
Co-assembly of sugar-based amphiphilic block polymers to achieve nanoparticles with tunable morphology, size, surface charge, and acid-responsive behavior†
Yen-Nan
Lin
 ab,
Lu
Su
a,
Justin
Smolen
a,
Richen
Li
a,
Yue
Song
a,
Hai
Wang
a,
Mei
Dong
a and
Karen L.
Wooley
*a
ab,
Lu
Su
a,
Justin
Smolen
a,
Richen
Li
a,
Yue
Song
a,
Hai
Wang
a,
Mei
Dong
a and
Karen L.
Wooley
*a
aDepartments of Chemistry, Chemical Engineering, Materials Science & Engineering, and the Laboratory for Synthetic-Biologic Interactions, Texas A&M University, College Station, Texas 77842, USA. E-mail: wooley@chem.tamu.edu
bCollege of Medicine, Texas A&M University, Bryan, Texas 77807, USA
First published on 11th October 2018
Abstract
The development of next-generation smart nanocarriers that can be tailored for specific applications requires precise control over physiochemical properties, yet modulation of nanostructures solely through synthetic routes is a time-consuming and labor-intensive process. In this work, co-assembly of two degradable glucose-based amphiphilic block polymers is demonstrated as a means to control nanoparticle size, surface charge, and stimuli-responsive properties, allowing optimization of these constructs for cytosolic drug delivery applications. Polymeric particles with varying weight fractions of carboxylate- and histamine-modified poly(DL-lactide)-b-poly(D-glucose carbonate)s (PDLLA-b-PDGC) were obtained with diameters ranging from ca. 30 nm to 3 μm and zeta potential values ranging from ca. −35 mV to −1.6 mV in nanopure water. Histamine moieties imparted pH-responsive behavior due to protonation below pH 7, whereas the carboxylates imparted colloidal stability and anionic character. Blending the acid- and histamine-functionalized polymers produced co-assemblies with different pH-dependent surface charge profiles. In particular, co-assemblies with 60 wt% histamine-modified PDLLA-b-PDGC (fhistamine = 0.6) swelled upon acidification from physiological pH (7.4) to endolysosomal pH (5.5), which is anticipated to enable drug release within endolysosomal compartments. The accessible procedures presented here for engineering highly tunable nanoparticles from glucose-based, functional, degradable polymers offer versatile strategies for accelerating the development and clinical implementation of such stimuli-responsive, tailored nanocarriers.
Introduction
Polymeric nanocarriers, especially those that respond to endogenous or exogenous stimuli, have tremendous potential in medicine.1–5 Tailoring polymeric nanocarriers to target specific tissues or mediate controlled release of therapeutics requires careful control over the physicochemical properties of these constructs.6–17 To this end, assembly of amphiphilic block copolymers has enabled the fabrication of nanomaterials with diverse morphologies and behaviors.4,18–26 However, synthesis and optimization of individual polymers for each unique situation is a time-consuming, expensive, and labor-intensive process, impeding the clinical translation of designer nanoparticles. It is imperative to reduce the burden associated with the development of smart nanomaterials to accelerate their development and clinical implementation.Due to the complexity of biological systems, several aspects need to be considered in the design of drug carriers.27 Nanocarrier size and surface charge significantly influence their biological interactions during the course of delivery.28 Particle size substantially affects the circulation time, clearance, and biodistribution of the carriers.6 Nanoparticles with diameters ranging from 20–200 nm are well poised to avoid renal clearance by filtration, in which particles <10 nm are filtered and cleared, reduce entrapment of hepatic and splenic fenestrations, in which particles >1 μm are cleared, and accumulate passively in tumor tissues via the enhanced permeability and retention (EPR) effect.6,29 Besides particle size, the surface charge also substantially impacts the pharmacokinetics and performance of drug carriers.30 During circulation in the blood stream, positively-charged particulates interact with plasma components and consequently facilitate aggregation, opsonization, and clearance.6 Carriers that passively deposit into tissues often enter cells via endocytosis;6 as endosomes mature into lysosomes, the lysosomal degradative enzymes can destroy encapsulated active drugs. Further, drugs such as paclitaxel and doxorubicin must reach cellular components outside of the endolysosomal compartments. Paclitaxel binds to beta-tubulin subunits of microtubules located in the cytosol, thereby preventing disassembly of the microtubules.31 Doxorubicin intercalates DNA and stabilizes the topoisomerase II complex located in the nucleus, preventing DNA replication needed for cell division.32,33 Since molecular targets of these drugs are located outside of the endolysosome, endolysosomal escape is crucial for these drugs. To facilitate cytosolic drug delivery, nanocarriers should exhibit optimal size, non-cationic surface charge, and colloidal stability during circulation, and then undergo charge-reversal to display cationic surfaces in response to acidification of endosomes (pH = 5.0–6.5) to induce rupture of endosomes and release of nanocarriers into the cytoplasm.3,34–38
Co-assembly of multiple polymeric components has been demonstrated to yield an array of functional composite nanoparticles with a range of properties without the need for synthesis of individual polymers for each nanoparticle.2,26,39,40 Co-assembly has also been applied to control the surface characteristics of DNA-conjugated nanocarriers, thereby tuning cellular uptake, nuclease resistance, and antisense activity.39 In addition, physical blending of different temperature-responsive polymers was employed to fine-tune the transition temperature of elastin-based materials.41 Advances in synthetic chemistry have enabled precise tuning of the physicochemical properties of natural product-based polymeric materials, which are particularly beneficial for biomedical applications owing to their biocompatibility.42–45 Co-assembly of sugar-based amphiphilic nonionic and cationic amphiphilic block polymers afforded nanocarriers for delivery of chemotherapeutics with tunable size, toxicity, and drug release kinetics.2 Taken together, these studies showcase the potential of co-assembly as a versatile approach to fine-tune the stimuli-responsive behavior and physicochemical properties of natural product-based nanocarriers.
Herein, co-assembly of acid- and histamine-modified hydrolytically-degradable glucose-based block copolymers was explored as a means to control the size, surface charge, and acid-responsive profile of polymer nanomaterials, enabling optimization for drug delivery and other potential applications. In designing the nanocarriers, poly(DL-lactide)-b-poly(D-glucose carbonate) (PDLLA-b-PDGC) building blocks were selected based upon their biocompatibility, functionality, and ability to assemble into versatile nanostructures with varying sizes, charges, and functions.2,46,47 The alkyne-containing PDGC block has been employed to enable facile post-polymerization modification via thiol–yne and copper(I)-catalyzed azide alkyne cycloaddition reactions, affording a variety of multi-functional polymeric systems.2,46,47 In addition, we recently demonstrated that poly(L-lactide)-b-poly(D-glucose carbonate) PLLA-b-PDGC degrades into low molar mass hydrophilic molecules,47 including lactic acid and carbon dioxide, which are anticipated to be easily eliminated to reduce systemic accumulation.47 Acid- and histamine-modified PDLLA-b-PDGC are anticipated to display complementary properties for drug delivery applications. Histamine-modification imparts pH-responsive properties to polymer systems, due to the presence of imidazole groups, which are expected to facilitate a neutral-to-positive charge transformation when pH values below the pKa of the imidazole conjugate acid (pKa = 6.95)48,49 are experienced, for instance in acidic endolysosomal environments. Besides charge switching, the buffering capacity of the imidazole groups close to endosomal pH is known to trigger hydrochloric acid influx into endosomes, leading to osmotic swelling and rupture of endolysosomes and cytosolic release of the contents through the proton-sponge effect.3,49,50 Such delivery mechanisms are especially beneficial for drugs having destinations in the cytoplasm (e.g., paclitaxel). While histamine-functionalized PDLLA-b-PDGC is anticipated to provide pH-responsive behavior and afford endosomal escape, these materials may also exhibit limited hydrophilicity, particularly at blood pH where the imidazoles are deprotonated, which may hinder the formation of colloidally-stable nanostructures. On the other hand, acid-functionalized PDLLA-b-PDGC is anticipated to impart colloidal stability and anionic character, but lack pH-responsive character in the physiologically-relevant pH range desired to promote endosomal rupture. Therefore, here we describe the co-assembly of the acid- and histamine-modified PDLLA-b-PDGC to achieve highly tunable nanomaterials, enabling facile optimization of smart sugar-based nanoparticles for drug delivery applications. This work represents fundamental advances in the fabrication of multi-functional nanostructures from glucose-derived acid-responsive amphiphilic block polymeric materials, with potential for these constructs to be useful in drug delivery applications.
