
On-surface synthesis of graphene clusters from a Z-bar-linkage precursor with quaterphenyl branches†
Zhen
Xu
 ,
Takahiro
Kojima
,
Wanyu
Wang
,
Kabya
Kaushik
,
Alex
Saliniemi
,
Takahiro
Nakae
and
Hiroshi
Sakaguchi
*
,
Takahiro
Kojima
,
Wanyu
Wang
,
Kabya
Kaushik
,
Alex
Saliniemi
,
Takahiro
Nakae
and
Hiroshi
Sakaguchi
*
Institute of Advanced Energy, Kyoto University, Uji, Kyoto 611-0011, Japan. E-mail: sakaguchi@iae.kyoto-u.ac.jp
First published on 24th January 2018
Abstract
The products of two-zone chemical vapor deposition on Au(111) from a Z-bar-linkage precursor with quaterphenyl-branches were investigated by scanning tunneling microscopy, Raman spectroscopy, and theoretical calculations. Annealing at 400 °C produced linear arrays of intramolecular-dehydrogenated precursors, and temperatures over 450 °C led to intermolecular dehydrogenation and to the formation of graphene clusters. This reaction pathway contrasts remarkably with the previous results reported for a Z-bar-linkage precursor with terphenyl branches, where homochiral polymerization proceeded and allowed the formation of acene-type graphene nanoribbons. The reason might originate from the conformation of the biradical form of the quaterphenyl-branched precursor, produced by debromination on Au(111) during 250 °C annealing. The quaterphenyl-branched precursor might favor a symmetric conformation with both the radicals pointing toward the Au(111) surface, whereas our previous results showed that the terphenyl-branched precursor might favor asymmetric (chiral) conformations, with one radical pointing toward and the other one away from the Au(111) surface. Steric hindrance of the symmetric conformations of the quaterphenyl-branched precursor is presumed to prohibit polymerization due to the strong affinity of the biradical to Au(111). Such symmetric precursor conformations lead to fused debrominated precursors via intramolecular dehydrogenation, and finally result in the conversion to graphene clusters via on-surface intermolecular fusion.
Introduction
Graphene nanoribbons (GNRs), narrow strips of graphene, are made of an sp2-hybridized carbon atom backbone and can display various edge structures.1 GNRs show adjustable electronic and optical properties which can be tuned by tailoring the width2–5 and edge structures,6–10 or by doping with heteroatoms.11–15 These properties make GNRs promising as functional components in electronic and optoelectronic devices.16–19 As a result, bottom-up fabrication of GNRs has focused on achieving precise control over the width, edge structures, and chemical composition. In this respect, metal-surface-assisted synthesis has proven to be an effective approach to fabricate atomically well-defined GNRs.20–26Although extensive studies have been carried out on the surface-assisted synthesis of GNRs, steering the on-surface reactions toward desired products is still a subject that needs to be addressed. The reaction pathways and underlying mechanisms for the growth of GNRs rely on the combination of the selected precursor and substrate.27–33 However, the geometric effects of the precursor on the GNR structure are not well understood. Recently, we reported a novel conformation-controlled catalysis involved in the growth of acene-type GNRs on Au(111) via two-zone chemical vapor deposition (2Z-CVD).24 The biradical form of the terphenyl-branched ‘Z-bar-linkage’ precursor adopts a chiral conformation with height asymmetry when deposited onto the Au(111) surface. The chiral conformations of the precursors form homochiral polymers that can be converted into densely packed acene-type GNRs via efficient dehydrogenation reactions.24 However, the effects of the branch length of the Z-bar-linkage precursor on the conformations on Au(111) have remained unknown.
Here, the length of the Z-bar-linkage precursor branch is changed by replacing the terphenyl with quaterphenyl (4′′′,5′′′′-dibromo-1,1′:4′,1′′:4′′,1′′′:2′′′,1′′′′:2′′′′,1′′′′′:4′′′′′,1′′′′′′:4′′′′′′,1′′′′′′′-octiphenyl, DBOP) in order to examine the adsorption conformations on Au(111), which influence the final products of the on-surface synthesis using 2Z-CVD. We found that the quaterphenyl-branched precursors favor height-symmetric conformations on Au(111), preventing polymerization and leading to intramolecular-fused debrominated precursors. Subsequent annealing led to the formation of graphene clusters via intermolecular dehydrogenation instead of acene-type GNRs.
