
Selenium embedded in MOF-derived N-doped microporous carbon polyhedrons as a high performance cathode for sodium–selenium batteries†
Siqi
Li
a,
Hai
Yang
a,
Rui
Xu
a,
Yu
Jiang
a,
Yue
Gong
 b,
Lin
Gu
b and
Yan
Yu
*ac
b,
Lin
Gu
b and
Yan
Yu
*ac
aDepartment of Materials Science and Engineering, University of Science and Technology of China, CAS Key Laboratory of Materials for Energy Conversion, Hefei, Anhui 230026, P. R. China. E-mail: yanyumse@ustc.edu.cn
bBeijing National Laboratory for Condensed Matter Physics The Institute of Physics, Chinese Academy of Sciences, Beijing 100190, China
cInstitute of Advanced Electrochemical Energy, Xi’an University of Technology, Xi’an 710048, China
First published on 19th June 2018
Abstract
Selenium cathodes have attracted much more attention due to their much higher electronic conductivity and comparable volumetric capacity when compared with sulfur cathodes. However, selenium cathodes still suffer from low utilization of active materials, high volume changes and the shuttle effect of polyselenides, resulting in rapid capacity fading. Herein, we prepared selenium–carbon composites as cathodes for sodium–selenium batteries to improve the utilization of selenium by embedding selenium in ZIF-8 derived N-doped microporous carbon polyhedrons (denoted as Se@N-MCPs). The N-MCPs could effectively accommodate the volume change of Se@N-MCPs, and alleviate the shuttle effect of polyselenides. The Se@N-MCP cathodes deliver an excellent discharge capacity of 612 mA h g−1 after 100 cycles at a current density of 0.1 A g−1 and a superior rate capability of 496 mA h g−1 at 5 A g−1 for Na–Se batteries. In addition, they also show a superior cycling life of ∼460 mA h g−1 at the current density of 1 A g−1 after 500 cycles with only 0.049% capacity decline per cycle.
Introduction
Lithium ion batteries (LIBs), one of the most promising rechargeable energy-storage devices, have been extensively applied in portable electronics, hybrid electric vehicles and electric vehicles.1,2 However, the limits of the natural abundance and uneven global distribution of lithium-sources prevent the application of LIBs in large-scale energy storage applications. For large-scale applications, it is urgent to develop a low-cost and sustainable energy storage system. Recently, sodium ion batteries (NIBs) have attracted more attention because of the low cost and abundant resources of sodium.3 Among the NIBs, Na–Se batteries have attracted more and more attention due to their high theoretical volumetric capacity (3253 mA h cm−3). In addition, Se shows many advantages, such as higher electronic conductivity (1 × 10−3 S m−1) and stable performance in carbonate-based electrolytes.4–6 All these properties demonstrated that Na–Se batteries are promising high-energy rechargeable batteries. However, bulk Se particles cannot be applied to Na–Se batteries directly because of their lower utilization of the active materials and poor electrochemical performance. The rapid capacity fading of Na–Se batteries is mainly attributed to the volume variation of Se and the shuttle effect of high-order polyselenides during the charge/discharge process.7 To mitigate these issues, researchers have fabricated various porous carbon/selenium composites to confine Se within the carbon matrix, thus preventing Se exposure to the electrolyte directly. These carbon materials with various morphologies include flexible carbon nanofibers,7–12 carbon nanosheets,13,14 carbon spheres,15,16 carbon nanotubes,4,17etc. Recently, Yang et al. reported a vacuum calcination approach to fabricate selenium/carbon composites.17 They used carbon-coated selenium wires as precursors prepared via a wet-chemical reaction and then obtained carbon/selenium tubes through a straightforward calcination process. The resultant selenium/carbon tubes with 53 wt% Se loading deliver a high reversible capacity of 601 and 509 mA h g−1 at 0.2C and 2C for sodium storage, respectively, based on the mass of Se. In order to obtain a higher Se loading, Ding et al. employed nanocellulose derived mesoporous carbon as precursors.14 After infiltration of Se into the mesoporous carbon matrix, they obtained a freestanding carbon/selenium composite film with 70 wt% Se loading. The cathode delivers a reversible capacity of 620 mA h g−1 with 82% retention over 300 cycles for lithium storage while it delivers 511 mA h g−1 with 98% retention after 150 cycles for sodium storage. In addition, they also compared the difference between the electrochemical kinetics with Li and Na including the transition from interfacial to diffusional control.Metal organic frameworks (MOFs) have attracted much attention due to their diversity of structure and composition, tunable porosity, and high specific surface area, thus exhibiting various types of applications in hydrogen storage,18 gas adsorption and separation,19 catalysis,20–22 electrochemical energy storage devices,23–28etc. One can expect that the confinement of Se in MOF-derived porous carbon could alleviate the dissolution of polyselenides. Recently, Xu et al. reported a ZIF-67 derived mesoporous carbon for fabricating carbon/selenium composites (denoted as Se/NPCPs) as cathodes for Na–Se batteries.29 The cathodes deliver a superior rate capability of 351.6 mA h g−1 and 307.8 mA h g−1 at 0.5C and 2C, respectively, along with a good cycling performance of only 0.05% decay per cycle after 1000 cycles at 2C. The weight content of nitrogen in the Se/NPCPs composites is 8.68 wt%. It has been demonstrated that the higher doping nitrogen amount could enhance the reversible sodium storage performance.24,30 To further increase the sodium storage performance, we selected ZIF-8 derived N-doped porous carbon (N-MCPs) that has a higher N-doping amount (12.88 wt%). We confined Se in the N-MCPs, exhibiting a high reversible capacity of 496 mA h g−1 at 5 A g−1 for Na–Se batteries.
Herein, we prepared carbon/selenium composites as cathodes for sodium–selenium batteries by impregnating selenium in ZIF-8 derived N-doped microporous carbon polyhedrons (denoted as Se@N-MCPs) via a melt-infusion method. This unique structure can offer various advantages: Firstly, confinement of selenium in N-MCPs can relieve the strain from volume changes during cycling and suppress the shuttle effect of polyselenides.10,11,29 Secondly, the nitrogen doped porous carbon can build up highly conductive pathways of electrons in the electrodes, thus enhancing the electrochemical activity and rate capability.15,25,29 Thirdly, the typical porous structure can ensure sufficient access of the electrolyte to selenium. Benefiting from the abovementioned advantages, the Se@N-MCP cathode delivers an excellent discharge capacity of 612 mA h g−1 after 100 cycles at a current density of 0.1 A g−1 and a superior rate capability of 496 mA h g−1 at 5 A g−1. In addition, it also shows a superior reversible capacity of 460 mA h g−1 at a current density of 1 A g−1 after 500 cycles with only 0.049% capacity decline per cycle. The as-prepared Se@N-MCP cathode materials demonstrated the viability of high performance Na–Se batteries in practical applications.
