DOI:
10.1039/C8QM00382C
(Research Article)
Mater. Chem. Front., 2018,
2, 2054-2062
Manipulating the positions of CH⋯N in acceptors of pyrimidine–pyridine hybrids for highly efficient sky-blue thermally activated delayed fluorescent OLEDs†
Received
5th August 2018
, Accepted 7th September 2018
First published on 12th September 2018
Abstract
Three novel TADF materials, 10-(6-(2,6-diphenylpyrimidin-4-yl)pyridin-3-yl)-9,9-diphenyl-9,10-dihydroacridine (DPAc-4PyPM), 9,9-diphenyl-10-(4-(2-phenyl-6-(pyridin-2-yl)pyrimidin-4-yl)phenyl)-9,10-dihydroacridine (DPAc-6PyPM) and 10-(4-(2,6-diphenylpyrimidin-4-yl)phenyl)-9,9-diphenyl-9,10-dihydroacridine (DPAc-TPPM) have been designed and synthesized. CH⋯N intramolecular hydrogen-bonding interactions were introduced into pyrimidine–pyridine hybrid acceptors, then the TADF properties can be tuned by skillful manipulation of the positions of CH⋯N in the acceptor moiety. Both DPAc-4PyPM and DPAc-6PyPM exhibit a higher PLQY than DPAc-TPPM. Simultaneously, the kRISC realized for DPAc-4PyPM is nearly 5 or 10 fold higher than that of its analogues. The optimized sky-blue device using DPAc-4PyPM as a dopant demonstrated the maximum external quantum efficiency, current efficiency and power efficiency of 24.34%, 53.89 cd A−1 and 36.79 lm W−1, respectively, which is comparable to most of the previously reported pyrimidine-based TADF OLEDs.
Introduction
Organic light emitting diodes (OLEDs) have gained continuous attention in academia and industry thanks to their potential for wearable displays and greener artificial lighting.1–10 Compared to fluorescent materials with a limited IQE of 25%, phosphors with the assistance of heavy-metals can harvest both singlet and triplet excitons and can theoretically approach 100% internal quantum efficiency (IQE).2,11 However, critical issues for phosphorescence OLEDs regarding high cost, toxicity and stability remain disadvantageous for application.12 Recently, pure organic thermally activated delayed fluorescence (TADF) molecules have emerged as high potential emissive materials capable of achieving 100% internal quantum efficiencies (IQEs) via efficient reverse intersystem crossing (RISC).13–16 Generally, a small singlet–triplet splitting (ΔEST) between the S1 and T1 states is required for TADF emitters to accelerate the RISC process. The widely used design principle is matching the donor (D) and acceptor (A) moieties in twisted structures, which can effectively separate the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), and achieve a small ΔEST.17 Unfortunately, a restricted orbital overlap is in favor of a minor ΔEST, but often leads to a low photoluminescence quantum yield (PLQY).18–20 In the past few years, numerous high-performance TADF molecules with judicious selection of different donor (D) and acceptor (A) constituents and careful management of their linking topologies have been designed and synthesized.16,21–30 However, the relatively long delayed lifetimes (τd) of TADF emitters would cause significant triplet-exciton involved quenching processes. As a result, it is necessary to obtain a high PLQY and kRISC for efficient TADF emitters by molecular engineering and to reveal their interaction.
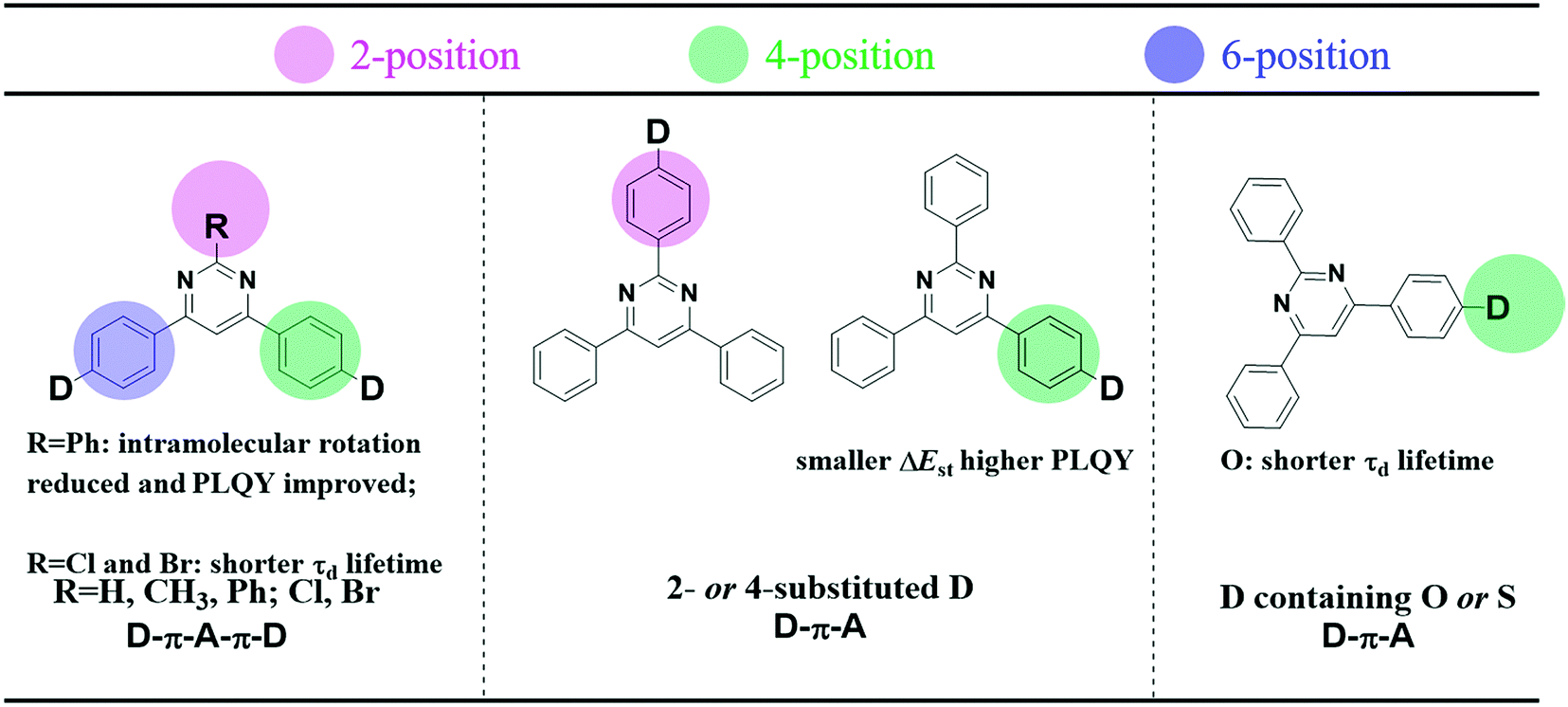
In comparison to the ubiquitous triazine acceptor, pyrimidine (PM) has advantages of facile functionalization and less stabilized unoccupied π orbitals.31 As shown in Fig. 1, a series of outstanding TADF emitters with a D–π–A–π–D configuration through symmetrical linking at the 4- and 6-position have been successively reported.31–37 The 2-substituted benzene and halogen of pyrimidine-based derivatives can reduce intramolecular rotation and shorten delayed fluorescence lifetimes (τd) improving device efficiency. Besides, the 4-phenyl substitution position of the donor units in 2,4,6-triphenylpyrimidine (2,4,6-TPPM) can induce a smaller ΔEST and a higher PLQY than its 2-phenyl substituted isomers.31,38 Recently, our group reported two novel pyrimidine derivatives with D–π–A configuration, and the heteroatom oxygen in the donor moiety offers a shorter τd than that of sulfur.20 Based on the above work, the effects of a substituent at the 2-position of pyrimidine (R = H, CH3, Ph; Cl, Br), linking topologies and heteroatom species in the donor moiety toward TADF properties have been systematically investigated (Fig. 1). Lately, three linear bipyrimidine core-based stereoisomers, 22bpmAc, 25bpmAc, and 55bpmAc, were synthesized and applied as green dopants in TADF devices. The two emitters with rigid skeletons due to the presence of intramolecular CH⋯N hydrogen-bonding interactions (25bpmAc and 55bpmA) showed higher PLQY relative to 22bpmAc by suppressing the nonradiative mechanism.39 As it is known, the dynamic equilibrium between the singlet excitons and triplet excitons is mainly established by the values of kF, kICkISC and kRISC.18,40 Because dynamic rate constants depend on the PLQY and lifetime (τ) (eqn (S1)–(S8)), the enhanced PLQY due to the introduction of CH⋯N intramolecular hydrogen-bonds will impact on kRISC.
