DOI:
10.1039/C8QM00366A
(Research Article)
Mater. Chem. Front., 2018,
2, 2254-2262
In situ synthesis of Cu2O–CuO–C supported on copper foam as a superior binder-free anode for long-cycle lithium-ion batteries†
Received
27th July 2018
, Accepted 7th October 2018
First published on 8th October 2018
Abstract
The present study reports a facile solvothermal method for a Cu-based metal–organic framework [Cu(BDC)]n (BDC = 1,4-benzenedicarboxylate) grown on porous copper foam, which is then in situ transformed into a multicomponent active Cu2O–CuO–C on copper foam for the first time by annealing under an air atmosphere. Scanning electron microscopy imaging indicates that the Cu2O–CuO–C composites were successfully immobilized on copper foam with high stability and good adhesion. Benefiting from the synergistic multidoping effects, the porous Cu2O–CuO–C/Cu with an optimized composition as an anode material for lithium-ion batteries (LIBs) exhibits a high discharge capacity (1321 mA h g−1) after 500 cycles at 0.1 A g−1. Moreover, this electrode presents superior cycling stability and rate capability. The introduction of the highly conductive copper foam substrate not only can act as a supporting substrate for immobilization of Cu2O–CuO–C composites but can also be regarded as an unimpeded highway for rapid charge transfer, resulting in the significant enhancement of lithium storage when tested as LIB anodes.
Introduction
Due to the rise of environmental issues and energy consumption, the utilization of new energy materials will become a trend for future generations. High energy density lithium ion batteries (LIBs) are one of the effective energy storage and conversion devices as they are a source of green energy and possess a good cycling life; they have been designed for use in electric vehicles, portable electronic products, aerospace, and so on.1–5 However, the technological and scientific development of LIBs has been challenging: commercial graphite as an anode cannot achieve high energy density on account of its low theoretical capacity of 372 mA h g−1.6,7 Therefore, development of substitute novel electrode materials is the main objective. Particularly, transition metal oxides (TMOs) have provoked massive interest as one category of promising anode candidates due to their low cost, surplus supply, and especially their high theoretical capacities.8–10 However, the major bottlenecks to arrest their practical application include low electronic conductivity and mild cycling performance originating from huge volume changes during lithium storage processes.11,12 As a consequence, more and more interest has been focused on searching for alternative anode materials with enhanced electrochemical performance to meet the increasing needs of compatible energy storage.
Interestingly, metal–organic frameworks (MOFs), obtained through bonding organic linkers and metal ions/clusters, possess high surface areas, shape controlled channels and tunable particle sizes.13,14 Recent reports highlighted that MOFs are also regarded as ideal template materials to fabricate metal oxide materials.15–19 Especially, a variety of mixed TMO composites derived from MOFs or MOF-hybrid materials are desirable in order to realize synergetic effects and maximize structural advantages. For instance, ZnO/NiO microspheres,20 concave cubic NiO–Co3O4@C,21 hierarchical porous ZnO/ZnCo2O4,22 multilayered Co3O4/NiO/C hybrids,23 and Co3O4/ZnO nanosheets,24 were previously developed with positive effects on enhancing electrochemical performance for lithium storage. Despite what continuous efforts have achieved, it still remains a significant challenge to solve the deeper issues associated with the low electrical conductivity and high surface resistance of TMO composites.
We have been working on the fabrication of novel transition metal oxide composites for electrochemical energy storage and conversion applications.25–27 In our previous research work, we prepared a ZnO/ZnCo2O4/CuCo2O4 nanostructure by annealing a ZIF-8 analogue through a metal-ion-exchange method. Different chemical compositions could be generated by changing the precursor ratios, which had a significant influence on the electrochemical performance.28 By one-step pyrolysis of a polymetallic ZIF analogue supported on reduced graphene oxide, a mixed transition metal oxide composite, Cu0.39Zn0.14Co2.47O4–ZnO, was successfully obtained due to the fact that RGO could serve as a platform for immobilization of the nanoparticles.29 As a continuation of our work, herein, we fabricated a novel multicomponent active metal oxide Cu2O–CuO–C/Cu through growth of Cu–MOF crystals on copper foam and thermal treatment as a template. Moreover, copper foam can serve as an effective platform for the growth of active materials. Such an intriguing arrangement can simplify the complicated electrode preparation procedures by direct utilization of copper foam as binders or conductive agent-free electrodes. In situ grown Cu2O–CuO–C composites on copper foam were used as LIB anode materials due to their advantages of unique structural characteristics and 3D conductive substrates.
Experimental section
Material characterization
Powder X-ray diffraction (PXRD) experiments were performed using an Advance diffractometer (Bruker D8). Thermogravimetric analyses (TGA) were conducted using a Netzsch Thermo Microbalance TG 209 F1 Libra. Raman spectra were recorded using a Renishaw inVia confocal Raman microscope. Sorption isotherms were studied at 77 K using a Belsorp max gas sorption analyzer. X-ray photoelectron spectroscopy (XPS) was performed using an ESCALAB 250Xi XPS spectrometer using Al Kα radiation. Transmission electron microscopy (TEM, JEM-2100HR, Japan) and field emission scanning electron microscopy (FESEM, TESCAN Maia 3, Czech) were used to characterize the surface morphology and architecture of materials. Inductively coupled plasma atomic emission spectroscopy (ICP-AES) analysis (Agilent 7500ce) was conducted to determine chemical composition.
Synthesis of Cu2O–CuO–C/Cu
Cu2O–CuO–C/Cu was synthesized via a two-step route including the preparation of Cu-BDC/Cu. Copper foam (70 mm × 70 mm × 0.5 mm) was ultrasonically immersed in acetone and hydrochloric acid solvent to eliminate possible oxide layers, subsequently rinsed with water and dried under air at room temperature for further use. Firstly, the pretreated copper foam was immersed in a mixture of 1,4-benzenedicarboxylic acid (H2BDC, 0.44 g) and Cu(NO3)2·3H2O (2.4 g) in a 60 mL solution of N,N′-dimethylformamide (DMF). Then the suspension was mixed thoroughly by sonication for 10 min. Subsequently the reaction kettle was incubated at 110 °C in an oven for 48 h. The generated Cu-BDC/Cu foam was collected by centrifugation and dried at 60 °C for 24 h. By annealing of the above prepared Cu-BDC/Cu foam at 300 °C in a tube furnace under an air atmosphere for 2 h, and finally cooling to room temperature, the porous three-dimensional Cu2O–CuO–C/Cu samples were generated.
Synthesis of Cu2O–CuO–C and CuO
Cu-BDC blue powder was obtained through a similar preparation procedure to the Cu-BDC/Cu composites. Meanwhile, Cu2O–CuO–C electrodes were fabricated under the same thermal treatment conditions as in the method for preparing Cu2O–CuO–C/Cu electrodes. The pure CuO phase was obtained through direct annealing of the above obtained Cu-BDC on copper foam at 800 °C for 6 h under an air atmosphere.