Results and discussion
Structural studies on the products of 2Z-CVD
The previously developed 2Z-CVD was employed for the on-surface synthesis.22 As depicted in Fig. 1, the system was evacuated using a rotary pump under argon (Ar) flow. 2Z-CVD is advantageous because it permits independent temperature control of two separate zones 1 and 2, and does not require ultra-high vacuum conditions. The generation of a high density of radicals has been hypothesized to occur in zone 1 by the collision of precursors with the hot wall. The polymerization of the precursor biradicals supplied to the Au(111) substrate has been assumed to occur in zone 2, and when annealed at higher temperature affords GNRs via dehydrogenation. This technique has successfully been used to fabricate a series of armchair-edged GNRs in high yield.22 In this study, the DBOP precursor was synthesized. DBOP was sublimated at 350 °C so that it could be supplied to the Au(111) substrate in zone 2, which was held at 250 °C, by passing through zone 1 (350 °C). The sample was subsequently annealed stepwise to higher temperatures of 400, 450, and 500 °C.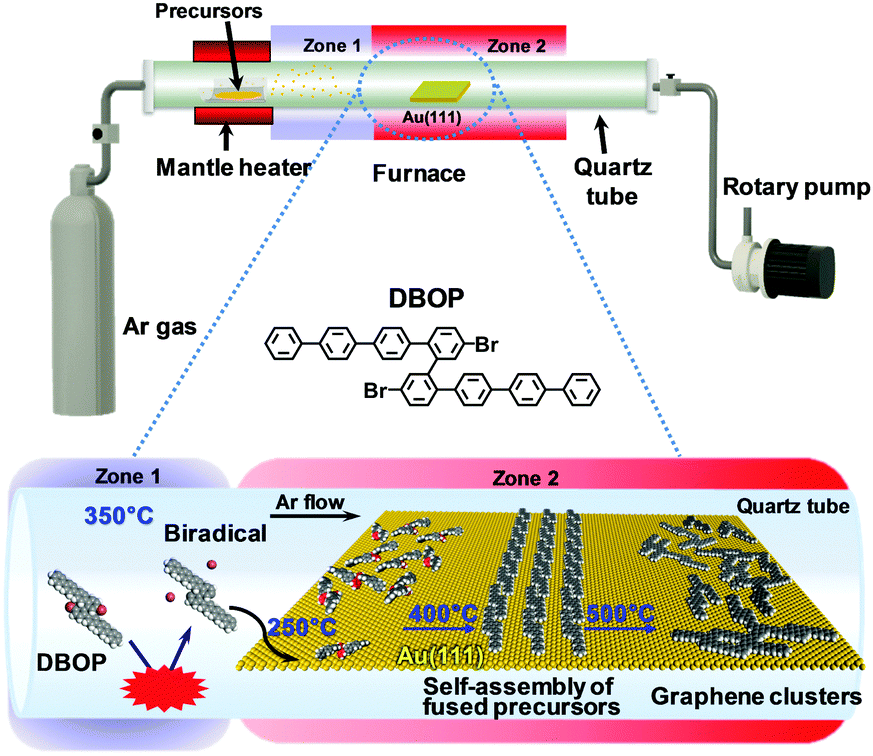 | ||
| Fig. 1 Schematic illustration depicting the growth of graphene clusters from DBOP by 2Z-CVD. The red benzene rings of the precursors in the space-filling models point toward the Au(111) surface. | ||
STM characterization of the samples was performed at room temperature under an Ar atmosphere. As shown in Fig. S1a (ESI†), disordered structures were observed at 250 °C. Upon annealing at 400 °C, well-ordered linear arrays of ‘S-shaped’ units were formed (Fig. 2a and Fig. S1b in the ESI†). Cross-sectional analysis of the individual S-shaped units revealed a central width of 1.11 nm and an arm length of 1.97 nm, which are in good agreement with the model (1.10 nm and 1.93 nm, respectively), assuming an intramolecular-fused, debrominated precursor molecule (Fig. 2b). The periodic distance between two adjacent quaterphenyl branches along the chain direction is 0.24 nm (Fig. S2 in the ESI†), smaller than the theoretically predicted spacing (0.34 nm) with C–Au bond formation (Fig. S2 in the ESI†), assuming a C–Au bond length of 0.24 nm.34 This result indicates no C–Au bond formation between debrominated DBOP molecules. This result also suggests that intramolecular dehydrogenation took place and flattened the conformation to an S-shape (Fig. 2a and Fig. S1b in the ESI†). The S-shaped molecules were self-assembled to form linear array structures. Further annealing the sample at 450 °C led to branched chain structures, suggesting that intermolecular dehydrogenation occurred with multiple cross-coupling modes between individual S-shaped molecules (Fig. 2c and Fig. S1c in the ESI†). The above reactions, intramolecular fusion and the following intermolecular C–C bond formation, provide a promising way to fabricate new low-dimensional carbon architectures without using halogen atoms by activating C–H bonds. Similar intermolecular C–C coupling was also observed for the quaterphenyl molecule on Cu(110) by activating meta C–H bonds of the terminal benzene.35,36 Further annealing at 500 °C enhanced the interchain-connected carbon nanostructures (Fig. 2d and Fig. S1d in the ESI†). Raman spectra of 2Z-CVD-grown samples measured at different temperatures show two significant points (Fig. 3). Firstly, the sample at 250 °C did not show a detectable Raman signature probably due to the small Raman scattering cross-section for debrominated DBOP presursors of polyphenylene structures.24 As the temperature was increased from 250 to 400 °C, a peak at 1594 cm−1 (G band) appeared. This could be due to the π-conjugation effects from intramolecular dehydrogenation and is in line with the STM results. Secondly, when increasing the temperature from 400 to 450 °C, the intensity of the D bands at 1333 cm−1 and 1259 cm−1 was remarkably enhanced. A further increase in the temperature to 500 °C showed a similar Raman signature as the 450 °C annealed sample.
