz, M. J. Iglesias, I. Fernández, C. M. Andújar Sánchez and G. R. Gómez, Chem. Rev., 2007, 107, 1580–1691 CrossRef PubMed; (c) L. Pouységu, D. Deffieux and S. Quideau, Tetrahedron, 2010, 66, 2235–2261 CrossRef; (d) S. P. Roche and J. A. Porco Jr., Angew. Chem., Int. Ed., 2011, 50, 4068–4093 CrossRef CAS PubMed; (e) C. X. Zhuo, W. Zhang and S. L. You, Angew. Chem., Int. Ed., 2012, 51, 12662–12686 CrossRef PubMed.
- X. Zhang, L. Han and S. L. You, Chem. Sci., 2014, 5, 1059–1063 RSC.
- J. Ruchti and E. M. Carreira, J. Am. Chem. Soc., 2014, 136, 16756–16759 CrossRef CAS PubMed.
- (a) B. M. Trost and D. A. Thaisrivongs, J. Am. Chem. Soc., 2008, 130, 14092–14093 CrossRef CAS PubMed; (b) J. Mao, J. Zhang, H. Jiang, A. Bellomo, M. Zhang, Z. Gao, S. D. Dreher and P. J. Walsh, Angew. Chem., Int. Ed., 2016, 55, 2526–2530 CrossRef CAS PubMed; (c) X. J. Liu and S. L. You, Angew. Chem., Int. Ed., 2017, 56, 4002–4005 CrossRef CAS PubMed.
- (a) A. Ramirez and S. Garcia-Rubio, Curr. Med. Chem., 2003, 10, 1891–1915 CrossRef CAS PubMed; (b) T. S. Kam, Alkaloids: Chem. Biol. Perspect., 1999, 14, 285–435 CAS; (c) H. Arai, Y. Hirasawa, A. Rahman, I. Kusumawati, N. C. Zaini, S. Sato, C. Aoyama, J. Takeo and H. Morita, Bioorg. Med. Chem., 2010, 18, 2152–2158 CrossRef CAS PubMed; (d) R. Eckermann and T. Gaich, Synthesis, 2013, 2813–2823 CAS; (e) J. M. Smith, J. Moreno, B. W. Boal and N. K. Garg, Angew. Chem., Int. Ed., 2015, 54, 400–412 CAS.
- Q. F. Wu, H. He, W. B. Liu and S. L. You, J. Am. Chem. Soc., 2010, 132, 11418–11419 CrossRef CAS PubMed.
- S. Z. Jiang, X. Y. Zeng, X. Liang, T. Lei, K. Wei and Y. R. Yang, Angew. Chem., Int. Ed., 2016, 55, 4044–4048 CrossRef CAS PubMed.
- X. Liang, S. Z. Jiang, K. Wei and Y. R. Yang, J. Am. Chem. Soc., 2016, 138, 2560–2562 CrossRef CAS PubMed.
- X. Liang, T. Y. Zhang, X. Y. Zeng, Y. Zheng, K. Wei and Y. R. Yang, J. Am. Chem. Soc., 2017, 139, 3364–3367 CrossRef CAS PubMed.
- (a) R. Takeuchi and S. Kezuka, Synthesis, 2006, 3349–3366 CrossRef CAS; (b) T. Ohmura and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 15164–15165 CrossRef CAS PubMed; (c) K. Tissot-Croset, D. Polet and A. Alexakis, Angew. Chem., Int. Ed., 2004, 43, 2426–2428 CrossRef CAS PubMed; (d) R. Weihofen, A. Dahnz, O. Tverskoy and G. Helmchen, Chem. Commun., 2005, 3541–3543 RSC; (e) R. Weihofen, O. Tverskoy and G. Helmchen, Angew. Chem., Int. Ed., 2006, 45, 5546–5549 CrossRef CAS PubMed; (f) O. V. Singh and H. Han, J. Am. Chem. Soc., 2007, 129, 774–775 CrossRef CAS PubMed; (g) C. Defieber, M. A. Ariger, P. Moriel and E. M. Carreira, Angew. Chem., Int. Ed., 2007, 46, 3139–3143 CrossRef CAS PubMed; (h) S. Spiess, C. Welter, G. Franck, J. P. Taquet and G. Helmchen, Angew. Chem., Int. Ed., 2008, 47, 7652–7655 CrossRef CAS PubMed; (i) L. M. Stanley and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 8971–8983 CrossRef CAS PubMed; (j) H. He, W. B. Liu, L. X. Dai and S. L. You, Angew. Chem., Int. Ed., 2010, 49, 1496–1499 CrossRef CAS PubMed.