Synthesis of CuO/Cu and Cu2O/Cu
CuO/Cu was fabricated by the annealing of a Cu-BDC sample at 800 °C for 2 h and subsequent attachment on copper foam. Pure Cu2O samples could not be obtained in our case. As a result, single metal oxide Cu2O was obtained from a commercial source and attached on the copper foam through a similar treatment procedure as used to generate the Cu2O/Cu sample.
Electrochemical measurements
Electrochemical measurements were performed using CR 2032 coin-type half cells with a Celgard 2400 membrane as the separator, Cu2O–CuO–C/Cu as the working electrode, and 1 M LiPF6 in diethyl carbonate and ethylene carbonate mixed solvent (v/v, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the electrolyte. Next, the active material was loaded on copper foams and cut into circular disks with a diameter of 12 mm. After that, coin cells were constructed in an Ar-filled glove box. A Land CT 2001A battery test system (Wuhan, China) was used to perform galvanostatic charge/discharge measurements from 0.01 to 3.0 V. Cyclic voltammetry and electrochemical impedance spectroscopy (EIS) tests were performed using a CHI-660E workstation.
:1) as the electrolyte. Next, the active material was loaded on copper foams and cut into circular disks with a diameter of 12 mm. After that, coin cells were constructed in an Ar-filled glove box. A Land CT 2001A battery test system (Wuhan, China) was used to perform galvanostatic charge/discharge measurements from 0.01 to 3.0 V. Cyclic voltammetry and electrochemical impedance spectroscopy (EIS) tests were performed using a CHI-660E workstation.
Results and discussion
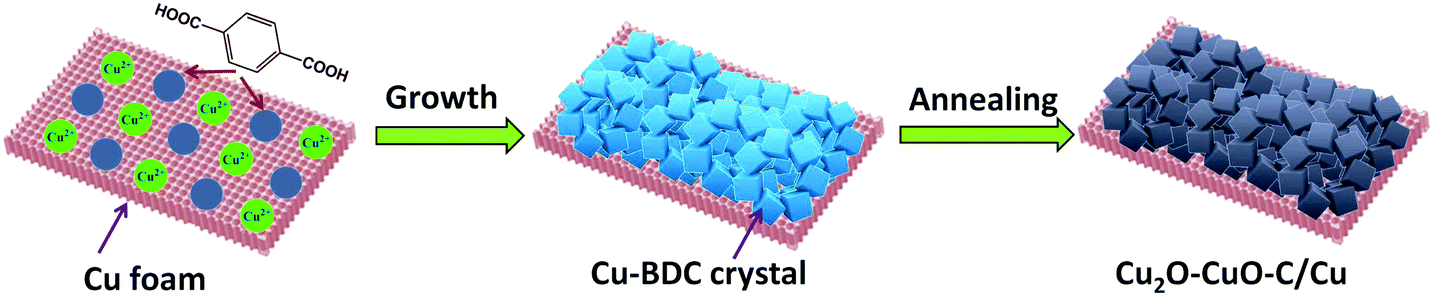
The synthesis strategy for Cu2O–CuO–C on Cu foam is depicted schematically in Fig. 1. Firstly, Cu-BDC MOF (BDC = 1,4-benzenedicarboxylate) crystals were directly grown on copper foam under solvothermal reaction. The powder X-ray diffraction (PXRD) pattern matched well with that of previously reported Cu-BDC crystal samples (Fig. S1, ESI†),30 demonstrating the successful preparation of Cu-BDC. The thermogravimetric analysis (TGA) curve showed that the samples displayed great weight loss in the range from 200 to 300 °C on account of the decomposition of the framework (Fig. S2, ESI†). The subsequent weight increasing step above 350 °C was related to the oxidation of copper foam, which is consistent with TGA analysis of pure copper foam in air flow (Fig. S3, ESI†). As a consequence, the precursor in this case was calcined at 300 °C in air to prevent Cu foam from spontaneously oxidizing, resulting in the generation of Cu2O–CuO–C/Cu as the optimized composition.
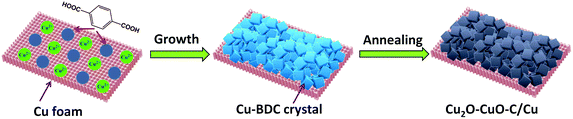 |
| | Fig. 1 Schematic illustration for preparing the Cu2O–CuO–C/Cu electrode. | |
Field emission scanning electron microscopy (FESEM) was applied to characterize the architecture and surface morphology of all of the fabricated samples. Obviously, Cu-BDC crystals were grown on the surface of the copper foam successfully (Fig. 2a and b). The high magnification SEM image indicates that the surface of the crystals is smooth and there is a relatively uniform cube shape with an approximate size of 3 μm (Fig. 2c). After annealing, Cu2O–CuO–C particles are still firmly attached on the copper foam and the shapes are basically maintained with relatively rough surfaces (Fig. 2d–f). The transmission electron microscopy (TEM) image shows that the microcrystals consist of many tiny nanoparticles and lots of pores are formed during thermal treatment (Fig. 2g). As displayed in the high-resolution TEM (HRTEM) image (Fig. 2h), lattice fringes with d-spacings of 0.246 nm and 0.253 nm could be indexed to the (111) lattice planes of Cu2O and (002) interplanar spacing of CuO, respectively, indicating the formation of mixed metal oxides. Homogeneous distribution of C, O, and Cu could be observed from the elemental mapping images in the material structure (Fig. 2i).
