
Designed synthesis of organosilica nanoparticles for enzymatic biodiesel production†
Mohammad
Kalantari
 ,
Meihua
Yu
*,
Manasi
Jambhrunkar
,
Yang
Liu
,
Yannan
Yang
,
Xiaodan
Huang
and
Chengzhong
Yu
*
,
Meihua
Yu
*,
Manasi
Jambhrunkar
,
Yang
Liu
,
Yannan
Yang
,
Xiaodan
Huang
and
Chengzhong
Yu
*
Australian Institute for Bioengineering and Nanotechnology, The University of Queensland, Brisbane, QLD 4072, Australia. E-mail: m.yu2@uq.edu.au; c.yu@uq.edu.au
First published on 20th April 2018
Abstract
Porous nanomaterials are of great significance in enzyme immobilization by addressing the intrinsic issues of the native form of enzymes, such as low enzymatic activity and reusability. In this work, we report the successful fabrication of benzene-bridged dendritic mesoporous organosilica nanoparticles (BDMONs) with highly enriched benzene groups in the pore channel wall by a delayed addition method. The developed BDMONs were explored as nano-carriers for lipase immobilization. This platform exhibited a specific activity 6.5 times higher than that of the free enzyme with an excellent reusability, and enhanced thermal and pH stability. It is demonstrated that both the hydrophobic benzene groups and dendritic large-pores are responsible for the superior performance of the immobilized lipase in comparison with dendritic mesoporous silica nanoparticles, ethane-bridged dendritic mesoporous organosilica nanoparticles, and benzene-bridged MONs without large-pores. In particular, the designed nanobiocatalyst functions better than the free enzyme in the transesterification of corn oil to produce biodiesel, showing 93% conversion in the first cycle while retaining 94% of the initial catalytic activity after 5 cycles.
Introduction
Lipase-mediated transesterification reaction is regarded as an extremely promising process to produce biodiesel with high-purity products under mild and environmentally friendly conditions.1 Yet, the application of native lipase is limited mainly by the poor catalytic performance.2 Over the past few decades, nanomaterials have played a key part in addressing critical problems in fundamental research and also a variety of promising applications, such as DNA detection,3 sensing heavy metals,4 supercapacitors,5 self-assembly of biomolecules,6 solar energy conversion,7 and LEDs.8 In particular, immobilization of lipase on desired nanomaterials has been developed as an elegant approach to improve the activity of the immobilized lipase.9 Through the interaction with a hydrophobic carrier such as hydrophobically modified silica,10 periodic mesoporous organosilica,11 graphene oxide,12,13 or carbon dots,14 the configuration of the lipase molecule is altered, making the active site more accessible and thus enhancing the activity. It is also well-documented that the activity of the immobilized lipase increases along with an increase of the hydrophobic property of the carrier.10 Meanwhile, the architecture of the catalyst carriers also plays a crucial role in the catalytic activity improvement.15 In this regard, dendritic mesoporous silica nanoparticles (DMSNs) with large central-radial pore channels and highly accessible internal surface areas have been recently developed as an excellent platform for a wide range of applications.16–18 Immobilized metal-based (e.g., ruthenium,19 tantalum hydride,20 and gold21) and enzyme-based (e.g. horseradish peroxidase22) catalysts on DMSNs have shown outstanding activity and stability attributed to their unique dendritic architecture. The open structure of DMSNs not only facilitates the accessibility of the substrate to the catalyst but also protects the catalyst species embedded in the pore channels against harsh conditions.23,24 Very recently, we reported the physical adsorption of lipase on octadecylalkyl modified DMSNs.25 High content C18 terminal chains on the pore surface of DMSNs created an ideal hydrophobic environment and induced interfacial activation of the lipase, leading to a specific activity 5.2 times higher than that of the free enzyme. However, the high content C18 end groups cause steric hindrance for chemical grafting of lipase to overcome leaching problems.26 Thus, simultaneous control over a highly hydrophobic wall composition, a modifiable external surface, and an easily accessible pore structure are of great importance to further improve the catalytic performance of chemically immobilized lipase.Mesoporous organosilica nanoparticles (MONs) with various organic bridge groups incorporated inside the silica framework have been successfully synthesized to enhance the functionality of silica materials.27 The surface of MONs can be modified with different functional moieties through the free hydroxyl groups on their pore surfaces to introduce functional moieties and host biomolecules.28,29 Benzene group grafted silica foam demonstrated a higher lipase activity compared with those modified with other organic groups,10 suggesting that a benzene-bridged carrier could be an ideal platform for lipase immobilization. The Shi group recently advanced hollow benzene-bridged MONs (BMONs) based on a hard-templating approach.30–32 The prepared particles are highly dispersed; however, the organic content (7 wt% carbon) is limited for hyperactivation of lipase. Later, Yang and co-workers developed a biphasic synthesis approach to synthesize small-sized BMONs with a relatively large pore size of 7.6 nm but a limited mesopore volume (<0.3 cm3 g−1).33 Therefore, it is an ongoing challenge to develop BMONs concurrently with a large-pore dendritic structure, high pore volumes and high hydrophobic contents for lipase immobilization.
Herein, we report the successful fabrication of monodispersed benzene-bridged DMONs with exceptional hydrophobic contents. Our success relies on a designed delayed addition strategy in which an organic precursor (1,2-bis(triethoxysilyl)benzene (BTEB)) is introduced 15 min after the addition of inorganic precursor tetraethyl orthosilicate (TEOS) into a basic aqueous solution, containing triethanolamine (TEA) as a catalyst, and cetyltrimethylammonium bromide (CTAB) and sodium salicylate (NaSal) as templating surfactants (Fig. 1A). This approach provides enough time to form primary dendritic mesoporous silica nanoparticles (Fig. 1B) which act as nucleation sites for subsequent co-condensation of silica sources derived from BTEB and the remaining TEOS, leading to the formation of benzene group enriched dendritic pore channels (Fig. 1C). In the conventional method, BTEB and TEOS are added into the reaction solution simultaneously, which results in the formation of aggregated nanoparticles with irregular shape, dendritic-like pore channels on the surface and much less benzene groups on the wall (Fig. 1D). After surfactant removal and post-modification with trimethoxysilane aldehyde (TMSA) (Fig. 1E), monodispersed BDMONs are covalently bound with lipases to avoid leaching of the enzymes (Fig. 1F), which show a superior catalytic activity (6.5 times higher than that of the free enzyme), outstanding reusability, enhanced thermal and pH stabilities. In addition, by comparison with the performance of dendritic mesoporous silica nanoparticles (DMSNs), BDMONs with small pores and ethane-bridged dendritic mesoporous organosilica nanoparticles (EDMONs), it is concluded that the extraordinary performance of the immobilized lipase on BDMONs relies on the increased hydrophobic content and uniform dendritic structure. This designed nanobiocatalysis shows great potential in the production of biodiesel with 93% conversion activity and high reusability (94% after 5 cycles).
