
The 4-pyridonyl group as a multifunctional electron donor in 1,8-naphthalimide-based photoluminescent and mechanically interlocked coordination compounds†
Chris S.
Hawes
 *ab,
Kevin
Byrne
c,
Wolfgang
Schmitt
c and
Thorfinnur
Gunnlaugsson
*b
*ab,
Kevin
Byrne
c,
Wolfgang
Schmitt
c and
Thorfinnur
Gunnlaugsson
*b
aSchool of Chemical and Physical Sciences, Keele University, Keele ST5 5BG, UK. E-mail: c.s.hawes@keele.ac.uk
bSchool of Chemistry and Trinity Biomedical Sciences Institute (TBSI), The University of Dublin, Trinity College Dublin, Dublin 2, Ireland
cSchool of Chemistry and Centre for Research on Adaptive Nanostructures and Nanodevices, The University of Dublin, Trinity College Dublin, Dublin 2, Ireland. E-mail: gunnlaut@tcd.ie
First published on 17th May 2018
Abstract
A new ligand system incorporating the 4-pyridonyl group as a substituent in 1,8-naphthalimide compounds is presented, with the pyridone group acting as both an electron donor for the internal charge transfer (ICT) fluorescence process and as a metal binding moiety. After establishing the solution-state photophysical properties of the new pyridyl and carboxyphenyl derivatives L1 and HL2, respectively, we have prepared and characterised four crystalline d-block metal complexes containing the 4-(4-pyridonyl)-1,8-naphthalimide residue. Complex 1, a discrete mononuclear Ag(I) complex, and complexes 2 and 3, both one-dimensional Zn(II) coordination polymers, all exhibit striking photoluminescence in the crystalline phase, the nature of which can be related to the solid-state behaviour of the pyridone substituent. Complex 4, a permanently porous Cu(II) material, shows no photoluminescence, but instead reveals a rare and intriguing inclined 2D → 3D polyrotaxane architecture, an unusual class of mechanically interlocked network in which the particular coordination chemistry of the pyridone group facilitates alternating looped and linear structural features.
Introduction
With the view of providing new means for controlling the strength and geometry of metal–ligand interactions, many elegant examples have recently emerged of the use of novel organic building blocks,1 in order to imbue additional functionality into coordination polymers or metal–organic frameworks (MOFs).2 These often take the form of functionalities not readily accessible from the typical late 3d or 4d metal ions present in such systems, and include Lewis basic sites for CO2 fixation, hydrogen bond donor groups for anion binding and organocatalysis, CO2 binding sites, redox-active groups, etc.3 Organic fluorophores present another interesting case of ligand-based functionality. Immobilising an organic fluorophore within a crystalline matrix, be it in the pure form or as a structural component of a coordination polymer, can have substantial influences on its emission behaviour.4 Aggregation processes tend to modulate emission properties through fluorophore π–π and n–π interactions, or quenching of non-radiative decay pathways involving molecular motions, etc.5 Additionally, immobilised photoactive ligands in the crystalline phase can exhibit strong responses to external stimuli by virtue of the high effective concentrations bought about by confinement phenomena.6In the study of MOFs and related materials, some classes of donor ligands are very strongly represented (e.g. carboxylates, imidazolates, pyridines, pyrazolates, phosphonates and nitriles/cyanides),7 while others are only very rarely encountered. Reports into the coordination chemistry of the 4-pyridonyl group, while not completely unknown, are surprisingly quite uncommon.8 As a relatively electron rich oxygen donor, with a minimal steric bulk at the coordination site, but without the tendency to adopt bridging coordination modes as the analogous phenolates, 4-pyridone-based ligands can be conceptually related to neutral nitrile ligands. For those systems, especially in the case of soft metal ions, many fascinating and ornate examples of metallosupramolecular materials have been reported, including examples of mechanically interlocked architectures.9 Such systems are of special interest as functional materials based on the possibility of reversible physical motions from low energy framework rearrangements, as well as their fascinating topological complexity.10 However, in comparison with nitriles, the harder oxygen donor present in 4-pyridone lends itself much more strongly towards coordination with 3d metal ions, themselves heavily favoured in supramolecular materials chemistry for their abundance and lability.11 Here, we report the synthesis of two new 4-pyridonyl ligands incorporating a photoluminescent 1,8-naphthalimide core. The photophysics of these compounds is investigated, and the structural chemistry of their Cu(II), Zn(II) and Ag(I) complexes is outlined, including their application in the formation of an intriguing mechanically interlocked polyrotaxane structure.
Results and discussion
Synthesis, structure and photophysical properties of L1 and HL2
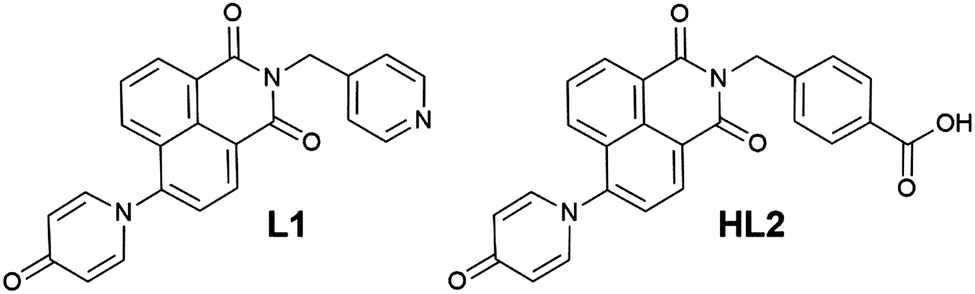
Based on our previously reported methodology for the synthesis of divergent 1,8-naphthalimide-derived ligands,12 the nitro-substituted precursors N-(4-picolyl)-4-nitro-1,8-naphthalimide and N-(4-carboxyphenylmethylene)-4-nitro-1,8-naphthalimide, respectively, were reacted with 4-hydroxypyridine in DMSO in the presence of potassium carbonate to give L1 and HL2, respectively (Scheme 1). In both cases, the N-4-pyridone substituted ligands were isolated in moderate to good yields and excellent purity after 3 hours of reaction at 110–115 °C. We did not observe any isolable quantities of the isomeric 4-pyridyl ether products. Although structural characterisation was primarily carried out using standard spectroscopic techniques and via structure determination of the corresponding metal complexes 1–4, a single crystal of HL2 as a DMF solvate HL2·DMF was prepared by recrystallization in hot DMF. It must be noted that crystals of HL2 were not prepared in any substantial quantity by this method; subsequent analysis and preparations were carried out on the amorphous HL2 phase isolated from the synthetic procedure. The structure of HL2·DMF, shown in Fig. 1, confirms the expected structure of the ligand species and shows a strong hydrogen bonding interaction (D⋯A distance 2.525(2) Å) between the carboxylic acid group and the pyridone carbonyl oxygen atom, in a head-to-tail fashion. The pyridone ring itself is non-coplanar with the naphthalimide group (interplanar angle 63°), and shows significant double bond localisation, with shorter C–C bonds for the pyridonyl 2- to 3-positions (crystallographically C21–C22 and C24–C25 1.354(2) and 1.351(2) Å, respectively) compared to the bonds for the 3- to 4-positions (crystallographically C22–C23 and C23–C24, 1.436(2) and 1.431(2) Å, respectively). The carbonyl group exhibits a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O distance of 1.270(2) Å. Unsurprisingly, face-to-face π–π interactions are observed between the naphthalimide groups in the structure of HL2; parallel groups associate at a mean interplanar distance of 3.56 Å on the concave face of the molecule.