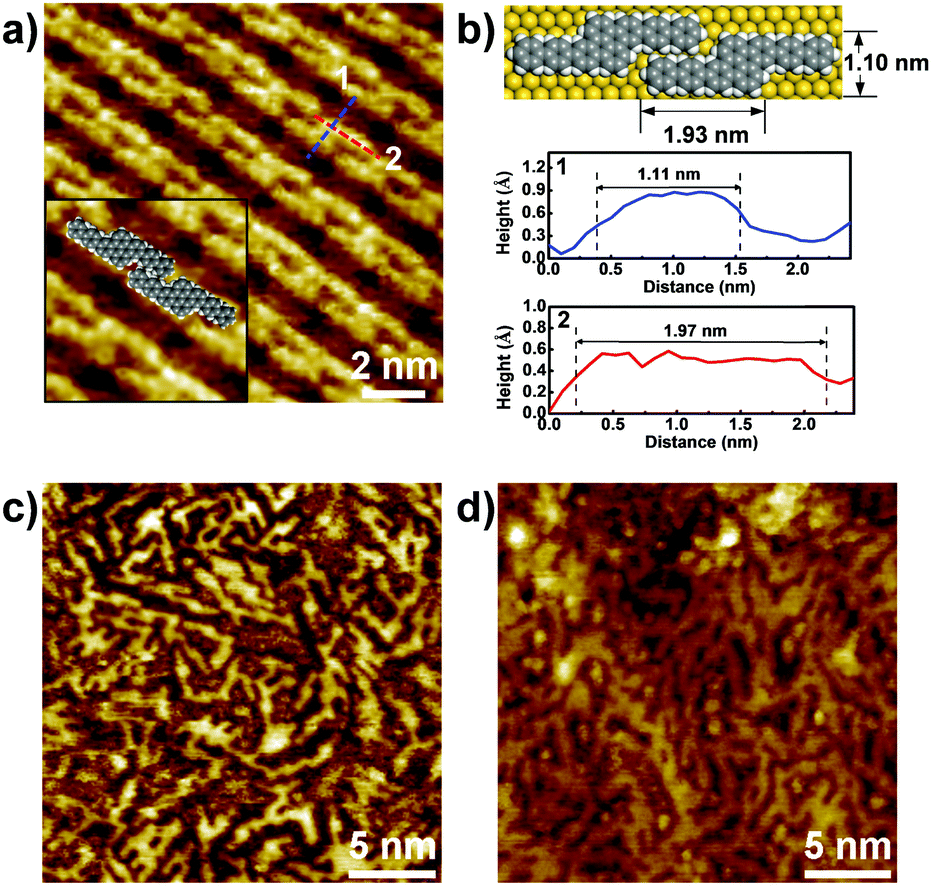 | ||
| Fig. 2 (a, c and d) STM images of samples annealed at 400 °C (the inset in (a) shows a magnified image with an overlaid model), 450 °C (c) and 500 °C (d). All samples were first grown by 2Z-CVD with a supply of 20 μg of DBOP precursor. (b) Space-filling model and cross-sectional analysis along the dotted lines in (a). | ||
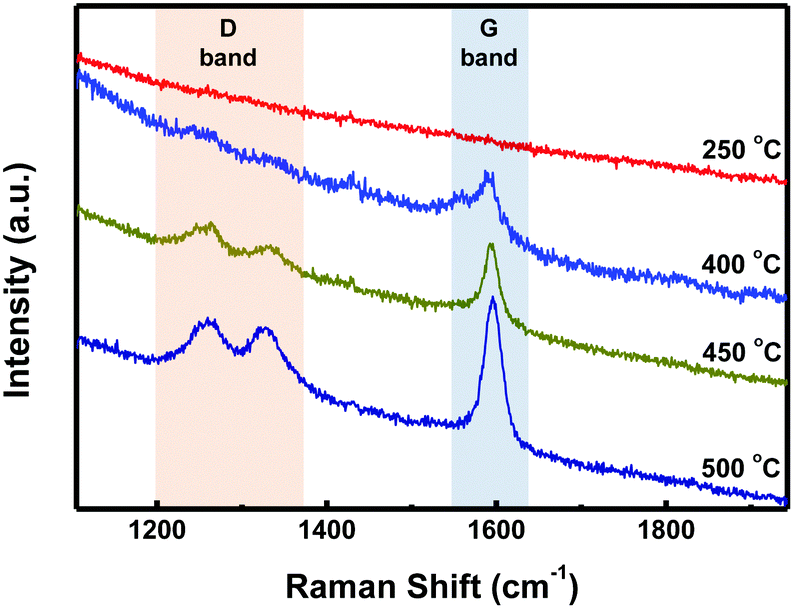 | ||
| Fig. 3 Raman spectra of 2Z-CVD-grown samples from DBOP annealed at different temperatures. | ||
Adsorption conformations of the precursor
In order to unravel the underlying mechanisms of the inhibited polymerization, conformations of the DBOP biradicals on Au(111) were computed by the quantum mechanics/molecular mechanics (QM/MM) approach according to our previous publication.24 Initial geometries of the DBOP biradicals on Au(111) were generated by the modification of previous results of optimized biradical geometries of 4′′,5′′′-dibromo-1,1′:4′,1′′:2′′,1′′′:2′′′,1′′′′:4′′′′,1′′′′′-sexiphenyl (DBSP) at the QM/MM level.24 The predicted conformations are parameterized by surface chirality and height asymmetry, as shown in Fig. 4. Here, surface chirality indicates the right (R)- or left (S)-handedness of the enantiomers for the molecules on a 2D surface, and height asymmetry indicates the relative height from Au(111) surface of the two radical-carbons in the centered biphenyl core. We considered height-symmetrical conformations (denoted as ‘dd’ for ‘down-down’ and ‘uu’ for ‘up-up’) which represent the two radical-carbons directed either toward or away from the surface. A height-asymmetrical