Experimental
Materials synthesis
In a typical synthesis process, ZIF-8 nanocrystals were firstly prepared according to the literature with slight modification.23 Solution A: 1 mmol of Zn(NO3)2·6H2O and 1.5 g PVP (k-30) were dissolved in 30 mL of methanol to form a clear solution under stirring. Solution B: 4 mmol of 2-methylimidazole (C4H6N2) was dissolved in 30 mL of methanol to form another clear solution under stirring. After that solution B was poured slowly into solution A under stirring for 1 h and then kept still for 24 h at ambient temperature. The obtained white precipitates were collected by centrifugation, washing with methanol at least three times and drying at 60 °C overnight. Finally, white ZIF-8 nanocrystals were obtained.Then the ZIF-8 nanocrystals were first heated up to 300 °C with a heating rate of 2 °C min−1 and kept for 2 h and then further heated up to 900 °C with a heating rate of 5 °C min−1 and kept for 6 h and then cooled down to room temperature under a N2 atmosphere in a tube furnace. To remove the Zn species, the as-prepared sample was immersed into HCl solution under stirring after calcination. After that, the ZIF-8 derived N-doped Microporous Carbon Polyhedrons (denoted as N-MCPs) were finally obtained.
The mixture of N-MCPs and selenium powder was ground in an agate mortar at different mass ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 2:1, and 3:1 respectively and then transferred into a sealed glass tube under an argon atmosphere. Subsequently, they were heated up to 260 °C with a heating rate of 2 °C min−1 and kept for 20 h, during which the selenium melted and penetrated into the pores of the N-MCPs and finally gave carbon/selenium composites (denoted as Se@N-MCPs).
:1, 2:1, and 3:1 respectively and then transferred into a sealed glass tube under an argon atmosphere. Subsequently, they were heated up to 260 °C with a heating rate of 2 °C min−1 and kept for 20 h, during which the selenium melted and penetrated into the pores of the N-MCPs and finally gave carbon/selenium composites (denoted as Se@N-MCPs).
Material characterization
XRD (TTR-III, Rigaku, Japan) measurements were made with Cu-Kα radiation. The Raman scattering spectra were recorded with a Renishaw System 2000 spectrometer. XPS experiments were performed using a Thermo-VG Scientific instrument. FESEM (JEOL, Tokyo, Japan) and TEM (JEM-2100F, JEOL, Japan) were employed to characterize the morphology of the materials. An ASAP 2020 Accelerated Surface Area and Porosimetry instrument was used to measure the materials’ nitrogen adsorption and desorption isotherms. The selenium content of the Se@N-MCPs was tested by a thermogravimetric analysis instrument (TGA Q5000IR, America) at a heating rate of 10 °C min−1 from room temperature to 800 °C under a nitrogen atmosphere.Electrochemical measurements
A homogeneous slurry was obtained by mixing the active material (Se@N-MCPs composites), acetylene black (AB), and carboxymethyl cellulose (CMC, water-soluble binder) with a weight ratio of 7:2:1 in an agate mortar and then it was cast on a carbon-coated aluminum foil and dried at 60 °C under vacuum overnight. There was ∼1.05 mg cm−2 active material (∼0.51 mg cm−2 Se) loaded on the electrode and the specific capacity was calculated based on the mass of Se. As a contrast, we prepared Se/AB electrodes by the same method. The electrochemical properties of the electrodes were evaluated by assembling CR2032 coin-type cells, which consist of selenium-based cathodes, glass fiber (Whatman) as separators, sodium foil as anodes, and 1 M NaClO4 in a mixed solvent of propylene carbonate with 5% FEC as electrolyte in an argon-filled glove box. The as-assembled batteries were tested on a battery test system (Neware BTS-610) at a voltage range from 0.5 to 3 V versus Na/Na+. The specific capacity mentioned in this paper was based on the mass of Se. For the cells, cyclic voltammetry measurements were conducted at a scan rate of 0.1 mV s−1 and EIS were tested in the frequency range from 100 kHz to 0.01 Hz using a CHI 660D electrochemical workstation (Chenhua Instrument Company, Shanghai, China).
Results and discussion
Fig. 1a shows a schematic illustration of the synthesis process for the obtained Se@N-MCP cathode materials. Firstly, ZIF-8 nanocrystals were prepared according to the literature.23 Secondly, N-doped carbon could be obtained after carbonization of ZIF-8 and etching of Zn by HCl solution. Finally, selenium powder was melted and penetrated into the pores of the N-MCPs by co-heating Se and N-MCPs in an Ar atmosphere, forming the final Se@N-MCPs. To optimize the electrochemical performance, mixtures with different weight ratios between Se and N-MCPs (Se:N-MCPs = 1:1, 2:1, and 3:1) were co-heated, and the obtained samples are marked as Se@N-MCPs, Se@N-MCPs (2:1), and Se@N-MCPs (3:1) respectively. To further confirm the advantages of Se@N-MCPs, we also prepared Se/AB composites by mixing Se powder and acetylene black with a mass ratio of 1:1. The thermogravimetric (TG) profiles (Fig. S1, ESI†) show that the content of Se distributed in the porous carbon polyhedrons was determined to be 48.55%, 59.23%, and 68.31% for Se@N-MCPs, Se@N-MCPs (2:1), and Se@N-MCPs (3:1), respectively. The Se@N-MCP electrode displays the best sodium storage performance among these three electrodes (Fig. S2, ESI†). It delivers a reversible capacity of 306 mA h g−1 after 100 cycles at 0.1 A g−1. Both Se@N-MCPs (2:1) and Se@N-MCPs (3:1) display inferior reversible capacities (234 mA h g−1 and 170 mA h g−1, respectively), which may be due to the unconnected carbon matrix. All the specific capacities abovementioned are calculated based on the mass of the composite. The N2 adsorption–desorption isothermal curves and corresponding pore size distribution of N-MCPs and Se@N-MCPs are displayed in Fig. 1b and c. Before infiltration of Se, the BET specific surface area of the N-MCPs reached up to 980.992 m2 g−1, providing sufficient space for Se loading. In addition, the image of the pore size distribution shows highly uniform micropores in the N-MCPs (pore diameter ∼0.548 nm). The pore volume of N-MCPs is 0.478 cm3 g−1, corresponding to a theoretical Se loading of 67 wt% (calculated based on the density of Se, 4.26 g cm−3).4 After the infiltration of Se into the N-MCPs, the BET specific surface area and pore volume of the Se@N-MCPs remarkably reduced to 16.175 m2 g−1 and 0.037 cm3 g−1, respectively, indicating that Se was successfully embedded into the micropores of N-MCPs. The pore size of the Se@N-MCPs (1.667 nm) is larger than that of the N-MCPs (0.548 nm), indicating that Se infiltrated into the pores of the N-MCPs. The nitrogen adsorption–desorption technique was carried out to investigate the Se@N-MCPs (2:1) and Se@N-MCPs (3:1) (Fig. S3, ESI†). With the content of Se increasing, the pore volume of Se@N-MCPs (2:1) and Se@N-MCPs (3:1) reduced to 0.022 cm3 g−1 and 0.003 cm3 g−1, respectively, as shown in Table S1 (ESI†). In addition, the pore sizes of the Se@N-MCPs (2:1) and Se@N-MCPs (3:1) are 1.543 nm and 1.610 nm, respectively, which are almost similar to that of Se@N-MCPs.