Experimental section
Materials
DL-Lactide was purchased from TCI America (Portland, OR) and purified by recrystallization from ethyl acetate. 1,5,7-Triazabicyclo[4.4.0]dec-5-ene (TBD) was used as received from TCI America (Portland, OR). Hydroxybenzotriazole (HOBt) and 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate (HBTU) were used as received from Chem-Impex International, Inc. (Wood Dale, IL). Dichloromethane (DCM) and N,N-dimethyl formamide (DMF) were dried using a solvent purification system (J. C. Meyer Solvent Systems, Inc., Laguna Beach, CA). Hydrochloric acid (HCl, 36.5–38.0 wt%) was purchased from Thermo-Fisher Scientific. Nanopure water (18.2 MΩ cm) was obtained from a Milli-Q water filtration system (Millipore Corp, USA). The alkyne-substituted glucose carbonate monomer, methyl-2-O-ethyloxycarbonyl-3-O-propargyloxycarbonyl-4,6-O-carbonyl-α-D-glucopyranoside (GC(EPC)), was synthesized according to a previously published procedure.46 3-(N-Morpholino)propanesulfonic acid (MOPS) was acquired from Sigma-Aldrich (St. Louis, MO). MOPS buffers were prepared according to the Cold Spring Harbor protocols. Briefly, MOPS (4.19 g), sodium acetate (410 mg), and disodium ethylenediaminetetraacetate dihydrate (Na2EDTA·2H2O, 372 mg) were dissolved in nanopure water (1 L), and the pH was adjusted using sodium hydroxide and hydrochloric acid solutions. All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO) and used without further purification unless otherwise noted. Spectra/Por dialysis membranes (MWCO 12–14 kDa) were purchased from Spectrum Laboratories, Inc. (Rancho Dominguez, CA).Synthesis of PDLLA290-b-PDGC20
A solution of DL-lactide (1.00 g, 6.94 mmol) and 4-methylbenzyl alcohol (0.10 mL, 24 mg mL−1 in DCM, 2.4 mg, 0.020 mmol) was prepared in anhydrous DCM (1.00 mL) and transferred to a vial equipped with a stir bar and a rubber septum in an argon-filled glovebox. The vial was then removed from the glovebox and connected via a needle inlet to a Schlenk line. A solution of TBD in DCM (0.10 mL, 14 mg mL−1, 0.010 mmol) was injected quickly into the vial of DL-lactide at −78 °C (acetone-dry ice bath). After stirring for 2 min, a solution of GC(EPC) (200 mg, 0.54 mmol) in DCM (1.00 mL) was added via syringe to the reaction mixture. The reaction was stirred for an additional 6 min at −78 °C and then quenched by addition of excess acetic acid. Precipitation from DCM into methanol three times and drying under vacuum yielded PDLLA290-b-PDGC20 as a white powder (952 mg, 79% yield). 1H NMR (500 MHz, CDCl3, ppm): δ 7.20 and 7.17 (ABq, J = 8 Hz), 5.36 (dd, J = 10, 10 Hz), 5.11–5.24 (m), 5.03 (d, J = 5 Hz), 4.89 (dd, J = 10, 10 Hz), 4.79–4.68 (m), 4.28 (s), 4.20 (dq, J = 8, 3 Hz), 4.01 (m), 3.42 (s), 2.59 (t, J = 3 Hz), 2.35 (s), 1.62–1.53 (m), 1.30 (t, J = 7 Hz). 13C NMR (126 MHz, CDCl3, ppm): δ 169.7, 154.1, 153.7, 153.6, 129.4, 128.5, 96.5, 77.3, 77.0, 76.3, 74.2, 73.6, 69.3, 69.1, 66.7, 65.8, 64.9, 56.0, 55.9, 16.8, 16.7, 14.2. FTIR: 3270, 2990, 2970, 1750, 1455, 1375, 1255, 1090, 1025, 905, 870, 750 cm−1. SEC (THF): Mn = 42.6 kDa, Đ = 1.10. TGA in Ar: 316–379 °C, 95% mass loss. Tg = 60 °C.Synthesis of PDLLA290-b-PDGC(COOH)20
A solution of PDLLA290-b-PDGC20 (600 mg, 0.21 mmol alkyne), 3-mercaptopropionic acid (2255 mg, 21 mmol), and 2,2-dimethoxy-2-phenylacetophenone (DMPA, 53.8 mg, 0.21 mmol) was prepared in anhydrous DMF (50 mL), deoxygenated under N2(g) for 30 min, and irradiated under UV light (365 nm) for 2 h. The reaction mixture was precipitated into diethyl ether twice and dissolved in DMF. The resulting copolymer solution was transferred to dialysis tubing (MWCO 6–8 kDa) and dialyzed against nanopure water at 4 °C for 3 d to remove DMF, excess thiol and photo-initiator. The solution was then lyophilized to give the anionic polymer as a white powder (620 mg, 93% yield). 1H NMR (500 MHz, DMSO-d6, ppm): δ 7.20 and 7.17 (ABq, J = 8 Hz), 5.32 (br), 5.20–5.08 (m), 4.98 (br), 4.85 (br), 4.77 (br), 4.32 (br), 4.19 (br), 4.11 (br), 3.31 (br), 3.09 (br), 2.70 (br), 2.48 (br), 1.43 (m), 1.18 (br). 13C NMR (126 MHz, DMSO-d6, ppm): δ 172.9, 169.1, 153.8, 153.4, 153.2, 95.6, 68.9, 68.6, 66.3, 64.5, 62.9, 54.9, 43.8, 34.8, 34.6, 33.6, 27.3, 25.9, 16.4, 13.9. FTIR: 3670-3090, 2995, 2940, 2850, 1750, 1665, 1455, 1380, 1265, 1185, 1130, 1090, 1050, 920, 865, 760 cm−1. TGA in Ar: 240–290 °C, 17% mass loss; 290–323 °C, 69% mass loss. Tg = 60 °C, 106 °C.Synthesis of PDLLA290-b-PDGC(His)20
A solution of PDLLA290-b-PDGC(COOH)20 (326 mg, 0.22 mmol acid), HOBt (44 mg, 0.33 mmol), and HBTU (130 mg, 0.34 mmol) was prepared in dimethyl sulfoxide (DMSO, 2.50 mL) and stirred at ambient temperature for 0.5 h. Histamine (97 mg, 0.87 mmol) and N,N-diisopropylethylamine (DIPEA, 42 mg, 0.33 mmol) were dissolved in DMSO (0.50 mL), then added to the reaction mixture and stirred for 24 h. The solution was transferred to dialysis tubing (MWCO 6–8 kDa) and dialyzed against nanopure water at 4 °C for 3 d to remove excess histamine and coupling agents. The solution was then lyophilized to give the histamine-modified polymer, PDLLA290-b-PDGC(His)20, as a white powder (281 mg, 80% yield). 1H NMR (500 MHz, DMSO-d6, ppm): δ 8.06 (br), 7.40 (br), 7.20 and 7.17 (ABq, J = 8 Hz), 5.33 (br), 5.20–5.08 (m), 4.99 (br), 4.86 (br), 4.78 (br), 4.34 (br), 4.19 (br), 4.11 (br), 3.99 (br), 3.31 (br), 3.08 (br), 2.77 (br), 2.72 (br), 2.48 (br), 2.32 (br), 1.44 (m), 1.19 (br). 13C NMR (126 MHz, DMSO-d6, ppm): δ 173.0, 169.2, 153.8, 153.5, 153.3, 134.6, 259.0, 68.9, 68.7, 64.5, 54.9, 43.8, 35.8, 34.6, 33.5, 27.9, 26.0, 16.5, 13.9. FTIR: 3670–3090, 2995, 2940, 2850, 1750, 1665, 1455, 1380, 1265, 1185, 1130, 1090, 1050, 920, 865, 760 cm−1. TGA in Ar: 240–290 °C, 17% mass loss; 290–323 °C, 69% mass loss. Tg = 58 °C.General procedure for co-assembly