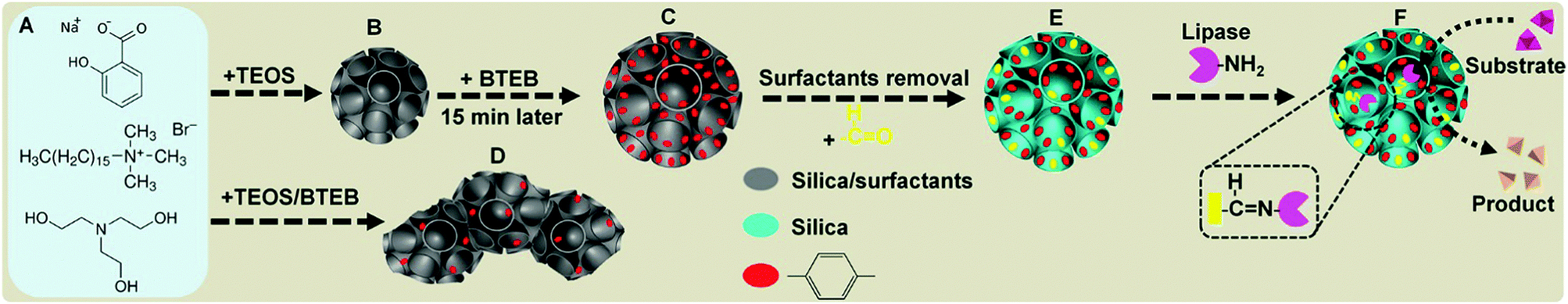 | ||
| Fig. 1 Schematic illustration of the proposed mechanism of the delayed addition method for synthesis of BDMONs and subsequent lipase immobilization on BDMONs. (A) Surfactants and catalyst; (B) primary dendritic mesoporous silica nanoparticles; (C) BDMONs with benzene group enriched dendritic pore channels; (D) aggregated BDMONs obtained by the conventional approach with simultaneous addition of BTEB and TEOS into the reaction solution; (E) BDMONs after surfactant removal and post-modification of TMSA; (F) BDMONs with covalently bonded lipase. | ||
Experimental
Chemicals
All chemicals were used as received without further purification. CTAB, TEA, NaSal, TEOS, BTEB, 1,2-bis(triethoxysilyl)ethane (BTEE), hydrogen chloride, toluene, ethanol, methanol, lipase from Candida rugosa, 4-nitrophenyl palmitate (pNPP) and corn oil were purchased from Sigma-Aldrich. TMSA was purchased from United Chemicals Technologies. Double distilled water obtained from a laboratory purification system was used for all experiments.Synthesis of BDMONs
In a typical synthesis, TEA (34 mg) was dissolved in distilled water (12.5 ml) at 80 °C. After intensive stirring for 30 min, CTAB (190 mg) and NaSal (42 mg) were added and the solution was kept stirring for 1 h. TEOS (1.34 ml) was added to the solution under stirring for 15 min, followed by the addition of BTEB (1.17 ml, molar ratio of BTEB to TEOS was 0.5). The reaction solution was kept stirring for 18 h. The precipitates were collected by centrifugation at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 min and then washed three times with ethanol to remove the residual reactants. The as-synthesized samples were extracted at 70 °C for 6 h in acidic ethanol (3 ml of 37% HCl in 50 ml of absolute ethanol) three times to remove the template CTAB. For comparison, other BDMONs were prepared with a molar ratio of BTEB to TEOS of 0.67, 1 or 1.5, while keeping the total silica sources molar and all other synthesis parameters constant. In another experiment, TEOS and BTEB with a molar ratio of 0.5 were added to the reaction solution at the same time. To further investigate the effect of shorter and longer time delays, BTEB was added to the reaction solution 5 min and 30 min after addition of TEOS while the molar ratio of BTEB to TEOS was 0.5 and all other synthesis parameters were kept unchanged. The final products were denoted as BDMONs-x-y, where x and y indicate the time gap between the addition of TEOS and BTEB and the molar ratio of BTEB to TEOS, respectively (please see Table S1, ESI†).
000 rpm for 15 min and then washed three times with ethanol to remove the residual reactants. The as-synthesized samples were extracted at 70 °C for 6 h in acidic ethanol (3 ml of 37% HCl in 50 ml of absolute ethanol) three times to remove the template CTAB. For comparison, other BDMONs were prepared with a molar ratio of BTEB to TEOS of 0.67, 1 or 1.5, while keeping the total silica sources molar and all other synthesis parameters constant. In another experiment, TEOS and BTEB with a molar ratio of 0.5 were added to the reaction solution at the same time. To further investigate the effect of shorter and longer time delays, BTEB was added to the reaction solution 5 min and 30 min after addition of TEOS while the molar ratio of BTEB to TEOS was 0.5 and all other synthesis parameters were kept unchanged. The final products were denoted as BDMONs-x-y, where x and y indicate the time gap between the addition of TEOS and BTEB and the molar ratio of BTEB to TEOS, respectively (please see Table S1, ESI†).
Synthesis of DMSNs
For the preparation of dendritic mesoporous silica nanoparticles (DMSNs), the as-synthesized BDMONs-15-1 sample was calcined at 600 °C in air for 6 h to remove organic contents and surfactants.Synthesis of BONs-SP
For the synthesis of small-pore BDMONs, the above procedure for preparation of BDMONs-15-1 was followed except that NaSal was not used in the reaction system.Synthesis of EDMONs-15-1
Ethane-bridged dendritic mesoporous organosilica nanoparticles (EDMONs) were synthesized following the procedure described above to prepare BDMONs-15-1 except that the same molar amount of BTEE was used as the organosilica precursor instead of BTEB and the amount of NaSal was increased to 84 mg.Material characterization
Transmission electron microscopy (TEM) and corresponding element mapping experiments were carried out on a HT7700-EXALENS transmission electron microscope operated at 200 kV. The samples for TEM were dispersed in toluene by sonication and then supported onto a holey carbon film on a copper grid. Field emission scanning electron microscopy (SEM) images were obtained on a JEOL 7800 microscope operated at 1 kV. For SEM measurements, the samples were dissolved in toluene and then dropped onto aluminium foil pieces and attached to a conductive carbon film on SEM mounts. The nitrogen adsorption–desorption isotherms were measured at 77 K on a nitrogen adsorption device (Micromeritics ASAP Tristar II 3020). The samples were degassed under vacuum for 18 h at 110 °C before analysis. The Brunauer–Emmet–Teller (BET) method was used to estimate the specific surface area. The pore distribution was derived from the adsorption branch of the isotherms using the Barret–Joyner–Halanda (BJH) method. Elemental analysis (EA) was carried out by a CHNS-O Analyzer (Flash EA1112 Series, Thermo Electron Corporation). X-ray photoelectron spectra (XPS) were collected on a Kratos Axis Ultra X-ray photoelectron spectrometer (PerkinElmer). Fourier transform infrared (FTIR) spectra were obtained using a Thermo Nicolet Nexus 6700 FTIR spectrometer equipped with a Diamond ATR (attenuated total reflection) Crystal. For each spectrum, 64 scans were collected at a resolution of 4 cm−1 over the range 400–4000 cm−1. To evaluate the inner pore surface hydrophobicity/hydrophilicity, the adsorption of water and hexane vapor was carried out at 25 °C.34 Briefly, the powdered sample (20 mg) was placed in a glass bottle without a lid and heated to 110 °C under a high vacuum of 0.9 mbar to remove all impurities for 3 h. After cooling the glass bottle (with a lid) to room temperature, the glass bottle (without a lid) was kept in contact with the water or hexane vapor for sufficient time at 25 °C. The adsorption capacity of the sample was estimated according to the mass change of the sample after the adsorption process using a digital microbalance (Model YP/1002, with a sensitivity of 0.1 mg). All adsorption experiments were performed in triplicate to ensure the reproducibility of the data. 29Si cross-polarization magic-angle spinning (CP-MAS) and 13C CP-MAS NMR spectra were recorded on a Bruker Avance III spectrometer with a 7T magnet, Zirconia rotor, 4 mm, rotated at 5 kHz.Time-dependent study of the formation of BDMONs-0-0.5 and BDMONs-15-0.5
TEM analysis was used at different reaction times to monitor the structural evolution of the particles. In a typical experiment, one drop of the reaction mixture was collected at different reaction times (15, 30, 45, and 60 min, 3, 6, and 18 h), centrifuged, and washed with ethanol prior to TEM analysis. For both samples, the time was counted after the addition of TEOS to the reaction mixture.Preparation of aldehyde-functionalized nanoparticles
In a typical experiment, particles (150 mg) were vacuum dehydrated at 120 °C for 6 h and then suspended in dry toluene (20 mL). The mixture was refluxed for 1 h at 110 °C, followed by the addition trimethoxysilane aldehyde (1.05 mmol). The product was collected after 18 h by centrifugation at 20000 rpm for 10 min and then washed with ethanol three times. The final samples were dried in a hood at room temperature overnight.
