
Highly degradable imine-doped mesoporous silica particles†
Leana
Travaglini
 a,
Pierre
Picchetti
a,
Ricardo
Totovao
a,
Eko Adi
Prasetyanto
ab and
Luisa
De Cola
*ac
a,
Pierre
Picchetti
a,
Ricardo
Totovao
a,
Eko Adi
Prasetyanto
ab and
Luisa
De Cola
*ac
aUniversité de Strasbourg, CNRS, ISIS UMR 7006, 8 allée Gaspard Monge, 67000 Strasbourg, France. E-mail: decola@unistra.fr
bFaculty of Medicine and Health Sciences, Atma Jaya Catholic University of Indonesia Jl, Pluit Raya 2, 14440 Jakarta, Indonesia
cInstitut für Nanotechnologie (INT) – Building 640, Karlsruhe Institute of Technology (KIT) – Campus Nord, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 31st October 2018
Abstract
The degradation of mesoporous silica particles (MSPs) in water is a key aspect that boosts their use especially in bio-related fields. Although MSP degradation in aqueous media has been proven, big efforts have been devoted to tuning silica dissolution in order to obtain functional materials whose degradation can be finely controlled and enhanced, to tackle the issue of bioaccumulation. In particular, the introduction of stimuli-responsive functional groups into the silica framework was proven to be a successful strategy. Yet, the fast dissolution of silica particles in aqueous media in the absence of external stimuli has to be fully addressed. In this context, we reported herein the preparation and thorough characterisation of MSPs containing imine groups embedded within the silica framework (Im-MSPs). Particles with different contents of imine groups have been investigated in order to assess the effect on the physicochemical properties and the Im-MSPs showed fast degradation in both acidic and neutral aqueous solutions, at a rate that depended on the pH value. Of special interest is their fast degradation at acidic pH, where instead MSPs are normally more stable. The described results unveil the potential of these particles in applications that require a fast degradation in aqueous media.
Introduction
Mesoporous silica particles (MSPs) have gained a great deal of attention in the last two decades due to their unique properties, such as large surface areas, high loading capacity, ease of functionalisation, cost-effective synthesis and biocompatibility.1 Due to the possibility of easily introducing organic functionalities and modifying both the mesostructure and overall morphology, MSPs can serve as versatile tools in tailoring materials able to fulfil specific functions.2–7 MSPs have been hence explored for a wide variety of applications ranging from catalysis, to sensing and drug delivery.8–13Amongst the features of MSPs, their degradation in aqueous media has played a key role in boosting their use especially in bio-related fields. In fact, MSPs degrade in aqueous media being hydrolysed to silicic acid Si(OH)4, which is non-toxic and can be easily cleared from the body.14,15 Nevertheless, the degradation rate depends on numerous factors, such as silica condensation degree, pore size, organic functionalisation, particle surface area and type of dispersion medium.15 Data reported so far on MSPs mainly describe the degradation in simulated biological fluids or in buffer solutions, and the complete dissolution of the particles is reported to occur in a time scale that ranges from a few days to several weeks depending on the types of both fluid and particles. Particularly challenging is the fast degradation at slightly acidic pH (5–6), since silica hydrolysis occurs at a lower rate compared to that at neutral pH.15–17
Big efforts have been devoted to tuning silica dissolution, in order to obtain functional materials whose degradation can be finely controlled and enhanced, to tackle the issue of bioaccumulation and incomplete release of active molecules.15 In this context, the incorporation within the silica framework of organic moieties cleavable in specific biological conditions was proven to be a successful strategy. In particular, organo-bridged silica-based particles doped with redox-responsive groups18–21 (i.e. disulphide bonds) sensitive to the reductive intracellular environment, redox and oxidation-responsive (e.g. di-selenide)22 or enzymatically–cleavable bonds (e.g. oxamides, amides) have been reported.23–25 Yet, the fast dissolution of silica particles in aqueous media in the absence of external stimuli has to be fully addressed, in particular at slightly acidic pH. Particles degrading at a high rate in water are not only of interest for specific delivery applications but could also be employed to template polymer nanomaterials or organic capsules preventing the use of harmful HF for their final removal.26 Lately, a carbamate-based alkoxysilicate has been used to enhance the degradation of non-porous silica particles in aqueous media,27 but no works have been reported to-date on MSPs.
To induce enhanced degradation of MSPs in water, in particular at acidic pH values, we decided to introduce a diimine-bearing organo-bridged alkoxysilane. The introduction of imine functional groups within the silica network was first reported by Bhaumik and co-workers,28 who prepared mesoporous organosilica materials for metal chemosensing by using a diimine-based linker. In this work we introduced within MSPs the silicate linker 1 shown in Fig. 1. The choice of compound 1 was dictated by the presence of two aromatic imine groups, which are highly reactive towards hydrolysis and therefore more effective in triggering the particle degradation in water. Microporous organosilica bulk material containing compound 1 has been previously reported and employed as an absorbent for metal cations.29 While we were writing this paper, a communication appeared on 50 nm mesoporous silica nanoparticles containing organosilane 1.30 However, we report a full detailed characterisation of 200 nm MSPs and of their behaviour at different pH values demonstrating how the kinetics of degradation can be controlled in different media.