O distance of 1.270(2) Å. Unsurprisingly, face-to-face π–π interactions are observed between the naphthalimide groups in the structure of HL2; parallel groups associate at a mean interplanar distance of 3.56 Å on the concave face of the molecule.
 | ||
| Scheme 1 Structure of ligands L1 and HL2. | ||
 | ||
| Fig. 1 (top) Structure of HL2 with heteroatom labelling scheme. (bottom) Extended structure of HL2 showing hydrogen bonding and π–π stacking interactions between adjacent chains. Selected hydrogen atoms, and lattice DMF molecules are omitted for clarity. | ||
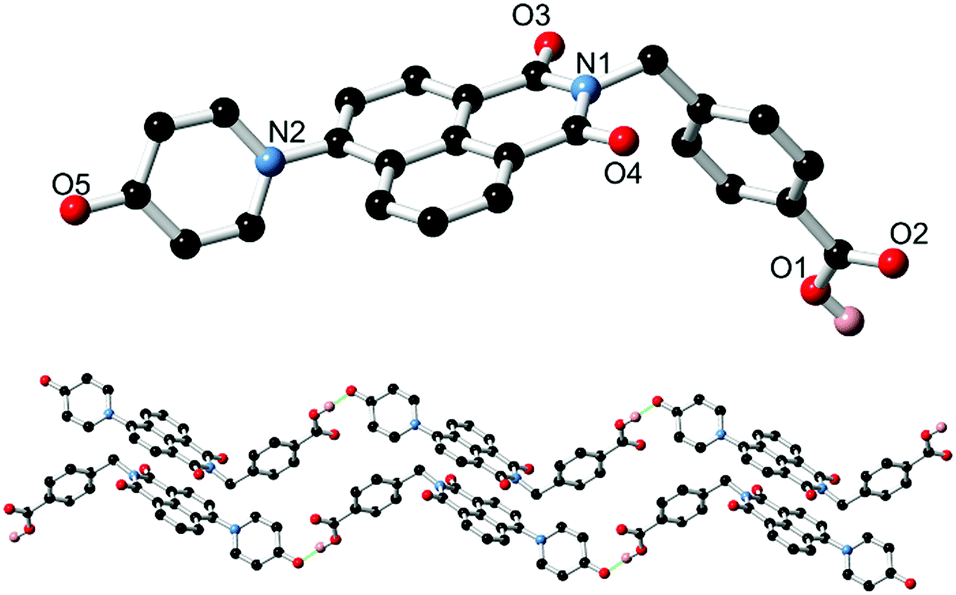
Based on the well-known propensity for 1,8-naphthalimide species containing electron-donor substituents at the 4-position to exhibit prominent photoluminescence,13 solutions of L1 and HL2 were examined for their fluorescence behaviour. In chloroform, L1 displays an absorption maximum at 351 nm corresponding to the typical naphthalimide internal charge transfer (ICT) transition, as shown in Fig. 2. Excitation at this wavelength gave blue-green emission with maxima of 453 nm. The photoluminescence quantum yield ΦPL for this process is estimated at 0.29 (±10%), measured by the comparative method with quinine sulfate/2 M H2SO4 as the reference standard. Both the absorbance and emission profiles of L1 were invariant to concentration, and the absorption and normalized excitation spectra are superimposable (ESI,† Fig. S10) suggesting no strong aggregation influences are at play under these conditions. Conversely, despite a similar absorbance profile (λmax = 357 nm), a solution of HL2 in DMSO exhibited no significant photoluminescence. These results are consistent with our previous findings regarding the poor photoluminescence of similar carboxylic acid-functionalised 1,8-naphthalimides,12 and may be exacerbated here by the seemingly strong hydrogen bond acceptor characteristics of the pyridine oxygen atom revealed (at least in the crystalline phase) from the structure of HL2·DMF.
 | ||
| Fig. 2 Overlaid absorption spectra for L1 and HL2 (black and blue, respectively) and emission spectrum (λex = 366 nm) for L1. All spectra were recorded at room temperature at 9 μM concentration in CHCl3 (L1) or DMSO (HL2). | ||
Synthesis, structure and photoluminescence of L1 complexes [Ag(L1)2]SbF6·3H2O 1 and poly-[ZnL1(NO3)2]·0.5H2O 2
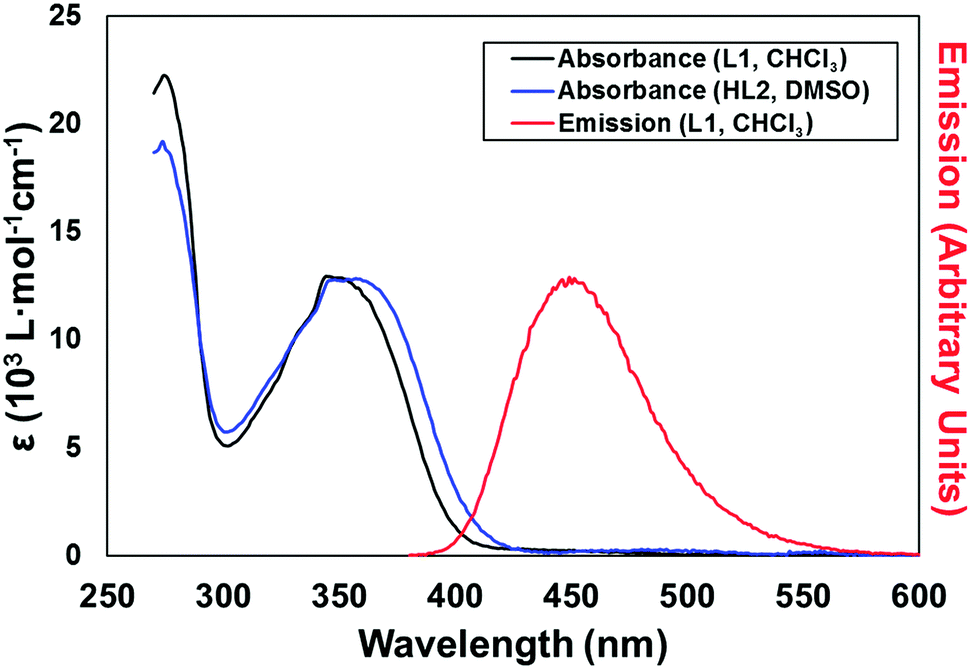
With the ligands L1 and HL2 in hand, we next turned our attention to the synthesis of coordination polymers containing the 4-(N-4-pyridonyl)-1,8-naphthalimide motif. As a neutral divergent donor species, we first reacted L1 with Ag(I), to gauge the potential of the broader naphthalimidopyridone motif as a bridging ligand. Reaction of L1 with AgSbF6 in methanol yielded a crop of yellow crystals of complex 1 after 3 days. These crystals exhibited green emission, with an emission maximum of 496 nm upon excitation at 360 nm; this is a bathochromic shift from the free ligand in solution, with the emission peak maximum reddened by ca. 1900 cm−1.The asymmetric unit of 1, in the monoclinic space group C2/c, contains one silver ion coordinated by the pyridyl nitrogen atoms of two unique L1 molecules in a linear two-coordinate arrangement, Fig. 3. Given its considerable Lewis basicity as a hydrogen bond acceptor, it is somewhat surprising that the pyridone oxygen atoms in the structure of 1 do not participate in strong coordination to the Ag ion. Instead, one of the two unique species engages in a weak O⋯Ag interaction at a distance of 3.030(5) Å, which is comparable to O⋯Ag contacts originating from the imide oxygen atoms at the same silver site and considerably longer than the Ag–N bonds (both 2.197(5) Å). As is often observed in 4-substituted 1,8-naphthalimides and related substituted naphthalenes,14 crystallographic disorder is evident on one of the two pyridone rings, which is distributed between the two equivalent naphthalimide 4-positions.
 | ||
| Fig. 3 (top) Structure of the cationic species of complex 1 with heteroatom labelling scheme; (bottom) extended structure of complex 1 showing anion⋯π contacts and π–π interactions between adjacent naphthalimide groups. Hydrogen atoms, backbone disorder and lattice solvent molecules are omitted for clarity. | ||
The extended structure of 1 is comprised of clip-shaped [Ag(L1)2] species which associate with one another through parallel head-to-tail π–π interactions, on one face each. The mean interplanar distance for these interactions are 3.39 and 3.37 Å for the two independent interactions, indicative of the type of substantial packing forces typically encountered in naphthalimide and naphthalenediimide π-systems.15 On the other face, two opposing naphthalimide groups define a cavity within which resides the hexafluoroantimonate anion. Substantial anion–π contacts are observed within this pocket, with the closest F⋯C contacts of 3.000(6) and 3.027(6) Å indicative of a significant interaction between the two species. The [Ag(L1)2] complexes further associate through the weak Ag⋯O interactions described above to form an extended network with interstitial methanol and water molecules.