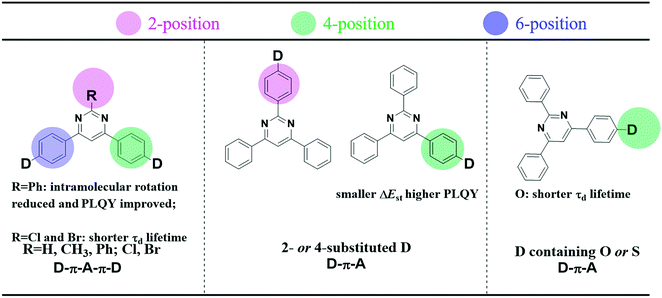 |
| | Fig. 1 (left) Effects of substituents at the 2-position of pyrimidine (R = H, CH3, Ph; Cl, Br), (middle) linking topologies and (right) heteroatom species in the donor moiety toward TADF properties. | |
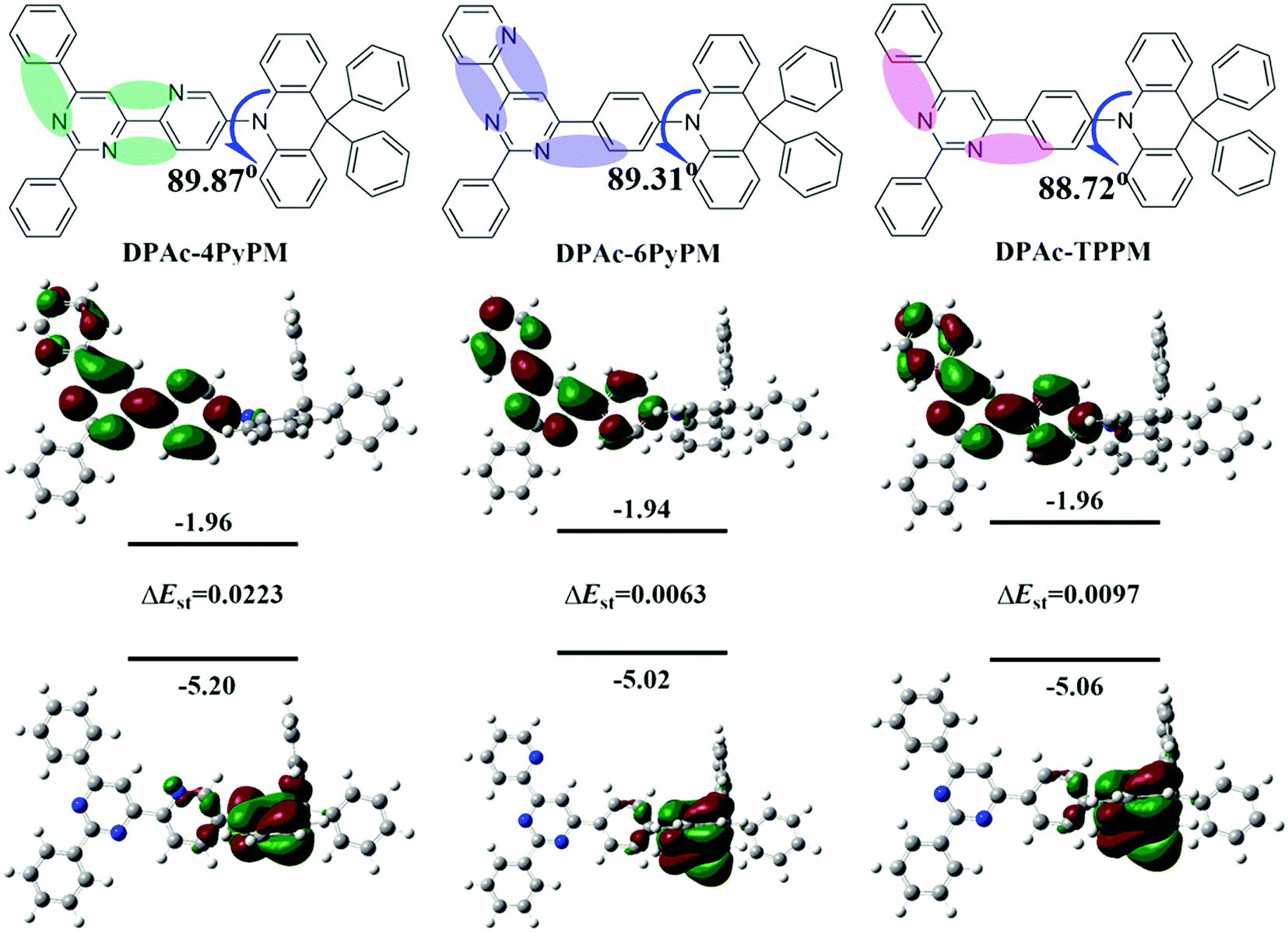
In order to further explore structure–property relationships of pyrimidine-based TADF emitters through tuning the positions of CH⋯N, the 4- and 6-positions of the pyrimidine ring were substituted with 2-pyridyl as acceptors (Fig. 2 and Fig. S1, ESI†). Then three novel TADF materials, 10-(6-(2,6-diphenylpyrimidin-4-yl)pyridin-3-yl)-9,9-diphenyl-9,10-dihydroacridine (DPAc-4PyPM), 9,9-diphenyl-10-(4-(2-phenyl-6-(pyridin-2-yl)pyrimidin-4-yl)phenyl)-9,10-dihydroacridine (DPAc-6PyPM) and 10-(4-(2,6-diphenylpyrimidin-4-yl)phenyl)-9,9-diphenyl-9,10-dihydroacridine (DPAc-TPPM) were designed and synthesized. Their electronic structures, crystallographic properties, thermal stabilities, electrochemical properties, photophysical properties and device performances were investigated. Both DPAc-4PyPM and DPAc-6PyPM with CH⋯N intramolecular hydrogen-bonding interactions displayed a higher photoluminescence quantum yield (PLQY) and external quantum efficiency (EQE) than their reference DPAc-TPPM. In particular, the sky-blue TADF OLED device based on DPAc-4PyPM displayed a maximum EQE, current efficiency, and power efficiency of 24.34%, 53.89 cd A−1and 36.79 lm W−1, respectively.
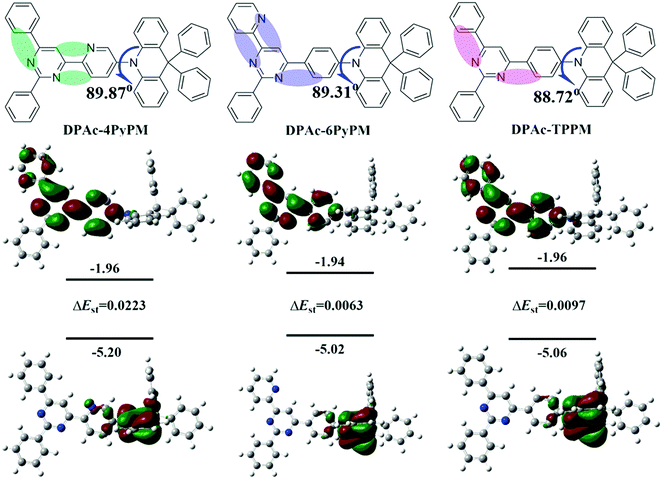 |
| | Fig. 2 Molecular structures (upper), and the HOMO/LUMO distributions evaluated by using the b3lyp/6-31g(d) level, and their singlet–triplet splitting (ΔEST) calculated with TD-b3lyp/6-31g(d). | |
Results and discussion
Molecular design and synthesis
The electronic cloud of the LUMO for TPPM distributes along the 4- and 6-positions of the phenyl and pyrimidine rings. Therefore, the pyridine was linked at the 4- and 6-positions of the pyrimidine ring to induce a distinct photophysical property change. Moreover, to ensure formation of intramolecular hydrogen-bonding interactions (CH⋯N) and further analyze the difference of TADF properties, the 4- and 6-positions of the pyrimidine ring were substituted by 2-pyridyl (Fig. 2 and Fig. S1, ESI†). The donor DPAc (9,9-diphenyl-9,10-dihydroacridine) with a rigid structure is widely used in molecular engineering for high-performance TADF.29,41 Combining all the above-mentioned considerations, three compounds (DPAc-4PyPM, DPAc-6PyPM and DPAc-TPPM) have been designed and synthesized. Density functional theory (DFT) calculations were utilized at the B3LYP/6-31G(d) level to investigate the geometric and electronic structures of DPAc-4PyPM, DPAc-6PyPM and DPAc-TPPM. The HOMO/LUMO distributions of the three compounds are displayed in Fig. 2. The HOMO is mainly distributed on the donor DPAc moiety, and the LUMO is spread at the acceptor of the pyrimidine–pyridine hybrid group except for the 2-substituted benzene in the pyrimidine ring. All three molecules exhibit large steric hindrance structures. For instance, the dihedral angles in the optimized structures between the donor unit connecting to the 4-position of pyrimidine–pyridine or the benzene hybrid plane are 89.87°, 89.31°, and 88.72° for DPAc-4PyPM, DPAc-6PyPM and DPPAc-TPPM, respectively. As is known, the value of ΔEST is proportional to the exchange interaction integral between the HOMO and the LUMO orbital wavefunction. We next calculated the S1 and T1 excited energies using time-dependent density functional theory (TD-DFT) at the B3LYP/6-31G(d) level in the gas phase based on the ground state geometries of the compounds. The theoretical ΔESTs are about 0.0223 eV for DPAc-4PyPM, 0.0063 eV for DPAc-6PyPM and 0.0097 eV for DPAc-TPPM, which indicate their potential to become efficient TADF materials.