Lipase immobilization
To covalently immobilize lipase on nanoparticles, a lipase solution (5 ml, 0.1 mg ml−1 in 100 mM sodium phosphate, pH 7.4) was added to an aldehyde functionalized particle solution (5 ml, 2 mg ml−1 in 100 mM sodium phosphate, pH 7.4) and shaken at 25 °C, 200 rpm. After 17 h, the suspension was centrifuged at 5000 rpm for 5 min and washed three times with sodium phosphate (100 mM, pH 7.4). The lipase concentration in the supernatants was determined by the Bradford method. The amount of immobilized enzyme was measured and calculated by subtracting the amount of lipase in the supernatant from the initial lipase content used for the immobilization.24Lipase activity assay
The hydrolysis of 4-nitrophenyl palmitate (pNPP) in sodium phosphate buffer (100 mM, pH 7.4) was used to measure the activities of the free and immobilized lipases.35 In a typical experiment, immobilized lipase solution in sodium phosphate buffer (100 mM, pH 7.4) was placed in a cuvette. After attainment of a constant absorbance at 410 nm, the pNPP substrate solution (150 μl, 10 mM in 2-propanol) was added and the increase of the absorbance at 410 nm was recorded using a UV-Vis spectrophotometric instrument (UV-2450, Shimadzu Company) to monitor the intensity observation, representative of the release rate of p-nitrophenol (pNP) for approximately ten minutes against a reference with the same volume and amount of buffer and enzyme solution. For the free lipase, the same amount of lipase was mixed with the reaction media under the assay conditions. The activity was calculated using the following equation:in which, ΔA, ΔAblank, V, and ε are the change of absorbance at 410 nm (a.u. min−1), change of absorbance without lipase (a.u. min−1), the volume of the assay (ml), and extinction coefficient (a.u. ml mol−1 min−1), respectively.36 The change of absorbance for a blank run without lipase was almost zero and the volume of the assay was identical for both tests. The relative activity was defined as the ratio of specific activity of immobilized lipase to the specific activity of free lipase.
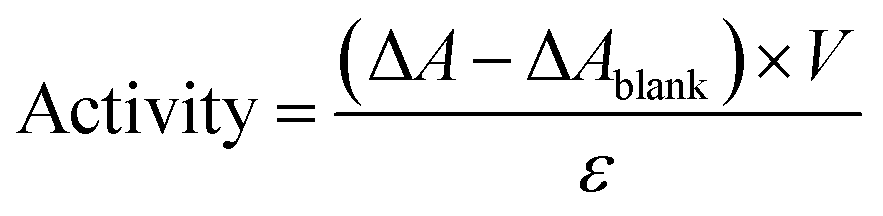
Reusability assay
The reusability of the immobilized lipases was studied by repeating the use of the immobilized enzymes to catalyse the hydrolysis reaction of pNPP under the activity assay conditions mentioned above. The duration of each cycle was 10 min. After each activity assay, the immobilized lipases were separated from the reaction system by centrifugation, washed three times with buffer solution to remove all of the substrates and products from the sample and applied in the next activity measurement with fresh substrates. The relative activity was defined as the ratio of the residual activity to the initial activity.Thermal stability assay
The effect of temperature on the activity of the free and immobilized lipases was studied by incubating in phosphate buffer (100 mM, pH 7.4) at temperatures ranging from 30 to 70 °C for 60 min followed by measuring the enzymatic activity. The relative activity was defined as the ratio of the residual activity to the initial value measured at 30 °C, pH 7.4 (the initial activity was defined as 100%).pH stability assay
The effect of pH on the activity of the free and immobilized enzymes was determined by incubating at room temperature in phosphate buffer (100 mM) at pH ranging from 3 to 9 for 10 min followed by determination of enzymatic activity. The relative activity was defined as the ratio of the residual activity to the initial value measured at 30 °C, pH 8 (the initial activity was defined as 100%).Biodiesel production
Free and immobilized lipases were used for biodiesel production by transesterification of corn oil with methanol. The reactions were conducted at 40 °C on a shaking incubator at 120 rpm. A typical reaction mixture consisted of corn oil (3 ml), 1.5 ml of distilled water, a weighed amount of the biocatalyst (containing 5 mg of lipase) or free lipase (5 mg), and a two-step addition of methanol with 250 μl of methanol in each step.37 After 24 h, the residual methanol in the reaction mixture was removed with the help of a rotary evaporator at 70 °C for 2 h, and then glycerol was separated from the reaction mixture by centrifugation at 10000 rpm for 10 min. The conversion of corn oil to methyl esters was measured by a DGU-20A3 high-performance liquid chromatography (HPLC) (Shimadzu, Tokyo, Japan) instrument equipped with an auto-injector and ultraviolet detector of 210 nm. The column was a Restek Viva C18 set at an isothermal oven temperature of 54 °C. The mobile phase was 100% acetonitrile and 0.05% of TFA with a flow of 0.15 mL min−1. Biodiesel yield was calculated by summing the areas of methyl esters present in the reaction mixture and on the basis of calibration curves built up with FAME standards.
Reusability assay for biodiesel production
To evaluate the reusability of immobilized lipase in repeated use, the immobilized lipase was separated by centrifugation and washed with buffer. The recovered immobilized enzymes were applied to the next batch of transesterification using fresh substrates. The assay conditions were the same as those mentioned above. The relative conversion was defined as the ratio of the residual conversion capability to the initial conversion capability.Results and discussion
Benzene-bridged dendritic mesoporous organosilica nanoparticles (BDMONs) were synthesized by a delayed method shown in Fig. 1. To further control organic group content, the BTEB to TEOS molar ratio (y) was adjusted from 0.5 to 1. The final products after surfactant extraction were denoted as BDMONs-x-y, where x and y indicate the time gap between the addition of TEOS and BTEB and the molar ratio of BTEB to TEOS, respectively (see details in the Experimental section and Table S1, ESI†).Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) were used to characterize the morphologies and structures of the synthesized particles. Low-magnification SEM images (Fig. 2a–c) of BDMONs-15-y reveal that all samples are spherical in shape with a uniform size of around 196, 195, and 198 nm (y = 0.5, 0.67, and 1, respectively). The opening pores on the surface of the particles and dendritic structures can also be clearly seen from high-magnification SEM images (Fig. 2e–g). From the TEM images of BDMONs-15-y (Fig. 2i–k), it can be conclusively observed that these particles have a dendritic pore structure and an excellent dispersity. In other synthesis conditions such as increasing the BTEB to TEOS molar ratio to 1.5 (Fig. S1, ESI†) or shorter/longer delay time (Fig. S2, ESI†), the dispersed dendritic structure with large opening pores was lost. Thus, the obtained samples were not included in the following studies. Nitrogen sorption analysis was carried out to explore the porosity of BDMONs-15-y. All nitrogen adsorption isotherms of BDMONs-15-y (Fig. 3a) show a type IV curve. The corresponding pore size distribution curves (Fig. 3b) exhibit a similar peak centered at 13.9 nm for BDMONs-15-0.5, BDMONs-15-0.67 and BDMONs-15-1. The BET surface area and the total pore volume are measured to be 552 m2 g−1 and 0.90 cm3 g−1 for BDMONs-0.5, 729 m2 g−1 and 1.11 cm3 g−1 for BDMONs-0.67, and 677 m2 g−1 and 1.09 cm3 g−1 for BDMONs-1(Table 1). To understand the influence of the BTEB addition time point on the particle structures, BDMONs-0-0.5 was prepared under the conventional approach by simultaneous addition of TEOS and organic precursors.28,31,32,38–40 BDMONs-0-0.5 show big clusters formed by the irregular shape of the particles (around 110 nm in size) with some dendritic-like pore channels on the surface (Fig. 2d, h, and l). The nitrogen sorption analysis of BDMONs-0-0.5 shows a broad pore size distribution curve centered at 14.0 nm (Fig. 3b). The BET surface area and the total pore volume of BDMONs-0-0.5 is 426 m2 g−1, and 0.91 cm3 g−1, respectively (Table 1).