 |
| | Fig. 2 (a–c) SEM images of the growth of Cu-BDC crystals on copper foam. (d–f) SEM images of Cu2O–CuO–C/Cu. (g) TEM image. (h) HRTEM lattice image. (i) Elemental mapping images of the Cu2O–CuO–C/Cu electrode. | |
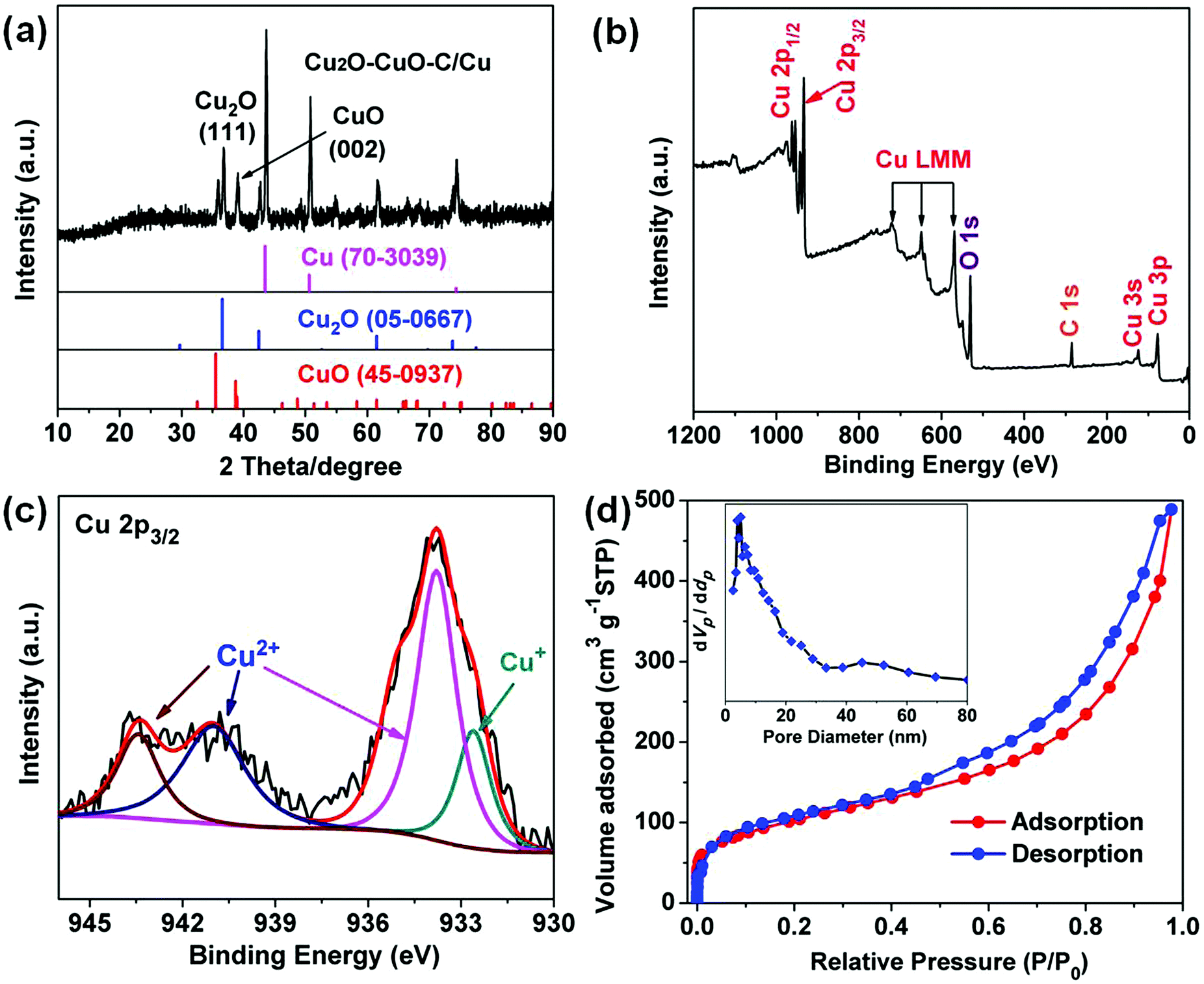
As depicted in Fig. 3a, the PXRD patterns indicate that all the diffraction peaks can be assigned to cubic Cu2O (JCPDS no. 05-0667), monoclinic CuO (no. 45-0937), and cubic Cu (no. 70-3039). No other impurities were observed, demonstrating the three-phase coexistence of the obtained products. The Raman spectrum of the carbon in the Cu2O–CuO–C/Cu material shows two strong characteristic peaks (Fig. S4, ESI†). The peak intensity of the G band (1596 cm−1) was enhanced compared to that of the D band (1348 cm−1), indicating a high graphitization degree.31,32 X-ray photoelectron spectroscopy (XPS) was performed to detect the structural composition and oxidation states of the obtained Cu2O–CuO–C/Cu solid samples. From the survey full spectrum, we could find the coexistence of Cu, C and O (Fig. 3b). For the Cu 2p3/2 spectrum, the main peak at 933.8 eV and the two satellite peaks (941.1 and 943.4 eV) can be attributed to Cu2+ in CuO (Fig. 3c), while the other peak at 932.6 eV can be assigned to Cu+ in Cu2O.33 Elemental analysis further confirmed the presence of carbon with a content of 10.3 wt%. Since the ratio of Cu2+:Cu+ could not be exactly determined by XPS analysis, we further used inductively coupled plasma atomic emission spectroscopy (ICP-AES) analysis and EDS data to confirm the composition of 75.6 wt% CuO and 14.1 wt% Cu2O in the Cu2O–CuO–C product (Fig. S5, ESI†). Compared with the type-I microporous property of the Cu-BDC/Cu precursor (Fig. S6, ESI†), the nitrogen adsorption–desorption isotherm in Fig. 3d depicts clearly typical mesoporous characteristics with a type IV isotherm. The Brunauer–Emmett–Teller (BET) surface area was 207.07 m2 g−1. Calculated using the Barrett–Joyner–Halenda (BJH) method, the pore-size distribution was found to be 4.3 and 45 nm. Such a fascinating porous structure is advantageous for Li-ion transfer and electrolyte diffusion, leading to the improvement of the electrochemical performance.