- (a) C. Gnamm, G. Franck, N. Miller, T. Stork, K. Brödner and G. Helmchen, Synthesis, 2008, 3331–3350 CAS; (b) A. Farwick, J. U. Engelhart, O. Tverskoy, C. Welter, Q. A. Umlauf, F. Rominger, W. J. Kerr and G. Helmchen, Adv. Synth. Catal., 2011, 353, 349–370 CrossRef CAS; (c) J. B. Xia, W. B. Liu, T. M. Wang and S. L. You, Chem. – Eur. J., 2010, 16, 6442–6446 CrossRef CAS PubMed.
- C. Gnamm, C. M. Krauter, K. Brodner and G. Helmchen, Chem. – Eur. J., 2009, 15, 2050–2054 CrossRef CAS PubMed.
- C. Gnamm, K. Brodner, C. M. Krauter and G. Helmchen, Chem. – Eur. J., 2009, 15, 10514–10532 CrossRef CAS PubMed.
- K. Y. Ye, H. He, W. B. Liu, L. X. Dai, G. Helmchen and S. L. You, J. Am. Chem. Soc., 2011, 133, 19006–19014 CrossRef CAS PubMed.
- (a) A. F. Parsons, Tetrahedron, 1996, 52, 4149–4174 CrossRef CAS; (b) M. G. Moloney, Nat. Prod. Rep., 1998, 15, 205–219 RSC; (c) M. G. Moloney, Nat. Prod. Rep., 2002, 19, 597–616 RSC.
- (a) M. M. Martinez and D. Hoppe, Org. Lett., 2004, 6, 3743–3746 CrossRef CAS PubMed; (b) M. E. Scott and M. Lautens, Org. Lett., 2005, 7, 3045–3047 CrossRef CAS PubMed; (c) J. F. Poisson, A. Orellana and A. E. Greene, J. Org. Chem., 2005, 70, 10860–10863 CrossRef CAS PubMed; (d) Y. Morita, H. Tokuyama and T. Fukuyama, Org. Lett., 2005, 7, 4337–4340 CrossRef CAS PubMed; (e) J. C. Anderson, J. M. A. O'Loughlin and J. A. Tornos, Org. Biomol. Chem., 2005, 3, 2741–2749 RSC; (f) D. M. Hodgson, S. Hachisu and M. D. Andrews, Org. Lett., 2005, 7, 815–817 CrossRef CAS PubMed; (g) S. K. Orellana, A. Pandey, A. E. Greene and J. F. Poisson, Org. Lett., 2006, 8, 5665–5668 CrossRef PubMed; (h) H. Sakaguchi, H. Tokuyama and T. Fukuyama, Org. Lett., 2007, 9, 1635–1638 CrossRef CAS PubMed; (i) Y. C. Jung, C. H. Yoon, E. Turos, K. S. Yoo and K. W. Jung, J. Org. Chem., 2007, 72, 10114–10122 CrossRef CAS PubMed; (j) J. M. Chalker, A. Yang, K. Deng and T. Cohen, Org. Lett., 2007, 9, 3825–3828 CrossRef CAS PubMed; (k) H. Sakaguchi, H. Tokuyama and T. Fukuyama, Org. Lett., 2008, 10, 1711–1714 CrossRef CAS PubMed.
- A. Farwick and G. Helmchen, Org. Lett., 2010, 12, 1108–1111 CrossRef CAS PubMed.
- (a) J. F. Gerster, S. R. Rohlfling and R. M. Winady, American Medicinal Chemistry Symposium, Toronto, 1982 Search PubMed; (b) I. Jacquemond-Collet, S. Hannedouche, N. Fabre, I. Fourasté and C. Moulis, Phytochemistry, 1999, 51, 1167–1169 CrossRef CAS; (c) J. H. Rokotoson, N. Fabre, I. Jacquemond-Collet, S. Hannedouche, I. Fourasté and C. Moulis, Planta Med., 1998, 64, 762–763 CrossRef PubMed; (d) P. J. Houghton, T. Z. Woldemariam, Y. Watanabe and M. Yates, Plant Med., 1999, 65, 250–254 CrossRef CAS PubMed; (e) I. Jacquemond-Collet, J. M. Bessière, S. Hannedouche, C. Bertrand, I. Fourasté and C. Moulis, Phytochem. Anal., 2001, 12, 312–319 CrossRef CAS PubMed.