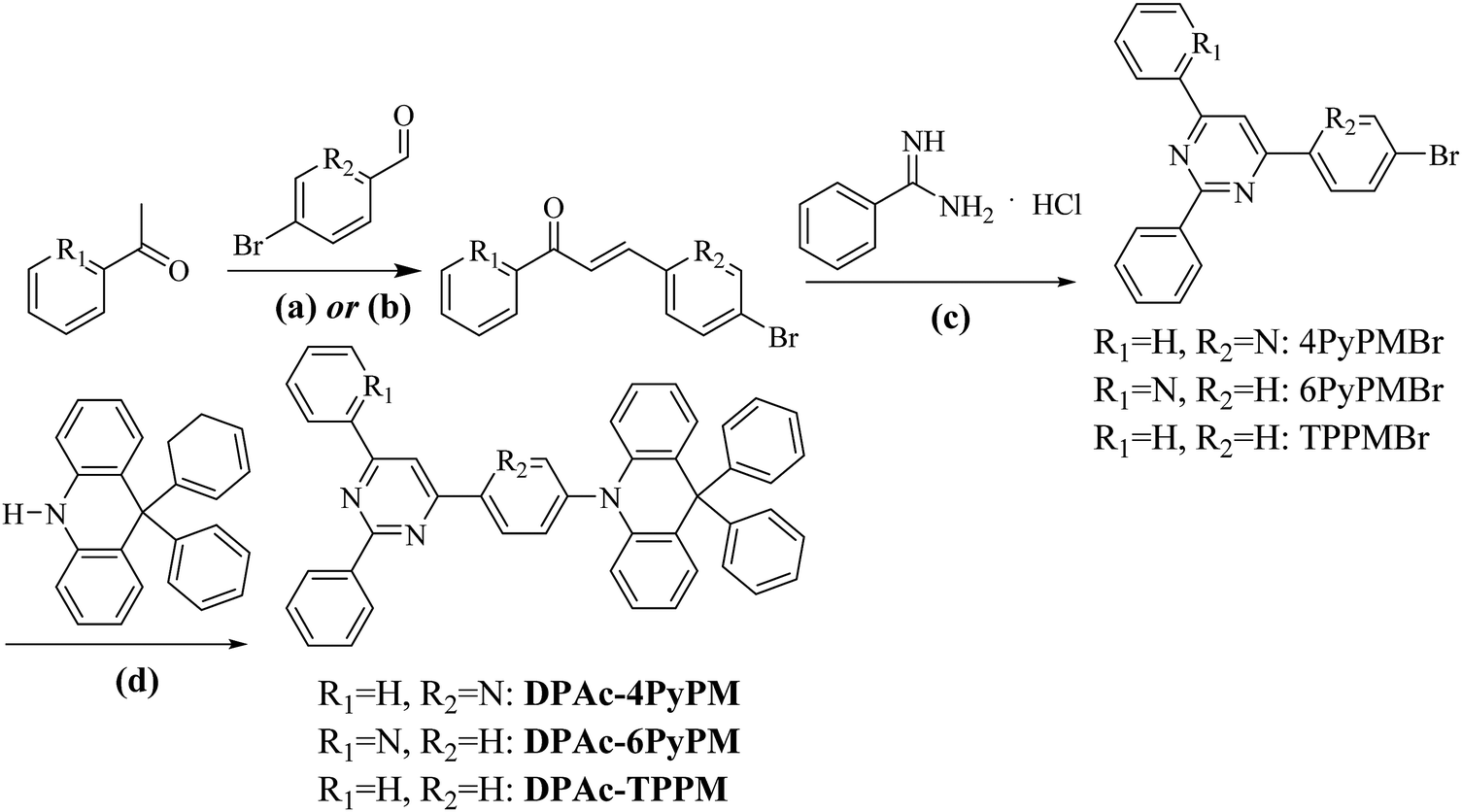
The synthetic routes and molecular structures are depicted in Scheme 1. Firstly, the pyridyl chalcones were synthesized through aldol condensation of the corresponding aldehyde and ketone (path a for R1 = H, R2 = N; path b for R1 = N, R2 = H). Then the above chalcone intermediates were allowed to react with benzamidine hydrochloride generating bromine-substituted acceptors of pyrimidine–pyridine or benzene hybrids (4PyPMBr, 6PyPMBr and TPPMBr). The final products were readily synthesized through the Buchwald–Hartwig amination of DPAc with the corresponding bromine-substituted pyrimidine–pyridine hybrids by employing a Pd(OAc)2/[(t-Bu)3PH]BF4 catalytic system. The detailed synthetic methods and analysis are listed in the experimental section. The target compounds were further purified by repeated temperature-gradient sublimation under vacuum conditions before measurements and device fabrication processes. The structures of these compounds were confirmed by 1H and 13C NMR spectroscopy, mass spectrometry, elemental analysis and single-crystal X-ray crystallographic analysis. It is worth noting that the central pyrimidinyl proton of 4PyPMBr and 6PyBr shifted downfield locating at 8.68 and 8.69 ppm, respectively, compared to that of TPPMBr at 7.97 ppm (Fig. S9, ESI†). This moving chemical shift illustrates that intramolecular CH⋯N bonding was formed between the pyrimidine and the nearby pyridyl groups.42
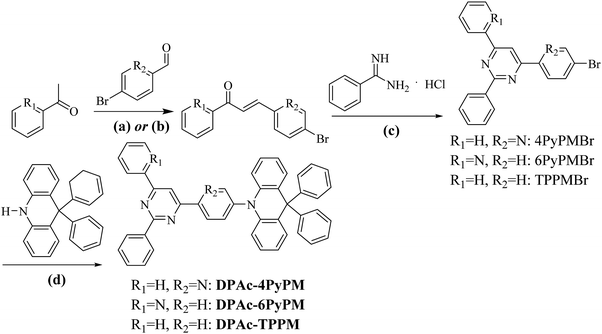 |
| | Scheme 1 Synthetic routes and chemical structures of the materials: (a) potassium hydroxide, methanol or (b) sodium hydroxide, ethanol, 0 → RT, overnight; (c) sodium hydroxide, ethanol, refluxed 8 h; and (d) Pd(OAc)2, [(t-Bu)3PH]BF4, t-BuONa, toluene, N2 110 °C 10 h. | |
X-ray single-crystal structure
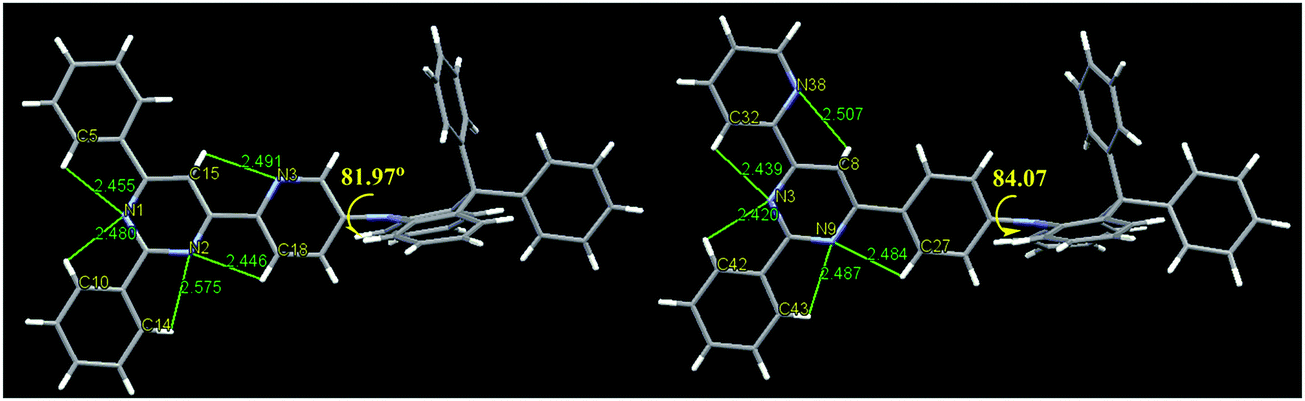
The single crystals obtained by slow evaporation of a solution in a mixture of dichloromethane and methanol. The crystal structure (Fig. 3) showed that the DPAc fragments are orthogonal to its connected pyridine or phenyl ring with a large dihedral angle (81.97° for DPAc-4PyPM and 84.07° for DPAc-6PyPM). This highly twisted structure, arising from the steric repulsion by the hydrogen atoms in the pyridine or phenyl and DPAc unit, contributes to effective spatial separation of the HOMO and LUMO and reduction of ΔEST. The multiple intramolecular CH⋯N interactions along the orientation of the electron cloud distribution for the LUMO were composed of C15H⋯N3, C18H⋯N2, and C5H⋯N1 for DPAc-4PyPM and C27H⋯N9, C32H⋯N3, and C8H⋯N38 for DPAc-6PyPM. These CH⋯N contacts could lock the intramolecular rotation and suppress nonradiative mechanisms.39 Similarly, for 2-phenyl, the involved intramolecular interactions (C10H⋯N1, C14H⋯N2 for DPAc-4PyPM and C42H⋯N3, C43H⋯N9 for DPAc-6PyPM) just rigidify the molecular structure and result in minor influences on the photophysical properties due to no electron cloud distributions.43 The packing diagrams from different axis directions for DPAc-4PyPM and DPAc-6PyPM are displayed in Fig. S2 (ESI†). Although the crystal structure of DPAc-TPPM wasn’t obtained, there is no doubt that DPAc-4PyPM and DPAc-6PyPM with one additional electron-deficient 2-pyridyl possess one more CH⋯N intramolecular interaction than that of DPAc-TPPM. Thus, the molecular rigidities of DPAc-4PyPM and DPAc-6PyPM would be stronger than that of DPAc-TPPM.