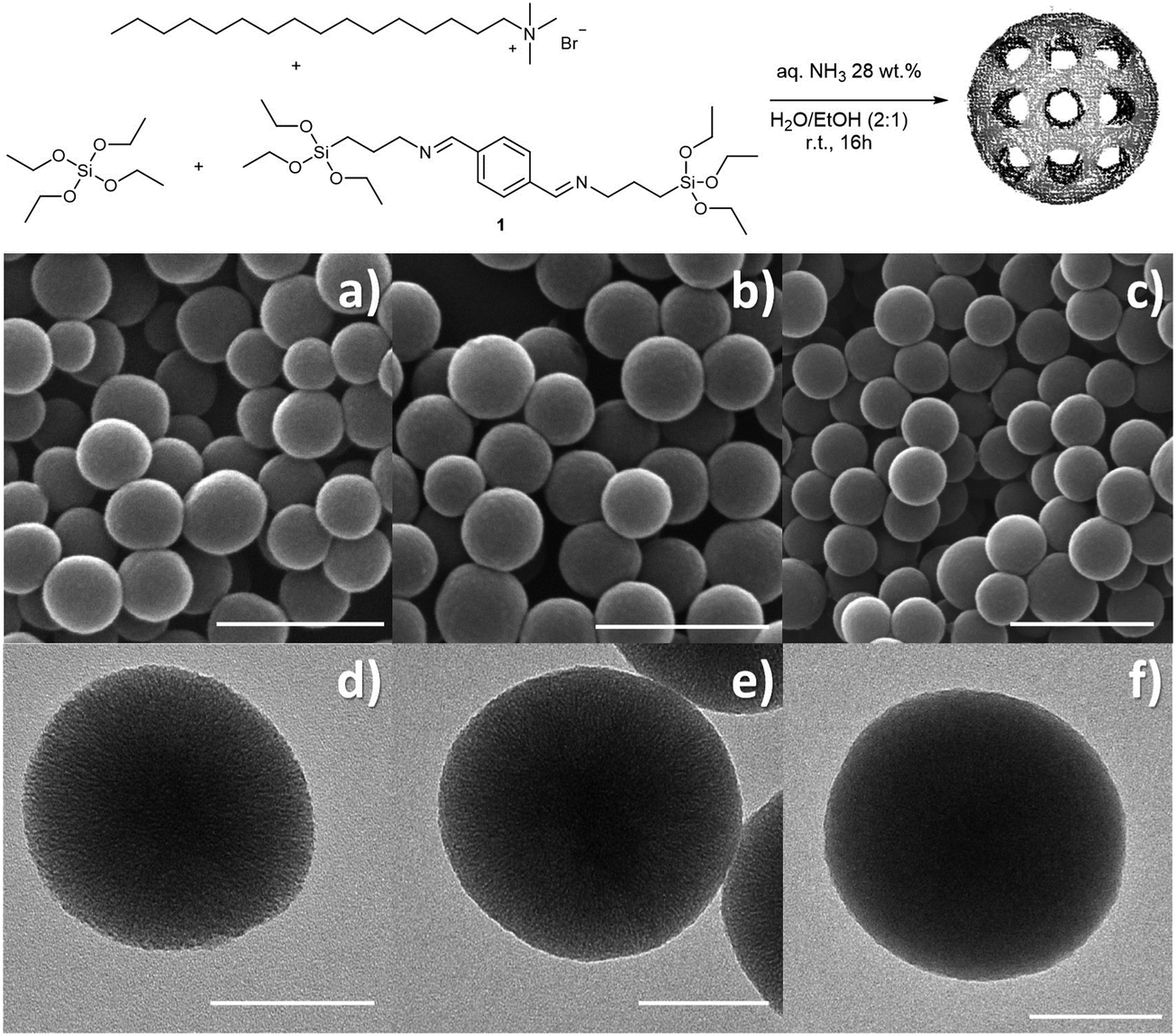 | ||
| Fig. 1 Schematic representation of the Im-MSP preparation (top); SEM images of (a) MSPs, (b) Im-MSPs-10% and (c) Im-MSPs-25%, scale bar = 500 nm; TEM images of (d) MSPs, (e) Im-MSPs-10% and (f) Im-MSPs-25%, scale bar = 100 nm. | ||
Hence, we reported the preparation and study of mesoporous organosilica particles containing the imine linker 1 embedded in the silica network, which we named Im-MSPs. The particles were prepared by co-condensing 1 with tetraethyl orthosilicate (TEOS) as schematically represented in Fig. 1. Im-MSPs with two different contents of imine-linker were prepared, in order to investigate the effect of the doping degree on the physicochemical properties of the particles, with a special focus on their degradability. The thorough characterization of the materials and investigation of their degradation are presented herein.
Experimental
Synthesis of mesoporous silica particles (MSPs)
CTAB (100 mg) and aq. NH3 (28 wt%, 710 μL) were dissolved in a H2O/EtOH mixture (60/30 v/v, 90 mL) and stirred (500 rpm) at r.t., TEOS (250 μL) was added and the mixture was stirred for 16 hours. The precipitate was recovered by centrifugation at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g and washed with H2O/EtOH (1:1, 2×) and EtOH (2×). Surfactant removal was performed by extraction in EtOH at 80 °C overnight.
000 × g and washed with H2O/EtOH (1:1, 2×) and EtOH (2×). Surfactant removal was performed by extraction in EtOH at 80 °C overnight.
Synthesis of diimine linker 1
The diimine-linker 1 was synthesised modifying a procedure reported in the literature.29 Terephthaldehyde (2.0 mmol, 268 mg) was dissolved in dry EtOH (11 mL) and stirred at r.t. under nitrogen atmosphere. (3-Aminopropyl)-triethoxysilane (APTES, 4.0 mmol, 936 μL) was added and the mixture was heated up to 80 °C and stirred for 2 h 30 min. The mixture was allowed to cool down and stirred for 10 min after the addition of anhydrous Na2SO4. The solid was removed by filtration and the organic solvent removed under reduced pressure to afford the pure compound 1 as a yellowish oil (944 mg, 87%). 1H NMR (400 MHz, CDCl3) δ 8.28 (2H, s, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–), 7.75 (4H, s, H aromatic), 3.82 (12H, q, 8.3 Hz, OCH2CH3), 3.63 (4H, t, 3.6 Hz, CH2–CHN), 1.81 (4H, m, CH2CH2CH2), 1.22 (18H, t, 1.2 Hz, OCH2CH3), 0.68 (4H, m, CH2Si); 13C (101 MHz, CDCl3) δ 160.8, 138.3, 128.5, 64.6, 58.6, 24.5, 18.5, 8.3. The NMR spectra are reported in the ESI.†
N–), 7.75 (4H, s, H aromatic), 3.82 (12H, q, 8.3 Hz, OCH2CH3), 3.63 (4H, t, 3.6 Hz, CH2–CHN), 1.81 (4H, m, CH2CH2CH2), 1.22 (18H, t, 1.2 Hz, OCH2CH3), 0.68 (4H, m, CH2Si); 13C (101 MHz, CDCl3) δ 160.8, 138.3, 128.5, 64.6, 58.6, 24.5, 18.5, 8.3. The NMR spectra are reported in the ESI.†
Im-MSP preparation
CTAB (100 mg) and aq. NH3 (28 wt%,710 μL) were dissolved in an ethanol/water mixture (29/60 v/v) and stirred at r.t. A solution TEOS/1 in EtOH (1 mL) (224.8 μL/60.5 mg and 187.4 μL/151.2 mg, for the Im-MSPs-10% and Im-MSPs-25%, respectively) was added and the mixture was stirred (500 rpm) at r.t. overnight. The precipitate was recovered by centrifugation at 20000 × g and thoroughly washed with distilled H2O/EtOH (1:1, 2×) and EtOH (2×). The organic template was removed by refluxing the particles in EtOH for 6 h. The material was recovered by centrifugation, re-dispersed in EtOH, sonicated for 5 minutes, and centrifuged again. The particles were then refluxed for a further 12 h in EtOH and centrifuged, and finally washed with EtOH (3×) and dried overnight under reduced pressure.