Having observed no coordination of the pyridone group in the crystalline phase in the presence of the soft Lewis acid Ag(I), we turned our attention to the harder Zn(II) ion. Reaction of L1 with zinc nitrate hexahydrate in acetonitrile gave pale yellow crystals over a period of several days. Like complex 1, complex 2 is photoluminescent in the solid state, but exhibits a hypochromic shift compared to the free ligand, with an emission maximum of 425 nm (λex = 360 nm), a blue shift of 1450 cm−1 compared to L1 in CHCl3 and interestingly, separated from the solid-state emission of 1 by 3350 cm−1 even despite containing the same fluorophore species.
The asymmetric unit of 2, solved in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and shown in Fig. 4, contains one molecule of L1, one zinc ion with two associated nitrato ligands, and a lattice acetonitrile molecule. L1 coordinates through both the pyridine nitrogen atom and the pyridone oxygen atom; the latter is bound at a Zn–O distance of 1.965(4) Å. The CO bond length of 1.280(6) Å and CO–Zn angle of 129.5(4)° are both consistent with coordination of the localised ketone resonance form. The adjacent C–C bond distances of 1.420(7) and 1.426(8) Å for the pyridonyl 3- to 4-positions (C20–C21 and C21–C22), compared to 1.364(7) and 1.356(8) Å for the remaining carbon–carbon bonds C22–C23 and C19–C20, are also consistent with bond localisation within the pyridyl ring, and are statistically equivalent to the values observed in the structure of HL2. Coordination through the conjugated pyridone moiety is consistent with the observed blue-shift in emission; reduction in the electron donor capabilities of the naphthalimide 4-substituent are known to induce blue-shifting of emission bands in related species.16
and shown in Fig. 4, contains one molecule of L1, one zinc ion with two associated nitrato ligands, and a lattice acetonitrile molecule. L1 coordinates through both the pyridine nitrogen atom and the pyridone oxygen atom; the latter is bound at a Zn–O distance of 1.965(4) Å. The CO bond length of 1.280(6) Å and CO–Zn angle of 129.5(4)° are both consistent with coordination of the localised ketone resonance form. The adjacent C–C bond distances of 1.420(7) and 1.426(8) Å for the pyridonyl 3- to 4-positions (C20–C21 and C21–C22), compared to 1.364(7) and 1.356(8) Å for the remaining carbon–carbon bonds C22–C23 and C19–C20, are also consistent with bond localisation within the pyridyl ring, and are statistically equivalent to the values observed in the structure of HL2. Coordination through the conjugated pyridone moiety is consistent with the observed blue-shift in emission; reduction in the electron donor capabilities of the naphthalimide 4-substituent are known to induce blue-shifting of emission bands in related species.16
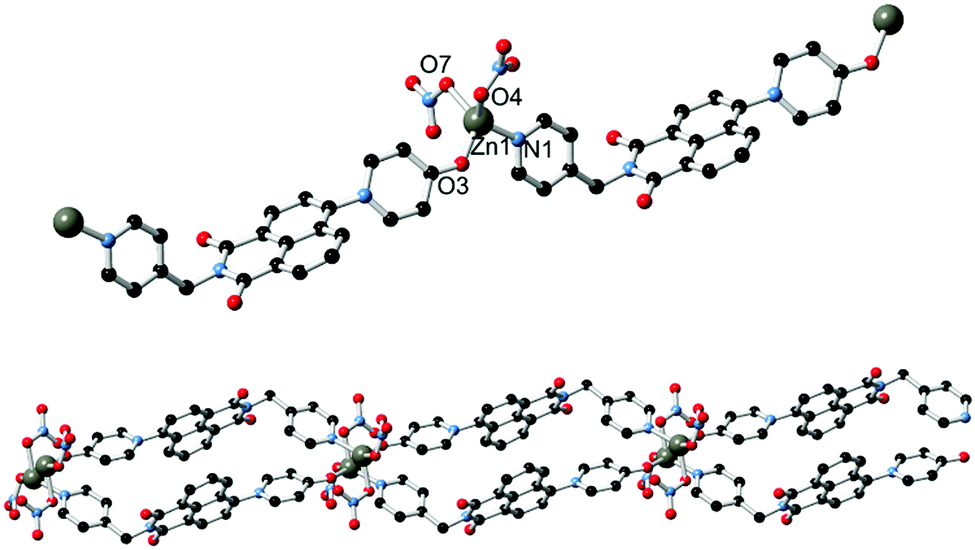 | ||
| Fig. 4 (top) The structure of the repeat unit of complex 2 with labelling scheme for coordinating atoms; (bottom) extended structure of complex 2 showing the interactions between adjacent chains. Hydrogen atoms, nitrate anion disorder and lattice solvent molecules are omitted for clarity. | ||
The complete structure of 2 is a one-dimensional coordination polymer in which each L1 molecule bridges two equivalent Zn(II) ions, whose tetrahedral coordination sphere is completed by two monodentate nitrato ligands. The interaction of adjacent chains is largely based on similar packing forces to those observed in complex 1. Parallel head-to-tail π–π interactions are present between adjacent naphthalimide groups on one face, separated by 3.46 Å interplanar distance and with antiparallel naphthyl groups slipped by 2.01 Å. The other face of the naphthalimide π system undergoes interactions with the nitrato ligands; although affected by crystallographic disorder, the minimum O⋯C distance of 3.096(7) Å for O7⋯C13 (the naphthalimide 5-position) is also indicative of anion⋯π type interactions. Beyond a single lattice acetonitrile molecule, the close packing of adjacent chains leaves no significant void volume within the structure of 2.
Synthesis and structure of poly-[Zn(L2)2]·1.5DMF·0.5H2O 3 and poly-[Cu3(L2)4(NO3)2]·13H2O·2DMF 4
The outcomes of reacting L1 with Ag(I) and Zn(II) are consistent with a preference of the pyridone donor group for harder metal ions, and with this in mind the divergent pyridone–carboxylate ligand HL2 was reacted with first-row transition metal ions with the intention of preparing coordination polymer materials of higher connectivity. Reaction of HL2 with zinc nitrate hexahydrate in DMF at 100 °C for four days yielded complex 3 as colourless crystals. The crystals exhibited blue photoluminescence under 360 nm irradiation; the emission maximum of 439 nm being comparable to that observed from the related complex 2. The structure of 3, solved and refined in the monoclinic space group C2/c, contains one molecule of deprotonated L2 and one tetrahedrally-coordinated zinc ion occupying a crystallographic special position, as well as one disordered partial-occupancy DMF molecule. The L2 species coordinates to the Zn(II) ion through both the pyridone oxygen atom and the carboxylate group, each in a monodenate mode (Fig. 5). The coordination geometry and structural parameters of the pyridone ring are consistent with those seen in complex 2, with the Zn1–O5 distance of 1.992(2) Å comparing well to that for the coordinating carboxylate (1.934(2) Å).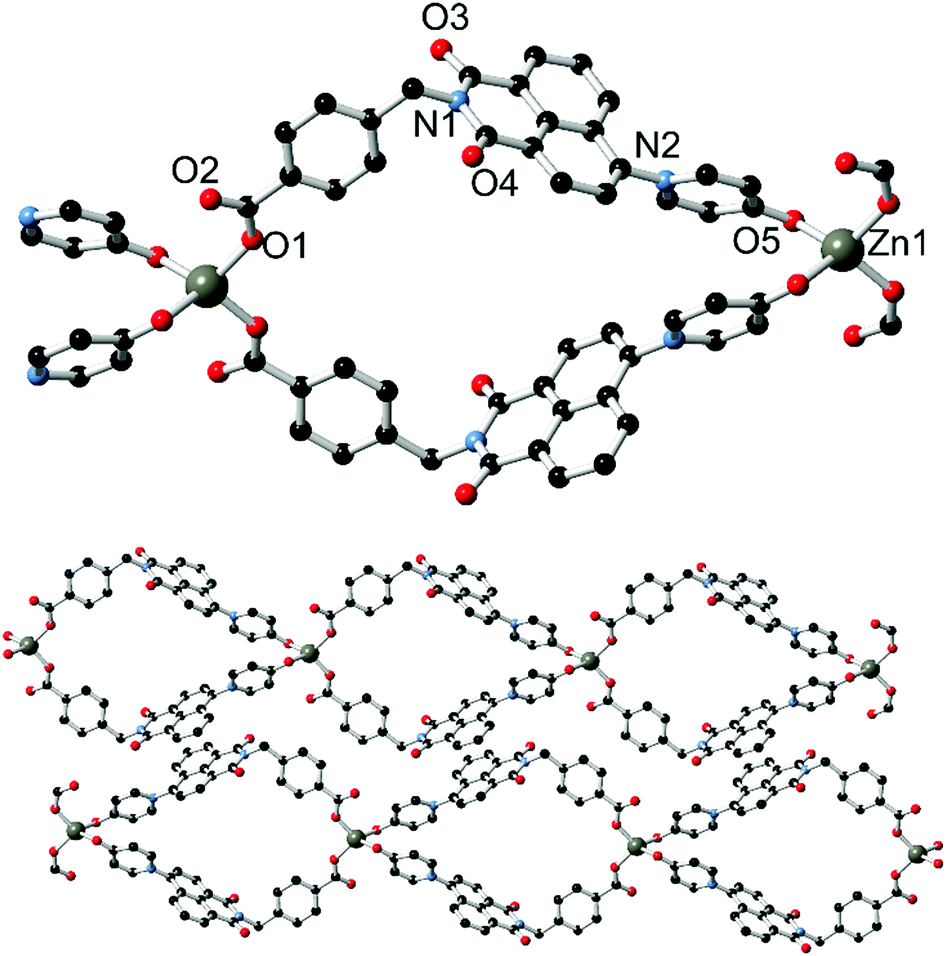 | ||