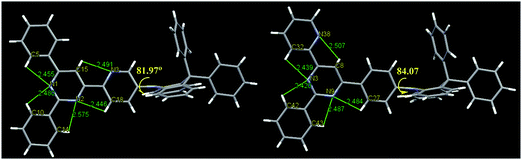 |
| | Fig. 3 (left) Crystal structures of DPAc-4PyPM (CCDC 1858076†) and (right) DPAc-6PyPM (1858077†). | |
Thermal and electrochemical properties
In Fig. S3 (ESI†), high decomposition temperatures (Td, corresponding to 5% weight loss) of 415 °C for DPAc-4PyPM, 401 °C for DPAc-6PyPM and 396 °C for DPAc-TPPM were measured by TGA. In the second heating run of the DSC measurement, the glass-transition temperature (Tg) of 166 °C was obtained for DPAc-6PyPM (Fig. S3, ESI†). It is regretful that no obvious glass transition temperatures (Tg) were observed for compounds DPAc-4PyPM and DPAc-TPPM (Fig. S3, ESI†). Good thermal stability of the compounds could ensure good film-forming properties in device fabrication and reduce phase separation. The electrochemical properties of these emitters are probed by cyclic voltammetry (CV). As shown in Fig. S4 (ESI†), the emitters presented clear oxidation behaviors, which can be assigned to the oxidation of the DPAc moiety. DPAc-4PyPM and DPAc-6PyPM presented lower reduction onset potentials than that of DPAc-TPPM, which can be attributed to the introduction of one additional electron-deficient pyridine. The energy levels were estimated with onset potentials using the energy level of ferrocene (Fc) (4.8 eV) as a reference and calibrated by comparing with Eonset (Fc/Fc+). Their HOMO levels were estimated to be −5.62 eV, −5.52 eV and −5.52 eV for DPAc-4PyPM, DPAc-6PyPM and DPAc-TPPM, respectively. The LUMO energy levels of DPAc-4PyPM, DPAc-6PyPM and DPAc-TPPM were found to be −2.73 eV, −2.52 eV and −2.48 eV, respectively, which were calculated using the equation ELUMO = EHOMO + Eoptg, where Eoptg is the optical band-gap obtained from the absorption spectra. The main physical properties are summarized in Table 1.
Table 1 Thermal and photophysical properties
| Compounds |
T
d/Tga (°C) |
λ
max,em
(nm) |
HOMO/LUMO/Egc (eV) |
E
S/ET/ΔEstd (eV) |
τ
p/τde (ns)/(μs) |
ϕ
PL
(%) |
k
RISC
(105 s−1) |
| N.A.: not available. Td was measured by TGA and Tg were measured by DSC. Measured in dilute toluene solution and the doped DPEPO films at room temperature. HOMO were measured from the CV; LUMO = HOMO + Eoptg, in which the optical energy gap (Eoptg) was derived from the absorption onset. Measured in doped DPEPO films at 77 K. Fitted according to Fig. 5b. The total PLQYs of emitters doped into DPEPO films under oxygen-free conditions at room temperature. Calculated using eqn (S1)–(S8). (Note: the x wt% emitters doped DPEPO, 10 wt% for DPAc-4PyPM and DPAc-6PyPM; 20 wt% DPAc-TPPM.) |
|
DPAc-4PyPM
|
415/N.A |
480/486 |
−5.62/−2.73/2.89 |
2.89/2.66/0.23 |
13.27/22.68 |
86 |
7.80 |
|
DPAc-6PyPM
|
401/166 |
460/468 |
−5.52/−2.52/3.00 |
3.00/2.80/0.20 |
11.19/16.26 |
83 |
1.33 |
|
DPAc-TPPM
|
396/N.A |
450/464 |
−5.52/−2.48/3.04 |
3.04/2.76/0.28 |
8.02/37.70 |
70 |
0.69 |
Photophysical and TADF properties
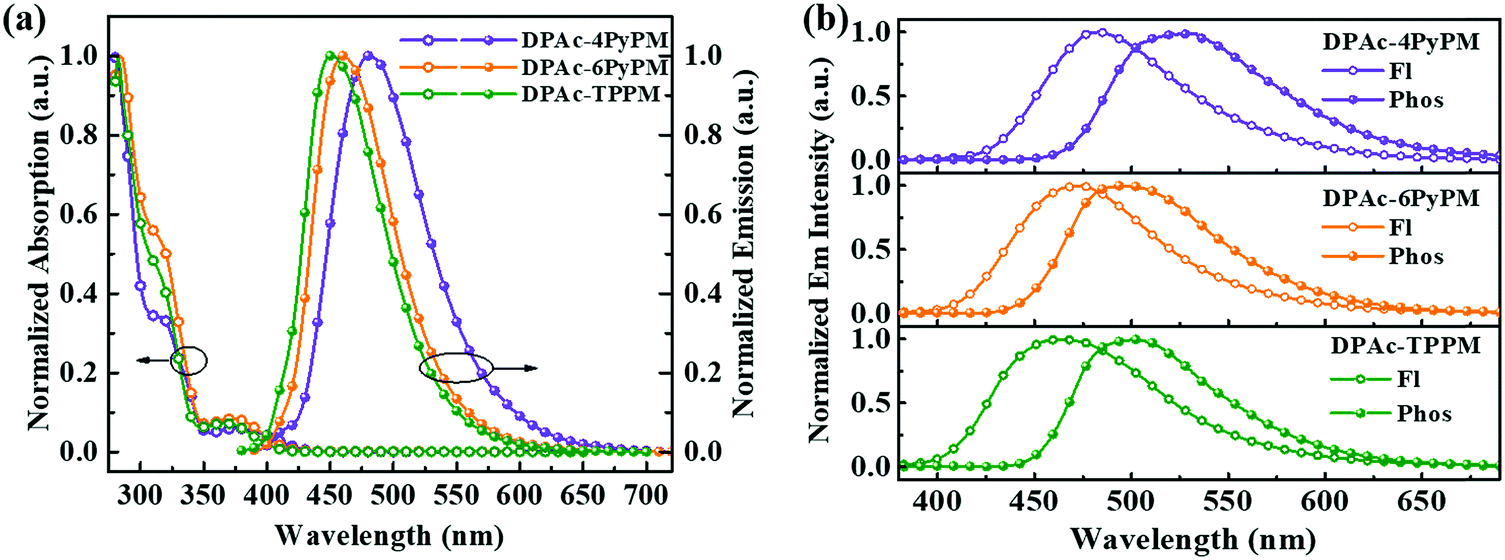
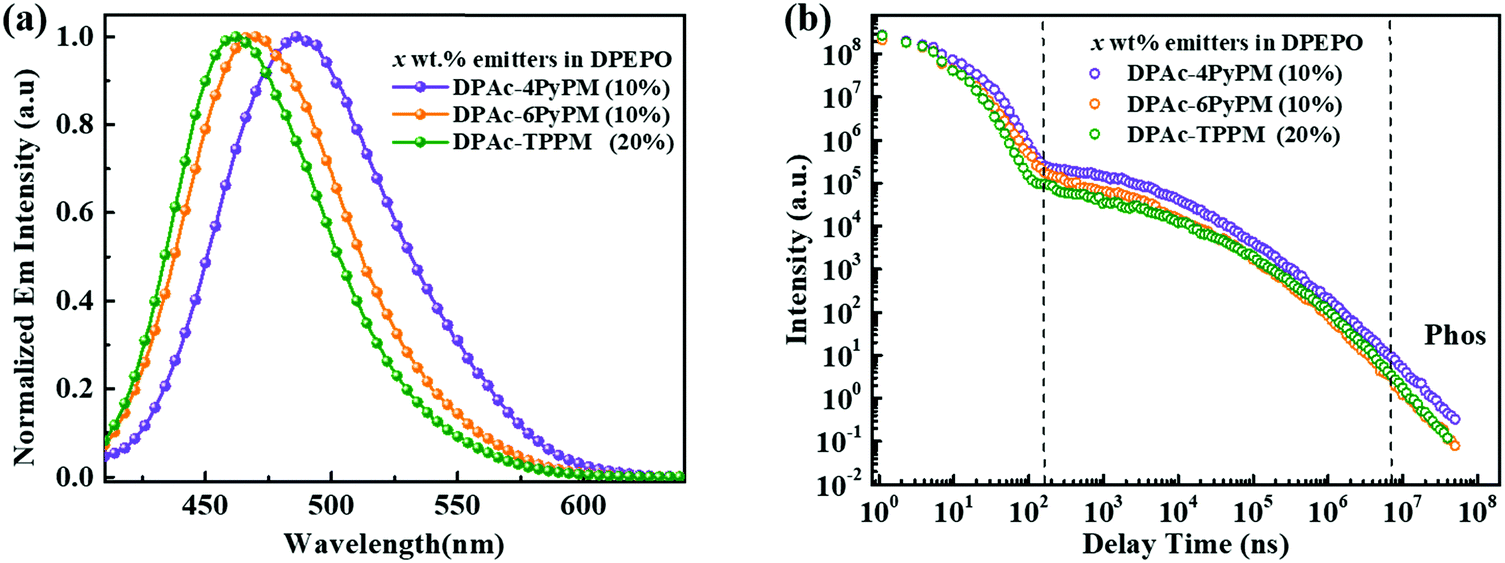
The ultraviolet-visible (UV-vis) absorption and photoluminescent (PL) spectroscopy spectra of the compounds measured in a dilute toluene solution at room temperature are shown in Fig. 4. While the stronger higher energy absorptions below 330 nm are attributed to the π–π* transitions of the conjugated aromatic units, the weak absorption of ≈350–390 nm can be attributed to the intramolecular charge-transfer (ICT) state from the DPAc unit to acceptors of pyrimidine–pyridine or benzene hybrids.44 The emission peaks of DPAc-4PyPM, DPAc-6PyPM, and DPAc-TPPM in dilute toluene solution are located at 480, 460, and 450 nm, respectively. Owing to nearly identical molecular configuration and electron cloud distributions, DPAc-4PyPM and DPAc-6PyPM showed a red-shift of 30 nm and 10 nm, respectively, arising from the additional electron-deficient pyridine which could enhance the ICT character. The PL spectra of these three emitters in a series of solvents with different polarities are shown in Fig. S5 (ESI†). On consideration of the comprehensive influences of structural relaxation (nonradiation energy loss) and the intramolecular charge transfer state (ICT) toward solvatochromic effects, the red shift values of emission are 112 nm, 130 nm, and 110 nm from