 | ||
| Fig. 1 (a) Schematic illustration of the preparation process for Se@N-MCPs. (b) N2 adsorption–desorption isothermal curves and (c) the corresponding pore-size distribution curves of N-MCPs and Se@N-MCPs. | ||
The ZIF-8 nanocrystals show the typical X-ray diffraction (XRD) pattern (Fig. S4, ESI†), identical to the corresponding simulated data in the literature.23Fig. 2a shows XRD patterns of pristine Se, N-MCPs, and Se@N-MCPs. The diffraction peaks of the pristine Se match with the trigonal-phase of Se, whereas after infiltration of Se into the N-MCPs, all diffraction peaks of Se in the XRD pattern of Se@N-MCPs disappeared, indicating that Se was uniformly dispersed in the N-MCPs.12 Both N-MCPs and Se@N-MCPs display a broad peak at 25° and a broad peak at 44° corresponding to the (002) and (101) planes of graphite, which indicates the amorphous nature of the as-prepared samples.15 The structural characteristics of pristine Se, the N-MCPs, and the Se@N-MCPs are demonstrated by the Raman spectra in Fig. 2b. Two broad peaks located at about 1580 cm−1 (G-band) and 1350 cm−1 (D-band) are observed from both the N-MCPs and Se@N-MCPs, corresponding to the E2g2 graphitic mode and the defect-induced mode, respectively.12 The intensity ratio (R = ID/IG) reflects the degree of graphitization of carbon. The R-value of the N-MCPs and the Se@N-MCPs is 1.02 and 1.01, respectively. The results above indicate that both of the as-prepared samples maintain the amorphous carbon, with no obvious change after the introduction of Se. For pristine Se, the peaks located at 142 cm−1 and 460 cm−1 represent Se12 with a ring structure while the peak located at 235 cm−1 represents chain-structured trigonal Se, indicating that the pristine Se is a mixture of ring-structured Se12 and chain-structured trigonal Se molecules.16 After infiltration of Se into the N-MCPs, the intensity of the characteristic peak of pristine Se remarkably reduced. Instead, a peak located at about 260 cm−1 corresponding to the Se8 ring appears and the chain-structure trigonal Se (235 cm−1) still exists with a lower intensity, indicating that a mixture of the Se8 ring and chain-structure trigonal Se is encapsulated into the pores of N-MCPs.16,31
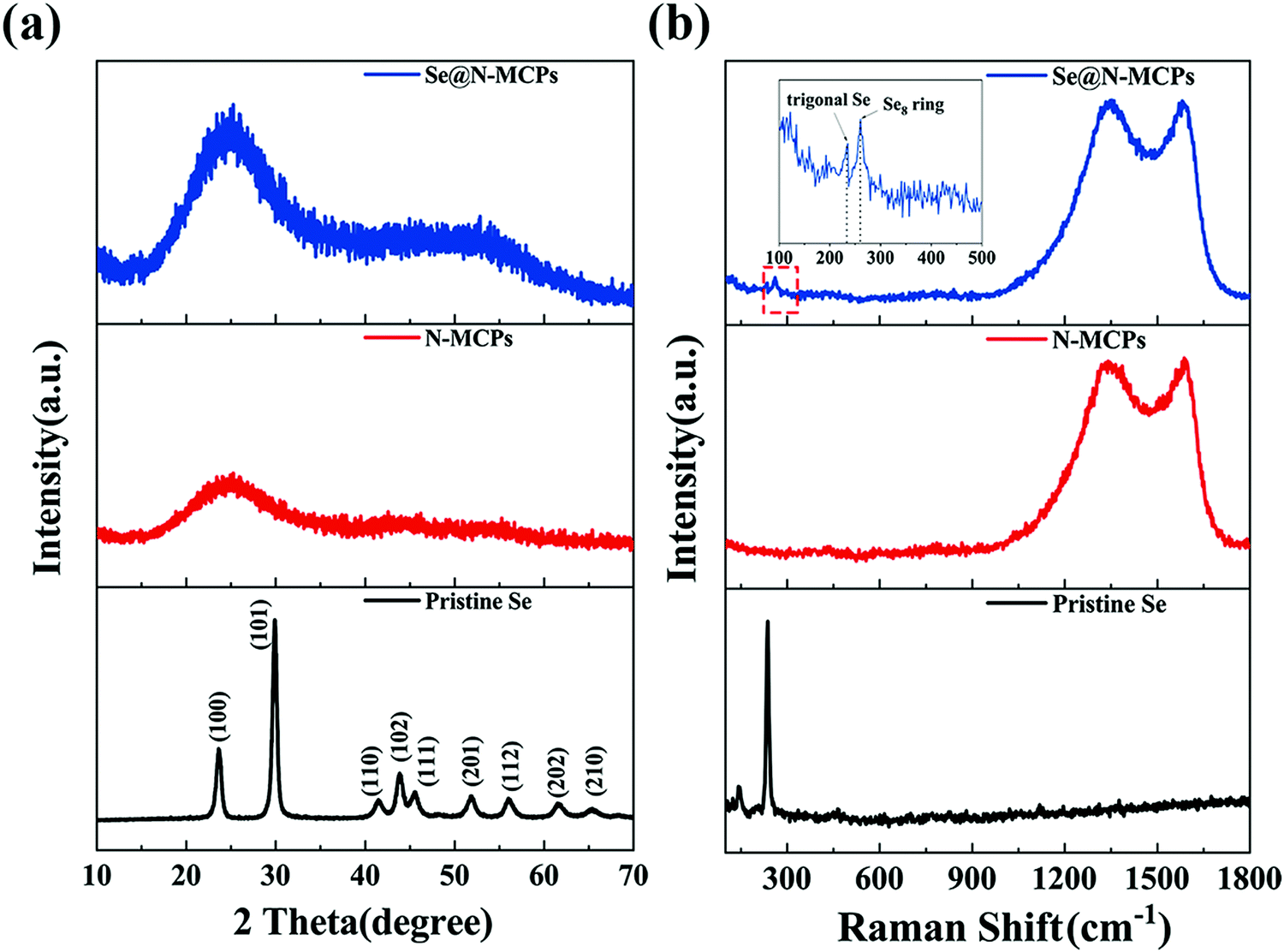 | ||
| Fig. 2 (a) XRD patterns of pristine Se, N-MCPs, and Se@N-MCPs. (b) Raman spectra of pristine Se, N-MCPs, and Se@N-MCPs. | ||
The morphology of the ZIF-8 nanocrystals was characterized by SEM and TEM images, exhibiting a polyhedron-like structure (Fig. S5a and b, ESI†). After carbonization and infiltration of Se, the morphology of the N-MCPs and the Se@N-MCPs retain the polyhedron-like structure. As shown in Fig. 3a, the SEM image of N-MCPs displays a uniform size of 120 nm. Fig. 3b displays the TEM image of the N-MCPs, exhibiting a highly porous structure with a uniform micropore distribution (pore diameter ∼0.548 nm). After impregnation of Se into the N-MCPs, the Se@N-MCPs (Fig. 3d and e) keep the original polyhedral uniform morphology with a smooth surface, indicating that selenium embedded into the pores of the carbon polyhedrons. However, no obvious lattice fringes appeared both in Fig. 3c and f, indicating a lower degree of graphitization of ZIF-8 derived porous carbon and that the amorphous state of Se existed within the N-MCPs, which was consistent with the results of the XRD and Raman spectrum. In agreement with the SEM and TEM results, the distribution of selenium in the N-MCPs was further characterized by high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM). The HAADF-STEM image (inset of Fig. 3g) and the corresponding element mappings of Se@N-MCPs (Fig. 3g–i) indicate the existence of C, N and Se elements, and all these elements are uniformly distributed throughout the porous carbon matrix.