| Fig. 5 Structure of complex 3, showing (top) a single loop with heteroatom labelling scheme, and (bottom) extended structure showing interactions between adjacent chains. Hydrogen atoms and lattice solvent molecules are omitted for clarity. | ||
The extended structure of 3 is a one-dimensional coordination polymer in which tetrahedral zinc ions are linked by two pairs of L2 molecules into a looped chain oriented parallel to the b direction. The loops, defined by the methylene bridge segregating the two donor types, exhibit directionality along the chain propagation axis based on the L2 orientation. This brings the two pyridone groups coordinating to each zinc within close proximity of one another; with centroid⋯centroid distance of 3.62 Å and a minimum C⋯C distance of 3.064(5) Å for C23–C23 (the pyridonyl carbonyl atom), the two pyridone rings exhibit a mean interplanar angle of 17.2° and are rotated to one another about the centroid–centroid vector by ca. 54°. The loops within the structure of 3 are approximately diamond shaped with interatomic dimensions of ca. 8 × 8 Å; this space is filled by a disordered lattice DMF molecule. Adjacent strands interact through the familiar parallel head-to-tail π–π stacking interactions between the planar naphthalimide groups, with naphthyl mean planes separated by 3.40 Å. The efficient packing involving these interactions leads to a dense extended structure, and no additional lattice solvent or void space was detected.
Reacting HL2 with copper nitrate under similar conditions, however, gave a complex belonging to a different structural class entirely. The dark green crystals of complex 4 posed difficulties in analysis by X-ray diffraction, and suffered from relatively poor diffraction intensity and intractable structural disorder. We noted that while the reaction of HL2 and copper nitrate hemipentahydrate in DMF reproducibly yielded complex 3 as a pure phase, individual crystals obtained by this method provided poor quality diffraction data. Gratifyingly, repeating the reaction using a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of N,N-dimethylacetamide (DMA) and ethanol as the solvent provided a small quantity of higher quality single crystals, combined with amorphous material. X-ray powder diffraction confirmed that the two crystalline materials showed equivalent lattice parameters and varied only by the identity of the diffuse lattice solvent. As such, while the structural model for complex 4 is derived from crystals grown in DMA/EtOH, bulk phase characterisation was carried out on the equivalent phase grown from DMF. Unsurprisingly, no photoluminescence was observed from the solid complex 4, which is ascribed to quenching by the d9 Cu(II) ions present within the structure.
:1 mixture of N,N-dimethylacetamide (DMA) and ethanol as the solvent provided a small quantity of higher quality single crystals, combined with amorphous material. X-ray powder diffraction confirmed that the two crystalline materials showed equivalent lattice parameters and varied only by the identity of the diffuse lattice solvent. As such, while the structural model for complex 4 is derived from crystals grown in DMA/EtOH, bulk phase characterisation was carried out on the equivalent phase grown from DMF. Unsurprisingly, no photoluminescence was observed from the solid complex 4, which is ascribed to quenching by the d9 Cu(II) ions present within the structure.
The diffraction data for 4 were solved and refined in the monoclinic space group C2/c, and the asymmetric unit contains two unique copper sites (one occupying a special position), two coordinating L2 species and one nitrato ligand. The structure of 4 is shown in Fig. 6. Substantial crystallographic disorder was encountered across the entirety of one of the two naphthalimide groups. The disorder mode is related to the 4-substituted disorder mode encountered in complex 1 but without co-incidence of any of the backbone naphthalimide atoms, due to anchoring through coordination at each end of the molecule. Significant quantities of diffuse electron density were also present; no sensible chemical model could be applied to these residuals and so the data were treated with the SQUEEZE routine to account for their scattering contribution.17
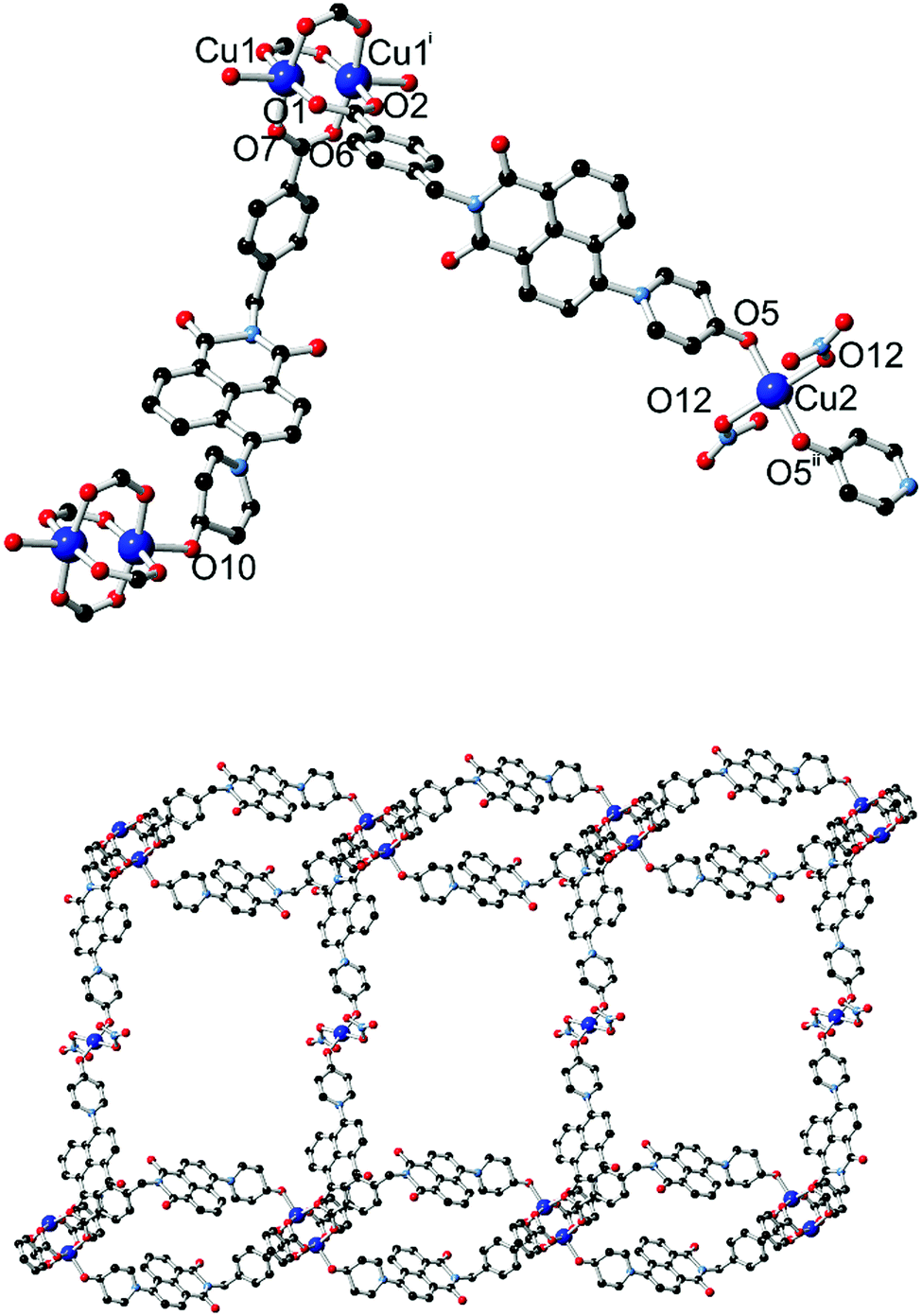 | ||
| Fig. 6 (top) The two distinct copper coordination environments within the structure of 4, with labelling scheme for coordinating heteroatoms. (bottom) Extended structure of a single layer of 4, consisting of linear bridges between looped L2-paddlewheel chains. Hydrogen atoms and backbone disorder are omitted for clarity. Symmetry codes used to generate equivalent atoms: (i) 1/2 − x, 3/2 − y, 1 − z; (ii) 2 − x, 1 − y, 2 − z. | ||
Two distinct copper environments are present in the structure of 4. A centrosymmetric copper paddlewheel motif involving Cu1 is present with four carboxylate donors in the equatorial positions, while the axial position of each copper site is filled by pyridone oxygen atom O10. Comprising four carboxylate groups and two pyridone groups, this assembly leaves two pyridone donor atoms free for additional interactions. The remaining copper site Cu2 adopts a four-coordinate square planar orientation with trans-oriented pyridone oxygen atoms and nitrato ligands. The first level of extended structure of 4, a 2-dimensional net, therefore consists of a one-dimensional looped chain of copper paddlewheels, reminiscent of complex 3, bridged laterally by coordination to the second unique copper site through the remaining pyridone oxygen atom. Each loop defines a cavity between parallel-oriented naphthalimide groups of 8.47 Å mean interplanar distance.