toluene (Tol) to N,N-dimethylformamide (DMF) were obtained for DPAc-4PyPM, DPAc-6PyPM and DPAc-TPPM, respectively.45 According to the onset wavelength of the fluorescence (Fl) and the phosphorescence (Phos) spectra of DPAc-4PyPM, DPAc-6PyPM and DPAc-TPPM in the DPEPO host, the ΔESTs are estimated to be 2.89/2.66 eV, 0.23 eV for DPAc-4PyPM, 3.00/2.80 eV, 0.20 eV for DPAc-6PyPM, and 3.04/2.76 eV, 0.28 eV for DPAc-TPPM, which are very close to the reported values in similar molecular system.33,44 The small ΔEST values support that the three TADF molecules can effectively harvest triplet excitons by the RISC process. The emission peaks and PLQYs of x wt% emitter-doped DPEPO films are 486 nm and 86% for DPAc-4PyPM (10 wt%), 468 nm and 83% for DPAc-6PyPM (10 wt%), and 464 nm and 70% for DPAc-TPPM (20 wt%), respectively (Fig. 5a and Table 1). As anticipated, the PLQYs for both 4 and 6-substituted pyridine in the pyrimidine ring were efficiently increased with participation of intramolecular CH⋯N interactions. As shown in Fig. 5b, three distinct time regions: one between 1 and 100 ns (prompt emission) and the other at later delay times (>100 ns) were obtained from the transient photoluminescence decay (PL) curves of x wt% dyes in DPEPO at room temperature.46,47 What's more, the intensity of the delayed components gradually increased with rising temperature from 80 K to 250 K (Fig. S6, ESI†), which indicates the presence of TADF. Then, at longer time delays, phosphorescence (PH) emission (after 107 ns) appears, which shows higher intensity at low temperatures, as expected for triplet emission.48 Calculating from their integral area of transient PL decay and PLQYs, it can be found that enhanced PLQYs comparing with DPAc-TPPM for DPAc-4PyPM and DPAc-6PyPM are delayed and prompt section, respectively (Table S1, ESI†). And then the kRISC of DPAc-4PyPM was significantly improved by about 5 and 10 fold compared with that of DPAc-6PyPM and DPAc-TPPM (Table 1), respectively. The related properties and details of the kinetic parameters are summarized Table 1 and Table S1 (ESI†). It is a rational deduction that the intramolecular CH⋯N interactions due to the introduction of 2-pyridine group at the 4-position of pyrimidine not only enhances PLQY but also significantly facilitates the reverse intersystem crossing (RISC) process.
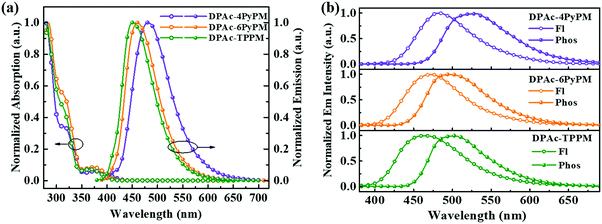 |
| | Fig. 4 (a) Absorption and emission spectra in toluene and (b) fluorescence and phosphorescence spectra of emitters in the DPEPO host at 77 K (10 wt% for DPAc-4PyPM and DPAc-6PyPM; 20 wt% for DPAc-TPPM). | |
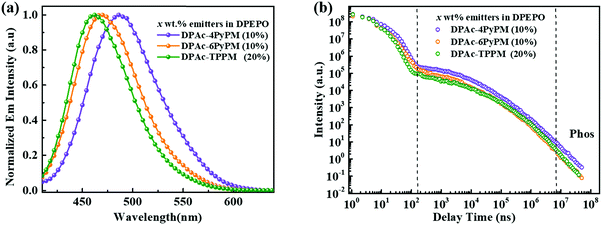 |
| | Fig. 5 (a) Fluorescence (Fl) spectra and (b) transient PL decay curve of delayed emission for x wt% emitters doped in DPEPO film (10 wt% for DPAc-4PyPM and DPAc-6PyPM; 20 wt% for DPAc-TPPM) at room temperature. | |
Electroluminescent properties
To evaluate the device performances of DPAc-4PyPM, DPAc-6PyPM and DPAc-TPPM, multilayer OLEDs were fabricated with doped films as the emitting layer (EML). The device configuration was ITO/MOO3 (10 nm)/NPB/mCP/DPEPO: x wt% emittets/DPEPO/TPBi (40 nm)/LiF (1 nm)/Al. MoO3 and LiF served as the hole- and electron-injecting layers, respectively. NPB (N,N′-bis(naphthalen-1-yl)-N,N′-bis(phenyl)-benzidine) and TPBi (1,3,5-tris(1-phenyl-1H-benzo[d]imidazol-2-yl)benzene) were used as the hole- and electron-transporting layers, respectively. And mCP (1,3-bis(9-carbazolyl)benzene) and DPEPO ((oxybis(2,1-phenylene))bis(diphenylphosphine oxide)) were employed as the exciton-blocking layers on either side of the emitting layer (EML). The doping concentration for each TADF emitter (10 wt% for DPAc-4PyPM, device A; 10 wt% for DPAc-6PyPM, device B; and 20 wt% for DPAc-TPPM, device C) and the thickness of the related functional layers were optimized, respectively. The specifically optimized device configuration and the chemical structures of the materials employed in TADF diodes were presented in Scheme S1 (ESI†). The energy level diagrams, current density–voltage–luminance characteristics (J–V–L), current and power efficiency (CE and PE) and external quantum efficiency (EQE) versus current density curves, and EL spectra of the devices are shown in Fig. 6.