Degradation study
The in vitro degradation of Im-MSPs was investigated in aqueous solutions at pH 5.2 and 7.4 and studied by means of scanning transmission electron microscopy (STEM), UV-Vis absorption spectroscopy and inductively coupled plasma atomic emission spectroscopy (ICP-AES). For comparison, all the experiments were performed also on the extracted silica model particle MSPs.Results and discussion
Synthesis and characterization of the particles
In designing our imine-doped MSPs (Im-MSPs), the first step was the optimisation of a protocol to obtain MSPs homogenous in size and characterised by high porosity. This optimised procedure allowed for the preparation of MSPs for comparison purposes and was further modified to prepare the hybrid Im-MSPs. The MSPs have been prepared by using a modified Stöber protocol31 (see Section 2.1 for synthesis details) using hexadecyltrimethylammonium bromide (CTAB) as the structure directing agent, tetraethyl orthosilicate (TEOS) as the silica source, ethanol as the co-solvent and aqueous ammonia as the basic catalyst. The reaction was performed at room temperature, under vigorous stirring for 16 h. The particles were recovered by centrifugation, washed thoroughly with a mixture of H2O/EtOH 2:1 (×3) and with EtOH (×2), dried and extracted in EtOH at 80 °C.
The morphological analysis conducted through scanning electron microscopy (SEM) showed the presence of homogenous spherical particles of ca. 220 nm in diameter (Fig. 1a and Fig. S1, ESI†). Dynamic light scattering (DLS) measurements on a dispersion of particles in ethanol confirmed both the size and low dispersity (hydrodynamic diameter Dh = 278 ± 68 nm, Fig. S1d, ESI†). Transmission electron microscopy (TEM, Fig. 1d and Fig. S1e, ESI†) showed porous particles, characterized by pores aligned towards the centre of the particles. The presence of an ordered mesoporous system was confirmed also by the small angle X-ray scattering pattern (SAXS, Fig. S2a, ESI†) recorded on the particles that showed a Bragg peak at q = 1.90 nm−1. A type IV isotherm, typical of mesoporous materials, was revealed by nitrogen sorption measurement (Fig. S2b, ESI†) that allowed for the calculation of a pore size distribution of 2.9 nm, total pore volume of 0.86 cm3 g−1 and BET surface area of 1243 m2 g−1.
Once the protocol for the model material had been established, hybrid particles containing two different amounts of imine-linker were prepared, in order to investigate the effect of the doping degree on the physicochemical properties of the particles and in particular on their degradability. The diimine-bearing alkoxysilane 1, synthesised upon reaction of terepthalaldehyde and APTES, was co-condensed with TEOS at two different molar ratios, namely TEOS:1 10:1 and 4:1. For the sake of clarity, the so-obtained two types of particles will be addressed as Im-MSPs-10% and Im-MSPs-25%, respectively, referring to the theoretical organic content based on the ratio between the two alkoxysilanes used during the synthesis. The SEM analysis performed on the recovered and washed materials (Fig. S3, ESI†) showed the presence of spherical particles comparable in size to MSPs, but also of objects of several microns characterized by a very rough surface and similar to collapsed spheres. The formation of these bigger objects was most likely due to the introduction of the hydrophobic organoalkoxysilane that inevitably induced a variation in terms of interactions and reaction rate. The Im-MSPs were therefore carefully purified by means of centrifugation/dispersion cycles (5000 × g 5 min, sonication 5 min, 5 cycles), in order to allow the bigger objects to sediment in the centrifuge tube while the smaller particles remain dispersed in supernatants. SEM was used to monitor the entire purification process, and the images acquired on the residual precipitate (Fig. S4, ESI†) showed the occurred enrichment in bigger objects. Conversely, the SEM micrographs on the purified Im-MSPs, taken after removal of the surfactant by extraction in EtOH at 80 °C, (Fig. 1b, c and Fig. S5, ESI†) showed the absence of big objects and the presence of homogenous spherical particles of size comparable to the model MSPs, as confirmed by the size distribution and DLS measurements reported in Fig. S5 (ESI†).
TEM analysis confirmed the morphology and clearly showed for Im-MSPs-10% a radial distribution of pores (Fig. 1e), indicating that the introduction of the organic linker, to this extent, does not affect the mesostructure. Conversely, in the case of Im-MSPs-25% even though the images revealed the presence of a porous system (Fig. 1f) a highly ordered array of pores could not be observed, suggesting that the introduction of a higher amount of organic linker had in this case affected the internal ordering. The SAXS patterns recorded on the Im-MSPs (Fig. 2a) were similar to that recorded on the model particles. The peaks were centred at q = 1.65 and 1.70 nm−1 for Im-MSPs-10% and Im-MSPs-25%, respectively, suggesting that the introduction of the organic linker has induced a slight increase of the distance between the pore centres. For Im-MSPs-25% the peak is less intense and slightly broader, indicative for a decrease of the order of the mesostructure, most likely induced by the higher content of the bulky and hydrophobic imine-based linker. The porosity of the particles was assessed by nitrogen sorption, which revealed for both the particles the IV type isotherm typical of mesoporous materials (Fig. 2b and c). For Im-MNPs-10% the related average pore width was found to be 2.7 nm, while the total pore volume was 0.52 cm3 g−1 and the BET surface area was 816 m2 g−1. Slightly lower values were instead obtained for Im-MNPs-25%, since the average pore diameter was found to be 2.4 nm, the total pore volume was 0.45 cm3 g−1 and the BET surface area was 638 m2 g−1. In accordance with SAXS analysis, the lower porosity observed for Im-MSPs-25% may be most likely attributable to the higher content of organic linker present in the particles.