 | ||
| Fig. 2 SEM (a–h) and TEM (i–l) images of BDMONs-15-0.5 (a, e and i), BDMONs-15-0.67 (b, f and j), BDMONs-15-1 (c, g and k), and BDMONs-0-0.5 (d, h and l). Scale bar is 100 nm. | ||
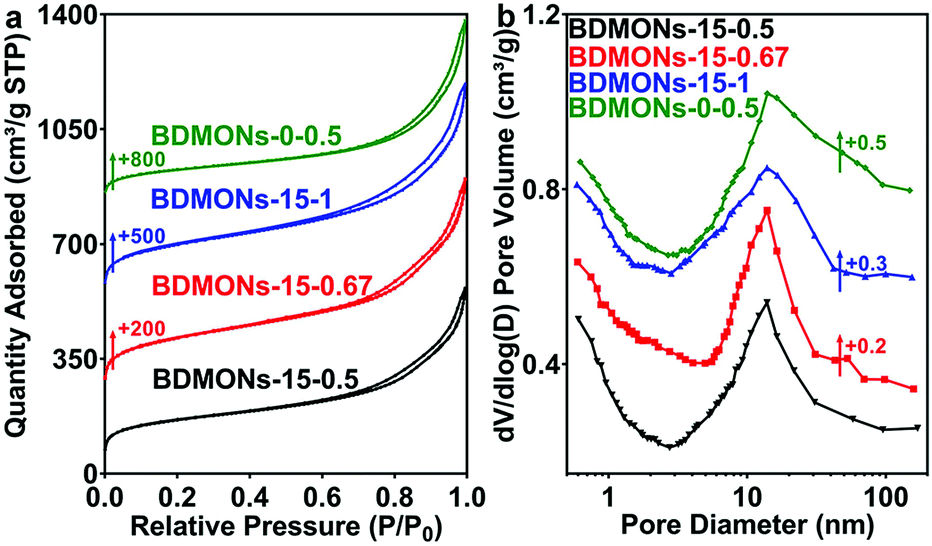 | ||
| Fig. 3 (a) N2 sorption isotherms and (b) pore size distribution curves of BDMONs-15-0.5, BDMONs-15-0.67, BDMONs-15-1, and BDMONs-0-0.5. | ||
| Sample | D p | d p | S BET | V P | C EA | C XPS | A |
|---|---|---|---|---|---|---|---|
| a Average particle size (nm). b Pore diameter calculated by BJH method from the adsorption branch (nm). c BET surface area (m2 g−1). d Total pore volume (cm3 g−1). e Carbon weight percentage estimated by EA. f Carbon weight percentage estimated by XPS. g Relative activity. | |||||||
| BDMONs-15-0.5 | 196 | 13.9 | 552 | 0.9 | 23.2 ± 0.3 | 38.8 ± 0.2 | 5.7 |
| BDMONs-15-0.67 | 195 | 13.9 | 729 | 1.11 | 27.4 ± 0.4 | 40.5 ± 0.4 | 6.3 |
| BDMONs-15-1 | 198 | 13.9 | 677 | 1.09 | 29.9 ± 0.3 | 40.3 ± 0.3 | 6.5 |
| BDMONs-0-0.5 | NA | 14.0 | 426 | 0.91 | 23.4 ± 0.2 | 22.4 ± 0.5 | 4.9 |
| DMSNs | 165 | 16.5 | 577 | 1.05 | 0 | 0 | 0.75 |
| BONs-SP | 76 | 3.6 | 478 | 1.09 | 29.8 ± 0.2 | 40.2 ± 0.3 | 5.4 |
| EDMONs-15-1 | 201 | 16.4 | 249 | 0.44 | 10.8 ± 0.1 | 15.0 ± 0.4 | 4.1 |
To gain information on the distribution of organic contents in BDMONs-x-y, the samples were characterized by elemental analysis (EA) and X-ray photoelectron spectroscopy (XPS). As expected, with an increase in the initial molar ratio of BTEB to TEOS, the total carbon content in the bulk sample measured from EA rises from 23.2 ± 0.3% for BDMONs-15-0.5 to 27.4 ± 0.4 and 29.9 ± 0.3 wt% for BDMONs-15-0.67 and BDMONs-15-1, respectively (Table 1). The carbon contents on the sample surface determined from XPS are 38.8 ± 0.2%, 40.5 ± 0.4%, and 40.3 ± 0.3% in BDMONs-15-y (y = 0.5, 0.67, and 1, respectively), higher than the values in the bulk samples. These results suggest that the BDMONs-15-y samples have enriched benzene groups on the surface. In contrast, the carbon content of BDMONs-0-0.5 measured by EA (23.4 ± 0.2%) and XPS (22.4 ± 0.5%) is very close but much lower than those of BDMONs-15-y (Table 1), indicative of the fairly uniform distribution of benzene groups for the BDMONs-0-0.5 sample but a lack of organic groups in the pore channels.
Water and hexane vapor adsorption experiments were carried out to estimate the surface hydrophobicity of the samples.34 The molar ratio of adsorbed water to hexane (λ) of the BDMONs-15-y sample is similar at 0.0083 ± 0.002, 0.0078 ± 0.002 and 0.0074 ± 0.002 for BDMONs-15-0.5, BDMONs-15-0.67 and BDMONs-15-1 respectively, demonstrating the hydrophobic nature of the mesopore surface for all samples (λ < 0.5).10 Compared to the BDMONs-15-y samples, BDMONs-0-0.5 shows an increased λ value, suggesting less hydrophobic groups. These results are consistent with carbon content results from XPS.
BDMONs-15-1 with a relatively higher carbon content was chosen for further characterization to confirm the organic groups in the framework. The dark-field scanning transmission electron microscopy (DF-STEM) and energy dispersive spectroscopy (EDS) elemental mapping of Si, O, and C elements in the framework of BDMONs-15-1 (Fig. 4a–d) evidently demonstrates the uniform distribution of C in the hybrid framework. In the 13C cross-polarization magic angle spinning (CPMAS) nuclear magnetic resonance NMR spectrum of BDMONs-15-1, distinct resonances at 132 ppm indicate the presence of benzene groups (Fig. 4e).28 The 29Si MAS NMR spectrum confirms the presence of both inorganic and organic moieties in BDMONs-15-1 (Fig. 4f). The two resonances at −105 and −114 ppm are assigned to the Q3 (Si(OSi)3(OH)) and Q4 (Si(OSi)4) sites, originated from TEOS, while the resonances at −66, −74, and −83 ppm correspond to T1 (C-Si(OSi)(OH)2), T2 (C-Si(OSi)2(OH)), and T3 (C-Si(OSi)3) sites, respectively, representing the successful integration of benzene groups into the framework (Fig. 4f).28 To gain insight on the organic content of BDMONs-15-1, the integrated peak area measurements on a percentage basis for Q and T signals were conducted. The ratio of [(T1 + T2 + T3)]/[(T1 + T2 + T3) + (Q1 + Q2)] of BDMONs-15-1 is calculated to be around 0.60, suggesting a high content of the incorporated organic groups.25 It can be concluded that the delayed addition approach plays a crucial role in the fabrication of BDMONs with desired high hydrophobic pore channels.