 | ||
| Fig. 3 (a) SEM, (b) TEM, and (c) HRTEM images of N-MCPs. (d) SEM, (e) TEM, and (f) HRTEM images of Se@N-MCPs. High angle annular dark-field STEM (HAADF-STEM) image and corresponding (g) C, (h) N, and (i) Se element mapping of Se@N-MCPs. | ||
The chemical compositions and surface electronic states of the Se@N-MCPs were further investigated by X-ray photoelectron spectroscopy (XPS). The XPS survey spectrum of the Se@N-MCPs is shown in Fig. 4a, indicating the chemical valence state of C, N and Se in the Se@N-MCPs. As shown in Fig. 4b, the high- resolution C 1s spectrum reveals three peaks, corresponding to C–C (284.6 eV), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N (285.4 eV), and C–N (286.7 eV), respectively, indicating the N-doping in the ZIF-8 derived porous carbon materials.29,32 The weight content of N in Se@N-MCPs is calculated to be about 12.88 wt% by comparing the intensity of the peak corresponding to element N with the intensity of all the peaks (Fig. 4a). Three main peaks located at 398.5 eV, 400.2 eV, and 401.1 eV were observed in the high-resolution N 1s spectrum (Fig. 4c), corresponding to pyridinic N (N-6), pyrrolic N (N-5), and graphitic N (N-4), respectively.32,33 The N-doped carbon matrix can enhance the electrical conductivity of the composites.29,33 Particularly, pyrrolic N can create abundant extrinsic defects and active sites favorable for electrochemical reactions.29,33Fig. 4d reveals the high-resolution spectra of Se 3d. The peaks located at 55.3 eV and 56.2 eV correspond to the Se 3d5/2 and 3d3/2, respectively.7 In addition, the peak at 59 eV is ascribed to the Se–O bonding.34 The formation of Se–O bonding might be due to a little oxygen being mixed into the argon atmosphere during sealing the glass tube and then a small amount of Se on the surface being oxidized into SeO2 after the infiltration of Se process. The content of SeO2 in the Se@N-MCPs is ∼2.15 wt%, according to the proportion of the peak area of Se–O in the high-resolution spectra of Se 3d and the content of Se in the Se@N-MCPs tested from TG profiles.
N (285.4 eV), and C–N (286.7 eV), respectively, indicating the N-doping in the ZIF-8 derived porous carbon materials.29,32 The weight content of N in Se@N-MCPs is calculated to be about 12.88 wt% by comparing the intensity of the peak corresponding to element N with the intensity of all the peaks (Fig. 4a). Three main peaks located at 398.5 eV, 400.2 eV, and 401.1 eV were observed in the high-resolution N 1s spectrum (Fig. 4c), corresponding to pyridinic N (N-6), pyrrolic N (N-5), and graphitic N (N-4), respectively.32,33 The N-doped carbon matrix can enhance the electrical conductivity of the composites.29,33 Particularly, pyrrolic N can create abundant extrinsic defects and active sites favorable for electrochemical reactions.29,33Fig. 4d reveals the high-resolution spectra of Se 3d. The peaks located at 55.3 eV and 56.2 eV correspond to the Se 3d5/2 and 3d3/2, respectively.7 In addition, the peak at 59 eV is ascribed to the Se–O bonding.34 The formation of Se–O bonding might be due to a little oxygen being mixed into the argon atmosphere during sealing the glass tube and then a small amount of Se on the surface being oxidized into SeO2 after the infiltration of Se process. The content of SeO2 in the Se@N-MCPs is ∼2.15 wt%, according to the proportion of the peak area of Se–O in the high-resolution spectra of Se 3d and the content of Se in the Se@N-MCPs tested from TG profiles.