A singular two-dimensional net in complex 4 contains very large rectangular windows, best described by the distances between adjacent paddlewheels of 37.44 × 18.49 Å and representing approximately two or one times the bridging length of the L2 species, respectively. Unsurprisingly, this volume is occupied by interpenetrating networks, however the mode of interpenetration is an unusual and unexpected 2D → 3D inclined polyrotaxane interpenetration mode.18 In this instance, the loops which connect adjacent copper paddlewheel nodes act as hosts for the square planar Cu2 node from an adjacent network, as shown in Fig. 7. The interactions between the two mechanically interlocked networks draws obvious comparisons to the anion⋯π interactions observed in complexes 1 and 2 above; though somewhat muddled by crystallographic disorder, a minimum O2N–O⋯C distance of 2.99(2) Å for O11⋯C35 (a coordinating nitrate oxygen atom to the naphthalimide 1(8)-position) is comparable to the contact distances observed for the anion⋯π contacts observed in those species. Further face-to-face π–π interactions also occur on the outer faces of these loops with adjacent networks.
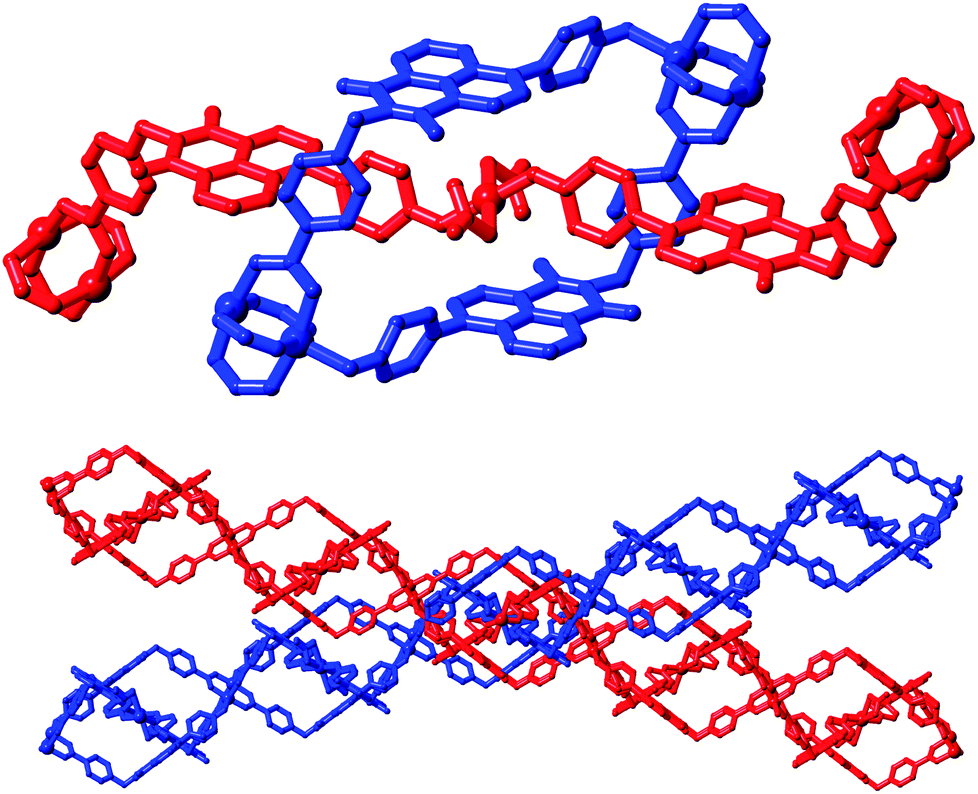 | ||
| Fig. 7 Interpenetration mode within complex 4; (top) the threading of a linear four-coordinate copper ion through a single loop from an independent network; (bottom) inclination between interlocked layers leading to the inclined 2D → 3D interpenetration mode. | ||
Guest exchange and gas adsorption study in complex 4
While most of the pore volume of a single network in complex 4 is occupied by interpenetration, a significant solvent-accessible volume remains in the structure, accounting for 32% of the unit cell volume. This free volume is localised in one-dimensional channels bordered by naphthalimide groups and the outer (inter-layer) corners of the copper paddlewheel sites, as shown in Fig. 8. Analysis by thermogravimetric analysis reveals a total volatile mass of approximately 21 wt%, lost in a gradual multi-step process with a plateau at 200 °C before the onset of thermal decomposition at 270 °C. In order to access the desolvated material under more mild conditions, the as-synthesised material was subjected to solvent exchange with methanol at room temperature for one week, and thermogravimetric analysis of this material revealed complete desolvation was achieved at 100 °C. Subjecting a sample of solvent-exchanged complex 4 to evacuation under dynamic vacuum at 100 °C for 48 hours, the material was tested for gas uptake performance using N2 and H2 probe gasses at 77 K, and CO2 at 278, 288 and 298 K. The adsorption isotherms for complex 4 are presented in Fig. 9. The N2 adsorption experiment revealed a typical type-I isotherm flattening above P/P0 = 0.01 at 60 cc(STP) g−1 adsorbed, up to a total capacity at P/P0 = 0.99 of 124 cc(STP) g−1. From these data, a BET surface area of 256 m2 g−1 was estimated. The H2 isotherm