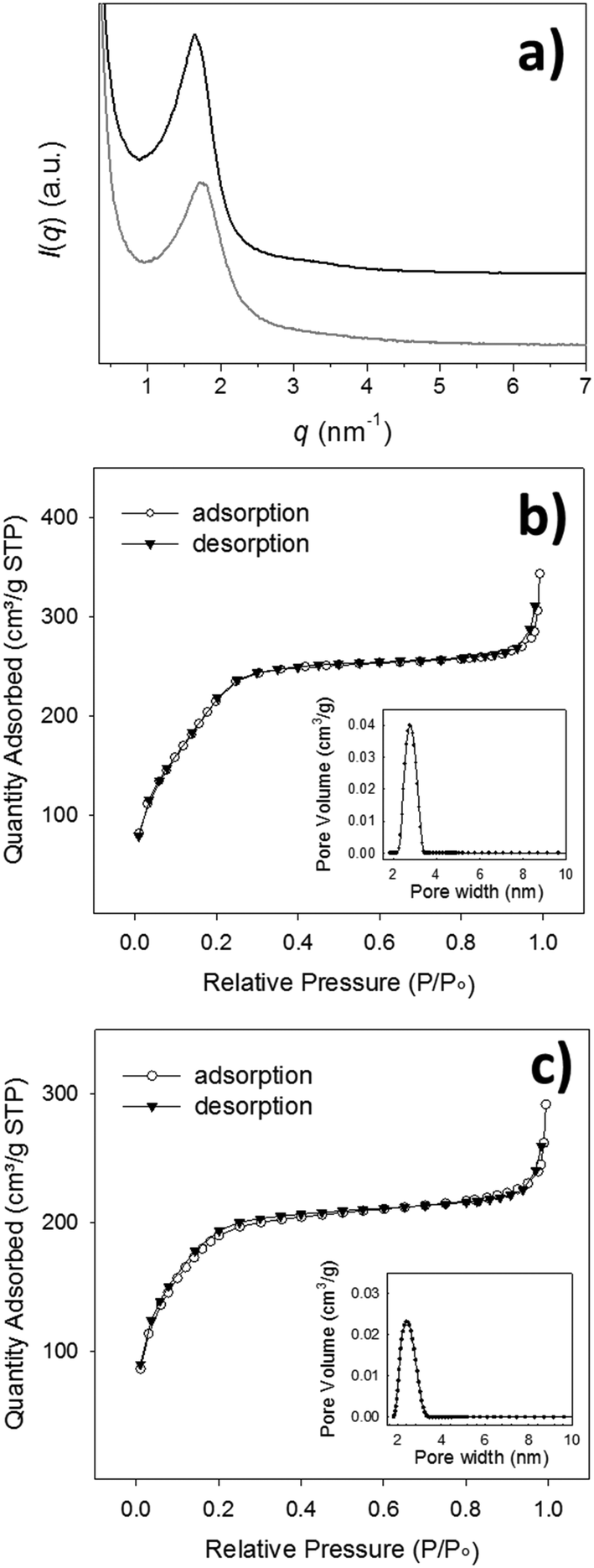 | ||
| Fig. 2 (a) SAXS patterns recorded on Im-MSPs-10% (black line) and Im-MSPs-25% (grey line); for the sake of clarity, the profiles have been given arbitrary displacements along the intensity axis. Adsorption/desorption isotherms obtained on (b) Im-MSPs-10% and (c) Im-MSPs-25%. The pore width distributions are reported in the insets. | ||
The quantitative analysis of the content of organic material embedded within the silica framework was determined by TGA on the Im-MSPs after surfactant removal (Fig. 3a). In a temperature range in which the model particles did not show any weight loss, Im-MSPs-10% and Im-MSPs-25% were found to contain 22 and 35 wt% of organic material, respectively. The effective incorporation of the imine-based linker was proven by attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR), X-ray photoelectron spectroscopy (XPS) and UV-Vis absorption spectroscopy. In fact, the ATR-FTIR transmittance spectra recorded on the particles (Fig. 3b) showed the typical band of CN stretching mode of imine groups at 1645 cm−1 confirming the presence of imine functional groups. In addition, the Csp3–H anti-symmetrical and symmetrical stretching mode bands at 2925 and 2855 cm−1, respectively, and band centred at 2981 cm−1 attributable to the Csp2–H stretching mode proved the presence of both the aliphatic chain and the aromatic system. As a further confirmation, XPS survey spectra (Fig. S6, ESI†) showed for both the Im-MSPs the presence of C(1s) and N(1s) signals at 285 and 399 eV. Moreover, in agreement with TGA results, a different organic content was detected in the two hybrid particles, as from the survey analysis the Si/N ratio was found to be 74:24 for Im-MSPs-10% and 65:35 for Im-MSPs-25%. The high-resolution scans for C(1s) and N(1s) clearly revealed the presence of the linker within the silica network. In particular, the deconvolution of the C(1s) peak (Fig. S8, ESI†) revealed for both the particles the presence of two components C-1 and C-2 at 284.1 and 284.8 eV, respectively. The component C-1 could be attributed to