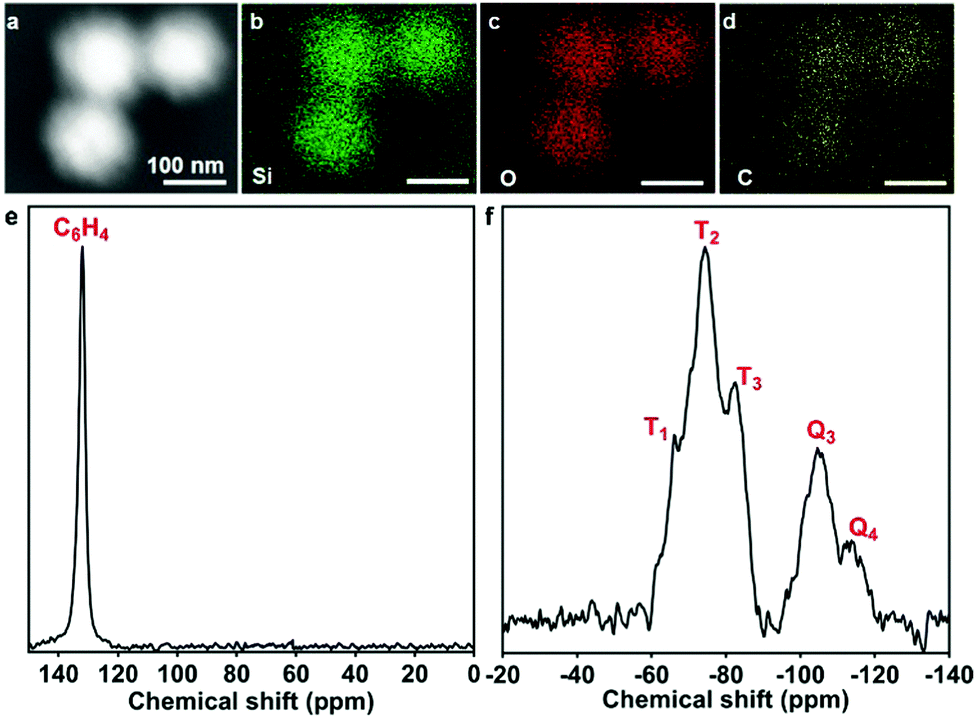 | ||
| Fig. 4 (a) Dark-field STEM image, (b–d) corresponding EDS mapping of Si, O and C elements in the framework of BDMONs-15-1; (e) 13C CPMAS and (f) 29Si HPDEC NMR spectra of BDMONs-15-1. | ||
To shed light on the key role of the addition time gap between the two silica sources on the formation of well-defined spherical and uniform particles, time-dependent experiments were conducted for the synthesis of BDMONs-0-0.5 and BDMONs-15-0.5 (see the Experimental part for details). TEM analysis was applied to monitor the structural evolution over the reaction time (Fig. S3, ESI†). For sample BDMONs-0-0.5, nonporous amorphous silica seeds (∼10 nm) are observed at 15 min reaction (Fig. S3a1, ESI†), which develop into aggregated larger particles (∼50 nm, Fig. S3b1, ESI†) at 30 min. By extending the reaction time to 1 h, small dendritic mesopores can be seen on the surface of the aggregated nanoparticles (Fig. S3c1, ESI†), and the dendritic-like pores are more distinct at the reaction time of 3 h (Fig. S3d1, ESI†). There is no significant change in structure afterward (Fig. S3e1 and f1, ESI†). For BDMONs-15-0.5, well-defined dendritic mesoporous silica nanoparticles have already formed with a particle size of around 100 nm and opening large pore size of ∼12 nm at the reaction time of 15 min before addition of BTEB (Fig. S3a2, ESI†). It is suggested that because of the high miscibility and strong electrostatic attraction between Sal− and CTA+ micelles,38 the hydrophobic part of Sal− moves into the hydrophobic region of the micelles and increases the packing parameter (g) which induces the micelle structural transition toward vesicular/lamellar structure and eventually form the central radial structure.41 After adding BTEB, the particle size steadily increases from 110 nm at 30 min (Fig. S3b2, ESI†) to 120 nm at 1 h (Fig. S3c2, ESI†) and the wall thickness tends to be thicker (around 5 nm vs. 7 nm) as reflected by the higher contrast under TEM observation, indicating the further deposition and assembly of organic/inorganic silica sources. By prolonging the reaction time, the dispersity of the dendritic particles improves and the particle size reaches 161 nm at 3 h (Fig. S3d2, ESI†), 173 nm at 6 h (Fig. S3e2, ESI†) and finally 196 nm at 18 h (Fig. S3f2, ESI†).
For the conventional method with BTEB and TEOS added to the reaction solution at the same time, it was witnessed that small oil drops were formed by BTEB because of its strongly hydrophobic nature as well as slow hydrolysis and condensation rate under basic conditions.42 TEOS is intrinsically hydrophobic, thus TEOS mixed with BTEB will preferentially stay inside the oil phase at the beginning of the reaction; supported by the observation that a longer time was required to form a white solution compared to the delayed synthesis approach with a similar amount of TEOS. As the reaction time prolongs, the hydrolysed organosilica species will form that have a lower charge density than inorganic silica species derived from TEOS and consequently hinder the interaction between negatively charged silica species and positively charged Sal−/CTA+ templates under basic conditions.38 That is the reason why crosslinked amorphous organosilica is formed without a defined mesostructure within the first 1 h. It takes a longer time to generate hydrolysed silica species with sufficient net negative charge to assemble with Sal−/CTA+ surfactants into porous hybrid structures, but with aggregated ill-defined morphology. In contrast, well-defined dendritic spherical structures are formed as early as 15 min after addition of TEOS in the delay method, which act as seeds for the accumulation of silica/silicate anions derived from small amounts of remaining TEOS and BTEB sources added at 30 min. The reported study on particle growth stated that as the hydrolysis and condensation time of silica sources increased, the final product tended to form larger and less homogeneous particles.43 It can be concluded that the pre-hydrolysis and condensation time of inorganic silica sources can be utilized to control the final hybrid organosilica shape and organic group distribution in the particles.
Lipase with a size of 3.3 × 4.2 × 5 nm,25 mostly used for biofuel production, was immobilized on BDMONs through a covalent method to avoid the leaching problems during the reuse process.44 Aldehyde groups were first grafted on the surface of the BDMONs (Fig. 1E), which were then reacted with the free amino groups of the lipase molecules to form covalent bonds (Fig. 1F). To examine the covalent binding of lipase on the aldehyde-functionalized BDMON samples, BDMONs-15-1 before and after modification and lipase immobilization was monitored using FT-IR analysis. As shown in Fig. S4 (ESI†), Aldehyde-BDMONs-15-1 displays peaks at 935 and 1060 cm−1 which are associated with stretching vibration modes of Si–OH and Si–O–Si, respectively.45 The peaks at 2980 and 1150 cm−1 are assigned to stretching vibration modes of C–H and Si–C of the benzene group, respectively.46 The characteristic peaks appearing at 1380 and 1600 cm−1 can be attributed to stretching vibration modes of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C.46 The peak at 1725 cm−1 is associated with the stretching vibrations of the CO bond in aldehyde groups, representing the successful aldehyde modification for the Aldehyde-BDMONs-15-1 sample.47 For pure lipase, the bands at 1530 and 1650 cm−1 are characteristic of the stretching vibrations of amide I and II bands, respectively.48 The FTIR spectrum of Lipase-Aldehyde-BDMONs-15-1 shows the absorption peak at 1680 cm−1 which is characteristic of the stretching vibration modes of the CN bond, authorizing the successful covalent attachment of lipase molecules on the particles via formation of amide bonds between aldehyde groups (Fig. 1F) on the surface of the particles and amino groups of the enzyme molecules.47 It should be noted that in this study a comparatively similar low lipase loading of 50 mg g−1 (see details in the Experimental section) was designed for all materials to simplify the assessment of the surface chemistry effect on the catalytic performance and stability of the immobilized lipase.
C.46 The peak at 1725 cm−1 is associated with the stretching vibrations of the CO bond in aldehyde groups, representing the successful aldehyde modification for the Aldehyde-BDMONs-15-1 sample.47 For pure lipase, the bands at 1530 and 1650 cm−1 are characteristic of the stretching vibrations of amide I and II bands, respectively.48 The FTIR spectrum of Lipase-Aldehyde-BDMONs-15-1 shows the absorption peak at 1680 cm−1 which is characteristic of the stretching vibration modes of the CN bond, authorizing the successful covalent attachment of lipase molecules on the particles via formation of amide bonds between aldehyde groups (Fig. 1F) on the surface of the particles and amino groups of the enzyme molecules.47 It should be noted that in this study a comparatively similar low lipase loading of 50 mg g−1 (see details in the Experimental section) was designed for all materials to simplify the assessment of the surface chemistry effect on the catalytic performance and stability of the immobilized lipase.