showed smooth adsorption and desorption branches with a maximum uptake of 79 cc(STP) g−1 at 1 atm. Complex 4 adsorbs CO2 with a maximum capacity of 53 cc(STP) g−1 (9.5 wt%) at 278 K and 1 atm, and estimation of the enthalpy of adsorption from the 278, 288 and 298 K isotherms provides a zero-loading enthalpy of adsorption of −22.5 kJ mol−1. Both the total capacities and enthalpy of adsorption are consistent with the moderate void volume and lack of defined CO2 interaction sites indicated by the structural model. The smooth adsorption and desorption branches suggest no structural rearrangements take place to accompany gas uptake, consistent with static interlocking of adjacent networks during the evacuation and adsorption process, although X-ray powder diffraction of the methanol exchanged material post-adsorption suggested structural changes following re-exposure to air, most likely related to a decomposition process (ESI,† Fig. S4).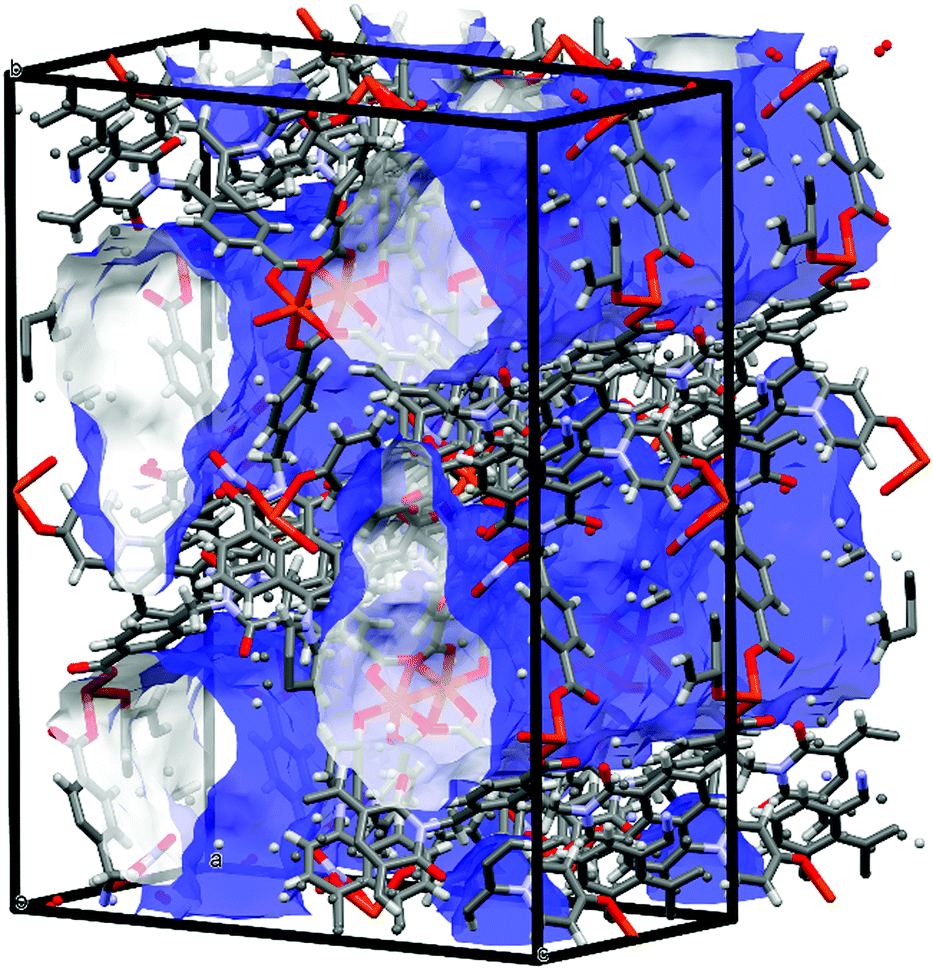 | ||
| Fig. 8 Internal pore volume in the structure of 4, rendered as a blue (external) and white (internal) surface (probe radius 1.2 Å). | ||
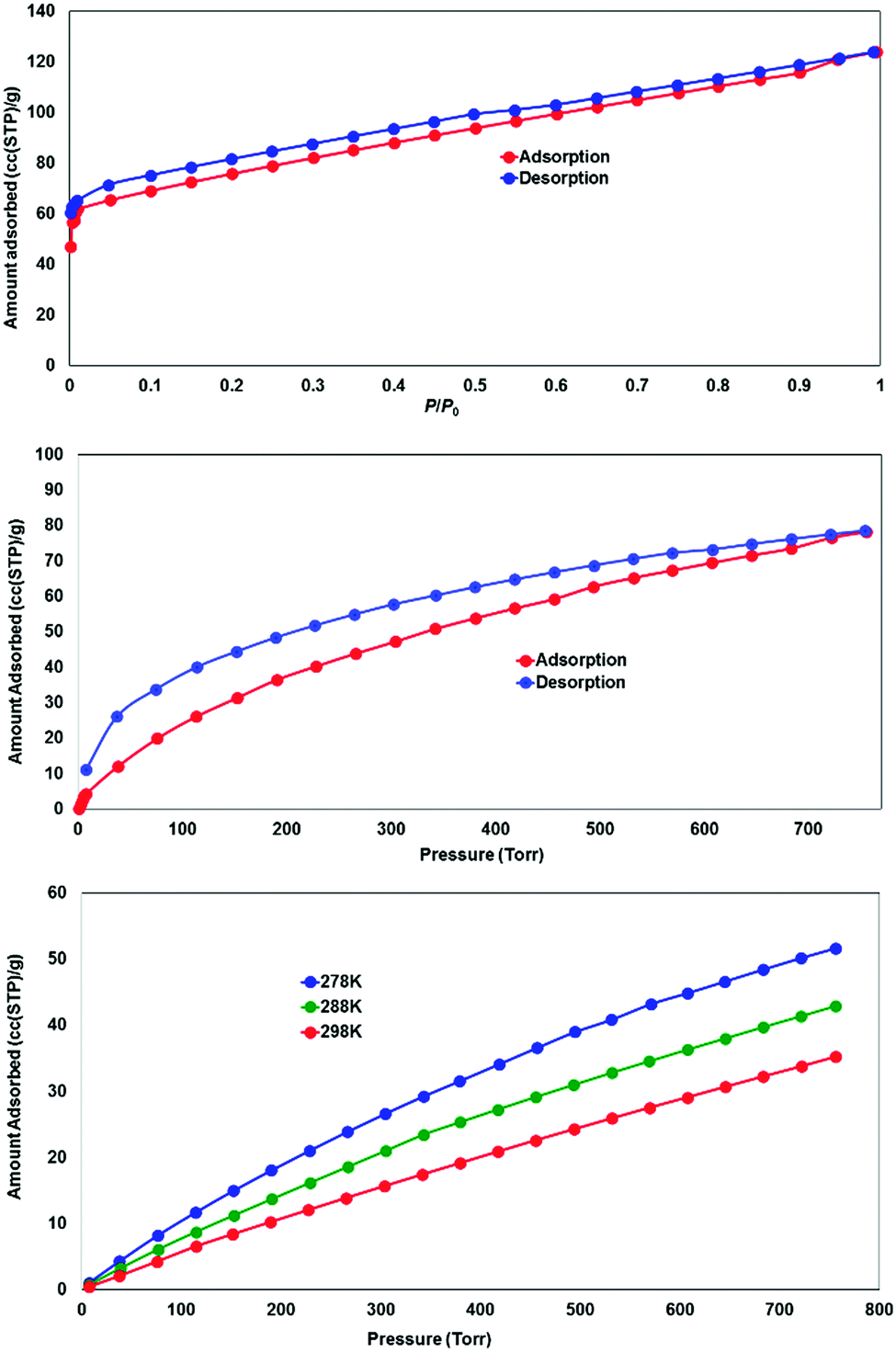 | ||
| Fig. 9 Gas adsorption performance for the solvent-exchanged complex 4, showing N2 adsorption at 77 K (top), H2 adsorption at 77 K (middle) and CO2 adsorption at 278, 288 and 298 K (bottom). | ||
Discussion
Broadly speaking, the findings reported above are consistent with the expectation of the pyridone substituent behaving as a relatively hard, neutral monodentate donor species, equitable with the pyridine nitrogen atom or carboxylate group for Zn(II) but less favoured in the case of Ag(I). The two coordination modes adopted by the pyridone group in complex 4 are of particular interest. In this case, with L2 acting as a heterotopic divergent ligand and in the presence of the well-known copper paddlewheel motif, axial coordination of a neutral monodentate donor is far from unprecedented.19 However, a crucial divergence between 4-pyridone and other terminal neutral monodentate donors (cf. aryl nitriles) is the M⋯OC angle. This angle, consistently in the range 117.4–129.5° for complexes 2–4 and the hydrogen bond (MH) in HL2, is consistent with coordination through an sp2 hybridised atom, contrasted to the typically near-linear coordination through nitrile donors.20 This angle, complementary to the tetrahedral pivot of the sp3 methylene bridge within the ligand molecule, allows for the formation of M2L2 loops between adjacent paddlewheels in the structure of 4 which are similar those present in complex 3 (albeit centrosymmetric in 4), and through which the mechanical interlocking is made possible. The prevalence of anion⋯π interactions in each of the complexes with inorganic anions is another interesting development, which may also have contributed to the tendency for encapsulating the bis-nitratocopper(II) moiety within the naphthalimide cavity in 4.