C-Si, Csp2, while the component C-2 to Csp3 and CN. In addition, the deconvolution of the N(1s) peak (Fig. S9, ESI†) revealed for both the particles the presence of two components N-1 and N-2. The major component N-1, centred at 399 eV, can be attributed to the nitrogen of imine groups (CN), while the minor component N-2, at ca. 401 eV is attributable to nitrogen of amines that may have probably been formed on the surface upon hydrolysis during the synthesis or the washings. The peak attribution has been performed according to the literature.32,33
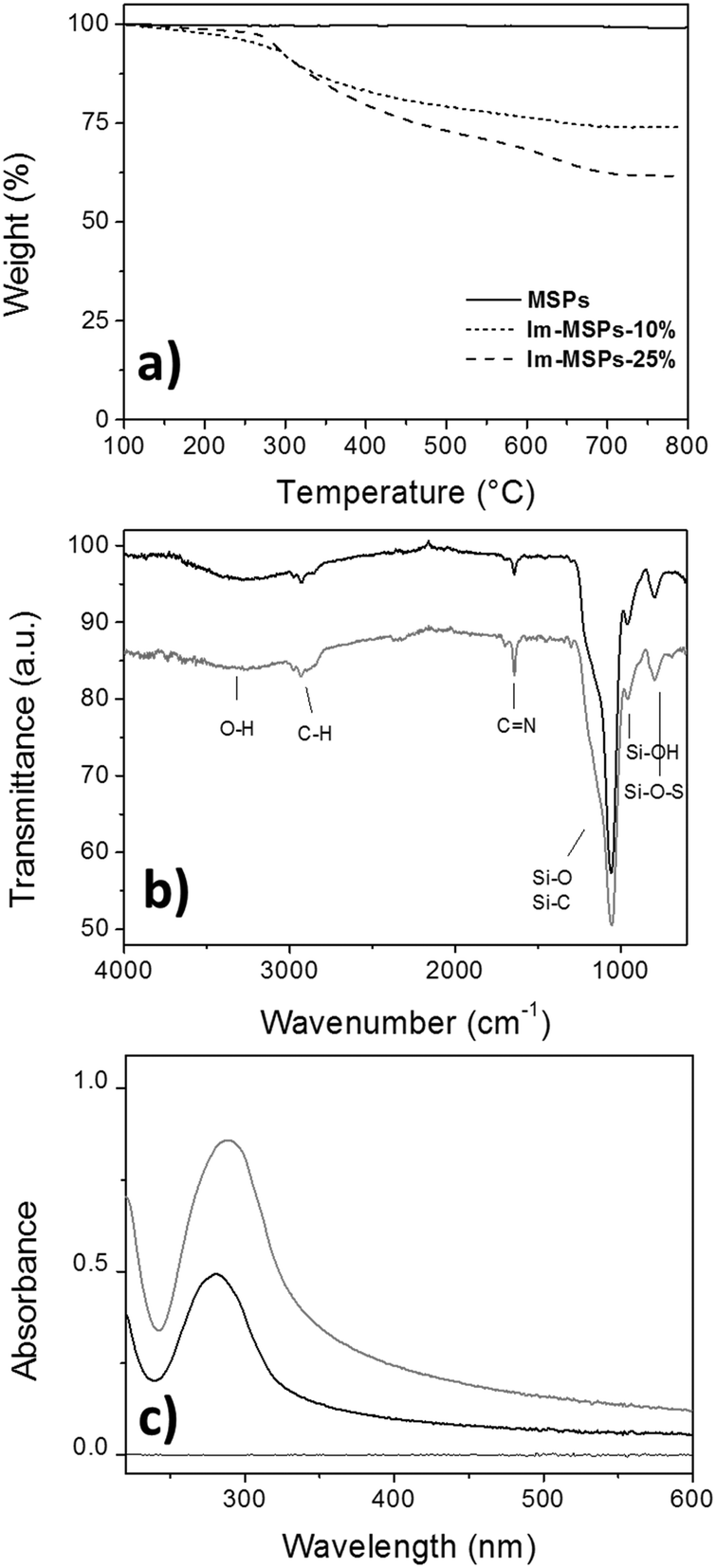 | ||
| Fig. 3 (a) TGA profiles of MSPs, Im-MSPs-10% and Im-MSPs-25%; (b) ATR-FTIR spectra of Im-MSPs-10% (black line) and Im-MSPs-25% (grey line); (c) UV-Vis absorption spectra of Im-MSPs-10% (black line) and Im-MSPs-25% (grey line) in EtOH (c = 0.05 mg mL−1), the samples were sonicated for 2 minutes before the measurement. | ||
Finally, the UV-Vis absorption spectra recorded on dispersions of the Im-MSPs in EtOH prepared at the same concentrations (c = 0.05 mg mL−1, Fig. 3c) showed the presence of the imine linker embedded within the silica framework. The band at ca. 280 nm observed for both the particles is attributable to the n–π* transition typical of the CN imine group. It is also possible to observe that the Im-MPSs-25% show a more intense band, in agreement with the different organic content of the two types of hybrid particles. If compared with the spectrum recorded on a solution of the organosilane 1 in EtOH (Fig. S10, ESI,†λmax = 275 nm), it is possible to observe a slight red shift (λmax = 281 nm for Im-MSPs-10% and λmax = 285 nm for Im-MSPs-25%), due to the embedment within the silica network. Moreover, the spectra of the hybrid particles showed a significant baseline shift, due to light scattering from the Im-MSPs dispersed in the medium. Notably, the high content of imine linker within the particles renders them more hydrophobic than silica model MSPs, increasing their tendency to form aggregates. This effect is more pronounced for the Im-MSPs-25%, explaining hence the bigger shift.