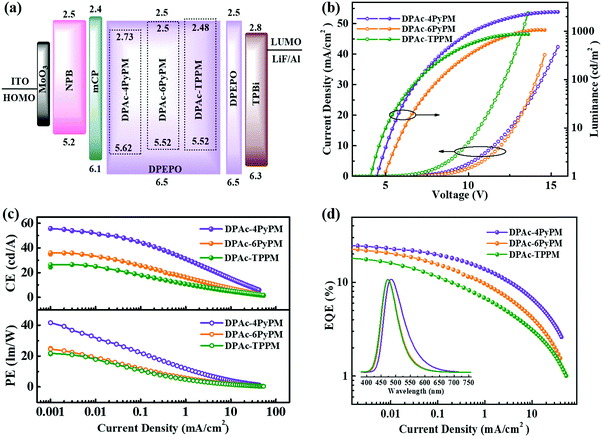 |
| | Fig. 6 (a) Energy-level diagram of the materials used in the devices. (b) Current density–voltage–luminance (J–V–L) characteristics. (c) Current efficiency (upper) and power efficiency (below) versus current density curves. (d) EQE versus current density curves. Inset: EL spectrum at 100 cd m−2. | |
Their EL spectra originated exclusively from the three TADF emitters without emission from the neighboring layers, indicative of a complete energy transfer from the host materials to the dopants. The devices of DPAc-4PyPM, DPAc-6PyPM and DPAc-TPPM exhibited sky blue EL emission with peaks at 484 nm, 472 nm and 472 nm, respectively, which are close to their PL spectra and this suggests that all of the devices have efficient carrier injection, transport and recombination in the EML. The main device data are summarized in Table S2 (ESI†). In order to further investigate the device mechanism, single-carrier devices with laddered doping ratios of 5, 10 and 20 wt% were also fabricated. The device structures and current density–voltage characteristics for single-carrier devices are presented in Fig. S7 (ESI†). The current density for both hole-only and electron-only devices increased with gradual rise in the doping concentration, which substantiated the charge trapping effect of the dopant.49 What's more, the variation of current density for all hole-only devices is more obvious than that of electron-only devices, which is attributed to the deeper hole-trapping sites originating from shallower HOMO energy levels. Although the structures of device A and B aren’t exactly the same, the same dopant concentration and approximative energy levels made them exhibit an analogous turn-on voltage (4.6 and 4.8 V). The DPAc-TPPM-based device with a higher dopant concentration can boost the trap-assisted recombination mechanism.50 Then device C showed a lower turn-on voltage of 4.2 V. With the absence of light out-coupling enhancement, the EL efficiencies with a maximum external quantum efficiency (EQE), current efficiency (CE), and power efficiency (PE) of 24.34%, 53.89 cd A−1 and 36.79 lm W−1 for the DPAc-4PyPM-based device, 22.42%, 35.85 cd A−1 and 23.45 lm W−1 for the DPAc-6PyPM-based device and 16.80%, 26.07 cd A−1 and 19.49 lm W−1 for the DPAc-TPPM-based device. The theoretical maximum EQE for TADF devices can be calculated using the following equation:51,52
where
γ refers to the radiative recombination factor, regarded as 0–1;
ηout stands for the out-coupling constant, commonly considered as 0.2–0.3. On the basis of experimental results of EQE
max,
ΦP and
ΦD, the
γηout values of the devices based on
DPAc-4PyPM,
DPAc-6PyPM and
DPAc-TPPM are estimated to be 0.32, 0.33 and 0.33 respectively. The values are slightly higher than the maximum theoretical limit of 0.30 for
γηout, which might originate from the improved horizontal orientation of the three emitters similar to spiroacridine-triazine hybrids with the homologous molecular configuration (Table S3 and Fig. S8, ESI
†).
41 Meanwhile, the high horizontal-dipole ratios in analogous molecules have also been confirmed.
42 Both the
DPAc-6PyPM-based device and
DPAc-TPPM-based device possessed nearly identical CIE coordinates of (0.15, 0.24). The EL efficiency of the
DPAc-6PyPM-based device is better than that of the
DPAc-TPPM-based device, which is mainly due to its higher PLQY and
kRISC resulting from intramolecular CH⋯N interactions. Furthermore, the
DPAc-4PyPM-based device shows smaller efficiency roll-off than the
DPAc-6PyPM-based device due to the highest
kRISC of
DPAc-4PyPM, which can facilitate the RISC process while reducing triplet-exciton-involved quenching, such as singlet–triplet annihilation (STA) and triplet–triplet annihilation (TTA) processes.
53,54
Conclusion
In summary, we designed and synthesized three TADF emitters (DPAc-4PyPM, DPAc-6PyPM and DPAc-TPPM) based on acceptors of pyrimidine–pyridine hybrids and a donor of DPAc. Increased structural rigidity was achieved by the presence of CH⋯N intramolecular hydrogen bonding interactions, which improved the PLQYs of DPAc-4PyPM and DPAc-6PyPM. The OLEDs using DPAc-4PyPM and DPAc-6PyPM as the emitters achieved a maximum EQE, CE and PE of 24.34%, 53.89 cd A−1 and 36.79 lm W−1, respectively, and 22.42%, 35.85 cd A−1and 23.45 lm W−1, respectively. Moreover, the kRISC of DPAc-4PyPM with CH⋯N at the 4-position of the pyrimidine ring realized nearly 5 and 10 fold improvements compared with the other analogues. And then the DPAc-4PyPM-based device exhibited the slowest efficiency roll-off. The results revealed that manipulating the positions of CH⋯N is a promising opportunity for the design and construction of highly efficient TADF materials.
Experiment
The experimental conditions and instruments used have been outlined in our previous studies.55 The chalcone derivative and TPPMBr were synthesized according to earlier reports.31 And 1H-NMR (600 MHz, CDCl3) spectroscopy data of TPPMBr were added in Fig. S9 (ESI†).
Synthesis of 4-(5-bromopyridin-2-yl)-2,6-diphenylpyrimidine (4PyPMBr)
To a stirring solution of potassium hydroxide (8.96 g, 160 mmol), methanol (20 mL), water (20 mL), and 1-(5-bromopyridin-2-yl)ethan-1-one (8.00 g, 40 mmol) were added at 0 °C. After 1 h, benzaldehyde (4.24 g, 40 mmol) was added to the solution. The reaction mixture was allowed to warm to room temperature and stirred overnight. Then the resultant mixture was cooled to −20 °C and kept for 2 h, filtered, and washed with icy ethanol to give (E)-3-(5-bromopyridin-2-yl)-1-phenylprop-2-en-1-one (yield = 10.0 g, 35 mmol, 87.5%). Afterwards, the above resulting crude product without purification, benzamidine hydrochloride (5.46 g, 35 mmol) and sodium hydroxide (4.2 g, 105 mmol) were dissolved in ethanol (150 mL). The mixture was then refluxed for 8 h. After cooling to room temperature, the formed precipitate was collected by filtration. The residue was extracted three times with dichloromethane and water. The combined organic layers were dried over anhydrous MgSO4. After filtration and evaporation, the crude product was purified by column chromatography on silica gel (petroleum ether/dichloromethane = 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) to afford 4PyPMBr as a white solid (yield = 6.22 g, 45.9%). 1H NMR (600 MHz, CDCl3) δ [ppm]: 8.80 (d, J = 1.9 Hz, 1H), 8.70 (dd, J = 7.8 Hz, 1.4 Hz, 3H), 8.68 (s, 1H), 8.63 (d, J = 8.4 Hz, 1H), 8.37 (dd, J = 7.7 Hz, 1.6 Hz, 2H), 8.03 (dd, J = 8.4 Hz, 2.2 Hz, 1H), 7.59–7.50 (m, 6H). MS (MALDI-TOF): calculated for C21H14BrN3, 387.04; found 387.58, [M]+.
:1, v/v) to afford 4PyPMBr as a white solid (yield = 6.22 g, 45.9%). 1H NMR (600 MHz, CDCl3) δ [ppm]: 8.80 (d, J = 1.9 Hz, 1H), 8.70 (dd, J = 7.8 Hz, 1.4 Hz, 3H), 8.68 (s, 1H), 8.63 (d, J = 8.4 Hz, 1H), 8.37 (dd, J = 7.7 Hz, 1.6 Hz, 2H), 8.03 (dd, J = 8.4 Hz, 2.2 Hz, 1H), 7.59–7.50 (m, 6H). MS (MALDI-TOF): calculated for C21H14BrN3, 387.04; found 387.58, [M]+.