In terms of the photoluminescence performance of the 4-pyridonylnaphthalimide fluorophore, it can be seen in Fig. 10 that only in the case of complex 1 was a substantial red shift observed in the solid state compared to the emission of L1 in solution. This contrasts with the normal behaviour of naphthalimides and other fluorophores in general which typically undergo red-shifting of their emission bands in the solid state, ascribed to the influence of fluorophore–fluorophore interactions, including J-aggregation.21 Complexes 1–3 all exhibit similar head-to-tail aggregation modes in the crystalline phase, and especially allowing for the intrinsic disorder and expected fluxionality at room temperature, the naphthalimide–pyridonyl torsion angles are all within similar ranges. The defining factor is most likely the coordination modes, where complex 1 almost exclusively involves coordination through the pyridyl group and includes only a weak contribution from the pyridonyl group. The presence of an sp3 methylene group segregating the coordinating pyridyl group from the fluorophore in the case of 1 would also be expected to minimise the influence of the heavy atom on the naphthalimide excited state in complex 1 when compared to fully conjugated systems. In contrast, complexes 2 and 3 involve strong coordination through the pyridone oxygen atom, most likely diminishing the capacity for electron donation from this group to contribute to the naphthalimide internal charge transfer (ICT) process. In the case of HL2, the presumed participation of the protonated carboxylic acid group in a non-radiative decay process is largely as expected for a compound of this type, particularly if intermolecular hydrogen bonding involving the pyridone oxygen atom can be invoked; when fully deprotonated in complex 3 the emission is comparable to that observed from L1 in the analogous zinc species 2, also consistent with minimal involvement of the imide substituent on the properties of the naphthalimide photoluminescence in these instances. We are currently building on this work and developing other functional coordination networks and MOF based on the use of pyridone–naphthalimide building blocks.
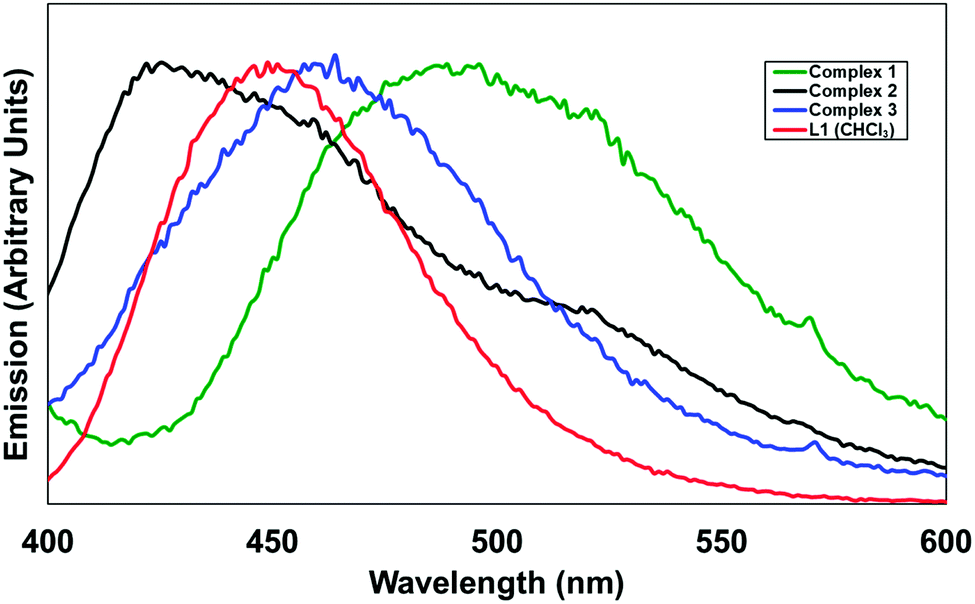 | ||
| Fig. 10 Solid-state emission spectra for complexes 1–3, compared with the emission of L1 in CHCl3 solution (red). λex = 366 nm (L1) and 380 nm for the solids. | ||
Conclusions
In this report, we have demonstrated the utility of the 4-pyridonyl group as both an electron donor and metal binding site for the development of 1,8-naphthalimide-based functional materials. The two pyridone-based ligands L1 and HL2 were used to generate four new discrete and polymeric coordination compounds, in which the structural properties and photoluminescence were related to the influence of 4-pyridone as a neutral monodentate oxygen donor ligand. As well as luminescent Ag(I) and Zn(II) complexes, an unusual mechanically interlocked coordination polymer 4 was prepared; gas adsorption experiments revealed microporosity, and the extended structure was rationalised based on the coordination preferences of the pyridone group. It is our view that the development of new multi-functional ligand species such as those demonstrated here will lead to even greater diversity of material properties and applications in the materials chemistry field in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the Irish Research Council (GOIPD 2015/446 to C. S. H.), Science Foundation Ireland (PI awards 13/IA/1865 to T. G. and 13/IA/1896 to W. S.), the European Research Council (CoG 2014 – 647719 to W. S.), the School of Chemical and Physical Sciences, Keele University, and the School of Chemistry, Trinity College Dublin.References
- G. Maurin, C. Serre, A. Cooper and G. Férey, Chem. Soc. Rev., 2017, 46, 3104–3107 RSC; Y. Cui, B. Li, H. He, W. Zhou, B. Chen and G. Qian, Acc. Chem. Res., 2016, 49, 483–493 CrossRef PubMed; A. G. Slater and A. I. Cooper, Science, 2015, 348, aaaS075 CrossRef PubMed.
- D. Zhao, D. J. Timmons, D. Yuan and H.-C. Zhou, Acc. Chem. Res., 2011, 44, 123–133 CrossRef PubMed.
- T. M. McDonald, W. R. Lee, J. A. Mason, B. M. Wiers, C. S. Hong and J. R. Long, J. Am. Chem. Soc., 2012, 134, 7056–7065 CrossRef PubMed; A. A. Tehrani, S. Abedi, A. Morsali, J. Wang and P. C. Junk, J. Mater. Chem. A, 2015, 3, 20408–20415 Search PubMed; J. M. Roberts, B. M. Fini, A. A. Sarjeant, O. K. Farha, J. T. Hupp and K. A. Scheidt, J. Am. Chem. Soc., 2012, 134, 3334–3337 CrossRef PubMed; D. M. D’Alessandro, Chem. Commun., 2016, 52, 8957–8971 RSC; Y. E. Cheon and M. P. Suh, Angew. Chem., Int. Ed., 2009, 48, 2899–2903 CrossRef PubMed.
- T. Xia, T. Song, Y. Cui, Y. Yang and G. Qian, Dalton Trans., 2016, 45, 18689–18695 RSC; H. He, E. Ma, J. Yu, Y. Cui, Y. Lin, Y. Yang, X. Chen, B. Chen and G. Qian, Adv. Opt. Mater., 2017, 5, 1601040 CrossRef; W. Cho, H. J. Lee, G. Choi, S. Choi and M. Oh, J. Am. Chem. Soc., 2014, 136, 12201–12204 CrossRef PubMed; M. Mitsui, K. Higashi, Y. Hirumi and K. Kobayashi, J. Phys. Chem. A, 2016, 120, 8317–8325 CrossRef PubMed; N. B. Shustova, B. D. McCarthy and M. Dincă, J. Am. Chem. Soc., 2011, 133, 20126–20129 CrossRef PubMed.