Degradation study of Im-MSPs
Once the properties and composition of the particles had been investigated, the evaluation of their degradation in aqueous solutions at both pH 7.4 and 5.2 was performed. Given the presence of the characteristic UV absorption band of imines at ca. 280 nm, the cleavage of the imine bond and the subsequent breakage of the hybrid particles were monitored by UV-Vis absorption spectroscopy. Im-MSPs were dispersed in acetate and PBS buffer solutions (pH = 5.2 and 7.4, respectively) at the same concentration of 0.1 mg mL−1 and stirred at room temperature. UV-Vis absorption spectra were recorded on the dispersions immediately after their preparation (t0) and after 3 and 6 hours and 1, 3 and 5 days. Before each measurement the samples were sonicated for 60 seconds to ensure a comparable degree of dispersion. As displayed in Fig. 4a, the spectrum recorded on Im-MSPs-10% after the addition of the pH 5.2 buffer solution and 60 sec sonication revealed the presence of an intense peak with maximum of absorption at 264 nm overlapped with the typical absorption band of the imine CN groups (287 nm). The first peak can be attributed to the absorption profile of terepthalaldehyde, which is produced upon hydrolysis of the imine bond, as confirmed by comparison with the absorption spectrum recorded on a solution of the bis-aldehyde in the same buffer solution. In fact, this spectrum clearly showed a profile characterized by an intense absorption at 264 nm and a weaker peak at 304 nm (Fig. S11, ESI†), attributable to the π–π* transitions of the PhCO grouping and to the n–π* transition of the carbonyl group, respectively.34
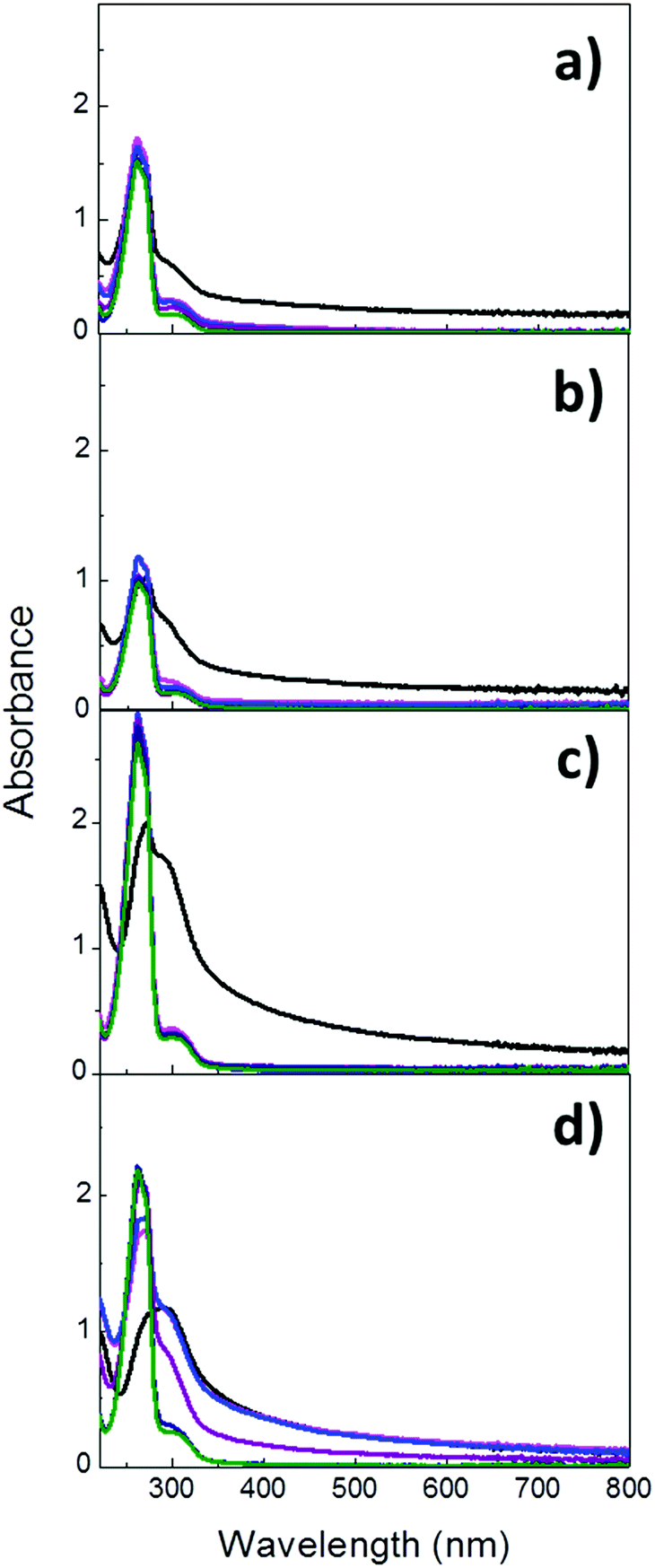 | ||
| Fig. 4 UV-Vis absorption spectra recorded on dispersions (c = 0.1 mg mL−1) of Im-MSPs-10% at (a) pH 5.2 (acetate buffer), (b) pH 7.4 (PBS buffer) and Im-MSPs-25% at (c) pH 5.2 (acetate buffer), and (d) pH 7.4 (PBS buffer) over a period of 5 days. The spectra have been recorded at t0 (black), 3 h (magenta), 6 h (blue), 1 d, (purple), 3 d (navy) and 6 d (green). | ||
This suggests that already after 1 min the imine bonds have been extensively hydrolysed. The spectra recorded in time showed that the absorption band due to the imine CN group is no longer observable already after 1 day, while only the bis-aldehyde absorption profile can be observed, and after 3 and 5 days its intensity does not vary, indicating the complete hydrolysis of imine groups already after 1 day. Moreover, the baseline shift observable in the first measurement is no longer present, explicable by a significant decrease of scattering due to the degradation of particles into smaller pieces. At pH 7.4 (Fig. 4b) a similar trend could be observed, but the spectrum recorded after the preparation of the dispersion showed a weaker contribution of the terephthalaldehyde and a stronger contribution of the imine signal. This was not surprising, considering that at neutral pH the hydrolysis of imine is slower. The absorption peak of the terephthalaldehyde reached its maximum value already after 1 day and remained constant up to 5 days. Moreover, its value is lower than that at acidic pH even though the particle concentrations were the same, in accordance with the fact that the imine hydrolysis occurs to a lesser extent at neutral pH. It has been also reported that analogous imines can form in neutral aqueous media,35 suggesting that the hydrolysis reaction is not complete under these conditions. The absorption band of the fewer imine groups still present is likely overlapping with the weaker band attributed to terephthalaldehyde. As regards the Im-MSPs-25%, a comparable behaviour was revealed but at both pH values after 1 minute the hydrolysis occurred to a lesser extent if compared to Im-MSPs-10%. In fact, at pH 5.2 (Fig. 4c) the bis-aldehyde peak at 264 nm could be clearly identified but it had not yet reached its maximum absorption value, contrarily to what had been observed for the Im-MSPs-10%. At pH 7.4 (Fig. 4d) instead the hydrolysis seems to proceed even slower, with the 264 nm peak appearing only as a shoulder of the imine peak and after 1 d a baseline shift attributable to scattering could still be observed. Nevertheless, in the first 24 hours the maximum of absorption was reached.
The different behaviour of the Im-MSPs-25% could be attributed to the higher organic content which renders the particles more hydrophobic and therefore more prone to aggregate in aqueous media, reducing the contact with water. In addition, the pores are smaller for these particles, as revealed by nitrogen absorption measurements, therefore influencing the surface area in contact with water and the diffusion of molecules within the pore channels. The degradation of the particles was also macroscopically obvious, since the scattering (Fig. S12 and S13, ESI†) from the dispersions decreased over time leading to clear solutions already after 6 hours in all cases except for the Im-MSPs-25% at pH 7.4, which turned to a clear solution after 1 d. This observation is in agreement with the initial presence and subsequent disappearance of baseline shifts in the UV spectra.
Deeper insights on the degradation of the particles at the microscopic level were gained by means of STEM investigation on aliquots (10 μL) of the dispersions taken at several time points (namely, t0, 3 h, 1 and 5 d). The analysis on Im-MSPs-10% at pH 5.2 (Fig. 5) clearly showed a structural breakdown already after 3 h, in accordance with the UV absorption spectrum revealing the presence of the hydrolysis product. After 1 day the particles could not be imaged in a straightforward manner. In the few regions of the grid where they were present, only smaller particles could be visualised, suggesting their dissolution from the exterior. This effect is even more pronounced after 5 days, where only amorphous and smaller particulate materials could be imaged (Fig. 5 and Fig. S14d, ESI†). A similar degradation process could be observed for the Im-MSPs-25% (Fig. 5c and Fig. S15, ESI†). The size distributions reported in Fig. S14 and S15 (ESI†) show this trend, demonstrating that the particles left in the mixture have a diameter down to four times smaller. However, it has to be noted that the size distributions are calculated considering only the spherical particles still present, not considering debris and small fragments of amorphous material. Conversely, at pH 7.4 the Im-MPSs degraded in a different fashion. As expected, and already suggested by the evolution in time of the UV absorption spectra, at pH 7.4 the organosilica particles degraded at a slower rate.36 The STEM images taken after 3 hours suggest that the particles degrade from the inside out (Fig. 5). In fact, many particles still show their spherical morphology but the contrast is no longer homogenous, highlighting the presence of a core in which there are areas denser than others, as can be clearly appreciated in the details of the images reported in the ESI† (Fig. S18). Moreover, it is possible to observe remains of particles similar to collapsed shells, suggesting that a slow etching from the inside is occurring. Over time this is more and more evident, and collapsed particles are present in the sample to a larger extent already after 1 d (Fig. 5c and d).