The hydrolytic reaction of pNPP was carried out as the model reaction to evaluate the enzymatic activity of the free and immobilized lipases, based on the catalytic activity curves of the tested samples (Fig. S5, ESI†) and activity calculation equation (see details in Experimental part). A relative activity of 5.7, 6.3, 6.5, and 4.9 is achieved for BDMONs-15-0.5, BDMONs-15-0.67, BDMONs-15-1, and BDMONs-0-0.5, respectively (Fig. 5a). Compared to the free lipase, all nanobiocatalyst systems display significantly enhanced activities, contributed by “interfacial activation” of lipase via the hydrophobic property of the BDMONs.9 The BDMONs-15-y samples show much higher catalytic activity than BDMONs-0-0.5, due to the increased hydrophobic benzene groups in the wall of the pore channels for lipase hydrophobic lid opening.10,11,25 In addition, BDMONs-15-y with dispersed properties may enhance the substrate diffusion into the dendritic mesopores and accessibility to enzymes, compared to aggregated BDMONs-0-0.5.15 The slight promotion of immobilized lipase activity with increasing molar ratio of BTEB to TEOS could be explained by the slightly increased benzene group contents. The relative activities of all BDMONs-15-y samples are higher than those in previous reports (see Table S2, ESI†) highlighting the superior performance of the designed BDMONs for hyperactivation of lipase.
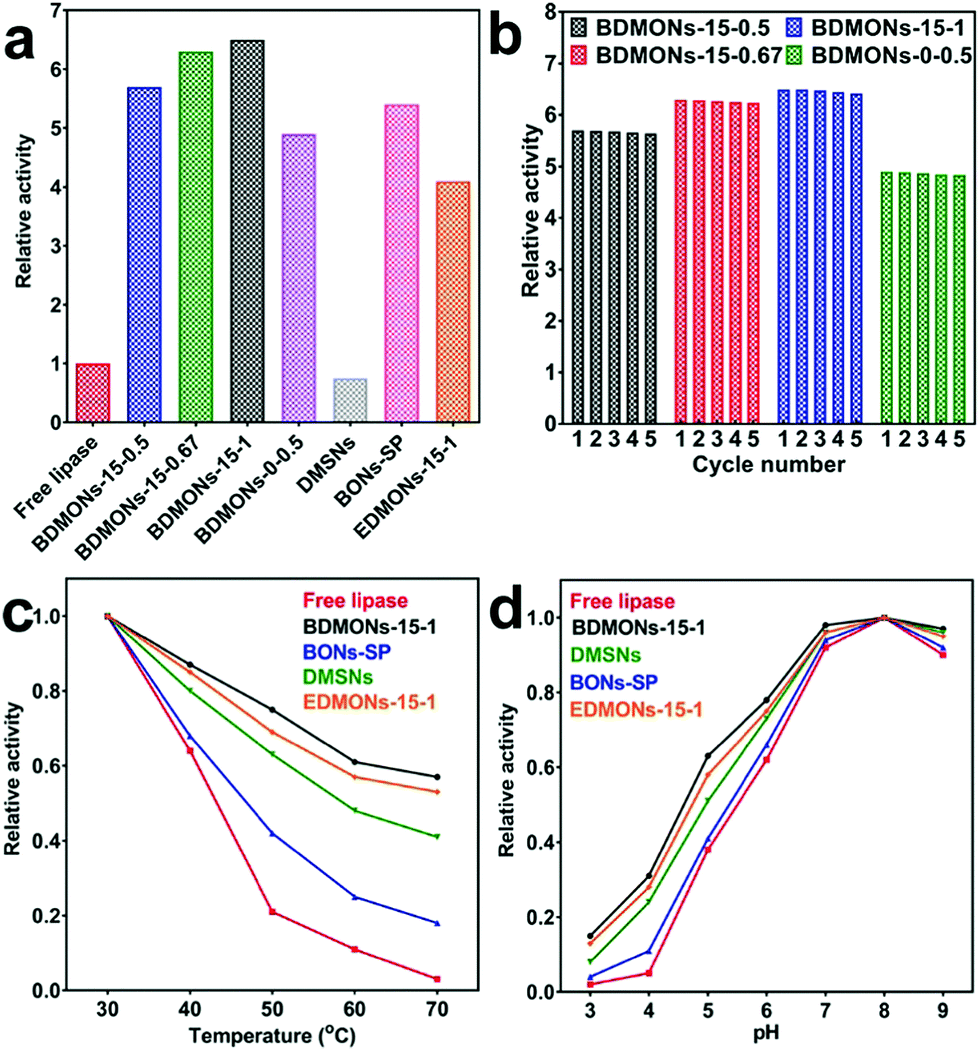 | ||
| Fig. 5 (a) Activity performance of free and immobilized lipases. The relative activity was defined as the ratio of the activity of immobilized lipase to the activity of free enzyme. (b) Reusability performance of immobilized lipases. The relative activity was defined as the ratio of the residual activity to the initial activity. (c) Thermal and (d) pH stability performance of free and immobilized lipases. For thermal stability, the relative activity is defined as the ratio of the residual activity to the initial value measured at 30 °C (the initial activity is defined as 100%). For pH stability, the relative activity is defined as the ratio of the residual activity to the initial value measured at pH 8 (the initial activity is defined as 100%). | ||
Reusability is the key advantage of the immobilized lipase over the free one.49 To evaluate the reusability, the samples were used in five cycles for the hydrolysis of pNPP. As presented in Fig. 5b, all samples show an exceptional reusability performance, retaining 99% of their initial activity after 5 cycles, highlighting the advantage of the covalent attachment method to eliminate the lipase leakage during the reuse process of these lipase immobilized nanocatalysts.
Thermal and pH stabilities are the critical requirement for industrial application of enzymes.44,49Fig. 5c presents the thermal stability performance of free lipase and lipase immobilized on BDMONs-15-1. The activity of free lipase sharply decreases for temperatures over 50 °C, maintaining only 3% of its original activity at 70 °C. On the contrary, immobilized lipase on BDMONs-15-1 shows a significantly improved thermal stability performance under the same conditions, 57% activity retention after incubation at 70 °C, suggesting that lipase molecules fitted within the pores of BDMONs-15-1 are more sheltered against higher temperatures.35 As shown in Fig. 5d, the pH stability curves of the free and immobilized enzymes display a similar trend and the same maximum activity at pH 8.0 (the relative activity was measured to be 6.52 times higher than that of the free enzyme). However, the immobilized lipase shows higher stability towards alkaline or acidic conditions compared to the free lipase (e.g. 0.97 vs. 0.9 at pH 9.0 and 0.15 vs. 0.02 at pH 3.0), contributed by the protection of enzymes immobilized within pores against harsh conditions.35
To understand the reasons for the superior performance of the lipase immobilized on BDMONs-15-1, several control carriers were prepared for comparison. BDMONs-15-1 was subjected to calcination to evaluate the effect of nanoenvironment hydrophobicity on the lipase activity. The calcined sample named as DMSNs possesses a similar structure as BDMONs-15-1 but with pure inorganic silica composition (Fig. S6 (ESI†) and Table 1). With a similar loading capacity of lipase (50 mg g−1), lipase immobilized on DMSNs shows a relative activity of only 0.75 (Fig. 5a and Table 1). The sharp decline of activity compared to that for BDMONs-15-1 is correlated with the conformational change of lipase in a hydrophilic nanoenvironment in which the active site is partially covered by the lid with reduced activity.10 In comparison with BDMONs-15-1, DMSNs demonstrate much weaker thermal and pH stability protection of the lipase (Fig. 5c and d), evidencing the crucial role of the hydrophobic microenvironment for enhancement of the catalytic activity and stability of immobilized lipase molecules under harsh conditions.