PDLLA290-b-PDGC(COOH)20 and PDLLA290-b-PDGC(His)20 were assembled in aqueous solution at predetermined weight ratios using a nanoprecipitation method.19,25,39 Briefly, the polymers (1 mg) were dissolved in acetone (500 μL), and then added drop-wise to aqueous solution (1 mL, nanopure water or MOPS buffer), followed by stirring in air for 24 h to allow evaporation of acetone. The final polymer concentration was adjusted to 1 mg mL−1 using nanopure water. The polymer assemblies were characterized in solution by dynamic light scattering (DLS) and zeta potential measurements, and in the dry state by transmission electron microscopy (TEM).Evaluation of the pH-responsive behavior of the co-assemblies
Block copolymers (2 mg) were first dissolved in acetone (1 mL) with a predetermined polymer weight ratio, and then added drop-wise to nanopure water (2 mL) followed by stirring in air for 24 h. The polymer co-assemblies were transferred to dialysis tubing (MWCO 6–8 kDa) and dialyzed against 1× MOPS buffer (pH = 5.5 or 7.4) for 12 h, and the resulting structures were characterized by DLS, zeta potential measurements, and TEM.Results and discussion
Polymer nanoparticles with controllable size, zeta potential, and pH-responsive behavior allow optimization towards specific drug delivery applications, such as drug release in the cytoplasm. Intravenous cytosolic drug carriers should exhibit the following criteria: (1) 20–200 nm diameter during blood circulation to minimize renal clearance, reduce reticuloendothelial clearance, and, for cancer drug delivery, passively accumulate into tumor tissue by the EPR effect; (2) non-cationic surfaces to impart colloidal stability during circulation; and (3) charge-reversal capabilities to promote escape from endosomal compartments into the cytoplasm upon acidification to ca. pH 5.0–6.5. To this end, alkyne-functionalized, sugar-based block polymers were synthesized by ring-opening polymerization and modified to afford acid- and histamine-modified block polymers for co-assembly into nanoparticles with tunable sizes, surface compositions and pH-responsive properties. Organocatalyzed sequential ring-opening polymerization of cyclic DL-lactide and bicyclic, glucose-based GC(EPC) yielded alkyne-containing block polymers. The polymers were modified to append carboxylates via photo-initiated thiol–yne click chemistry, and the acid-functionalized polymers were further modified by amidation to afford histamine-modified polymers. Histamine was selected due to its pH-responsiveness and endosomal escape capability, which is desirable for cytosolic delivery.3,6,11 Co-assembly of these polymers in different weight ratios enabled fabrication of nanoparticles with tunable size, surface charge, and pH-responsive profiles well suited for cytosolic drug delivery.The functional and degradable diblock PDLLA290-b-PDGC20, was synthesized by organocatalyzed sequential ring-opening polymerization of cyclic DL-lactide, followed by bicyclic GC(EPC) at −78 °C in DCM with 4-methylbenzyl alcohol as the initiator and TBD as the organocatalyst, according to previously-reported procedures.2,46,47 The degree of polymerization and compositions of the lactide and glucose carbonate blocks were controlled by varying the monomer–initiator ratio. The number-average degree of polymerization and corresponding Mn (49.2 kDa) were determined by end group analysis using 1H NMR spectroscopy (Fig. S2, ESI†). SEC revealed PDLLA290-b-PDGC20 to have a narrow molar mass distribution, with dispersity (Đ < 1.20) and Mn = 42.6 kDa determined relative to polystyrene standards (Fig. S1, ESI†).
The anionic PDLLA290-b-PDGC(COOH)20, was prepared by post-polymerization modification of PDLLA290-b-PDGC20via photo-initiated thiol–yne click reaction with a large excess of 3-mercaptopropionic acid (100 molar equivalents relative to alkyne groups). 3-Mercaptopropionic acid was selected due to its hydrophilicity and anionic character, which is desirable for preventing aggregation in physiological environments. In addition, the carboxylic acid groups allowed further polymer modification, which was particularly appealing for installation of histamine moieties that are incompatible with thiol–yne reactions. Successful addition of 3-mercaptopropionic acid to PDLLA290-b-PDGC20 was supported by 1H NMR spectroscopic analysis of the polymer in DMSO-d6, noting the appearance of proton resonances at δ 3.09 and 2.70 ppm (Fig. S4, ESI†), and by FTIR spectroscopy, with the appearance of peaks characteristic of O–H stretching in the carboxylic acid between 3600 and 2300 cm−1 after modification (Fig. S6, ESI†).
The pH-responsive PDLLA290-b-PDGC(His)20, was prepared by post-polymerization modification of PDLLA290-b-PDGC(COOH)20via amidation with an excess of histamine (4 molar equivalents relative to carboxylic acid groups) (Scheme 1). Histamine was conjugated via amidation with the carboxylic acid groups of PDLLA290-b-PDGC(COOH)20 since photo-initiated thiol–yne click chemistry was not suitable for histamine due to the presence of the radical-sensitive imidazole groups.51,52 Successful conjugation of histamine to PDLLA290-b-PDGC(COOH)20 was supported by 1H NMR spectroscopic analysis of the polymer in DMSO-d6, noting the appearance of imidazole proton resonances at δ 8.06 and 7.40 ppm (Fig. S5, ESI†). FTIR spectroscopy further revealed the disappearance of O–H stretching in carboxylic acid between 3600 and 2300 cm−1 after modification (Fig. S6, ESI†).