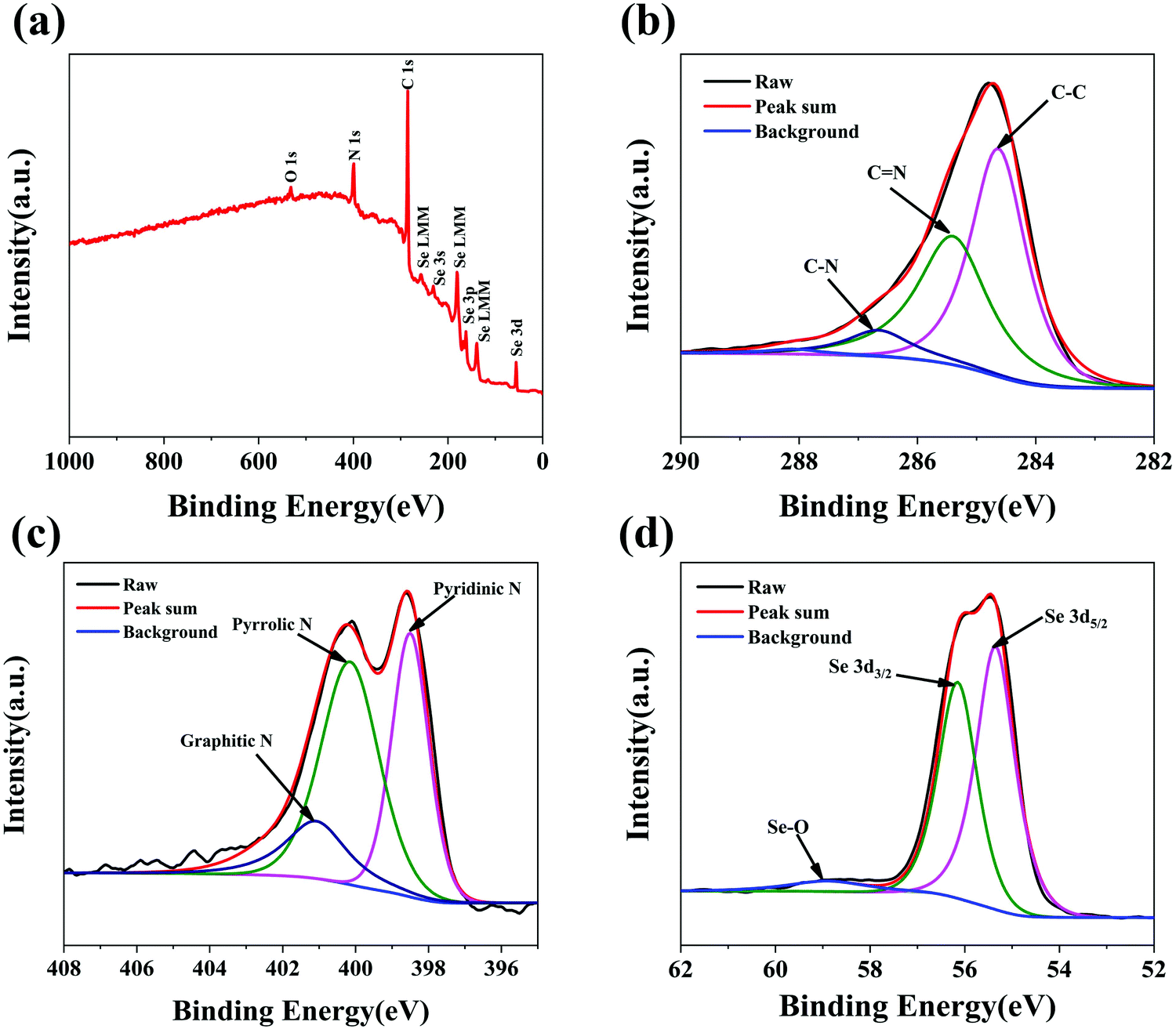 | ||
| Fig. 4 (a) XPS survey spectrum of Se@N-MCPs. High-resolution XPS spectrum of (b) C 1s, (c) N 1s and (d) Se 3d of Se@N-MCPs. | ||
In order to investigate the electrochemical performance of the Se@N-MCPs, we employed Se@N-MCPs as cathodes for Na–Se batteries. Fig. 5a reveals the cyclic voltammogram (CV) curves of the first three cycles of the Se@N-MCPs electrodes at a scan rate of 0.1 mV s−1 between 0.5 V and 3 V. During the first discharge process, there is only one cathodic peak located at 0.96 V, indicating the conversion of Se into Na2Se directly.17 After the first cycle, the single cathodic peak splits into two peaks located at 1.45 V and 1.23 V, respectively, indicating that the conversion of Se into Na2Se is a two-step reaction in the following cycles.4,17 The two cathodic peaks indicate that the conversion is a step-wise process with an intermediate phase, which might be Na2Se2.4,17 The single anodic peak is located at 1.68 V, indicating a direct phase change conversion from Na2Se to Se. After the second cycle, the CV curves overlapped demonstrating the good electrochemical stability of the Se@N-MCP electrodes. Furthermore, a galvanostatic discharge–charge test was performed at a current density of 0.1 A g−1 within the voltage window of 0.5–3 V, as shown in Fig. 5b. The charge/discharge profiles are consistent with the peaks’ position in CV curves. The Se@N-MCP electrodes exhibit an excellent sodium storage capacity, delivering initial charge and discharge capacities of 645 mA h g−1 and 944 mA h g−1, respectively, corresponding to an initial Coulombic efficiency of 68.3%. The initial capacity loss is mainly attributed to the formation of the solid electrolyte interface (SEI) layer and some irreversible reactions of Na within the carbon matrix at high potentials.13 After the first cycle, the Coulombic efficiency approaches 100% in the following cycles, as shown in Fig. 5c. The Se@N-MCPs electrodes reveal excellent cycling stability with a capacity of 612 mA h g−1 after 100 cycles at a current density of 0.1 A g−1, demonstrating that the encapsulation of Se in N-MCPs could effectively suppress the shuttle effect of polyselenides. The capacity contribution from the N-MCPs is almost negligible at the same current density (0.1 A g−1) (Fig. S6, ESI†), indicating that the high sodium storage capacity in Se@N-MCP electrodes is essentially contributed from Se. As a comparison, the CV curves of Se/AB electrodes (Fig. S7a, ESI†) show a broad peak located at 1.13 V in the first cycle and the intensity of the broad peak reduced gradually in the following cycles. Fig. S7b (ESI†) shows the galvanostatic charge/discharge curves of the Se/AB, which is consistent with the CV curves. The Se/AB electrodes only deliver a reversible capacity of about 18 mA h g−1 after 100 cycles at a current density of 0.1 A g−1 (Fig. S7c, ESI†). The Se@N-MCP electrodes also display an excellent rate capability (Fig. 5d), delivering a discharge capacity of 645, 609, 570, 548, 526, and 496 mA h g−1 at 0.1, 0.2, 0.5, 1, 2, and 5 A g−1, respectively. The capacity remains 622 mA h g−1 when the current density is tuned back to 0.1 A g−1, indicating the reversibility of sodium storage in the Se@N-MCPs. Furthermore, the Se@N-MCP electrode exhibits a superior long-term cycling performance of 460 mA h g−1 at a current density of 1 A g−1 after 500 cycles with only 0.049% capacity decline per cycle, indicating the excellent electrochemical stability of Se@N-MCPs (Fig. 5e). The rate capability (Fig. S7d, ESI†) and long-term cycling performance (Fig. S7e, ESI†) of the Se/AB electrodes were measured under the same conditions. However, the Se/AB electrodes exhibit poor sodium storage performance, which may be attributed to the dissolution of polyselenides into the electrolyte, resulting in loss of active materials and a poor cycling performance. Furthermore, visual inspection on the color change of the electrolyte in the Se@N-MCPs, Se@N-MCPs (2:1), Se@N-MCPs (3:1) and Se/AB cells after cycling was done. By contrast, the color change on the separators of the Se/AB cell is the most obvious among all the cells (Fig. S8d, ESI†) and the Se@N-MCP cell only shows a little color change (Fig. S8a, ESI†), demonstrating that the confinement of Se in N-MCPs could alleviate the dissolution of polyselenides into the electrolyte, improve the utility of active materials, thus enhancing the cycling stability of the Se@N-MCP electrodes. With the content of Se in the composites increasing, the color obviously turns dark on the separators of Se@N-MCPs (2:1) and Se@N-MCPs (3:1), which could be due to the lower adsorption of polyselenides, as shown in Fig. S8b and c (ESI†). Meanwhile, the Se@N-MCP electrodes display the best long-term cycling performance, as shown in Table S2 (ESI†).