To explore the contribution of dendritic large pores on the performance of the immobilized lipase, benzene-bridged organosilica nanoparticles with a small pore of 3.6 nm (Fig. S7 (ESI†) and Table 1) were synthesized without the addition of co-surfactant NaSal while keeping the other synthesis parameters the same as for BDMONs-15-1 (see Experimental section for more details). The final product after surfactant extraction was denoted as BONs-SP. The relative activity is measured to be 5.4 (Fig. 5a and Table 1), lower than that for BDMONs-15-1 with similar carbon content (Table 1), indicating the advantageous effect of immobilization of lipase inside the hydrophobic dendritic large pores over immobilization on the external surface of BONs-SP to induce lid opening.25 As shown in Fig. 5c and d, BONs-SP improves the thermal and pH stability of the immobilized lipase but is not comparable to BDMONs-15-1. These results suggest that a dendritic structure is beneficial for superior protection of the immobilized lipase against harsh conditions. In terms of activity promotion, hydrophobic benzene groups obviously contribute more than a dendritic structure.
To verify the advantages of benzene groups in the silica framework over other organic groups (such as ethane groups), EDMONs-15-1 was prepared simply by substituting BTEB by BTEE (see Experimental section for more details). EDMONs-15-1 also possesses a dendritic structure, spherical morphology, and relatively bigger pore size of 16.4 nm (Fig. S8 (ESI†) and Table 1). The measured bulk and surface carbon contents (10.8 ± 0.1 wt% and 15.0 ± 0.4 wt%, respectively) were lower than those for BDMONs-15-1, as the same molar amount of BTEE contributes less carbon amount than BTEB. The relative activity of lipase immobilized on EDMONs-15-1 is measured to be 4.1 (Fig. 5a), much lower than that for BDMONs-15-1 (see Table 1), suggesting that our intentional choice of BTEB as the organosilica precursor plays a key role to create an exceptional organic content to open the lipase lid wider than BTEE for further improved activity. The thermal and pH stability performance of the EDMONs-15-1 sample is improved compared to the free enzyme but is weaker than that for BDMONs-15-1 (Fig. 5c and d). The above experiments collectively reveal that the superior performance of BDMONs-15-1 in promoting lipase activity and stability arises from its unique features such as dendritic structure, dispersity, and more importantly tailored high hydrophobic content.
Immobilized lipase on BDMONs-15-1 shows the best performance among the samples, and hence was further used to catalyse the transesterification reaction of corn oil. As expected, BDMONs-15-1 exhibits significantly higher conversion of soybean oil to biodiesel (93%, Fig. 6) in comparison to the free enzyme (14%). Fig. 6 also represents the excellent reusability performance of the immobilized lipase in BDMONs-15-1, retaining 94% of its original conversion capability after running 5 cycles, which is attributed to the full recovery of the biocatalysts as well as the chemical attachment of lipase on the particles, inhibiting enzyme leakage during the experiments.50,51
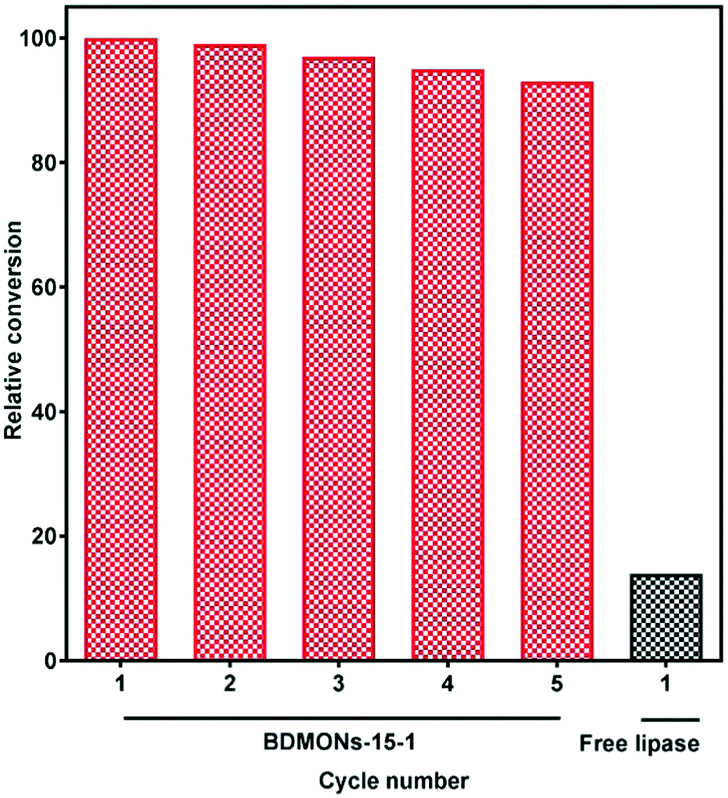 | ||
| Fig. 6 Reusability performance of lipase immobilized on BDMONs-15-1 for biodiesel production. | ||
Conclusions
In summary, an innovative delayed addition method has been developed to fabricate monodispersed BDMONs with enriched benzene groups in the channel wall for lipase immobilization. It is demonstrated that the exceptional hydrophobic content and unique dendritic large pores of BDMONs are essential keys to create immobilized lipase with superior activity, excellent reusability and enhanced thermal/pH stability for biodiesel production. The advanced nanobiocatalysis system obtained from this work is of great promise in biocatalysis applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the Australian Research Council. We also thank Australian National Fabrication Facility-Queensland Node (ANFFQ), and the Australian Microscopy and Microanalysis Research Facility at the Centre for Microscopy and Microanalysis, The University of Queensland for technical assistance.References
- A. F. Lee, J. A. Bennett, J. C. Manayil and K. Wilson, Chem. Soc. Rev., 2014, 43, 7887–7916 RSC.
- M. C. R. Franssen, P. Steunenberg, E. L. Scott, H. Zuilhof and J. P. M. Sanders, Chem. Soc. Rev., 2013, 42, 6491–6533 RSC.
- N. Parvin, Q. Jin, Y. Wei, R. Yu, B. Zheng, L. Huang, Y. Zhang, L. Wang, H. Zhang, M. Gao, H. Zhao, W. Hu, Y. Li and D. Wang, Adv. Mater., 2017, 29, 1606755 CrossRef PubMed.
- C.-L. Hsu, C.-W. Lien, S. G. Harroun, R. Ravindranath, H.-T. Chang, J.-Y. Mao and C.-C. Huang, Mater. Chem. Front., 2017, 1, 893–899 RSC.
- K. Zhu, Y. Wang, J. A. Tang, S. Guo, Z. Gao, Y. Wei, G. Chen and Y. Gao, Mater. Chem. Front., 2017, 1, 958–966 RSC.
- S. K. Brar, P. Singh, M. Bajaj, A. Deep, N. Wangoo and R. K. Sharma, Mater. Chem. Front., 2017, 1, 449–454 RSC.
- Y. Yang, Q. Jin, D. Mao, J. Qi, Y. Wei, R. Yu, A. Li, S. Li, H. Zhao, Y. Ma, L. Wang, W. Hu and D. Wang, Adv. Mater., 2017, 29, 1604795 CrossRef PubMed.
- Y. Cui, X. Bu, H. Zou, X. Xu, D. Zhou, H. Liu, X. Zhang, Y. Liu, H. Sun, J. Jiang and H. Zhang, Mater. Chem. Front., 2017, 1, 387–393 RSC.
- Y. Zhang, J. Ge and Z. Liu, ACS Catal., 2015, 5, 4503–4513 CrossRef.
- Q. Jin, G. Jia, Y. Zhang, Q. Yang and C. Li, Langmuir, 2011, 27, 12016–12024 CrossRef CAS PubMed.
- Z. Zhou, R. N. K. Taylor, S. Kullmann, H. Bao and M. Hartmann, Adv. Mater., 2011, 23, 2627–2632 CrossRef CAS PubMed.