 | ||
| Fig. 5 STEM images taken over time on dispersions of Im-MSPs-10% (c = 0.1 mg mL−1) in (a) AcOH/AcONa aqueous buffer (pH 5.2) and in (b) PBS buffer (pH 7.4) and on Im-MSPs-25% (c = 0.1 mg mL−1) (c) in AcOH/AcONa aqueous buffer (pH 5.2) and in (d) PBS buffer (pH 7.4). Scale bar = 1 μm. | ||
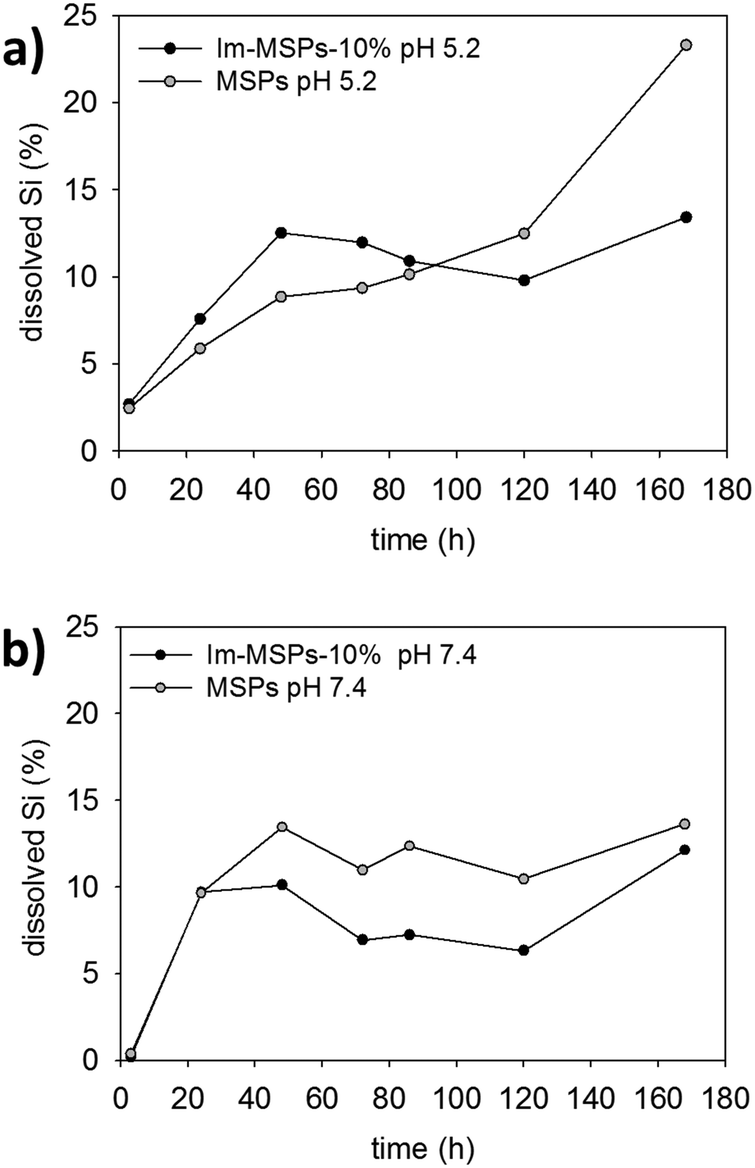 | ||
| Fig. 6 Percentage at each time point of Si dissolved calculated from ICPS-AES data for Im-MSPs-10% and MSPs at 25 °C in buffer solutions at (a) pH 5.2 and (b) pH 7.4. | ||
In order to better assess the effect of the diamine linker on the particle degradation, control experiments using MSPs were performed. Similarly, the MSPs were dispersed in acetate and phosphate buffer solutions, and aliquots of the dispersions analysed by STEM after 1 and 5 days. At pH 5.2 the STEM analysis (Fig. S19, ESI†) revealed a good stability of the particles in the time range of the observation, the particles not showing any obvious variation in morphology. The size distribution calculated from the micrographs revealed however a decrease (ca. 20 nm) of the mean particle size, indicative of a slow erosion of MSPs due to hydrolysis, whereas, at pH = 7.4 the particles were degraded to a significant extent, and after 5 days only degraded particles and small debris could be imaged. The difference in contrast for the degraded particles imaged suggests dissolution from the inside. The degradation from the interior may be attributed to the presence of a thin layer of denser silica on the surface of particles probably leading to higher resistance towards silica dissolution.37–39 This difference is attributed to a hardening of the outer layer, possibly due to silica deposition or further cross-linking of the silica matrix in contact with the species in solution. The particles were in fact synthesised in conditions that could favour a dissimilar condensation degree in the outer shell and a core with low condensation. Specifically, the particles were prepared at room temperature in the presence of a large amount of EtOH and were not calcined. At acidic pH this phenomenon was not observed since silica hydrolysis is known to be slower and silica particles are more stable in slightly acidic solutions.15,17,34
Quantitative information and comparison between the particles were obtained by measuring the content of dissolved silica in solution with inductively coupled plasma atomic emission spectroscopy (ICP-AES). We performed the experiments on MSPs and Im-MSPs-10%, the particles with the lowest content of imine, to assess the effect on the breakdown of even a low content of diimine-linker. The particles were dispersed in the buffer solutions at both pH 5.2 and 7.4 (c = 0.1 mg mL−1) in a dialysis tube and dialysed at 25 °C against the buffer solution (ESI† for details). The dialysis solutions were replaced after 3 hours, 1, 2, 3, 4, 5 and 7 days and the collected solutions analysed by ICP-AES. In Fig. 6 the percentages of dissolved Si at different time points are reported for Im-MSPs-10% and MSPs at pH 5.2 and 7.4. At acidic pH (Fig. 6a) the amount of Si detected in the solutions is higher for the Im-MSPs over the first 5 days indicating that the presence of the imine-based linker has a significant effect on the degradation of the particles, while for MSPs the Si content is always lower until the fifth day and only afterwards it increases, in accordance with the slow hydrolysis of silica occurring at acidic pH values. Instead, at neutral pH (Fig. 6b) the dissolution of MSPs is higher over the entire range of time compared to Im-MSPs. It must be noted though that such a comparison is done assuming that the behaviour of the two different particles is the same inside the dialysis tubes. Nevertheless, we cannot exclude the hypothesis that hydrophobicity of the oganosilica particles could influence the ability of the hydrolysed species to diffuse through the dialysis membrane. For the sake of completeness, the raw data and the total percentage of Si at each time point are reported in Fig. S21 and S22 (ESI†), respectively.
The behaviour of Im-MSPs-10% at neutral pH cannot be explained only in terms of imine cleavage or silica hydrolysis, since the imine groups are extensively cloven within the first 24 hours and that MSPs are hydrolysed to a larger extent. Such degradation behaviour is most probably due to the complex interplay between the hydrolysis of the organic moieties and silica. The slower dissolution, compared to MSPs, could be in fact due to the presence of organic hydrophobic moieties. In fact, it has been recently reported that organo-bridged MSPs tend to degrade undergoing a very complex dissolution/re-deposition process in neutral and basic conditions that leads to the formation of hollow particles or collapsed shells whose surface is enriched by the organosilane moiety that re-condenses on the particle surface.40,41 Moreover, this would be in agreement with the UV-Vis absorption data at pH 7.4 which suggest a non-complete hydrolysis of the imines.
Conclusions
In summary, we prepared and fully characterised mesoporous organosilica particles containing diimine-based organic moieties embedded within the silica network. The particles were proven to be highly degradable, with degradation occurring at different rates and following different mechanisms according to the pH value. The fast degradation occurring at slightly acidic pH is of special interest, since normally silica particles are particularly stable in acidic media. Our Im-MSPs are a promising example of material to be employed in those applications requiring both a fast release and fast particle degradation in aqueous media.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by L’Oréal. Prof. L. De Cola acknowledges AXA Research funds for financial support. The authors thank Dr S. Silvestrini for XPS measurements.References
- I. I. Slowing, J. L. Vivero-Escoto, B. G. Trewyn and V. S. Y. Lin, J. Mater. Chem., 2010, 20, 7924–7937 RSC.
- L. Travaglini and L. De Cola, Chem. Mater., 2018, 30, 4168–4175 CrossRef CAS.
- V. Giglio, S. Varela-Aramburu, L. Travaglini, F. Fiorini, P. H. Seeberger, L. Maggini and L. De Cola, Chem. Eng. J., 2018, 340, 148–154 CrossRef CAS.
- X. Du, X. Li, L. Xiong, X. Zhang, F. Kleitz and S. Z. Qiao, Biomaterials, 2016, 91, 90–127 CrossRef CAS PubMed.