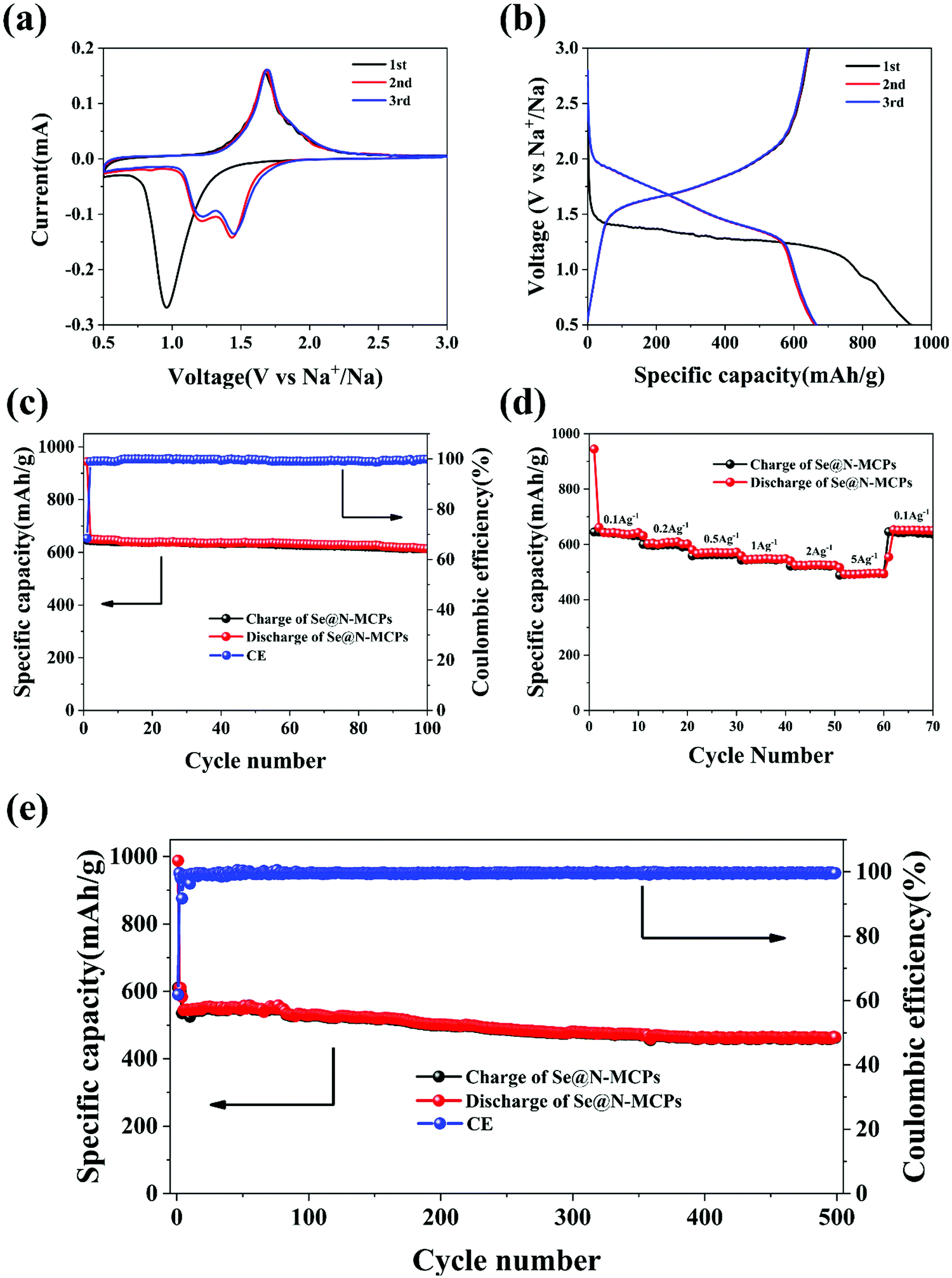 | ||
| Fig. 5 (a) Cyclic voltammograms of the Se@N-MCP electrodes at a scan rate of 0.1 mV s−1 in the voltage window from 0.5 V to 3 V versus Na+/Na. (b) Galvanostatic charge/discharge profiles of the Se@N-MCP electrodes at a current density of 0.1 A g−1 between 0.5 V and 3 V versus Na+/Na. (c) Cycling performances of the Se@N-MCP electrodes at a current density of 0.1 A g−1 and the corresponding CE during cycling. (d) Rate capability of the Se@N-MCP electrodes at various current densities. (e) Long-term cycling performance of the Se@N-MCP electrodes at a current density of 1 A g−1. | ||
To further elaborate the electrochemical kinetics of the Se@N-MCP electrodes, we investigated their current response behaviors at various scan rates from 0.1 mV s−1 to 2 mV s−1, as shown in Fig. 6a. The CV scan test is well suited to research rapid charge/discharge kinetics. The two cathodic peaks (denoted as P1 and P2) related to the two sequential reactions exhibited a difference in their dependence on the scan rate. The intensity of the peak current for P1 and P2 was almost similar at 0.1 mV s−1 (0.13 vs. 0.11 mA). With the scan rate increasing to 1 mV s−1, P1 became a dominant peak, while P2 remained a minor one (2.02 vs. 0.75 mA). At the scan rate of 2 mV s−1, P2 in the CV curves almost disappeared and P1 remained. To further analyze the difference in kinetics, we used a power-law equation, i = avb (equivalent to log(i) = log(a) + blog(v)), where i and v are the peak current and scan rate, and a and b are adjustable parameters.14,17 Generally speaking, a b-value of 0.5 denotes a diffusion-limited process, while a b-value of 1 denotes an interface-limited process, the interface potentially being solid–liquid or solid–solid.14 With the b-value closer to 1, the electrochemical behavior is more capacitive, indicating a faster kinetics process. Fig. 6b displays that the b-value was fitted to be 0.895, 0.875, and 0.841. This indicates that the conversion of Se into the intermediate phase is faster than that of the intermediate phase into Na2Se, while the conversion of Na2Se into Se is the slowest one.