Synthesis of 4-(4-bromophenyl)-2-phenyl-6-(pyridin-2-yl)pyrimidine (6PyPMBr)
The corresponding chalcone, (E)-3-(4-bromophenyl)-1-(pyridin-2-yl)prop-2-en-1-one, was synthesized according to path b. The compound 6PyPMBr was prepared as a white solid using identical procedures (70%). 1H NMR (600 MHz, CDCl3) δ [ppm]: 8.72 (ddd, J = 11.1 Hz, 9.3 Hz, 2.7 Hz, 5H), 8.69 (s, 1H), 8.25 (d, J = 8.5 Hz, 2H), 7.91 (td, J = 7.8 Hz, 1.5 Hz, 1H), 7.67 (d, J = 8.5 Hz, 2H), 7.59–7.49 (m, 3H), 7.48–7.39 (m, 1H). MS (MALDI-TOF): calculated for C21H14BrN3, 387.04; found 387.62, [M]+.
Synthesis of DPAc-4PyPM
A mixture of 4PyPMBr, 4-(5-bromopyridin-2-yl)-2,6-diphenylpyrimidine (1.32 g, 3.4 mmol), 9,9-diphenyl-9,10-dihydroacridine (1.18 g, 3.57 mmol), t-BuONa (0.816 g, 8.5 mmol), tri-tert-butylphosphonium tetrafluoroborate (59 mg, 0.204 mmol) and palladium acetate (15 mg, 0.068 mmol) was dissolved in dry toluene (20 mL), and purged three times by a nitrogen/vacuum cycle. The solution was refluxed for 12 h under N2. Then, the resultant mixture was cooled to room temperature, filtered, and washed with brine. The organic layer was dried and concentrated by rotary evaporation. The product was purified by column silica gel chromatography (petroleum ether/ethyl acetate = 50:1) and recrystallized to afford a light green product (1.52 g, 70%). 1H NMR (400 MHz, CDCl3) δ [ppm]: 8.93 (d, J = 8.3 Hz, 1H), 8.76 (d, J = 8.6 Hz, 3H), 8.53–8.32 (m, 3H), 7.70 (dd, J = 8.2 Hz, 1.9 Hz, 1H), 7.57 (d, J = 5.2 Hz, 6H), 7.28 (t, J = 8.2 Hz, 6H), 7.12 (dt, J = 8.3 Hz, 4.2 Hz, 2H), 7.07–6.86 (m, 8H), 6.52 (d, J = 8.1 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ [ppm]: 165.37, 164.40, 162.86, 153.94, 152.06, 145.98, 141.85, 139.96, 139.11, 137.96, 137.28, 131.00, 130.80, 130.75, 130.40, 130.23, 128.93, 128.56, 128.45, 127.77, 127.47, 127.11, 126.51, 123.22, 121.02, 114.28, 110.78, 56.89. HRMS (APCI, m/z): [M + H]+ cal for C46H32N4 640.2627, found 641.2699. Elemental analysis calcd (%) C46H32N4: C, 86.22; H, 5.03, N, 8.74; found: C, 86.22; H, 5.04; N, 8.73.
Synthesis of DPAc-6PyPM
The procedure is similar to that used for DPAc-4PyPM. For this compound, we used 6PyPMBr (4-(4-bromophenyl)-2-phenyl-6-(pyridin-2-yl)pyrimidine) instead of 4PyPMBr. The resulting crude product was purified by silica gel column chromatography (petroleum ether/dichloromethane = 10:1) and recrystallized to give a white solid (67%). 1H NMR (600 MHz, CDCl3) δ [ppm]: 8.85–8.73 (m, 5H), 8.56 (d, J = 8.3 Hz, 2H), 7.96 (t, J = 7.8 Hz, 1H), 7.64–7.53 (m, 3H), 7.48 (dd, J = 6.8 Hz, 5.2 Hz, 1H), 7.33–7.27 (m, 8H), 7.14–7.00 (m, 6H), 6.97–6.88 (m, 4H), 6.54 (d, J = 8.2 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ [ppm]: 164.42, 164.38, 163.89, 154.52, 149.48, 146.43, 143.30, 142.02, 137.92, 137.33, 137.17, 131.75, 130.83, 130.44, 130.11, 129.75, 129.69, 128.57, 128.45, 127.68, 126.96, 126.33, 125.43, 121.98, 120.38, 114.16, 110.85, 56.81. HRMS (APCI, m/z): [M + H]+ cal for C46H32N4 640.2627, found 641.2732. Elemental analysis calcd (%) C46H32N4: C, 86.22; H, 5.03, N, 8.74; found: C, 86.21; H, 5.04; N, 8.74.
Synthesis of DPAc-TPPM
The procedure is similar to that used for DPAc-4PyPM. For this compound, we used TPPMBr (4-(4-bromophenyl)-2,6-diphenylpyrimidine) instead of 4PyPMBr. The resulting crude product was purified by silica gel column chromatography (petroleum ether/dichloromethane = 15:1) and recrystallized to give a white solid (73%). 1H NMR (600 MHz, CDCl3) δ [ppm]: 8.75 (d, J = 6.7 Hz, 2H), 8.46 (d, J = 8.2 Hz, 2H), 8.39–8.27 (m, 2H), 8.07 (s, 1H), 7.68–7.49 (m, 6H), 7.32–7.26 (m, 8H), 7.12–7.06 (m, 2H), 7.03 (d, J = 7.2 Hz, 4H), 6.98–6.85 (m, 4H), 6.53 (d, J = 8.2 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ [ppm]: 165.04, 164.69, 163.99, 146.42, 143.27, 142.00, 138.00, 137.45, 137.41, 131.86, 130.96, 130.81, 130.44, 130.13, 129.78, 129.53, 129.01, 128.51, 127.68, 127.33, 126.95, 126.34, 120.41, 114.13, 110.44, 60.90. HRMS (APCI, m/z): [M + H]+ cal for C47H33N3 639.2674, found 640.2756. Elemental analysis calcd (%) C47H33N3: C, 88.23; H, 5.20; N, 6.57; found: C, 88.25; H, 5.19; N, 6.56.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research work was supported by the NSFC/China (51573065, 51727809), China Postdoctoral Science Foundation (2017M620321), and the science and technology support program of Hubei Province (2015BAA075). We are thankful to SCTS/CGCL HPCC of HUST for providing computing resources and technical support. The Analytical and Testing Center at Huazhong University of Science and Technology is acknowledged for characterization of new compounds. The Center of Micro-Fabrication and Characterization (CMFC) of Wuhan National laboratory for Optoelectronics (WNLO) of HUST is acknowledged for fabrication of the OLED devices. In particular, we are thankful for the instrument support from The Organic Electroactive Materials (OEM) group, headed by Professor Andrew Monkman, Department of Physics, Durham University.
References
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913 CrossRef CAS.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- C.-G. Zhen, Y.-F. Dai, W.-J. Zeng, Z. Ma, Z.-K. Chen and J. Kieffer, Adv. Funct. Mater., 2011, 21, 699–707 CrossRef CAS.
- T.-H. Han, Y. Lee, M.-R. Choi, S.-H. Woo, S.-H. Bae, B. H. Hong, J.-H. Ahn and T.-W. Lee, Nat. Photonics, 2012, 6, 105–110 CrossRef CAS.
- I. S. Park, K. Matsuo, N. Aizawa and T. Yasuda, Adv. Funct. Mater., 2018, 28 Search PubMed.
- T.-L. Wu, M.-J. Huang, C.-C. Lin, P.-Y. Huang, T.-Y. Chou, R.-W. Chen-Cheng, H.-W. Lin, R.-S. Liu and C.-H. Cheng, Nat. Photonics, 2018, 12, 235–240 CrossRef CAS.
- X. Liang, Z. P. Yan, H. B. Han, Z. G. Wu, Y. X. Zheng, H. Meng, J. L. Zuo and W. Huang, Angew. Chem., Int. Ed. Engl., 2018, 57, 11316–11320 CrossRef CAS PubMed.
- H. Liu, J. Zeng, J. Guo, H. Nie, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed. Engl., 2018, 57, 9290–9294 CrossRef CAS PubMed.
- J. Guo, X.-L. Li, H. Nie, W. Luo, S. Gan, S. Hu, R. Hu, A. Qin, Z. Zhao, S.-J. Su and B. Z. Tang, Adv. Funct. Mater., 2017, 27, 1606458 CrossRef.
- J. Huang, H. Nie, J. Zeng, Z. Zhuang, S. Gan, Y. Cai, J. Guo, S.-J. Su, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2017, 56, 12971–12976 CrossRef CAS PubMed.
- B. Minaev, G. Baryshnikov and H. Agren, Phys. Chem. Chem. Phys., 2014, 16, 1719–1758 RSC.
- H. Zhao, Z. Wang, X. Cai, K. Liu, Z. He, X. Liu, Y. Cao and S.-J. Su, Mater. Chem. Front., 2017, 1, 2039–2046 RSC.
- K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253–258 CrossRef CAS.