- M. Mathesh, B. Luan, T. O. Akanbi, J. K. Weber, J. Liu, C. J. Barrow, R. Zhou and W. Yang, ACS Catal., 2016, 6, 4760–4768 CrossRef CAS.
- A. Rezaei, O. Akhavan, E. Hashemi and M. Shamsara, Chem. Mater., 2016, 28, 3004–3016 CrossRef CAS.
- S. Sarkar, K. Das and P. K. Das, Langmuir, 2016, 32, 3890–3900 CrossRef CAS PubMed.
- J. C. S. D. Santos, O. Barbosa, C. Ortiz, A. Berenguer-Murcia, R. C. Rodrigues and R. Fernandez-Lafuente, ChemCatChem, 2015, 7, 2413–2432 CrossRef.
- V. Polshettiwar, D. Cha, X. Zhang and J. M. Basset, Angew. Chem., Int. Ed., 2010, 49, 9652–9656 CrossRef CAS PubMed.
- D. Shen, J. Yang, X. Li, L. Zhou, R. Zhang, W. Li, L. Chen, R. Wang, F. Zhang and D. Zhao, Nano Lett., 2014, 14, 923–932 CrossRef CAS PubMed.
- Y.-J. Yu, J.-L. Xing, J.-L. Pang, S.-H. Jiang, K.-F. Lam, T.-Q. Yang, Q.-S. Xue, K. Zhang and P. Wu, ACS Appl. Mater. Interfaces, 2014, 6, 22655–22665 CAS.
- A. Fihri, M. Bouhrara, U. Patil, D. Cha, Y. Saih and V. Polshettiwar, ACS Catal., 2012, 2, 1425–1431 CrossRef CAS.
- V. Polshettiwar, J. Thivolle-Cazat, M. Taoufik, F. Stoffelbach, S. Norsic and J.-M. Basset, Angew. Chem., Int. Ed., 2011, 50, 2747–2751 CrossRef CAS PubMed.
- Z. Chen, C. Zhao, E. Ju, H. Ji, J. Ren, B. P. Binks and X. Qu, Adv. Mater., 2016, 28, 1682–1688 CrossRef CAS PubMed.
- C. Lei, C. Xu, A. Nouwens and C. Yu, J. Mater. Chem. B, 2016, 4, 4975–4979 RSC.
- X. Du and S. Z. Qiao, Small, 2015, 11, 392–413 CrossRef CAS PubMed.
- A. Maity and V. Polshettiwar, ChemSusChem, 2017, 10, 3866–3913 CrossRef CAS PubMed.
- M. Kalantari, M. Yu, Y. Yang, E. Strounina, Z. Gu, X. Huang, J. Zhang, H. Song and C. Yu, Nano Res., 2017, 10, 605–617 CrossRef CAS.
- N. García, E. Benito, J. Guzmán and P. Tiemblo, J. Am. Chem. Soc., 2007, 129, 5052–5060 CrossRef PubMed.
- Y. Chen and J. Shi, Adv. Mater., 2016, 28, 3235–3272 CrossRef CAS PubMed.
- M. Wu, Q. Meng, Y. Chen, Y. Du, L. Zhang, Y. Li, L. Zhang and J. Shi, Adv. Mater., 2015, 27, 215–222 CrossRef CAS PubMed.
- M. Wu, Q. Meng, Y. Chen, L. Zhang, M. Li, X. Cai, Y. Li, P. Yu, L. Zhang and J. Shi, Adv. Mater., 2016, 28, 1963–1969 CrossRef CAS PubMed.
- Y. Chen, P. Xu, H. Chen, Y. Li, W. Bu, Z. Shu, Y. Li, J. Zhang, L. Zhang, L. Pan, X. Cui, Z. Hua, J. Wang, L. Zhang and J. Shi, Adv. Mater., 2013, 25, 3100–3105 CrossRef CAS PubMed.
- Y. Chen, Q. Meng, M. Wu, S. Wang, P. Xu, H. Chen, Y. Li, L. Zhang, L. Wang and J. Shi, J. Am. Chem. Soc., 2014, 136, 16326–16334 CrossRef CAS PubMed.
- K. Zhu, M. Wu, H. Lai, C. Guo, J. Li, Y. Wang, Y. Chen, C. Wang and J. Shi, Biomaterials, 2016, 74, 188–199 CrossRef CAS PubMed.
- Y. Yang, Y. Niu, J. Zhang, A. K. Meka, H. Zhang, C. Xu, C. X. C. Lin, M. Yu and C. Yu, Small, 2015, 11, 2743–2749 CrossRef CAS PubMed.
- H. Wang, M. Tang, L. Han, J. Cao, Z. Zhang, W. Huang, R. Chen and C. Yu, J. Mater. Chem. A, 2014, 2, 19298–19307 CAS.
- M. Kalantari, M. Kazemeini, F. Tabandeh and A. Arpanaei, J. Mater. Chem., 2012, 22, 8385–8393 RSC.
- V. R. R. Marthala, M. Friedrich, Z. Zhou, M. Distaso, S. Reuss, S. A. Al-Thabaiti, W. Peukert, W. Schwieger and M. Hartmann, Adv. Funct. Mater., 2015, 25, 1832–1836 CrossRef CAS.
- X. Wang, P. Dou, P. Zhao, C. Zhao, Y. Ding and P. Xu, ChemSusChem, 2009, 2, 947–950 CrossRef CAS PubMed.
- Y. Yang, S. Bernardi, H. Song, J. Zhang, M. Yu, J. C. Reid, E. Strounina, D. J. Searles and C. Yu, Chem. Mater., 2016, 28, 704–707 CrossRef CAS.
- Y. Yang, J. Wan, Y. Niu, Z. Gu, J. Zhang, M. Yu and C. Yu, Chem. Mater., 2016, 28, 9008–9016 CrossRef CAS.
- Y. Yang, Y. Lu, P. L. Abbaraju, J. Zhang, M. Zhang, G. Xiang and C. Yu, Angew. Chem., Int. Ed., 2017, 56, 8446–8450 CrossRef CAS PubMed.
- C. Xu, M. Yu, O. Noonan, J. Zhang, H. Song, H. Zhang, C. Lei, Y. Niu, X. Huang, Y. Yang and C. Yu, Small, 2015, 11, 5949–5955 CrossRef CAS PubMed.
- H. Zou, R. Wang, X. Li, X. Wang, S. Zeng, S. Ding, L. Li, Z. Zhang and S. Qiu, J. Mater. Chem. A, 2014, 2, 12403–12412 CAS.
- K. S. Rao, K. El-Hami, T. Kodaki, K. Matsushige and K. Makino, J. Colloid Interface Sci., 2005, 289, 125–131 CrossRef CAS PubMed.
- P. Adlercreutz, Chem. Soc. Rev., 2013, 42, 6406–6436 RSC.
- J. Zhang, S. Karmakar, M. Yu, N. Mitter, J. Zou and C. Yu, Small, 2014, 10, 5068–5076 CAS.
- J. Croissant, X. Cattoën, M. Wong Chi Man, P. Dieudonné, C. Charnay, L. Raehm and J.-O. Durand, Adv. Mater., 2015, 27, 145–149 CrossRef CAS PubMed.
- P. Esmaeilnejad-Ahranjani, M. Kazemeini, G. Singh and A. Arpanaei, Langmuir, 2016, 32, 3242–3252 CrossRef CAS PubMed.
- J. Cui, Y. Zhao, R. Liu, C. Zhong and S. Jia, Sci. Rep., 2016, 6, 27928 CrossRef CAS PubMed.
- A. Liese and L. Hilterhaus, Chem. Soc. Rev., 2013, 42, 6236–6249 RSC.
- A. K. Vahidi, Y. Yang, T. P. N. Ngo and Z. Li, ACS Catal., 2015, 5, 3157–3161 CrossRef CAS.
- M. Kalantari, M. Kazemeini and A. Arpanaei, Biochem. Eng. J., 2013, 79, 267–273 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Further characterization of the materials. See DOI: 10.1039/c8qm00078f |
| This journal is © the Partner Organisations 2018 |