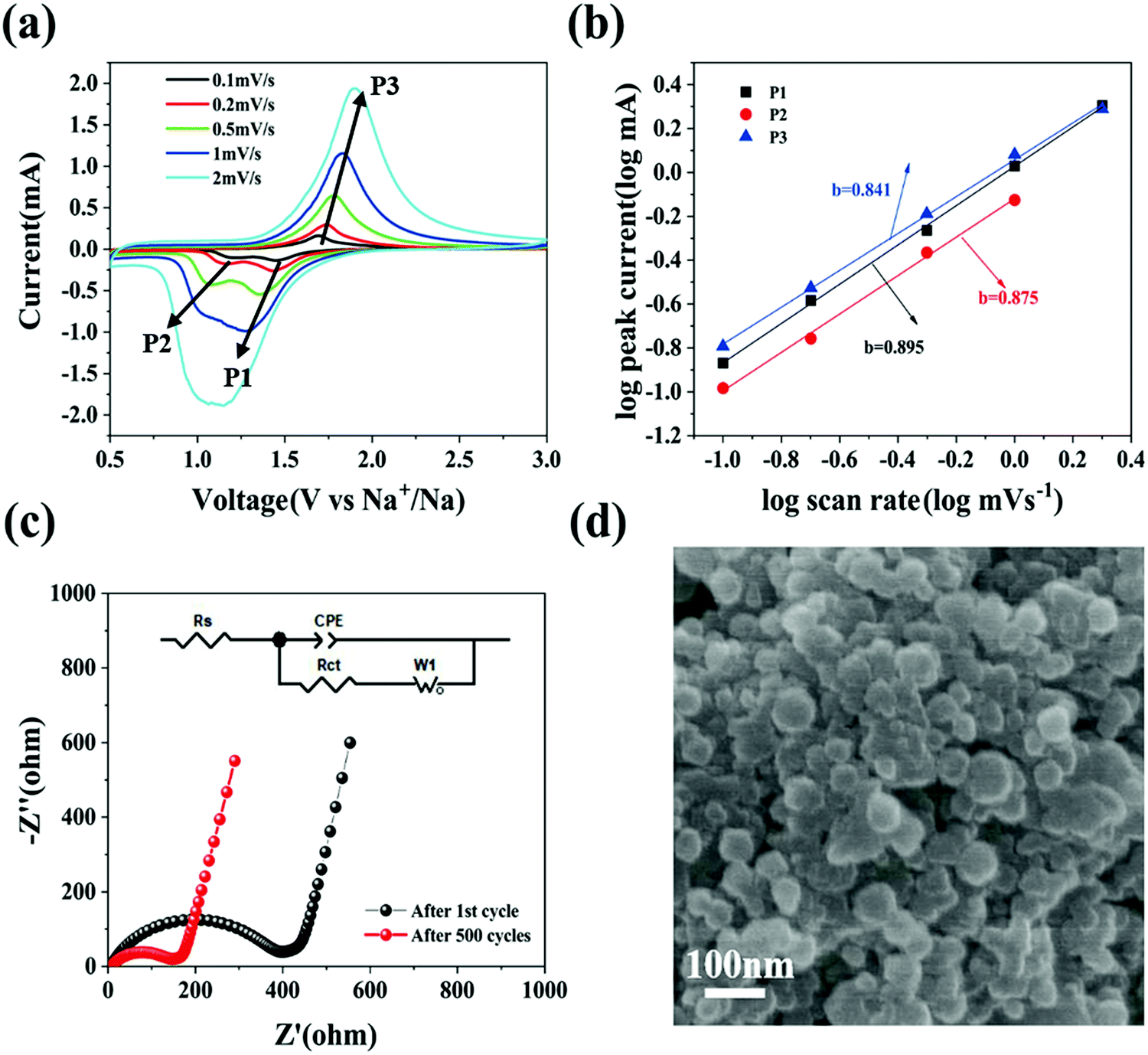 | ||
| Fig. 6 Characterization of Se@N-MCP electrodes: (a) cyclic voltammograms at various scan rates. (b) b-Value determination based on logarithmic peak currents versus scan rates. (c) Nyquist plots of the cell between after the 1st cycle and after 500 cycles at 1 A g−1. (d) SEM image after 500 cycles at 1 A g−1. | ||
Electrochemical impedance spectroscopy (EIS) was conducted to investigate the impedance evolution of the Se@N-MCP electrodes. Nyquist plots are simulated with the equivalent circuit inset after the 1st cycle and after the 500th cycle at 1 A g−1, as shown in Fig. 6c. Both curves contain a semicircle at the high frequency region and a straight line at the low frequency region. The charge transfer resistance (Rct) of the Se@N-MCPs after the 1st cycle is 373 Ω, which is much larger than that of Se@N-MCPs after 500 cycles (135 Ω), indicating that the porous structure promotes electrolyte infiltration into the electrode, thus shortening the transport path of the sodium ions.10,25,35 For comparison, AC impedance spectroscopy measurements (Fig. S9, ESI†) on the cells after three cycles for the Se@N-MCP, Se@N-MCP (2:1), Se@N-MCP (3:1), and Se/AB electrodes were investigated. The Se@N-MCPs show the lowest charge transfer resistance (Rct = 195 Ω), while the Rct of the Se@N-MCPs (2:1), Se@N-MCPs (3:1), and Se/AB are 278 Ω, 484 Ω, and 964 Ω, respectively, indicating that N-MCPs offer a highly conductive pathway for electrons in the electrodes and facilitate rapid charge transfer. By contrast, the Se/AB electrodes display the highest charge transfer resistance, because of the lower electronic conductivity. To further investigate the structural stability of Se@N-MCPs, the SEM image of Se@N-MCPs after 500 cycles at 1 A g−1 is shown in Fig. 6d. The morphology of the Se@N-MCPs remains after long-term cycling except for some residual electrolytes and SEI on the surface, indicating that the unique pore structure of the N-MCP matrix makes a contribution to alleviating the volume variation of Se during cycling, thus enhancing the cycling stability of the Se@N-MCP electrodes for Na–Se batteries. The pore volume of the N-MCPs and Se@N-MCPs is 0.478 and 0.037 cm3 g−1, respectively (Table S1, ESI†). After sodiation, the pore volume of Na2Se@N-MCPs is 0.2977 cm3 g−1 which is lower than that of N-MCPs ∼0.478 cm3 g−1, indicating that the micropores in the N-MCPs are sufficient for accommodating the volume change.29
Conclusion
In summary, we confined selenium in ZIF-8 derived N-doped microporous carbon polyhedrons (Se@N-MCPs) as cathodes for high performance Na–Se batteries. The N-MCPs show high electrical conductivity, uniform pore size distribution, and high specific surface area, thus resulting in improved sodium storage performance. The Se@N-MCP cathodes deliver an excellent discharge capacity of 612 mA h g−1 after 100 cycles at a current density of 0.1 A g−1 and a superior rate capability of 496 mA h g−1 at 5 A g−1 for the Na–Se batteries. In addition, they also exhibit a superior cycling life of 460 mA h g−1 at a current density of 1 A g−1 after 500 cycles with only 0.049% capacity decline per cycle. This facile strategy could be extended to prepare other energy storage materials, which can realize efficient ionic and electronic transport.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was financially supported by the National Key R&D Program of China (Grant No. 2018YFB0905400), the National Natural Science Foundation of China (No. 51622210, No. 51522212), and the Fundamental Research Funds for the Central Universities (WK3430000004).References
- B. C. Melot and J. M. Tarascon, Acc. Chem. Res., 2013, 46, 1226–1238 CrossRef PubMed.