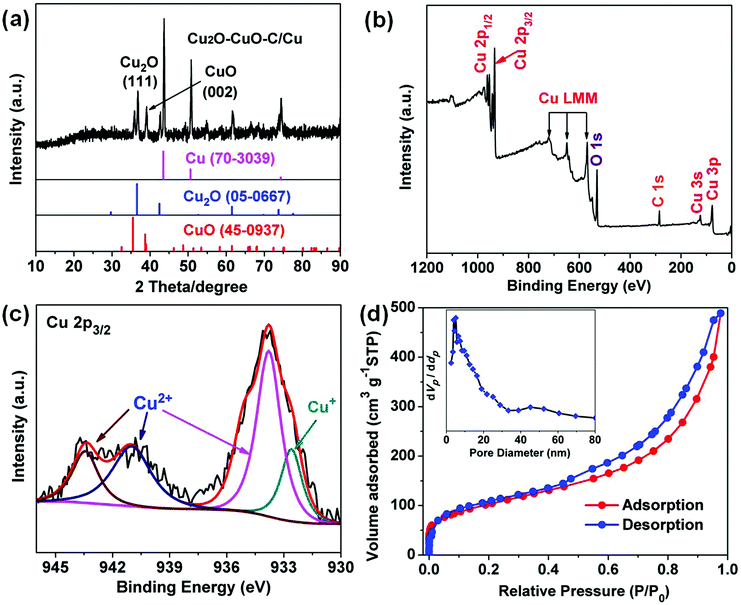 |
| | Fig. 3 Cu2O–CuO–C/Cu (a) XRD pattern. (b) Full XPS spectrum. (c) High-resolution XPS spectrum of Cu 2p3/2. (d) Nitrogen adsorption–desorption isotherm with pore size distribution (inset). | |
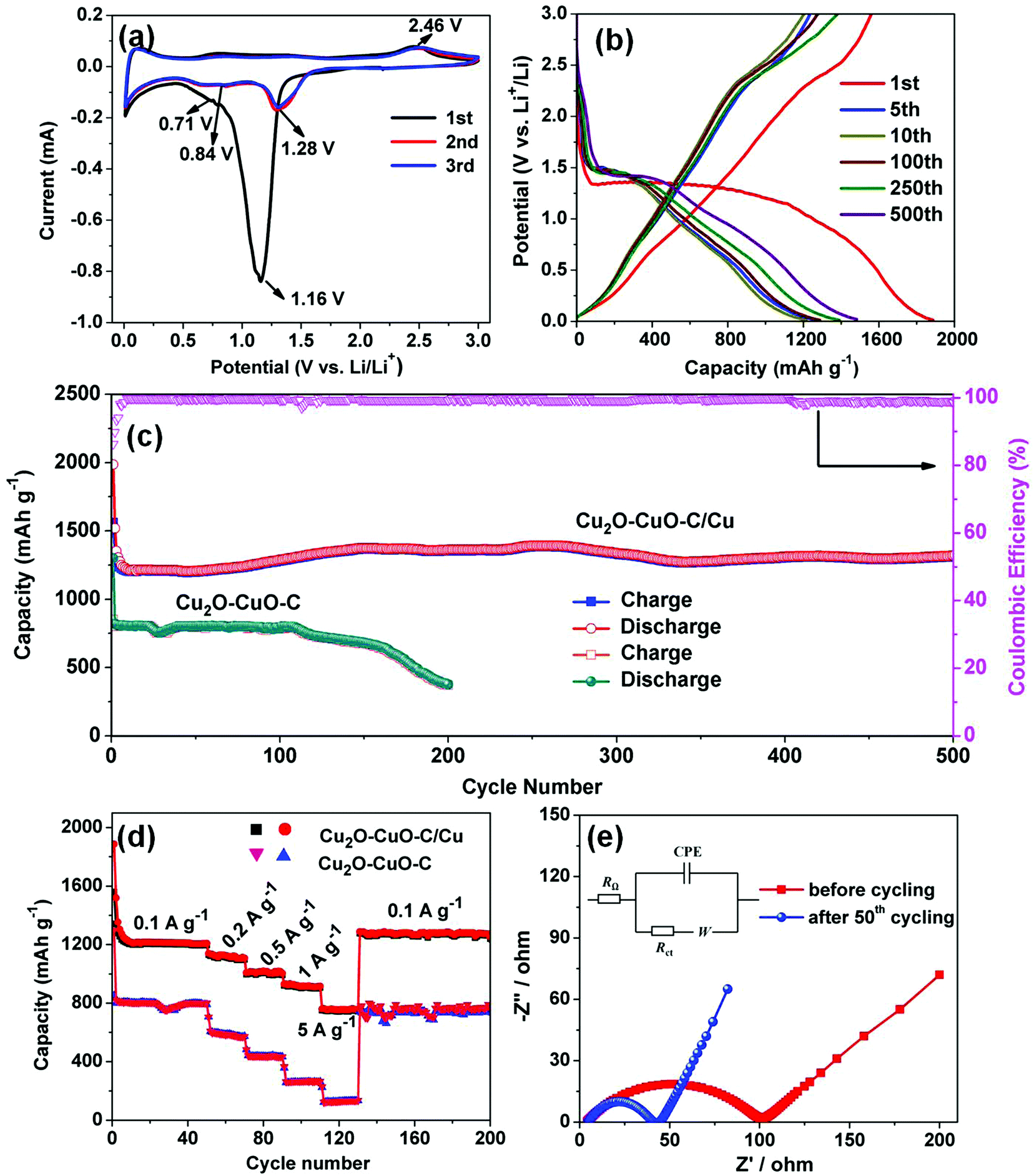
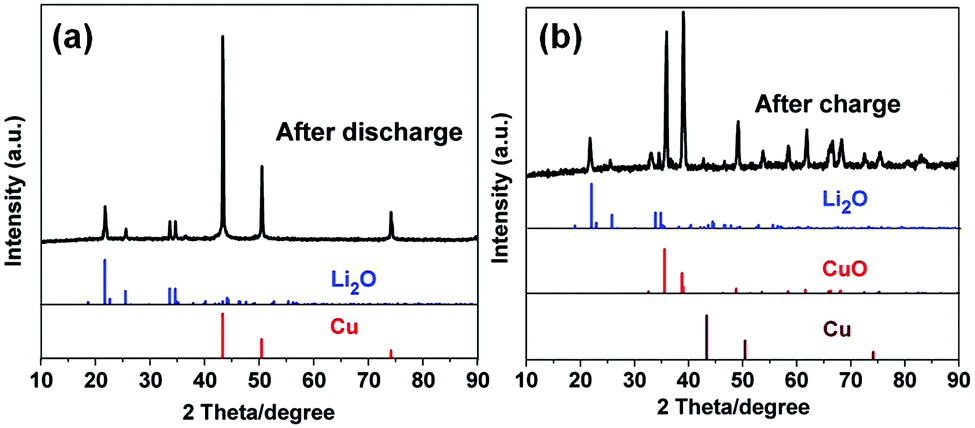
Motivated by the unique structural characteristics, the Cu2O–CuO–C/Cu electrode was investigated as an anode material for LIBs. Without any binders or conductive agents, we directly used the Cu2O–CuO–C/Cu composites as the working electrode. Firstly, cyclic voltammetry (CV) was performed. In the first cathodic sweep (Fig. 4a), a broad peak at 1.16 V was indexed to the reduction of CuO into Cu2O (eqn (1)).34 The peak at 0.71 V is assigned to the decomposition of Cu2O to Cu (eqn (2)), together with the formation of a solid electrolyte interphase (SEI) layer.35 In the initial anodic scan, one well-defined peak was found at 2.46 V, corresponding to the re-oxidation of Cu to CuO (eqn (3)).36 The main reduction was shifted to 1.28 V and 0.84 V in the subsequent cycles, indicating the occurrence of an irreversible phase transformation after the first cycle.37 Evidently, from the second cycle onwards, the CV curves are almost overlapped, confirming the reversibility efficiency. To better understand the lithium storage mechanism, PXRD patterns of the electrode after 50 cycles at 0.1 A g−1 were also recorded. After the discharge process (Fig. 5a), the diffraction peaks of Cu (JCPDS no. 70-3039) and Li2O (JCPDS no. 76-0768) can be observed simultaneously. After the charge process (Fig. 5b), the diffraction peaks belong to Cu2O and CuO. Cu peaks were observed, originating from the copper foam substrate. The presence of extra Li2O peaks derives from the electrode following the initial irreversible reaction.38 Considering all of this, the reaction mechanisms are proposed as follows:
| | | 2CuO + 2Li+ + 2e− → Cu2O + Li2O | (1) |
| | | Cu2O + 2Li+ + 2e− → 2Cu + Li2O | (2) |
| | | Cu + Li2O → CuO + 2Li+ + 2e− | (3) |
Galvanostatic charge/discharge at 0.1 A g
−1 was carried out from 0.01–3.0 V (
Fig. 4b). In the first discharge process, the electrode exhibited a distinct long voltage plateau in the range of 1.32–1.10 V and a sloping voltage in the range of 1.08–0.67 V, which belongs to a multi-step reduction from CuO to Cu
2O and further transformation into Cu and Li
2O, which is well consistent with the CV analysis. From the 2nd cycle, the discharge plateau increased to 1.35 V, maybe associated with the irreversible structural transformation, polarization, and new phase formation after the first insertion.