- A. Endo, M. Ogasawara, A. Takahashi, D. Yokoyama, Y. Kato and C. Adachi, Adv. Mater., 2009, 21, 4802–4806 CrossRef CAS PubMed.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29 Search PubMed.
- Y. Im, M. Kim, Y. J. Cho, J.-A. Seo, K. S. Yook and J. Y. Lee, Chem. Mater., 2017, 29, 1946–1963 CrossRef CAS.
- X. Cai, X. Li, G. Xie, Z. He, K. Gao, K. Liu, D. Chen, Y. Cao and S.-J. Su, Chem. Sci., 2016, 7, 4264–4275 RSC.
- D. Zhang, M. Cai, Y. Zhang, D. Zhang and L. Duan, Mater. Horiz., 2016, 3, 145–151 RSC.
- Q. Zhang, S. Xiang, Z. Huang, S. Sun, S. Ye, X. Lv, W. Liu, R. Guo and L. Wang, Dyes Pigm., 2018, 155, 51–58 CrossRef CAS.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- T. Y. Huang, W. Jiang and L. Duan, J. Mater. Chem. C, 2018, 6, 5577–5596 RSC.
- J. H. Kim, J. H. Yun and J. Y. Lee, Adv. Opt. Mater., 2018, 1800255 CrossRef.
- J. J. Guo, Z. J. Zhao and B. Z. Tang, Adv. Opt. Mater., 2018, 6, 1800264 CrossRef.
- X. Cao, D. Zhang, S. Zhang, Y. Tao and W. Huang, J. Mater. Chem. C, 2017, 5, 7699–7714 RSC.
- D. Zhang, X. Song, M. Cai, H. Kaji and L. Duan, Adv. Mater., 2018, 30, 1705406 CrossRef PubMed.
- C. Duan, J. Li, C. Han, D. Ding, H. Yang, Y. Wei and H. Xu, Chem. Mater., 2016, 28, 5667–5679 CrossRef CAS.
- Q. Q. Liang, C. M. Han, C. B. Duan and H. Xu, Adv. Opt. Mater., 2018, 6, 1800020 CrossRef.
- W. Zeng, H. Y. Lai, W. K. Lee, M. Jiao, Y. J. Shiu, C. Zhong, S. Gong, T. Zhou, G. Xie, M. Sarma, K. T. Wong, C. C. Wu and C. Yang, Adv. Mater., 2018, 30 Search PubMed.
- X. Chen, Z. Yang, Z. Xie, J. Zhao, Z. Yang, Y. Zhang, M. P. Aldred and Z. Chi, Mater. Chem. Front., 2018, 2, 1017–1023 RSC.
- P. Ganesan, R. Ranganathan, Y. Chi, X. K. Liu, C. S. Lee, S. H. Liu, G. H. Lee, T. C. Lin, Y. T. Chen and P. T. Chou, Chemistry, 2016, 23, 2858–2866 CrossRef PubMed.
- R. Komatsu, H. Sasabe, Y. Seino, K. Nakao and J. Kido, J. Mater. Chem. C, 2016, 4, 2274–2278 RSC.
- I. S. Park, J. Lee and T. Yasud, J. Mater. Chem. C, 2016, 4, 7911–7916 RSC.
- K. Wu, T. Zhang, L. Zhan, C. Zhong, S. Gong, N. Jiang, Z. H. Lu and C. Yang, Chemistry, 2016, 22, 10860–10866 CrossRef CAS PubMed.
- Y. P. Xiang, Y. B. Zhao, N. Xu, S. L. Gong, F. Ni, K. L. Wu, J. J. Luo, G. H. Xie, Z. H. Lu and C. L. Yang, J. Mater. Chem. C, 2017, 5, 12204–12210 RSC.
- B. W. Li, Z. Y. Li, T. P. Hu, Y. Zhang, Y. Wang, Y. P. Yi, F. Y. Guo and L. C. Zhao, J. Mater. Chem. C, 2018, 6, 2351–2359 RSC.
- R. Komatsu, T. Ohsawa, H. Sasabe, K. Nakao, Y. Hayasaka and J. Kido, ACS Appl. Mater. Interfaces, 2017, 9, 4742–4749 CrossRef CAS PubMed.
- K. Nakao, H. Sasabe, R. Komatsu, Y. Hayasaka, T. Ohsawa and J. Kido, Adv. Opt. Mater., 2017, 5, 1600843 CrossRef.
- H. J. Park, S. H. Han, J. Y. Lee, H. Han and E. G. Kim, Chem. Mater., 2018, 30, 3215–3222 CrossRef CAS.
- L. Gan, K. Gao, X. Cai, D. Chen and S. J. Su, J. Phys. Chem. Lett., 2018, 9, 4725–4731 CrossRef CAS PubMed.
- T. A. Lin, T. Chatterjee, W. L. Tsai, W. K. Lee, M. J. Wu, M. Jiao, K. C. Pan, C. L. Yi, C. L. Chung, K. T. Wong and C. C. Wu, Adv. Mater., 2016, 28, 6976–6983 CrossRef CAS PubMed.
- P. Ganesan, D.-G. Chen, J.-L. Liao, W.-C. Li, Y.-N. Lai, D. Lo, C.-H. Chang, C.-L. Ko, W.-Y. Hung, S.-W. Liu, G.-H. Lee, P.-T. Chou and Y. Chi, J. Mater. Chem. C, 2018 10.1039/c8tc03645d.
- Q. Zhang, B. Wang, J. Tan, G. Mu, W. Yi, X. Lv, S. Zhuang, W. Liu and L. Wang, J. Mater. Chem. C, 2017, 5, 8516–8526 RSC.
- I. S. Park, H. Komiyama and T. Yasuda, Chem. Sci., 2017, 8, 953–960 RSC.
- X. L. Lv, W. Z. Zhang, D. X. Ding, C. M. Han, Z. Huang, S. P. Xiang, Q. Zhang, H. Xu and L. Wang, Adv. Opt. Mater., 2018, 6, 1800165 CrossRef.
- F. B. Dias, J. Santos, D. R. Graves, P. Data, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, T. Palmeira, M. N. Berberan-Santos, M. R. Bryce and A. P. Monkman, Adv. Sci., 2016, 3, 1600080 CrossRef PubMed.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- P. L. Dos Santos, J. S. Ward, M. R. Bryce and A. P. Monkman, J. Phys. Chem. Lett., 2016, 7, 3341–3346 CrossRef CAS PubMed.
- S. L. Gong, Y. L. Chang, K. L. Wu, R. White, Z. H. Lu, D. T. Song and C. L. Yang, Chem. Mater., 2014, 26, 1463–1470 CrossRef CAS.
- W. Zhang, J. Jin, Z. Huang, X. Lv, S. Zhuang and L. Wang, J. Mater. Chem. C, 2017, 5, 4636–4644 RSC.
- J. Lee, K. Shizu, H. Tanaka, H. Nomura, T. Yasuda and C. Adachi, J. Mater. Chem. C, 2013, 1, 4599 RSC.
- Y. P. Xiang, S. L. Gong, Y. B. Zhao, X. J. Yin, J. J. Luo, K. L. Wu, Z. H. Lu and C. L. Yang, J. Mater. Chem. C, 2016, 4, 9998–10004 RSC.
- K. Masui, H. Nakanotani and C. Adachi, Org. Electron., 2013, 14, 2721–2726 CrossRef CAS.
- Y. Zhang and S. R. Forrest, Phys. Rev. Lett., 2012, 108, 267404 CrossRef PubMed.
- J. J. Jin, W. Z. Zhang, B. Wang, G. Y. Mu, P. Xu, L. Wang, H. Huang, J. S. Chen and D. G. Ma, Chem. Mater., 2014, 26, 2388–2395 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1: the strategy of constructing CH⋯N intramolecular hydrogen-bonding interactions; Fig. S2: packing diagrams from different axis directions; Fig. S3(a): the TGA and (b) DSC thermograms; Fig. S4(a): cyclic voltammogram; Fig. S5: fluorescence spectra in different solvents at room temperature and phosphorescence (Phos) spectra in 2-methyl-tetrahydrofuran at 77 K and their ΔEst values; Fig. S6: the temperature-dependent transient PL decay spectra of x wt% emitters in DPEPO film; Table S1: detailed kinetic parameters; Scheme S1: the structure of the optimized device and chemical structures of the materials employed in the TADF diodes; Fig. S7: current density–voltage characteristics of hole-only and electron-only devices with different concentrations; Table S2: recently reported TADF OLEDs based on acceptors of pyrimidine derivatives; Table S3: optimized molecular structure and transition dipole moment; Fig. S8: the total orientations of the transition dipole moments; Fig. S9: 1H NMR spectra (600 MHz) displaying the aromatic protons and signal of central pyrimidine unit. CCDC 1858076 and 1858077. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8qm00382c |
|
| This journal is © the Partner Organisations 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*