conformation (denoted as ‘ud’ for ‘up-down’) represents two radical-carbons positioned at different heights from the surface. Because the R and S conformers should show the same energy, we focus only on the R conformer in the calculation process. The calculation results (Fig. 4 and Table S1 in the ESI†) indicate the height-symmetrical Rdd conformer to have the lowest formation energy in the singlet state. Its formation energy is 35.31 kcal mol−1 lower than that of the height-asymmetrical Rud conformer, and 172.70 kcal mol−1 lower than that of the Ruu conformer. Thereby, the Rdd conformer was considered to be the dominant species as it is stabilized by such a remarkable energy difference. Contrasting greatly with DBOP, the previously reported DBSP showed the most energetically stable conformer to be Rud.24 This is because Rud of the DBSP showed a linear structure of two terphenyl branches, whereas Rdd showed a remarkably distorted ‘double-arch’ structure of two terphenyl branches. In the present case, the radical–Au(111) interaction and the resulting molecular distortion are presumed to play a key role in the determination of the total energy. The two radical-carbons in the DBOP's Rdd conformer strongly bind to Au(111). The longer branch-length of the quaterphenyl in DBOP might alleviate the torsion in the branches, while strong torsion takes place in the shorter branch of the terphenyl in DBSP. The strong radical–Au(111) interaction of DBOP, with decreased torsion of the branches in Rdd, might be the reason for the lowest formation energy. Thus, the conformation differences between DBOP and DBSP on Au(111) might originate from a balance between the radical–Au(111) interactions and the molecular torsion.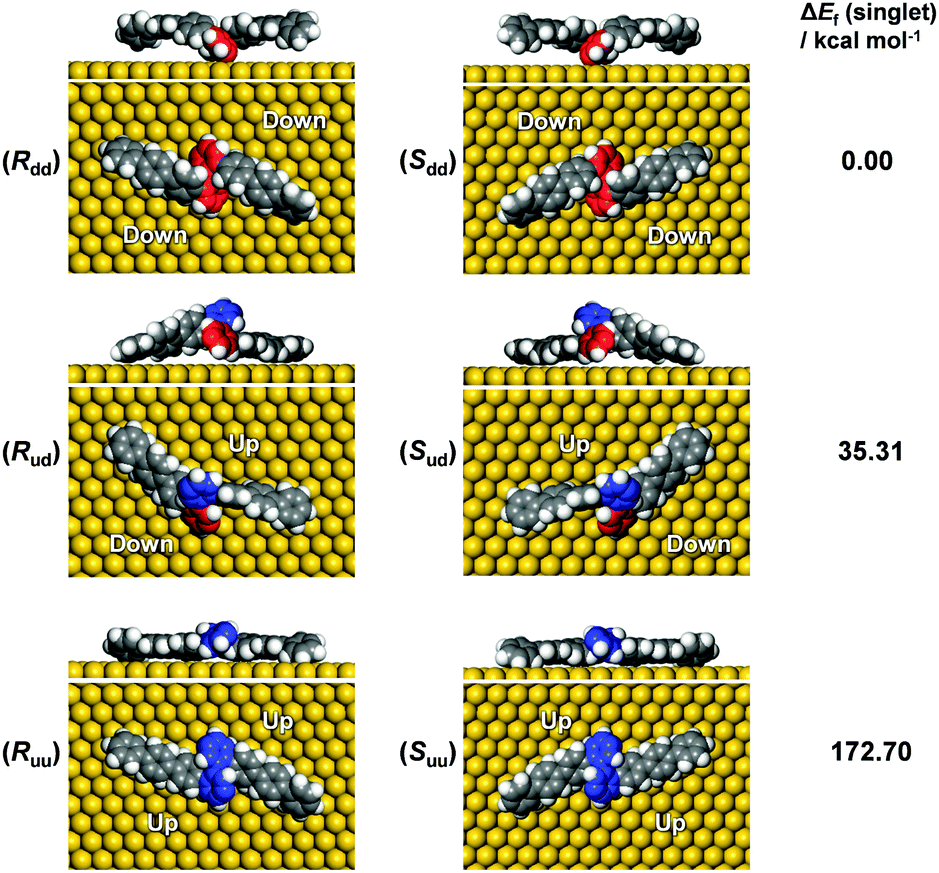 | ||