39 The electrode gave an initial charge capacity of 1559 mA h g
−1 and discharge capacity of 1986 mA h g
−1. The large capacity loss (coulombic efficiency of 78.4%) is probably due to the irreversible conversion involved in the decomposition of the electrolyte and inevitable formation of a SEI layer. Meanwhile, the coulombic efficiency approaches 100% after the initial few cycles, indicating good cyclability of the Cu
2O–CuO–C/Cu electrode. Obviously, gradually increased capacity can be ascribed to a gradual activation process, which is common for anode electrodes.
40 At the end of the 500th cycle, an excellent capacity of 1321 mA h g
−1 can be retained (
Fig. 4c), indicating remarkable long-term cycling stability. To evaluate the rate performance, the Cu
2O–CuO–C/Cu electrode was cycled at different current densities as displayed in
Fig. 4d. The average rate capacities were found to be 1124, 1007 and 930 mA h g
−1 at current densities of 0.2, 0.5 and 1 A g
−1, respectively. Moreover, an unexpected capacity of 753 mA h g
−1 was still maintained even at an ultrahigh rate of 5 A g
−1. Interestingly, when the current density was restored to 0.1 A g
−1, the electrode almost retrieved its original capacity, indicating wonderful rate capability.
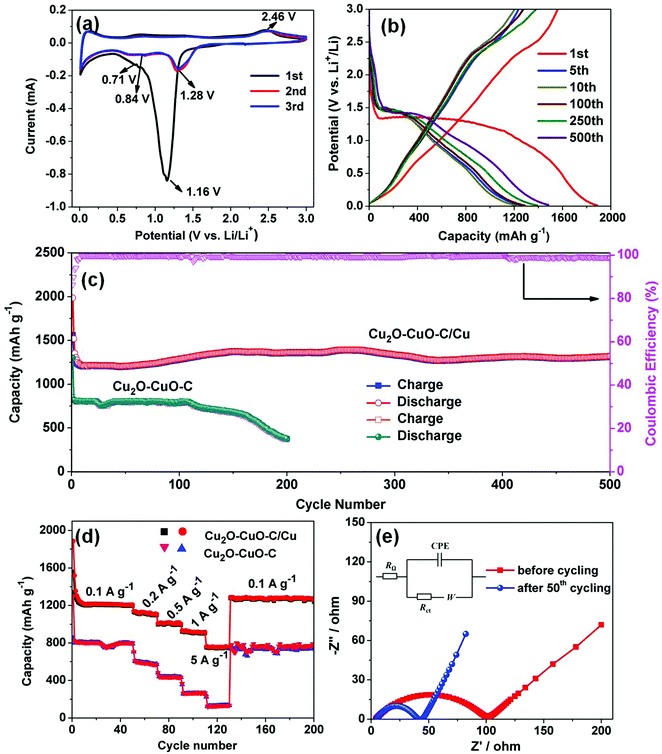 |
| | Fig. 4 (a) CV profiles of the Cu2O–CuO–C/Cu electrode at a scan rate of 0.1 mV s−1. (b) Charge/discharge curves at 0.1 A g−1. (c) Comparison of the cycling performance of Cu2O–CuO–C/Cu and Cu2O–CuO–C electrodes (the pink curve shows the coulombic efficiency of the Cu2O–CuO–C/Cu anode). (d) Rate capabilities at various current densities. (e) Nyquist plots of the Cu2O–CuO–C/Cu electrode after 50 cycles over the frequency range between 100 kHz and 0.01 Hz. | |
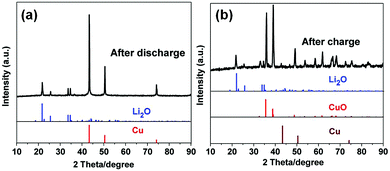 |
| | Fig. 5 XRD patterns of the Cu2O–CuO–C/Cu electrode after the initial cycles. | |
To further investigate the origin of the superior cycling stability, electrochemical impedance spectra (EIS) of the electrode before and after cycling were also recorded (Fig. 4e). Obviously, both of the Nyquist plots exhibit a depressed semicircle in the medium frequency region, corresponding to charge transfer resistance (Rct), together with a slanted line in the range of the low-frequency region related to Warburg impedance. The equivalent electrical circuit is also presented in the inset of Fig. 4e. RΩ represents the ohmic resistance, and Rct and W are designated the charge transfer resistance and Warburg impedance, respectively. In addition, CPE is associated with double layer capacitance.41 It was observed that the Cu2O–CuO–C/Cu electrode after 50 cycles showed a much lower charge transfer resistance (40 Ω) than the Rct (101 Ω) before cycling, implying fast charge transfer kinetics during the lithium storage process.42,43
Table 1 shows a comparison of the electrochemical performance with other previously related CuO/Cu2O anode materials. Obviously, the Cu2O–CuO–C/Cu electrode presents the more excellent performance among these materials. Moreover, this observed excellent capacity of the Cu2O–CuO–C/Cu electrode is greatly higher than the theoretical values of Cu2O (375 mA h g−1) and CuO (674 mA h g−1). As we know, electrode materials are involved in two types of energy storage mechanism: (i) pseudocapacitive charge storage, and (ii) diffusion-controlled processes.48 We believe that the extra capacity mainly results from pseudocapacitance contributions. Herein, we carried out a kinetic analysis according to the CV curves at different sweep rates with similar shapes from 0.2 to 1.2 mV s−1 (Fig. 6a). The pseudocapacitive behavior can be analyzed in accordance with the power law between the scan rate (ν) and peak current (i), as shown in the following eqn (4) and (5):
| | | log(i) = blog(ν) + loga | (5) |
where
a and
b are changeable constants.
49,50 By calculation of the slope of the log(
i) against log(
ν) plot, the
b value can be determined. According to the obtained
b value, two processes can be classified: the capacitive process (
b = 1) and the diffusion-controlled process (
b = 0.5).