- (a) W. B. Wang, S. M. Lu, P. Y. Yang, X. W. Han and Y. G. Zhou, J. Am. Chem. Soc., 2003, 125, 10536–10537 CrossRef CAS PubMed; (b) W. J. Tang, S. F. Zhu, L. J. Xu, Q. L. Zhou, Q. H. Fan, H. F. Zhou, K. Lam and A. S. C. Chan, Chem. Commun., 2007, 613–615 RSC; (c) Z. J. Wang, G. J. Deng, Y. Li, Y. M. He, W. J. Tang and Q. H. Fan, Org. Lett., 2007, 9, 1243–1246 CrossRef CAS PubMed; (d) F. R. Gou, W. Li, X. Zhang and Y. M. Liang, Adv. Synth. Catal., 2010, 352, 2441–2444 CrossRef CAS; (e) S. M. Lu, Y. Q. Wang, X. W. Han and Y. G. Zhou, Angew. Chem., Int. Ed., 2006, 45, 2260–2263 CrossRef CAS PubMed; (f) D. W. Wang, W. Zeng and Y. G. Zhou, Tetrahedron: Asymmetry, 2007, 18, 1103–1107 CrossRef CAS; (g) O. Hara, T. Koshizawa, K. Makino, I. Kunimune, A. Namiki and Y. Hamada, Tetrahedron, 2007, 63, 6170–6181 CrossRef CAS; (h) T. Wang, L. G. Zhuo, Z. Li, F. Chen, Z. Ding, Y. He, Q. H. Fan, J. Xiang, Z. X. Yu and A. S. C. Chan, J. Am. Chem. Soc., 2011, 133, 9878–9891 CrossRef CAS PubMed.
- G. Satyanarayana, D. Pflästerer and G. Helmchen, Eur. J. Org. Chem., 2011, 6877–6886 CrossRef CAS.
- J. W. Daly, B. Witkop, T. Tokuyama, T. Nishikawa and I. L. Karle, Helv. Chim. Acta, 1977, 60, 1128–1140 CrossRef CAS PubMed.
- Z. P. Yang, Q. F. Wu, W. Shao and S. L. You, J. Am. Chem. Soc., 2015, 137, 15899–15906 CrossRef CAS PubMed.
- Y. Ito, E. Nakajo, M. Nakatsuka and T. Saegusa, Tetrahedron Lett., 1983, 24, 2881–2884 CrossRef CAS.
- (a) H. Chen and J. F. Hartwig, Angew. Chem., Int. Ed., 1999, 38, 3391–3393 CrossRef CAS; (b) M. K. Tse, J. Y. Cho and M. R. Smith, Org. Lett., 2001, 3, 2831–2833 CrossRef CAS PubMed; (c) S. Shimada, A. S. Batsanov, J. A. K. Howard and T. B. Marder, Angew. Chem., Int. Ed., 2001, 40, 2168–2171 CrossRef CAS; (d) C. N. Iverson and M. R. Smith, J. Am. Chem. Soc., 1999, 121, 7696–7697 CrossRef CAS; (e) A. Ros, R. Fernández and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229–3243 RSC; (f) T. Ishiyama and N. Miyaura, J. Organomet. Chem., 1999, 680, 3–11 CrossRef.
- T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390–391 CrossRef CAS PubMed.
- (a) T. Ishiyama, Y. Nobuta, J. F. Hartwig and N. Miyaura, Chem. Commun., 2003, 2924–2925 RSC; (b) T. Ishiyama, J. Takagi, Y. Yonekawa, J. F. Hartwig and N. Miyaura, Adv. Synth. Catal., 2003, 345, 1103–1106 CrossRef CAS; (c) T. Ishiyama, J. Takagi, J. F. Hartwig and N. Miyaura, Angew. Chem., Int. Ed., 2002, 41, 3056–3058 CrossRef CAS.
- D. F. Fischer and R. Sarpong, J. Am. Chem. Soc., 2010, 132, 5926–5927 CrossRef CAS PubMed.
- J. M. Murphy, X. B. Liao and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 15434–15435 CrossRef CAS PubMed.
- X. B. Liao, L. M. Stanley and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 2088–2091 CrossRef CAS PubMed.
- S. Q. Zhou and Y. X. Jia, Org. Lett., 2014, 16, 3416–3418 CrossRef CAS PubMed.
- Y. Feng, D. Holte, J. Zoller, S. Umemiya, L. R. Simke and P. S. Baran, J. Am. Chem. Soc., 2015, 137, 10160–10163 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2018 |