 | ||
| Scheme 1 Synthesis and aqueous assembly of acid-(PDLLA290-b-PDGC(COOH)20) and histamine-modified (PDLLA290-b-PDGC(His)20) PLA-b-PDGC. The size and morphology of the nanostructures can be tuned by varying the weight ratio of the acid- and histamine-modified polymers and assembly medium. | ||
To gauge the properties of nanoparticles formed from the acid- and histamine-modified block polymers, PDLLA290-b-PDGC(COOH)20 and PDLLA290-b-PDGC(His)20 were first assembled individually in aqueous solution using a nanoprecipitation method.19,25,39 Briefly, the polymers were dissolved in acetone (500 μL, 2 mg mL−1), and then added drop-wise to nanopure water (1 mL), followed by stirring in air for 24 h to allow for evaporation of acetone. The final polymer concentration was then adjusted to 1 mg mL−1 using nanopure water. DLS revealed that upon assembly in nanopure water (pH = ca. 6), PDLLA290-b-PDGC(COOH)20 formed nanoparticles with hydrodynamic diameters (Dh(number)) < 50 nm, while PDLLA290-b-PDGC(His)20 formed micron-sized aggregates (Fig. 1 and Fig. S9, ESI†). The differences between the two polymeric particles were attributed to the different functional side chains affording varying degrees of electrostatic stabilization in nanopure water, with anionic carboxylates on PDLLA290-b-PDGC(COOH)20 and neutral or cationic imidazoles on PDLLA290-b-PDGC(His)20. The aggregation of the histamine-functionalized polymers into larger particles is attributed to the lower hydrophilic character imparted by the imidazole-containing pendant groups compared to the carboxylate-containing pendant groups in nanopure water at pH = ca. 6. Amphiphilic polymers with less hydrophilic character tend to assemble into larger particle sizes to minimize the energetically unfavorable contact of hydrophobic regions with water molecules, since larger particles have smaller surface area-to-volume ratios.53 PDLLA290-b-PDGC(COOH)20 nanoparticles displayed anionic surfaces with zeta potentials of ca. −36 mV, whereas electrophoretic light scattering of PDLLA290-b-PDGC(His)20 aqueous assemblies could only be conducted after centrifugation of large aggregates with the remaining particles displaying neutral surfaces with zeta potentials of ca. −1.6 mV in nanopure water (Fig. 2 and Fig. S9, ESI†), consistent with the aggregation observed for PDLLA290-b-PDGC(His)20 (Fig. S9, ESI†). Although neither PDLLA290-b-PDGC(COOH)20 nor PDLLA290-b-PDGC(His)20 alone was anticipated to yield assemblies suitable for cytosolic drug delivery, we hypothesized that blending the two polymers would enable the fabrication of co-assemblies with highly tunable particle sizes and surface charges to tailor degradable, smart sugar-based nanoparticle drug carriers.
 | ||
| Fig. 1 (A) Hydrodynamic diameter and (B) polydispersity index of polymer co-assemblies, measured by DLS, as a function of the weight fraction of PDLLA290-b-PDGC(His)20 (fhistamine) in nanopure water. | ||
 | ||
| Fig. 2 Zeta potential of the co-assemblies in nanopure water as a function of fhistamine. | ||
Co-assemblies of PDLLA290-b-PDGC(COOH)20 and PDLLA290-b-PDGC(His)20 were fabricated in aqueous solution (1 mg mL−1 in nanopure water) at predetermined weight fractions using a similar nanoprecipitation method as for the single-component assemblies,19,25,39 affording particles with a variety of sizes and surface compositions. As depicted in Fig. S9 (ESI†), DLS data revealed unimodal size distributions of all nanocarriers, suggesting the incorporation of PDLLA290-b-PDGC(COOH)20 and PDLLA290-b-PDGC(His)20 into the same nanostructures. The size of the co-assemblies increased from Dh(number) = 30 ± 10 nm to Dh(number) = 120 ± 30 nm (Fig. 1 and Fig. S9, ESI†) as the weight fraction of the histamine-modified PDLLA290-b-PDGC(His)20 (fhistamine) increased from 0 to 0.6, owing to the decrease in hydrophilic content of the assemblies in nanopure water (pH = ca. 6). Co-assemblies with fhistamine = 0.6 were found to be at a threshold composition, above which the particle diameter increased from nanoscale to microscale (Fig. 1 and Fig. S9, ESI†).
An initial postulate as to the mechanism of assembly and rationale for the variations in particle sizes is based upon several factors. PDLLA290-b-PDGC(COOH)20 is more hydrophilic compared to PDLLA290-b-PDGC(His)20 in nanopure water, due to the presence of the carboxylate side-chains instead of imidazole side-chains. The increased hydrophilicity is supported by the ability to form stable supramolecular assemblies comprised of only PDLLA290-b-PDGC(COOH)20 in water, whereas stable co-assemblies could only be formed when the weight fraction of PDLLA290-b-PDGC(His)20 was maintained at ≤60% (fhistamine ≤ 0.60). With the greater hydrophilicity and water solubility for the PDGC(COOH)20 coronal segments, the hydrophilic volume fraction is larger in PDLLA290-b-PDGC(COOH)20 assemblies compared to co-assemblies containing PDLLA290-b-PDGC(His)20. Furthermore, acid–base attractive interactions between the carboxylic acids of the PDGC(COOH)20 segments and the histamines of the PDGC(His)20 segments would further reduce the hydrophilic volume fraction in the co-assemblies. Reduction in hydrophilic volume fraction would change the average molecular shape of the polymer chain in aqueous solution, resulting in different morphology of the assemblies in order to minimize the total free energy of the system.54,55 TEM images of Fig. S9 (ESI†) suggest that assemblies of PDLLA290-b-PDGC(COOH)20 are primarily micelles, whereas the larger co-assembled particles may be vesicular. Micelle-to-vesicle transformations upon reduction of hydrophilic volume fraction is consistent with other polymeric assemblies reported in the literature.56,57
Polymer co-assembly further allowed fabrication of particles with controllable particle surface charge depending on the feed ratio of two sugar-based copolymers. Particles with zeta potentials ranging from ca. −36 to −1.6 mV were formed simply by altering the fhistamine (Fig. 2 and Fig. S9, ESI†). When fhistamine ≤ 0.6, the zeta potential of the co-assemblies remained relatively constant, as the overall particle surfaces were dominated by the hydrophilic anionic carboxylates of PDLLA290-b-PDGC(COOH)20. As fhistamine increased, the surface density of the acid groups decreased, yielding a corresponding decrease in the magnitude of the negative zeta potential (Fig. 2 and Fig. S9, ESI†). Interparticle repulsion decreased with the decreasing magnitude of zeta potential, consistent with the marked increase in co-assembly diameter at fhistamine > 0.6.