| Fig. 4 Optimized conformations of biradical forms of the precursor DBOP on Au(111) with the corresponding formation energies (ΔEf) in the singlet state by the QM/MM approach. Colors of the benzene indicate the relative heights from the Au(111) surface; red, pointing toward the surface; blue, away from the surface. | ||
As outlined above, altering the arm length of the Z-bar linkage precursor resulted in a completely different reaction pathway compared with that of DBSP.24 Thus, a precursor biradical adsorption conformation dependent reaction pathway was concluded, and is shown in Scheme 1. In the case of DBSP, the precursor biradicals adopt favorable chiral Rud and Sud conformations with height asymmetry on Au(111), and form homochiral polymers. Annealing the polymers at higher temperatures leads to stepwise dehydrogenation, and finally, acene-GNRs are formed at 500 °C via complete dehydrogenation. However, DBOP displays a vastly different pathway than DBSP, as the DBOP biradicals favor height-symmetric chiral Rdd and Sdd conformations on Au(111). Neither of these two kinds of conformers (Fig. 4) can polymerize due to strong radical–Au(111) interactions, which results in steric hindrance (Fig. S3 in the ESI†) during the lifetime of the radicals. Annealing the sample at 400 °C leads to self-assembled arrays of intramolecular-fused debrominated precursors. Further annealing at 450 and 500 °C results in intermolecular connected graphene clusters.