Table 1 Comparison of the electrochemical properties of various Cu2O–CuO materials as anode materials for LIBs
| Sample |
Rate capacity |
Cycle performance |
Ref. |
| Capacity (mA h g−1) |
Current density (A g−1) |
Capacity (mA h g−1) |
Current density (mA g−1) |
Cycle number |
| Cu/Cu2O/CuO-NS-NWs |
39.3 |
10 |
345 |
100 |
50 |
34
|
| Cu2O–CuO–RGO |
224 |
5 |
842.5 |
100 |
80 |
35
|
| CuO–Cu2O/graphene |
183 |
5 |
487 |
200 |
60 |
44
|
| CuO/Cu2O/Cu |
215.1 |
1 |
534.1 |
100 |
200 |
45
|
| CuO/Cu2O–GPC (MOF-derived) |
210.4 |
3 |
887.3 |
60 |
200 |
36
|
| CuO/Cu2O@CeO2 (MOF-derived) |
250 |
2 |
592.3 |
200 |
100 |
46
|
| CuO/Cu2O (MOF-derived) |
130 |
1.6 |
740 |
100 |
250 |
47
|
| Cu2O–CuO–C/Cu (MOF-derived) |
753 |
5 |
1321 |
100 |
500 |
This work |
 |
| | Fig. 6 Electrochemical kinetic analysis of the Cu2O–CuO–C/Cu electrode. (a) CV curves at various sweep rates. (b) b-value determination using the relationship between scan rate and peak current. (c) Separation of the capacitive contribution at 1.0 mV s−1 (shaded region). (d) Contribution percentages of the capacitive contribution for various sweep rates. | |
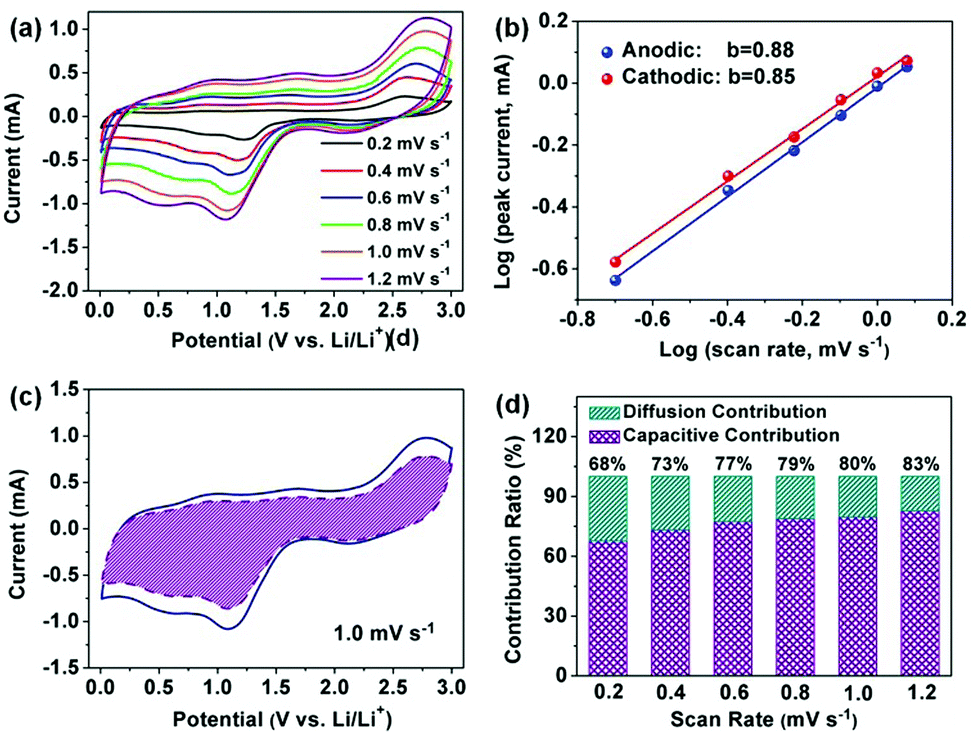
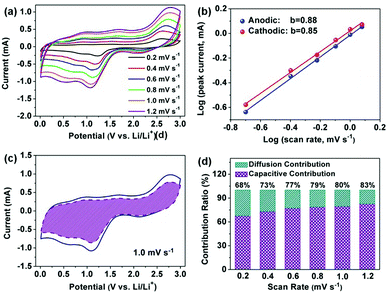
A good linear relationship can be observed as depicted in Fig. 6b, and the value of b for the main cathodic peaks (I) and anodic peaks (II) was calculated to be 0.85 and 0.88, respectively, suggesting fast kinetics from the dominant pseudocapacitive effects. Additionally, the ratio of the capacitive contributions can be quantitatively calculated by separating the diffusion-controlled process (k1ν1/2) and capacitive behavior (k2ν) according to the following equations.51,52
where
k1 and
k2 are adjustable parameters, and can be quantified at a given potential by plotting
i/
ν1/2 against
ν1/2. As a result, utilizing
eqn (7), 76% of the total current (shaded region) from the dominant capacitive contribution was found at a scan rate of 1.0 mV s
−1 (
Fig. 6c). Obviously, the pseudocapacitive contribution gradually increases when the scan rate rises (
Fig. 6d). All these results are responsible for the higher capacity of the Cu
2O–CuO–C/Cu electrode compared to the theoretical value, indicating the pseudocapacitive process as the majority of the charge storage mechanism, which facilitates the fast charge storage and high rate capacity.
On the basis of the above discussed results, we trust that the advantageous structural and compositional features also contribute to the remarkable rate capability and outstanding cycling stability. (1) Compared with single metal oxides, two active metal oxide composites attached on Cu foam can provide a higher electrical conductivity because of a low activation energy of electron transfer and offer richer redox chemical kinetics.53,54 For comparison, we also increased the calcination temperature up to 800 °C to obtain pure CuO phase as the final product. The PXRD patterns in Fig. S7 (ESI†) further confirm the complete conversion of Cu2O/CuO/Cu to CuO. The CuO electrode exhibited good cycling stability; however, a relatively inferior specific capacity of 367 mA h g−1 was obtained (Fig. S8, ESI†). Moreover, the advantages of Cu2O–CuO dual active components can be directly proved by comparing the corresponding electrochemical performance of single metal oxide CuO or Cu2O attached on Cu foam, respectively. Unsurprisingly, both of the CuO/Cu foam and Cu2O/Cu foam electrodes exhibited much inferior capability compared to that of the mixed metal oxide Cu2O–CuO/Cu (Fig. S9 and S10, ESI†). These results indicate that the synergistic effects between different components greatly contribute to the high capacity. (2) A large BET surface area and porosity not only can provide rapid access of the electrolyte into the electrode, but also facilitate more contact areas between the electrolyte and material.55 Meanwhile, the nanoparticles can create a shorter distance for lithium-ion diffusion, making it possible to charge faster or to draw high current in a short time, which is vital for the rate capability.56,57 (3) For comparison, a control experiment was carried out to better understand the role of the copper foam substrate. As presented in Fig. S11 (ESI†), the Cu foam after the hydrothermal process (without the MOF source) and sintering at 300 °C shows negligible capacity, indicating no contributions to the total capacity. However, highly conductive substrate copper foam can act as one highway for fast charge transfer during cycling. Moreover, we also investigated the electrochemical performance of the Cu2O–CuO–C electrode. As depicted in Fig. 4c, the capacity of the obtained Cu2O–CuO–C drops quickly at a current of 0.1 A g−1 and retains 382 mA h g−1 after 200 cycles. At a high rate of 5 A g−1, only 154 mA h g−1 can be maintained, revealing a worse capability compared to that of the Cu2O–CuO–C/Cu electrode. Additionally, according to eqn (1) and (2), extra Li2O can be provided by the initial irreversible reaction. In addition to the Cu decomposed from Cu2O, the Cu from the copper foam substrate can probably participate in the oxidation reaction during the subsequent charging process since extra Li2O is provided as shown in Fig. 5b, which will partially offset the irreversible loss in each cycle and contribute to the high capacity. This phenomenon can be verified by the almost disappearance of the diffraction peaks of Cu for the Cu2O–CuO–C/Cu electrode after charge cycles (Fig. 5b) compared to those of the original Cu2O–CuO–C/Cu electrode (Fig. 3a). These results imply that the copper foam can support all the Cu2O–CuO–C particles for immobilization and prompt all active substances involved in the reaction at the same time.