- Y. Nishi, J. Power Sources, 2001, 100, 101–106 CrossRef.
- J. Y. Hwang, S. T. Myung and Y. K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC.
- S. Xin, L. Yu, Y. You, H. P. Cong, Y. X. Yin, X. L. Du, Y. G. Guo, S. H. Yu, Y. Cui and J. B. Goodenough, Nano Lett., 2016, 16, 4560–4568 CrossRef PubMed.
- A. Abouimrane, D. Dambournet, K. W. Chapman, P. J. Chupas, W. Weng and K. Amine, J. Am. Chem. Soc., 2012, 134, 4505–4508 CrossRef PubMed.
- C. P. Yang, Y. X. Yin and Y. G. Guo, J. Phys. Chem. Lett., 2015, 6, 256–266 CrossRef PubMed.
- H. Wang, Y. Jiang and A. Manthiram, Adv. Energy Mater., 2018, 8, 1701953 CrossRef.
- D. Ma, Y. Li, J. Yang, H. Mi, S. Luo, L. Deng, C. Yan, P. Zhang, Z. Lin, X. Ren, J. Li and H. Zhang, Nano Energy, 2018, 43, 317–325 CrossRef.
- H. Wang, S. Li, Z. Chen, H. K. Liu and Z. Guo, RSC Adv., 2014, 4, 61673–61678 RSC.
- B. Yuan, X. Sun, L. Zeng, Y. Yu and Q. Wang, Small, 2018, 14, 1703252 CrossRef PubMed.
- L. Zeng, X. Wei, J. Wang, Y. Jiang, W. Li and Y. Yu, J. Power Sources, 2015, 281, 461–469 CrossRef.
- L. Zeng, W. Zeng, Y. Jiang, X. Wei, W. Li, C. Yang, Y. Zhu and Y. Yu, Adv. Energy Mater., 2015, 5, 1401377 CrossRef.
- J. Ding, H. Zhou, H. Zhang, T. Stephenson, Z. Li, D. Karpuzov and D. Mitlin, Energy Environ. Sci., 2017, 10, 153–165 RSC.
- J. Ding, H. Zhou, H. Zhang, L. Tong and D. Mitlin, Adv. Energy Mater., 2018, 8, 1701918 CrossRef.
- B. Kalimuthu and K. Nallathamby, ACS Appl. Mater. Interfaces, 2017, 9, 26756–26770 CrossRef PubMed.
- C. Luo, Y. Xu, Y. Zhu, Y. Liu, S. Zheng, Y. Liu, A. Langrock and C. Wang, ACS Nano, 2013, 7, 8003–8010 CrossRef PubMed.
- X. Yang, H. Wang, D. Y. W. Yu and A. L. Rogach, Adv. Funct. Mater., 2018, 28, 1706609 CrossRef.
- N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef PubMed.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- W. Wang, X. Xu, W. Zhou and Z. Shao, Adv. Sci., 2017, 4, 1600371 CrossRef PubMed.
- L. Oar-Arteta, T. Wezendonk, X. Sun, F. Kapteijn and J. Gascon, Mater. Chem. Front., 2017, 1, 1709–1745 RSC.
- F. Zheng, Y. Yang and Q. Chen, Nat. Commun., 2014, 5, 5261 CrossRef PubMed.
- W. Li, S. Hu, X. Luo, Z. Li, X. Sun, M. Li, F. Liu and Y. Yu, Adv. Mater., 2017, 29, 1605820 CrossRef PubMed.
- Z. Li and L. Yin, Nanoscale, 2015, 7, 9597–9606 RSC.
- Y. Chen, W. Zhang, X. Jiang, Y. V. Kaneti, D. Tang, X. Wang, A. A. Alshehri, J. You, Y. Yamauchi and M. Hu, Mater. Chem. Front., 2018, 2, 520–529 RSC.
- A. Eftekhari and Z. Fan, Mater. Chem. Front., 2017, 1, 1001–1027 RSC.
- B. Liu, H. Shioyama, H. Jiang, X. Zhang and Q. Xu, Carbon, 2010, 48, 456–463 CrossRef.
- Q. Xu, T. Liu, Y. Li, L. Hu, C. Dai, Y. Zhang, Y. Li, D. Liu and M. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 41339–41346 CrossRef PubMed.
- T. Yang, T. Qian, M. Wang, X. Shen, N. Xu, Z. Sun and C. Yan, Adv. Mater., 2016, 28, 539–545 CrossRef PubMed.
- S. N. Yannopoulos and K. S. Andrikopoulos, J. Chem. Phys., 2004, 121, 4747–4758 CrossRef PubMed.
- K. Yuan, T. Hu, Y. Xu, R. Graf, L. Shi, M. Forster, T. Pichler, T. Riedl, Y. Chen and U. Scherf, Mater. Chem. Front., 2017, 1, 278–285 RSC.
- S. K. Park, J. S. Park and Y. C. Kang, ACS Appl. Mater. Interfaces, 2018, 10, 16531–16540 CrossRef PubMed.
- X. Li, J. Liang, Z. Hou, W. Zhang, Y. Wang, Y. Zhu and Y. Qian, Adv. Funct. Mater., 2015, 25, 5229–5238 CrossRef.
- Y. Jiang, Z. Yang, W. Li, L. Zeng, F. Pan, M. Wang, X. Wei, G. Hu, L. Gu and Y. Yu, Adv. Energy Mater., 2015, 5, 1402104 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00177d |
| This journal is © the Partner Organisations 2018 |