- J. G. Croissant, F. Yevhen, A. Abdulaziz and N. M. Khashab, Adv. Healthcare Mater., 2018, 7, 1700831 CrossRef PubMed.
- S.-H. Wu, C.-Y. Mou and H.-P. Lin, Chem. Soc. Rev., 2013, 42, 3862–3875 RSC.
- A. Noureddine and C. J. Brinker, Chem. Eng. J., 2018, 340, 125–147 CrossRef CAS.
- D. Tarn, C. E. Ashley, M. Xue, E. C. Carnes, J. I. Zink and C. J. Brinker, Acc. Chem. Res., 2013, 46, 792–801 CrossRef CAS PubMed.
- J. G. Croissant, X. Cattoen, M. Wong Chi Man, J.-O. Durand and N. M. Khashab, Nanoscale, 2015, 7, 20318–20334 RSC.
- I. I. Slowing, B. G. Trewyn, S. Giri and V. S. Y. Lin, Adv. Funct. Mater., 2007, 17, 1225–1236 CrossRef CAS.
- S. Wu, Z. Li, J. Han and S. Han, Chem. Commun., 2011, 47, 11276–11278 RSC.
- D. Moelans, P. Cool, J. Baeyens and E. F. Vansant, Catal. Commun., 2005, 6, 307–311 CrossRef CAS.
- S. P. Hadipour Moghaddam, J. Saikia, M. Yazdimamaghani and H. Ghandehari, ACS Appl. Mater. Interfaces, 2017, 9, 21133–21146 CrossRef CAS PubMed.
- H. Yamada, C. Urata, Y. Aoyama, S. Osada, Y. Yamauchi and K. Kuroda, Chem. Mater., 2012, 24, 1462–1471 CrossRef CAS.
- J. G. Croissant, Y. Fatieiev and N. M. Khashab, Adv. Mater., 2017, 29, 1604634 CrossRef PubMed.
- G. B. Alexander, W. M. Heston and R. K. Iler, J. Phys. Chem., 1954, 58, 453–455 CrossRef CAS.
- K. Braun, A. Pochert, M. Beck, R. Fiedler, J. Gruber and M. Lindén, J. Sol-Gel Sci. Technol., 2016, 79, 319–327 CrossRef CAS.
- K. Möller and T. Bein, Chem. Mater., 2017, 29, 371–388 CrossRef.
- L. Maggini, I. Cabrera, A. Ruiz-Carretero, E. A. Prasetyanto, E. Robinet and L. De Cola, Nanoscale, 2016, 8, 7240–7247 RSC.
- J. Croissant, X. Cattoën, M. W. C. Man, A. Gallud, L. Raehm, P. Trens, M. Maynadier and J.-O. Durand, Adv. Mater., 2014, 26, 6174–6180 CrossRef CAS PubMed.
- S. Quignard, S. Masse, G. Laurent and T. Coradin, Chem. Commun., 2013, 49, 3410–3412 RSC.
- D. Shao, M. Li, Z. Wang, X. Zheng, Y.-H. Lao, Z. Chang, F. Zhang, M. Lu, J. Yue, H. Hu, H. Yan, L. Chen, W.-F. Dong and K. W. Leong, Adv. Mater., 2018, 1801198 CrossRef PubMed.
- L. Maggini, L. Travaglini, I. Cabrera, P. Castro-Hartmann and L. De Cola, Chem. – Eur. J., 2016, 22, 3697–3703 CrossRef CAS PubMed.
- Y. Fatieiev, J. G. Croissant, K. Julfakyan, L. Deng, D. H. Anjum, A. Gurinov and N. M. Khashab, Nanoscale, 2015, 7, 15046–15050 RSC.
- J. G. Croissant, Y. Fatieiev, K. Julfakyan, J. Lu, A.-H. Emwas, D. H. Anjum, H. Omar, F. Tamanoi, J. I. Zink and N. M. Khashab, Chem. – Eur. J., 2016, 22, 14806–14811 CrossRef CAS PubMed.
- S. Yasuhiro, K. Tae-Wan, R. Ryong and T. Osamu, Angew. Chem., Int. Ed., 2004, 43, 5231–5234 CrossRef PubMed.
- Z. Gao, S. P. Hadipour Moghaddam, H. Ghandehari and I. Zharov, RSC Adv., 2018, 8, 4914–4920 RSC.
- D. Chandra, T. Yokoi, T. Tatsumi and A. Bhaumik, Chem. Mater., 2007, 19, 5347–5354 CrossRef CAS.
- M. Paul, N. Pal, P. R. Rajamohanan, B. S. Rana, A. K. Sinha and A. Bhaumik, Phys. Chem. Chem. Phys., 2010, 12, 9389–9394 RSC.
- L. Liu, C. Kong, M. Huo, C. Liu, L. Peng, T. Zhao, Y. Wei, F. Qian and J. Yuan, Chem. Commun., 2018, 54, 9190–9193 RSC.
- M. Grün, I. Lauer and K. K. Unger, Adv. Mater., 1997, 9, 254–257 CrossRef.
- Y.-C. Chiang, C.-Y. Lee and H.-C. Lee, Mater. Chem. Phys., 2007, 101, 199–210 CrossRef CAS.
- J. Kim, D. Jung, Y. Park, Y. Kim, D. W. Moon and T. G. Lee, Appl. Surf. Sci., 2007, 253, 4112–4118 CrossRef CAS.
- M. S. Baymak, K. L. Vercoe and P. Zuman, J. Phys. Chem. B, 2005, 109, 21928–21929 CrossRef CAS PubMed.
- C. Godoy-Alcántar, A. K. Yatsimirsky and J.-M. Lehn, J. Phys. Org. Chem., 2005, 18, 979–985 CrossRef.
- F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry: Structure and mechanisms, Plenum Press, 1984 Search PubMed.
- P. J. Kempen, S. Greasley, K. A. Parker, J. L. Campbell, H. Y. Chang, J. R. Jones, R. Sinclair, S. S. Gambhir and J. V. Jokerst, Theranostics, 2015, 5, 631–642 CrossRef CAS PubMed.
- Z. Teng, X. Su, Y. Zheng, J. Sun, G. Chen, C. Tian, J. Wang, H. Li, Y. Zhao and G. Lu, Chem. Mater., 2013, 25, 98–105 CrossRef CAS.
- Y. J. Wong, L. Zhu, W. S. Teo, Y. W. Tan, Y. Yang, C. Wang and H. Chen, J. Am. Chem. Soc., 2011, 133, 11422–11425 CrossRef CAS PubMed.
- X. Du, W. Li, B. Shi, L. Su, X. Li, H. Huang, Y. Wen and X. Zhang, J. Colloid Interface Sci., 2018, 528, 379–388 CrossRef CAS PubMed.
- E. Yamamoto, S. Uchida, A. Shimojima, H. Wada and K. Kuroda, Chem. Mater., 2018, 30, 540–548 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00438b |
| This journal is © the Partner Organisations 2019 |