Conclusions
In summary, we have successfully reported the novel in situ synthesis of a Cu2O–CuO–C/Cu electrode on copper foam for the first time using a facile two-step strategy, including the direct growth of a Cu-based MOF on Cu foam and subsequent thermal annealing in air. The prepared Cu2O–CuO–C/Cu as a superior binder-free anode shows high reversible capacity and excellent cycling performance, resulting from the uniquely elaborate synergistic effects of the multicomponents and the introduction of the highly conductive substrate copper foam for rapid charge transfer. We expect that this new and effective approach may shed light onto the fabrication of promising electrode materials derived from MOFs for energy storage.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21671071 and 21471061), the Great Scientific Research Project of Guangdong Ordinary University (No. 2016KZDXM023), the Innovation Team Project of Guangdong Ordinary University (No. 2015KCXTD005), the Science and Technology Planning Project of Guangdong Province (2017B090917002, 2017A010104015 and 2015B010135009), and the Open Project of State Key Laboratory of Supramolecular Structure and Materials (sklssm201812).
Notes and references
- S. Gao, Y. Lin, X. Jiao, Y. Sun, Q. Luo, W. Zhang, D. Li, J. Yang and Y. Xie, Nature, 2016, 529, 68–71 CrossRef CAS PubMed.
- X. Zhang, Y. Zheng, X. Liu, W. Lu, J. Dai, D. Y. Lei and D. R. MacFarlane, Adv. Mater., 2015, 27, 1090–1096 CrossRef CAS PubMed.
- Y. Yao, N. Xu, D. D. Guan, J. T. Li, Z. C. Zhuang, L. Zhou, C. W. Shi, X. Liu and L. Q. Mai, ACS Appl. Mater. Interfaces, 2017, 9, 39425–39431 CrossRef CAS PubMed.
- Z. H. Liu, D. D. Guan, Q. Yu, L. Xu, Z. C. Zhuang, T. Zhu, D. Zhao, L. Zhou and L. Q. Mai, Energy Storage Mater., 2018, 13, 112–118 CrossRef.
- C. J. Tang, Y. N. Liu, C. Xu, J. X. Zhu, X. J. Wei, L. Zhou, L. He, W. Yang and L. Q. Mai, Adv. Funct. Mater., 2018, 28, 1704561 CrossRef.
- G. Xia, D. Liu, F. Zheng, Y. Yang, J. Sua and Q. Chen, J. Mater. Chem. A, 2016, 4, 12434–12441 RSC.
- G. Xia, J. Su, M. Li, P. Jiang, Y. Yang and Q. Chen, J. Mater. Chem. A, 2017, 5, 10321–10327 RSC.
- L. Hou, L. Lian, L. Zhang, G. Pang, C. Yuan and X. Zhang, Adv. Funct. Mater., 2015, 25, 238–246 CrossRef CAS.
- B. Liu, X. Zhang, H. Shioyamaa, T. Mukaia, T. Sakaia and Q. Xu, J. Power Sources, 2010, 195, 857–861 CrossRef CAS.
- K. Zhang, X. Han, Z. Hu, X. Zhang, Z. Tao and J. Chen, Chem. Soc. Rev., 2015, 44, 699–728 RSC.
- Y. Han, P. Qi, S. Li, X. Feng, J. Zhou, H. Li, S. Su, X. Li and B. Wang, Chem. Commun., 2014, 50, 8057–8060 RSC.
- M. R. Palacin, Chem. Soc. Rev., 2009, 38, 2565–2575 RSC.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C. Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- Q. Yang, Q. Xu and H.-L. Jiang, Chem. Soc. Rev., 2017, 46, 4774–4808 RSC.
- H. Li, M. Liang, W. Sun and Y. Wang, Adv. Funct. Mater., 2016, 26, 1098–1103 CrossRef CAS.
- Z.-Y. Sui, P.-Y. Zhang, M.-Y. Xu, Y.-W. Liu, Z.-X. Wei and B. H. Han, ACS Appl. Mater. Interfaces, 2017, 9, 43171–43178 CrossRef CAS PubMed.
- J.-L. Niu, G.-X. Hao, J. Lin, X.-B. He, P. Sathishkumar, X.-M. Lin and Y.-P. Cai, Inorg. Chem., 2017, 56, 9966–9972 CrossRef CAS PubMed.
- T. Jiang, F. Bu, X. Feng, I. Shakir, G. Hao and Y. Xu, ACS Nano, 2017, 11, 5140–5147 CrossRef CAS PubMed.
- G. Huang, L. Zhang, F. Zhang and L. Wang, Nanoscale, 2014, 6, 5509–5515 RSC.
- J. Li, D. Yan, S. Hou, T. Lua, Y. Yao, D. H. C. Chu and L. Pan, Chem. Eng. J., 2018, 335, 579–589 CrossRef CAS.
- G. Huang, D. Yin, F. Zhang, Q. Li and L. Wang, Inorg. Chem., 2017, 56, 9794–9801 CrossRef CAS PubMed.
- X. Xu, K. Cao, Y. Wang and L. Jiao, J. Mater. Chem. A, 2016, 4, 6042–6047 RSC.
- Y. Wang, M. Kong, Z. Liu, C. Lin and Y. Zeng, J. Mater. Chem. A, 2017, 5, 24269–24274 RSC.
- G. Fang, J. Zhou, Y. Cai, S. Liu, X. Tan, A. Pan and S. Liang, J. Mater. Chem. A, 2017, 5, 13983–13993 RSC.
- J.-L. Niu, G.-X. Hao, J. Lin, X.-B. He, P. Sathishkumar, X.-M. Lin and Y.-P. Cai, Inorg. Chem., 2017, 56, 9966–9972 CrossRef CAS PubMed.