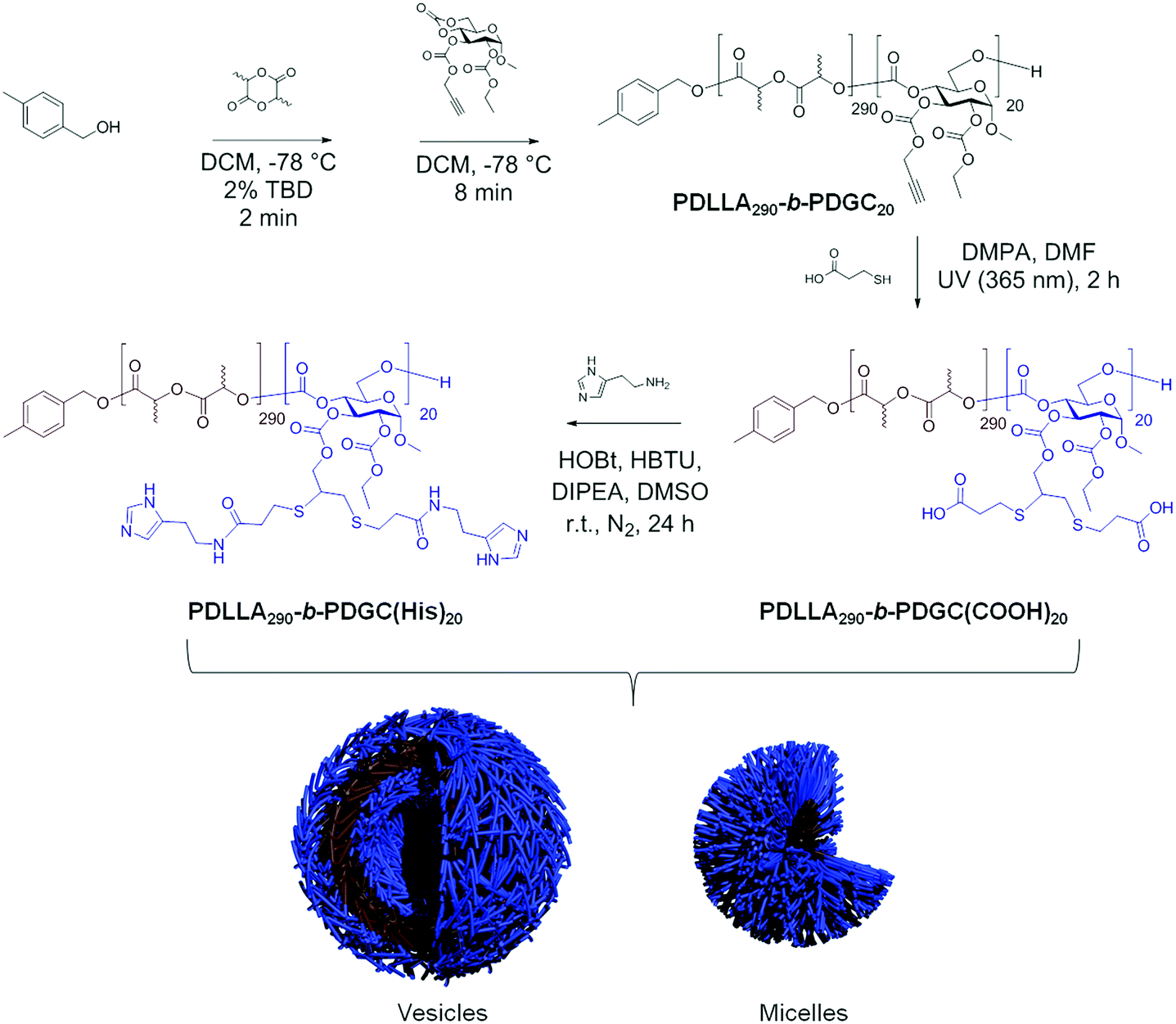
The histamine moieties within the co-assemblies imparted highly tunable pH-dependent charge-switching profiles to the polymeric particles. While the surface charge of PDLLA290-b-PDGC(COOH)20 assemblies remained relatively constant with zeta potentials of ca. −35 mV, PDLLA290-b-PDGC(His)20 assemblies underwent a cationic-to-anionic surface charge transformation (ca. +22 mV to −15 mV) as pH was increased from 5 to 8, with an apparent isoelectric point (pH(I)) of ca. 7 (Fig. 3A and Fig. S10, ESI†). The pH-dependent charge transformation of PDLLA290-b-PDGC(His)20 was in agreement with the reported pKa of the imidazole conjugate acid (pKa = 6.95).48 Interestingly, the apparent isoelectric point of PDLLA290-b-PDGC(His)20 was ca. 7, where PDLLA290-b-PDGC(His)20 was expected to be partially positive due to protonation of ca. 50% of the imidazoles at pH 7. The neutralization may result from the surface charge balance between protonated PDLLA290-b-PDGC(His)20 and adsorbed anionic hydroxyl ions on the surface of the assemblies. Since oxygen atoms of water molecules preferentially orient toward the hydrophobic particle surface,58,59 hydrogen atoms of the interfacially-adsorbed water molecules can facilitate adsorption of anionic hydroxyl ions (from self-ionization of water), leading to negative surface zeta potentials for hydrophobic polymeric particles.58,59
 | ||
| Fig. 3 pH-Responsive properties of acid- and histamine-modified, sugar-based block copolymers and their co-assemblies: (A) pH-dependent zeta potential of nanoparticles comprised of PDLLA290-b-PDGC(COOH)20, PDLLA290-b-PDGC(His)20, and co-assemblies (fhistamine = 0.6) in MOPS buffer (20 mM); (B) acid-induced charge reversal of PDLLA290-b-PDGC(COOH)20 and PLA290-b-PDGC(His)20 co-assemblies. | ||
Polymeric co-assemblies exhibited different pH-dependent charge-switching profiles compared to particles comprised only of PDLLA290-b-PDGC(COOH)20 or PDLLA290-b-PDGC(His)20. Aqueous co-assemblies with fhistamine from 0.6–0.7 underwent anionic-to-cationic surface charge transformations as the solution pH was decreased from 7.4 to 5.5, (Fig. 3B and Fig. S11, ESI†). The pH-dependent zeta potential change upon acidification was more pronounced for polymer co-assemblies (fhistamine = 0.6, ca. +55 mV) compared to PDLLA290-b-PDGC(His)20 assemblies (ca. +35 mV) (Fig. 3A and Fig. S10, ESI†). In addition, polymer co-assemblies (fhistamine = 0.6) underwent charge-reversal within a narrower pH range of 1.3 (i.e., from pH 5.5 to 6.8), compared to the wider pH range of 2 (from pH 6 to 8) required for the transformation of PDLLA290-b-PDGC(His)20 assemblies. Additionally, the isoelectric point of the polymer co-assemblies (fhistamine = 0.6) was lower (pH(I) = ca. 6) than that of the PDLLA290-b-PDGC(His)20 assemblies (pH(I) = ca. 7). The tunability of the pH-responsive properties by this co-assembly procedure was further demonstrated in co-assemblies with varying fhistamine. Co-assemblies with higher fhistamine exhibited less pronounced charge transformations compared to co-assemblies with lower fhistamine, with surface compositions dominated by the carboxylates of the PDLLA290-b-PDGC(COOH)20 at pH 7.4. Therefore, the overall surface charge of the co-assemblies could be modulated both by pH and by the relative ratio of carboxylate and imidazole functionalities in the nanostructures (Fig. 3B and Fig. S11, ESI†).