- N. L. C. Leung, N. Xie, W. Yuan, Y. Liu, Q. Wu, Q. Peng, Q. Miao, J. W. Y. Lam and B. Z. Tang, Chem. – Eur. J., 2014, 20, 15349–15353 CrossRef PubMed.
- T. Murase, S. Sato and M. Fujita, Angew. Chem., Int. Ed., 2007, 119, 1101–1103 CrossRef; N. Vallavoju and J. Sivaguru, Chem. Soc. Rev., 2014, 43, 4084–4101 RSC; C. Deraedt and D. Astruc, Coord. Chem. Rev., 2016, 324, 106–122 CrossRef.
- T.-H. Chen, I. Popov, W. Kaveevivitchai and O. Š. Miljanić, Chem. Mater., 2014, 26, 4322–4325 CrossRef.
- D. M. L. Goodgame, D. A. Grachvogel, A. J. P. White and D. J. Williams, Inorg. Chem., 2001, 40, 6180–6185 CrossRef PubMed; Y.-X. Zhu, Z.-W. Wei, M. Pan, H.-P. Wang, J.-Y. Zhang and C.-Y. Su, Dalton Trans., 2016, 45, 943–950 RSC; Q.-Y. Yang, M. Pan, S.-C. Wei, K. Li, B.-B. Du and C.-Y. Su, Inorg. Chem., 2015, 54, 5707–5716 CrossRef PubMed; D. M. Goodgame, D. A. Grachvogel and D. J. Williams, J. Chem. Soc., Dalton Trans., 2002, 2259–2260 RSC; Y.-B. Shu, X.-L. Tang and W.-S. Liu, Inorg. Chem. Front., 2014, 1, 226–230 RSC.
- K. D. Shimizu and J. Rebek, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 4257–4260 CrossRef; I. Ino, J. C. Zhong, M. Munakata, T. Kuroda-Sowa, M. Maekawa, Y. Suenaga and Y. Kitamori, Inorg. Chem., 2000, 39, 4273–4279 CrossRef PubMed; A. K. Mishra and S. Verma, Inorg. Chem., 2010, 49, 8012–8016 CrossRef PubMed; H.-J. Kim, S.-K. Kang, Y.-K. Lee, C. Seok, J.-K. Lee, W.-C. Zin and M. Lee, Angew. Chem., Int. Ed., 2010, 49, 8471–8475 CrossRef PubMed.
- K. Zhu, C. A. O’Keefe, N. Vukotic, R. W. Schurko and S. J. Loeb, Nat. Chem., 2015, 7, 514–519 CrossRef PubMed; J. E. M. Lewis, M. Galli and S. M. Goldup, Chem. Commun., 2017, 53, 298–312 RSC; V. N. Vukotic, C. A. O’Keefe, K. Zhu, K. J. Harris, C. To, R. W. Schurko and S. J. Loeb, J. Am. Chem. Soc., 2015, 137, 9643–9651 CrossRef PubMed; G. Gholami, G. Baggi, K. Zhu and S. J. Loeb, Dalton Trans., 2017, 46, 2462–2470 RSC; A. Coskun, M. Hmadeh, G. Barin, F. Gándara, Q. Li, E. Choi, N. L. Strutt, D. B. Cordes, A. M. Z. Slawin, J. F. Stoddart, J.-P. Sauvage and O. M. Yaghi, Angew. Chem., Int. Ed., 2012, 51, 2160–2163 CrossRef PubMed; H. Deng, M. A. Olson, J. F. Stoddart and O. M. Yaghi, Nat. Chem., 2010, 2, 439–443 CrossRef PubMed; Q. Li, C.-H. Sue, S. Basu, A. K. Shveyd, W. Zhang, G. Barin, L. Fang, A. A. Sarjeant, J. F. Stoddart and O. M. Yaghi, Angew. Chem., Int. Ed., 2010, 49, 6751–6755 CrossRef PubMed; Q. Li, W. Zhang, O. Š. Miljanić, C. B. Knobler, J. F. Stoddart and O. M. Yaghi, Chem. Commun., 2010, 46, 380–382 RSC; J. E. M. Lewis, P. D. Beer, S. J. Loeb and S. M. Goldup, Chem. Soc. Rev., 2017, 46, 2577–2591 RSC; K. Zhu and S. J. Loeb, in Organizing Mechanically Interlocked Molecules to Function Inside Metal-Organic Frameworks, Molecular Machines and Motors, ed. A. Credi, S. Silvi and M. Venturi, Topics in Current Chemistry, Springer, Cham, 2014, vol. 354 CrossRef PubMed; N. C. Frank, D. J. Mercer and S. J. Loeb, Chem. – Eur. J., 2013, 19, 14076–14080 CrossRef PubMed; C. Lincheneau, B. Jean-Denis and T. Gunnlaugsson, Chem. Commun., 2014, 50, 2857–2860 RSC; J. P. Byrne, S. Blasco, A. B. Aletti, G. Hessman and T. Gunnlaugsson, Angew. Chem., Int. Ed., 2016, 55, 8938–8943 CrossRef PubMed.
- S. R. Batten, S. M. Neville and D. R. Turner, Coordination Polymers: Design, Analysis and Application, Royal Society of Chemistry, Cambridge, 2009 Search PubMed.
- C. S. Hawes, K. Byrne, W. Schmitt and T. Gunnlaugsson, Inorg. Chem., 2016, 55, 11570–11582 CrossRef PubMed; C. S. Hawes, A. D. Lynes, K. Byrne, W. Schmitt, G. Ryan, M. E. Möbius and T. Gunnlaugsson, Chem. Commun., 2017, 53, 5898–5992 RSC.
- S. Banerjee, E. B. Veale, C. M. Phelan, S. A. Murphy, G. M. Tocci, L. J. Gillespie, D. O. Frimannsson, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2013, 42, 1601–1618 RSC; E. Calatrava-Pérez, S. A. Bright, S. Achermann, C. Moylan, M. O. Senge, E. B. Veale, D. C. Williams, T. Gunnlaugsson and E. M. Scanlan, Chem. Commun., 2016, 52, 13086–13089 RSC.
- A. B. Carter, R. J. Laverick, D. J. Wales, S. O. Akponasa, A. J. Scott, T. D. Keene and J. A. Kitchen, Cryst. Growth Des., 2017, 17, 5129–5144 Search PubMed; J. I. Lovitt, C. S. Hawes, A. D. Lynes, B. Haffner, M. E. Möbius and T. Gunnlaugsson, Inorg. Chem. Front., 2017, 4, 296–308 RSC; J. A. Kitchen, P. Martinho, G. G. Morgan and T. Gunnlaugsson, Dalton Trans., 2014, 43, 6468–6479 RSC.
- D. L. Reger, A. Debreczeni, J. J. Horger and M. D. Smith, Cryst. Growth Des., 2011, 11, 4068–4079 Search PubMed; L. J. McCormick and D. R. Turner, CrystEngComm, 2013, 15, 8234–8236 RSC; N. S. S. Kumar, M. D. Gujrati and J. N. Wilson, Chem. Commun., 2010, 46, 5464–5466 RSC.
- Z. Xu, Y. Xiao, X. Qian, J. Cui and D. Cui, Org. Lett., 2005, 7, 889–892 CrossRef PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef PubMed.
- J. Yang, J.-F. Ma and S. R. Batten, Chem. Commun., 2012, 48, 7899–7912 RSC.
- M. Köberl, M. Cokoja, W. A. Herrmann and F. E. Kühn, Dalton Trans., 2011, 40, 6834–6859 RSC; S. Bureekaew, S. Amirjalayer and R. Schmid, J. Mater. Chem., 2012, 22, 10249–10254 RSC.
- B. N. Storhoff and H. C. Lewis Jr., Coord. Chem. Rev., 1977, 23, 1–29 CrossRef.
- S. Mukherjee and P. Thilagar, Phys. Chem. Chem. Phys., 2014, 16, 20866–20877 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, additional crystallographic discussion, NMR, UV-visible and fluorescence spectra, additional gas adsorption data, thermogravimetric analysis plots and X-ray powder diffraction data. CCDC 1831226–1831230. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8qm00182k |
| This journal is © the Partner Organisations 2018 |