- H.-J. Peng, G.-X. Hao, Z.-H. Chu, C.-L. He, X.-M. Lin and Y.-P. Cai, J. Alloys Compd., 2017, 727, 1020–1026 CrossRef CAS.
- H.-J. Peng, G.-X. Hao, Z.-H. Chu, J. Lin, X.-M. Lin and Y.-P. Cai, Cryst. Growth Des., 2017, 17, 5881–5886 CrossRef CAS.
- J.-L. Niu, C.-H. Zeng, H.-J. Peng, X.-M. Lin, P. Sathishkumar and Y.-P. Cai, Small, 2017, 13, 1702150 CrossRef PubMed.
- J.-L. Niu, H.-J. Peng, C.-H. Zeng, X.-M. Lin, P. Sathishkumar, Y.-P. Cai and A.-W. Xu, Chem. Eng. J., 2018, 336, 510–517 CrossRef CAS.
- G. H. Dang, Y. T. H. Vu, Q. A. Dong, D. T. Le, T. Truong and N. T. S. Phan, Appl. Catal., A, 2015, 491, 189–195 CrossRef CAS.
- Y. Chen, X. Li, K. Park, J. Song, J. Hong, L. Zhou, Y. W. Mai, H. Huang and J. B. Goodenough, J. Am. Chem. Soc., 2013, 135, 16280–16283 CrossRef CAS PubMed.
- F. Zheng, Y. Yang and Q. Chen, Nat. Commun., 2014, 5, 5261 CrossRef CAS PubMed.
- H. Li, Z. Su, S. Hu and Y. Yan, Appl. Catal., B, 2017, 207, 134–142 CrossRef CAS.
- Y. Zhao, Y. Zhang, H. Zhao, X. Li, Y. Li, L. Wen, Z. Yan and Z. Huo, Nano Res., 2015, 8, 2763–2776 CrossRef CAS.
- L. Sun, Q. Deng, Y. Li, L. Deng, Y. Wang, X. Ren and P. Zhang, Electrochim. Acta, 2016, 222, 1650–1659 CrossRef CAS.
- B. Ramaraju, C.-H. Li, S. Prakash and C.-C. Chen, Chem. Commun., 2016, 52, 946–949 RSC.
- A.-Y. Kim, M. K. Kim, K. Cho, J.-Y. Woo, Y. Lee, S.-H. Han, D. Byun, W. Choi and J. K. Lee, ACS Appl. Mater. Interfaces, 2016, 8, 19514–19523 CrossRef CAS PubMed.
- Q. Wang, B. Yu, X. Li, L. Xing and X. Xue, J. Mater. Chem. A, 2016, 4, 425–433 RSC.
- Y. H. Xu, G. Q. Jian, Y. H. Liu, Y. J. Zhu, M. R. Zachariah and C. S. Wang, Nano Energy, 2014, 3, 26–35 CrossRef CAS.
- Y. Sun, X. Hu, W. Luo, F. Xia and Y. Huang, Adv. Funct. Mater., 2013, 23, 2436–2444 CrossRef CAS.
- L. A. Riley, S. H. Lee, L. Gedvilias and A. C. Dillon, J. Power Sources, 2010, 195, 588–592 CrossRef CAS.
- Z. Xie, Z. He, X. Feng, W. Xu, X. Cui, J. Zhang, C. Yan, M. A. Carreon, Z. Liu and Y. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 10324–10333 CrossRef CAS PubMed.
- L. Hu, X.-M. Lin, J.-T. Mo, J. Lin, H.-L. Gan, X.-L. Yang and Y.-P. Cai, Inorg. Chem., 2017, 56, 4289–4295 CrossRef CAS PubMed.
- X. Zhou, J. Shi, Y. Liu, Q. Su, J. Zhang and G. Du, J. Alloys Compd., 2014, 615, 390–394 CrossRef CAS.
- S. Liu, H. Hou, X. Liu, J. Duan, Y. Yao and Q. Liao, Ionics, 2016, 23, 1075–1082 CrossRef.
- L. Wang, X. Wang, Z. Meng, H. Hou and B. Chen, J. Mater. Sci., 2017, 52, 7140–7148 CrossRef CAS.
- L. Hu, Y. Huang, F. Zhang and Q. Chen, Nanoscale, 2013, 5, 4186–4190 RSC.
- D. Chao, P. Liang, Z. Chen, L. Bai, H. Shen, X. Liu, X. Xia, Y. Zhao, S. V. Savilov, J. Lin and Z. X. Shen, ACS Nano, 2016, 10, 10211–10219 CrossRef CAS PubMed.
- D. Wang, W. Zhou, R. Zhang, X. Huang, J. Zeng, Y. Mao, C. Ding, J. Zhang, J. Liu and G. Wen, J. Mater. Chem. A, 2018, 6, 2974–2983 RSC.
- X. Xu, K. Cao, Y. Wang and L. Jiao, J. Mater. Chem. A, 2016, 4, 6042–6047 RSC.
- X. Hu, C. Li, X. Lou, Q. Yang and B. Hu, J. Mater. Chem. A, 2017, 5, 12828–12837 RSC.
- H. Li, J. Lang, S. Lei, J. Chen, K. Wang, L. Liu, T. Zhang, W. Liu and X. Yan, Adv. Funct. Mater., 2018, 28, 1800757 CrossRef.
- X. Xu, K. Cao, Y. Wang and L. Jiao, J. Mater. Chem. A, 2016, 4, 6042–6047 RSC.
- C. Yuan, H. B. Wu, Y. Xie and X. W. Lou, Angew. Chem., Int. Ed., 2014, 53, 1488–1504 CrossRef CAS PubMed.
- Z. Bai, Y. Zhang, Y. Zhang, C. Guo and B. Tang, J. Mater. Chem. A, 2015, 3, 5266–5269 RSC.
- X. S. Zhou, L. J. Wan and Y. G. Guo, Adv. Mater., 2013, 25, 2152–2157 CrossRef CAS PubMed.
- F. Zou, X. Hu, Z. Li, L. Qie, C. Hu, R. Zeng, Y. Jiang and Y. Huang, Adv. Mater., 2014, 26, 6622–6628 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: XRD patterns, TGA curves, Raman spectrum, EDS profile, and nitrogen adsorption–desorption isotherm. See DOI: 10.1039/c8qm00366a |
|
| This journal is © the Partner Organisations 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Jia
Lin
a,
Jiliang
Niu
a,
Jinji
Lan
a,
R. Chenna Krishna
Reddy
a,
Yuepeng
Cai
*a,
Jincheng
Liu
b and
Gang
Zhang
c
*a,
Jia
Lin
a,
Jiliang
Niu
a,
Jinji
Lan
a,
R. Chenna Krishna
Reddy
a,
Yuepeng
Cai
*a,
Jincheng
Liu
b and
Gang
Zhang
c