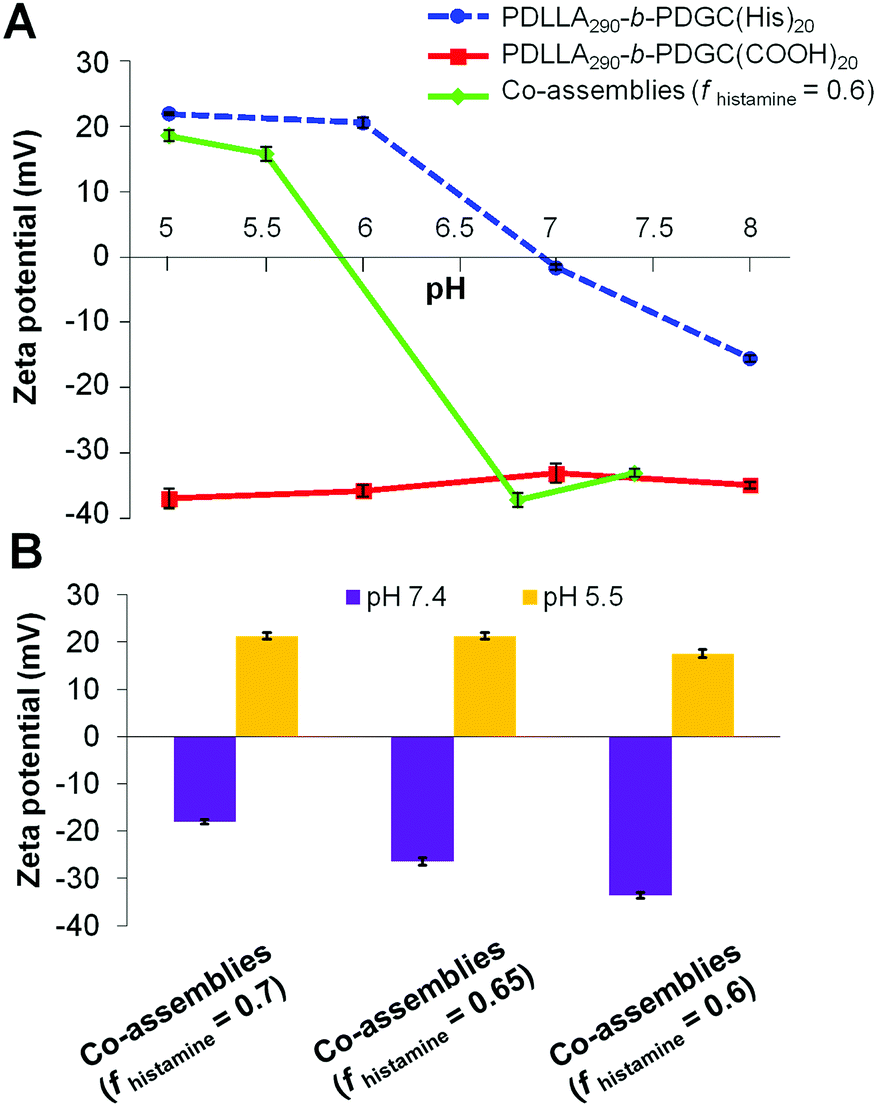
In addition to tuning particle size and pH-dependent surface charge, co-assembly altered the morphology of the polymeric nanoparticles as a function of pH and composition. PDLLA290-b-PDGC(COOH)20 aqueous assemblies remained relatively constant in size (Fig. 4 and Fig. S13, ESI†), whereas PDLLA290-b-PDGC(His)20 assemblies decreased in size and the co-assemblies (fhistamine = 0.6) increased in size with decreasing pH (Fig. 4, 5 and Fig. S13, ESI†). At pH = 5.5, a majority of the imidazole groups were protonated and PDLLA290-b-PDGC(His)20 assemblies exhibited cationic surfaces, imparting hydrophilicity and affording colloidally-stable nano-sized assemblies. The imidazole groups of PDLLA290-b-PDGC(His)20 were deprotonated at pH = 7.4, increasing the hydrophobic character and, consequently, the diameter of the assemblies, which eventually aggregated into micron-sized precipitates.
 | ||
| Fig. 4 Hydrodynamic diameter of assemblies comprised of PDLLA290-b-PDGC(COOH)20, PDLLA290-b-PDGC(His)20, and co-assemblies (fhistamine = 0.6) in MOPS buffer (20 mM) at pH = 5.5 and 7.4. | ||
 | ||
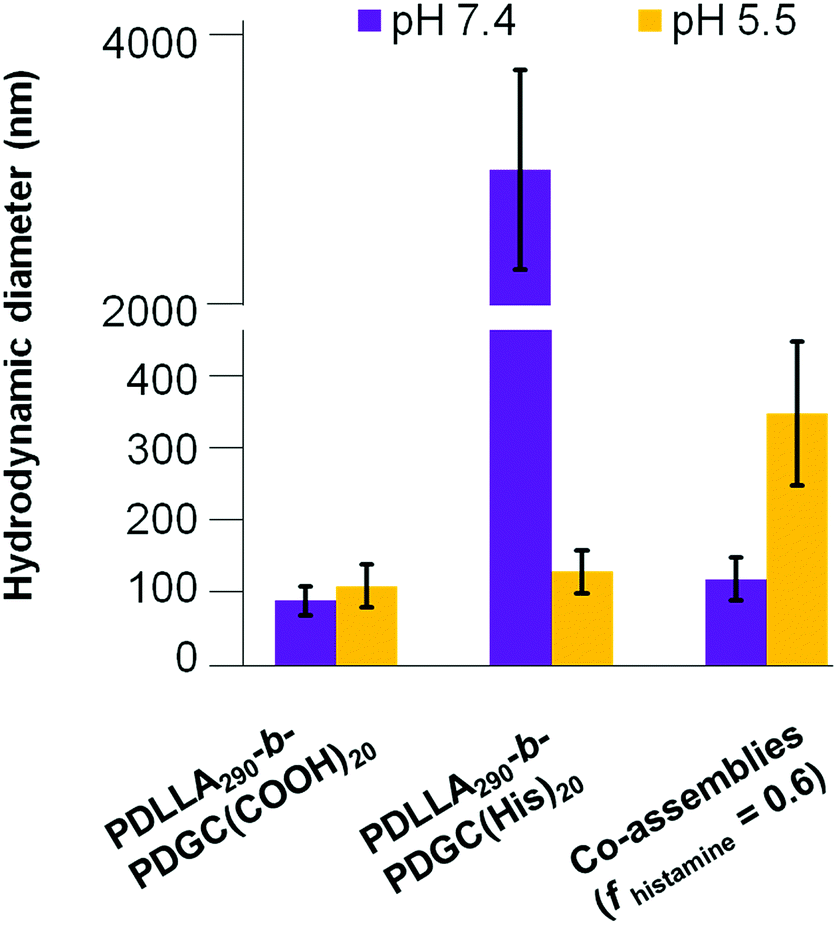
| Fig. 5 Morphological characterization of co-assemblies (fhistamine = 0.6) at pH 7.4 and pH 5.5 using DLS and TEM. TEM samples were negatively stained by 1 wt% phosphotungstic acid aqueous solution. | ||
In contrast, co-assemblies (fhistamine = 0.6) exhibited acid-triggered swelling behavior. We speculated that, at pH 7.4, unprotonated imidazole groups were primarily localized within the hydrophobic core, whereas the acid groups dominated the surface of the particles. With decreasing pH and imidazole protonation, the hydrophilicity of the protonated imidazole groups resulted in swelling of the co-assemblies. Interestingly, PDLLA290-b-PDGC(COOH)20 formed different morphologies when assembled in nanopure water vs. MOPS buffer (pH 7.4). The PDLLA290-b-PDGC(COOH)20 formed micelles ca. 30 nm in diameter in nanopure water but formed vesicles ca. 90 nm in diameter in MOPS buffer (pH 7.4, Fig. S9 and S13, ESI†). The difference may arise from charge shielding by the buffer ions, reducing the electrostatic repulsion between carboxylates, thereby reducing the hydrophilic volume fraction and giving rise to different morphologies compared to those formed in nanopure water. Such results suggested the potential tunability in morphology by altering the salt composition of the aqueous solution.
Despite the successful charge-switching ability of the imidazole-containing pH-responsive PDLLA290-b-PDGC(His)20, the micron-size self-assemblies (Fig. S13, ESI†) at physiological pH of 7.4 would reduce the circulation time and diminish the EPR effect. It was, therefore, imperative to reduce particle size while maintaining the charge-switching capability of the imidazole-containing polymer. TEM images of co-assemblies (fhistamine = 0.6) at pH 7.4 showed vesicular structures with average diameters of 121 ± 35 nm (Fig. 5), in good agreement with the DLS results (Dh(number) = 120 ± 30 nm). These multicomponent polymeric co-assemblies (fhistamine = 0.6) are well poised for cytosolic delivery, being within the optimal size range (20–200 nm) to minimize renal clearance, reduce reticuloendothelial clearance, and affording passive accumulation in tumor tissue by the EPR effect. The co-assemblies displayed carboxylate-dominated anionic surfaces (with zeta potentials of ca. −34 mV) at pH 7.4 and underwent charge-reversal to imidazole-dominated cationic surfaces (ca. +15 mV) at endolysosomal pH (5.5) (Fig. 3). During the charge-reversal process, the hydrophobic-to-hydrophilic transformation associated with the protonation of imidazole moieties swelled the polymeric co-assemblies, as shown in the increase in Dh(number) from 120 nm to 350 nm with decreasing pH (Fig. 5). TEM images of co-assemblies (fhistamine = 0.6) also indicated swelling in acidic environments, by an increase in particle diameter from 121 ± 35 nm at pH 7.4 to 197 ± 77 nm at pH 5.5. This acid-induced swelling was further associated with a broadening of the size distribution and the observation of non-spherical morphologies, as evidenced by discrepancy between DLS and TEM size measurements at endolysosomal pH. In conjunction with the endosomal escape capabilities expected from these co-assemblies, the acid-triggered swelling is expected to facilitate cytosolic delivery of therapeutics.3,6,11,50,60
Conclusions
In summary, polymer co-assembly has been shown to be a versatile approach to coincidentally tune the size, shape and surface charge of nanoscopic supramolecular assemblies in water, to optimize the stimuli-responsive properties of nanostructures. Sequential ring opening polymerizations of lactide and an alkynyl-bearing glucose-carbonate monomer afforded reactive block polymers that were conveniently transformed into amphiphilic derivatives having carboxylic acid or histamine side chain groups. Assembly and co-assembly of these block polymers yielded nanoparticles with highly tunable size, zeta potential, and pH-responsive behaviors, while bypassing tedious and time-consuming synthesis. The tailorability derived from co-assembly enables the facile optimization of nanomaterials for different biomedical applications. This work represents a fundamental advance in the design and preparation of glucose-derived acid-responsive amphiphilic block polymeric materials, with potential for these constructs to be useful in a broad range of drug delivery applications. For instance, it is anticipated that the acid-induced swelling of the assemblies will facilitate acid-triggered drug release in endolysosomal environments (pH 4.5–6.5). Ongoing studies are determining the fundamental behaviors of these co-assembled polymer mixtures in vitro, with an aim to pursue practical strategies for their utilization in biomedical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the National Science Foundation (CHE-1610311, DMREF-1629094, and DMR-1507429) and the Robert A. Welch Foundation through the W. T. Doherty-Welch Chair in Chemistry (A-0001). Use of the TAMU/LBMS and the Texas A&M University Laboratory for Synthetic-Biologic Interactions are also acknowledged. Dr Rachel Letteri is acknowledged for great insights, guidance, and detailed editings.References
- X. Zhang, L. Han, M. Liu, K. Wang, L. Tao, Q. Wan and Y. Wei, Mater. Chem. Front., 2017, 1, 807–822 RSC.
- L. Su, R. Li, S. Khan, R. Clanton, F. Zhang, Y.-N. Lin, Y. Song, H. Wang, J. Fan, S. Hernandez, A. S. Butters, G. Akabani, R. MacLoughlin, J. Smolen and K. L. Wooley, J. Am. Chem. Soc., 2018, 140, 1438–1446 CrossRef CAS PubMed.
- R. Shrestha, M. Elsabahy, S. Florez-Malaver, S. Samarajeewa and K. L. Wooley, Biomaterials, 2012, 33, 8557–8568 CrossRef CAS PubMed.
- H. Cabral, N. Nishiyama and K. Kataoka, Acc. Chem. Res., 2011, 44, 999–1008 CrossRef CAS PubMed.
- N. Nishiyama, Y. Bae, K. Miyata, S. Fukushima and K. Kataoka, Drug Discovery Today: Technol., 2005, 2, 21–26 CrossRef CAS PubMed.
- M. Elsabahy and K. L. Wooley, Chem. Soc. Rev., 2012, 41, 2545–2561 RSC.
- A. Blanazs, S. P. Armes and R. J. Anthony, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- J. Zou, F. Zhang, S. Zhang, S. F. Pollack, M. Elsabahy, J. Fan and K. L. Wooley, Adv. Healthcare Mater., 2014, 3, 441–448 CrossRef CAS PubMed.
- D. Li, L. Sun, Y. Zhang, M. Yu, J. Guo and C. Wang, Mater. Chem. Front., 2017, 1, 521–529 RSC.
- Y. H. Lim, G. S. Heo, S. Cho and K. L. Wooley, ACS Macro Lett., 2013, 2, 785–789 CrossRef CAS PubMed.
- D. Putnam, C. A. Gentry, D. W. Pack and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 1200–1205 CrossRef CAS.
- H. Wang, L. Su, R. Li, S. Zhang, J. Fan, F. Zhang, T. P. Nguyen and K. L. Wooley, ACS Macro Lett., 2017, 6, 219–223 CrossRef CAS.
- F. Zhang, J. A. Smolen, S. Zhang, R. Li, P. N. Shah, S. Cho, H. Wang, J. E. Raymond, C. L. Cannon and K. L. Wooley, Nanoscale, 2015, 7, 2265–2270 RSC.
- F. Zhang, S. Zhang, S. F. Pollack, R. Li, A. M. Gonzalez, J. Fan, J. Zou, S. E. Leininger, A. Pavía-Sanders, R. Johnson, L. D. Nelson, J. E. Raymond, M. Elsabahy, D. M. P. Hughes, M. W. Lenox, T. P. Gustafson and K. L. Wooley, J. Am. Chem. Soc., 2015, 137, 2056–2066 CrossRef CAS PubMed.
- S. Zhang, H. Wang, Y. Shen, F. Zhang, K. Seetho, J. Zou, J.-S. A. Taylor, A. P. Dove and K. L. Wooley, Macromolecules, 2013, 46, 5141–5149 CrossRef CAS PubMed.
- S. Zhang, J. Zou, M. Elsabahy, A. Karwa, A. Li, D. A. Moore, R. B. Dorshow and K. L. Wooley, Chem. Sci., 2013, 4, 2122–2126 RSC.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS PubMed.
- I. T. H. Epps and R. K. O'Reilly, Chem. Sci., 2016, 7, 1674–1689 RSC.
- H. Cui, Z. Chen, S. Zhong, K. L. Wooley and D. J. Pochan, Science, 2007, 317, 647–650 CrossRef CAS PubMed.
- Y. Chen, K. Zhang, X. Wang, F. Zhang, J. Zhu, J. W. Mays, K. L. Wooley and D. J. Pochan, Macromolecules, 2015, 48, 5621–5631 CrossRef CAS.
- J. C. Brendel and F. H. Schacher, Chem. – Asian J., 2018, 13, 230–239 CrossRef CAS PubMed.
- F. H. Schacher, P. A. Rupar and I. Manners, Angew. Chem., Int. Ed., 2012, 51, 7898–7921 CrossRef CAS PubMed.
- J. Zhu, S. Zhang, F. Zhang, K. L. Wooley and D. J. Pochan, Adv. Funct. Mater., 2013, 23, 1767–1773 CrossRef CAS.
- U. Tritschler, S. Pearce, J. Gwyther, G. R. Whittell and I. Manners, Macromolecules, 2017, 50, 3439–3463 CrossRef CAS.
- X. Yan, M. Delgado, A. Fu, P. Alcouffe, S. G. Gouin, E. Fleury, J. L. Katz, F. Ganachaud and J. Bernard, Angew. Chem., Int. Ed., 2014, 126, 7030–7033 CrossRef.
- X. Yan, R. Ramos, E. Hoibian, C. Soulage, P. Alcouffe, F. Ganachaud and J. Bernard, ACS Macro Lett., 2017, 6, 447–451 CrossRef CAS.
- K. C. L. Black, A. Ibricevic, S. P. Gunsten, J. A. Flores, T. P. Gustafson, J. E. Raymond, S. Samarajeewa, R. Shrestha, S. E. Felder, T. Cai, Y. Shen, A.-K. Löbs, N. Zhegalova, D. H. Sultan, M. Berezin, K. L. Wooley, Y. Liu and S. L. Brody, Biomaterials, 2016, 98, 53–63 CrossRef CAS PubMed.
- A. Varela-Moreira, Y. Shi, M. H. A. M. Fens, T. Lammers, W. E. Hennink and R. M. Schiffelers, Mater. Chem. Front., 2017, 1, 1485–1501 RSC.
- M. Elsabahy, G. S. Heo, S.-M. Lim, G. Sun and K. L. Wooley, Chem. Rev., 2015, 115, 10967–11011 CrossRef CAS PubMed.
- M. Elsabahy, S. Zhang, F. Zhang, Z. J. Deng, Y. H. Lim, H. Wang, P. Parsamian, P. T. Hammond and K. L. Wooley, Sci. Rep., 2013, 3, 3313 CrossRef PubMed.
- M. A. Jordan and L. Wilson, Nat. Rev. Cancer, 2004, 4, 253 CrossRef CAS PubMed.
- F. A. Fornari, J. K. Randolph, J. C. Yalowich, M. K. Ritke and D. A. Gewirtz, Mol. Pharmacol., 1994, 45, 649–656 CAS.
- R. L. Momparler, M. Karon, S. E. Siegel and F. Avila, Cancer Res., 1976, 36, 2891–2895 CAS.
- Y. Lee, T. Ishii, H. J. Kim, N. Nishiyama, Y. Hayakawa, K. Itaka and K. Kataoka, Angew. Chem., Int. Ed., 2010, 49, 2552–2555 CrossRef CAS PubMed.
- Y. Lee, K. Miyata, M. Oba, T. Ishii, S. Fukushima, M. Han, H. Koyama, N. Nishiyama and K. Kataoka, Angew. Chem., 2008, 120, 5241–5244 CrossRef.
- Y. Lee, T. Ishii, H. Cabral, H. J. Kim, J.-H. Seo, N. Nishiyama, H. Oshima, K. Osada and K. Kataoka, Angew. Chem., Int. Ed., 2009, 48, 5309–5312 CrossRef CAS PubMed.
- Y. Lee, S. Fukushima, Y. Bae, S. Hiki, T. Ishii and K. Kataoka, J. Am. Chem. Soc., 2007, 129, 5362–5363 CrossRef CAS PubMed.
- F. Pittella, M. Zhang, Y. Lee, H. J. Kim, T. Tockary, K. Osada, T. Ishii, K. Miyata, N. Nishiyama and K. Kataoka, Biomaterials, 2011, 32, 3106–3114 CrossRef CAS PubMed.
- D. Wang, X. Lu, F. Jia, X. Tan, X. Sun, X. Cao, F. Wai, C. Zhang and K. Zhang, Chem. Mater., 2017, 29, 9882–9886 CrossRef CAS.
- Y. Anraku, A. Kishimura, M. Oba, Y. Yamasaki and K. Kataoka, J. Am. Chem. Soc., 2010, 132, 1631–1636 CrossRef CAS PubMed.
- F. Fernandez-Trillo, J. C. M. van Hest, J. C. Thies, T. Michon, R. Weberskirch and N. R. Cameron, Chem. Commun., 2008, 2230–2232 RSC.
- R. Xiao, E. L. Dane, J. Zeng, C. J. McKnight and M. W. Grinstaff, J. Am. Chem. Soc., 2017, 139, 14217–14223 CrossRef CAS PubMed.
- G. L. Gregory, E. M. Lopez-Vidal and A. Buchard, Chem. Commun., 2017, 53, 2198–2217 RSC.
- K. Mikami, A. T. Lonnecker, T. P. Gustafson, N. F. Zinnel, P.-J. Pai, D. H. Russell and K. L. Wooley, J. Am. Chem. Soc., 2013, 135, 6826–6829 CrossRef CAS PubMed.
- Y.-Y. T. Tsao and K. L. Wooley, J. Am. Chem. Soc., 2017, 139, 5467–5473 CrossRef CAS PubMed.
- L. Su, S. Khan, J. Fan, Y.-N. Lin, H. Wang, T. P. Gustafson, F. Zhang and K. L. Wooley, Polym. Chem., 2017, 8, 1699–1707 RSC.
- Y. Song, Y. Chen, L. Su, R. Li, R. A. Letteri and K. L. Wooley, Polymer, 2017, 122, 270–279 CrossRef CAS.
- H. Walba and R. W. Isensee, J. Org. Chem., 1961, 26, 2789–2791 CrossRef CAS.
- J. Shi, J. G. Schellinger, R. N. Johnson, J. L. Choi, B. Chou, E. L. Anghel and S. H. Pun, Biomacromolecules, 2013, 14, 1961–1970 CrossRef CAS PubMed.
- J.-B. Du, Y. Cheng, Z.-H. Teng, M.-L. Huan, M. Liu, H. Cui, B.-L. Zhang and S.-Y. Zhou, Mol. Pharmaceutics, 2016, 13, 1711–1722 CrossRef CAS PubMed.
- I. Dogan, Spectrosc. Lett., 1992, 25, 1–11 CrossRef CAS.
- J. Llano and L. A. Eriksson, J. Phys. Chem. B, 1999, 103, 5598–5607 CrossRef CAS.
- L. R. Pratt and A. Pohorille, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 2995–2999 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- D. E. Discher and F. Ahmed, Annu. Rev. Biomed. Eng., 2006, 8, 323–341 CrossRef CAS PubMed.
- L. Zhang and A. Eisenberg, J. Am. Chem. Soc., 1996, 118, 3168–3181 CrossRef CAS.
- L. Zhang and A. Eisenberg, Polym. Adv. Technol., 1998, 9, 677–699 CrossRef CAS.
- J. K. Beattie, Lab Chip, 2006, 6, 1409–1411 RSC.
- K. G. Marinova, R. G. Alargova, N. D. Denkov, O. D. Velev, D. N. Petsev, I. B. Ivanov and R. P. Borwankar, Langmuir, 1996, 12, 2045–2051 CrossRef CAS.
- H. Deng, J. Liu, X. Zhao, Y. Zhang, J. Liu, S. Xu, L. Deng, A. Dong and J. Zhang, Biomacromolecules, 2014, 15, 4281–4292 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00353j |
| This journal is © the Partner Organisations 2018 |