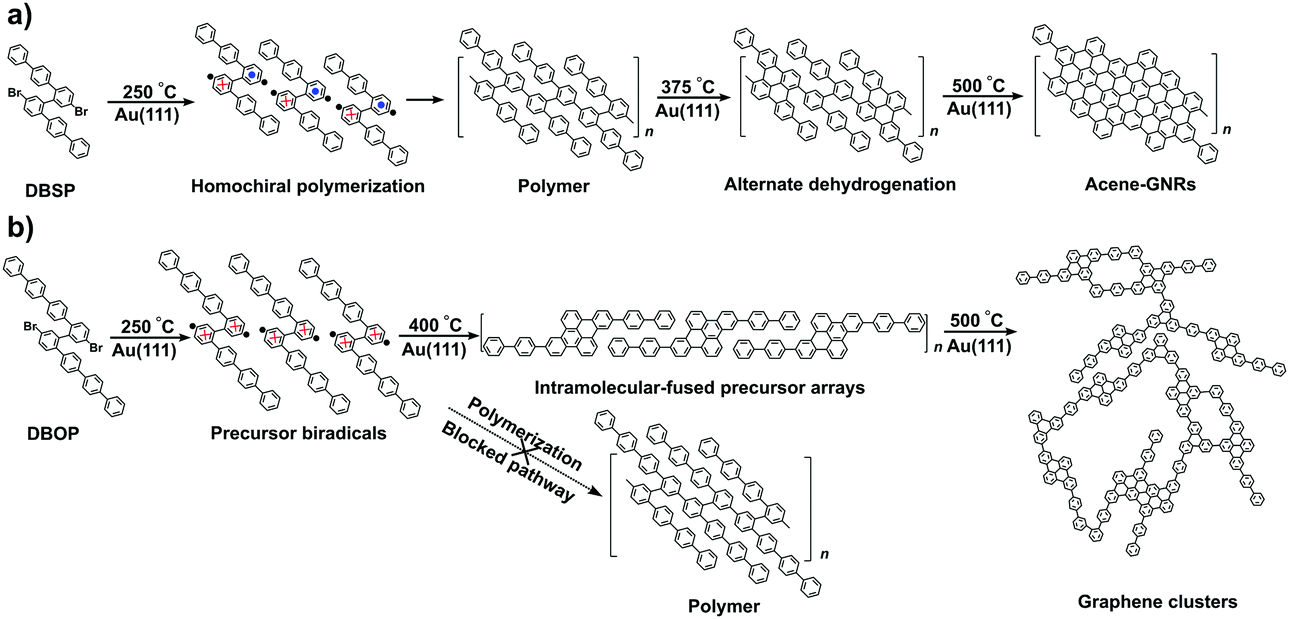 | ||
| Scheme 1 Comparison of two different reaction pathways of Z-bar-linkage precursors DBSP (a) and DBOP (b) via 2Z-CVD. Blue dots (red cross) in the chemical structures indicate the benzene rings pointing away from the Au(111) surface (toward the Au(111) surface). The black dots represent the radicals. | ||
Conclusions
In this study, the products formed on Au(111) from the Z-bar-linkage precursor DBOP via 2Z-CVD were examined. The results were compared with an analogue precursor DBSP under similar CVD conditions. The previously reported DBSP showed the formation of homochiral polymers at 250 °C, which converted into densely packed acene-type GNRs at 500 °C through stepwise dehydrogenation. In great contrast with DBSP, DBOP shows a different reaction pathway. The polymerization of the DBOP biradicals is hindered, subsequently leading to self-assembled arrays of intramolecular-fused debrominated precursors at 400 °C, and forming graphene clusters at 450 and 500 °C. Theoretical calculations revealed that the DBOP biradicals favored the Rdd and Sdd conformations with height symmetry on Au(111). The inhibited polymerization of DBOP biradicals might result from the steric hindrance caused by strong radical–Au(111) interactions. This conformation effect can be especially useful for the rational design of precursors for surface-assisted synthesis of carbon-based materials.Experimental
Two-zone chemical vapor deposition (2Z-CVD)
The 2Z-CVD system is composed of a quartz tube (ϕ 26 mm, length 86 cm) as the reactor, a rotary pump which can evacuate the system to below 7 × 10−4 Torr, a two-zone electric furnace with temperature controllers, an Ar gas flow system with a mass-flow controller, and a mantle heater for precursor sublimation. Ar gas was fed into the quartz tube at a flow rate of 500 sccm, resulting in a vacuum of 1 Torr. Before the CVD process, the quartz tube was annealed at 1000 °C for 20 min to remove impurities that can deactivate precursor radicals. The precursor (20 μg, prepared by casting 200 μL of precursor solution, 0.1 mg mL−1 in toluene) in a quartz boat was placed in the quartz tube before zone 1. An Au(111)-deposited mica substrate was placed in zone 2 of the quartz tube. The CVD process was conducted in two sequential steps: firstly, zone 1 (the path of the precursors) and zone 2 were heated to 350 and 250 °C, respectively, and the precursors were sublimated using the mantle heater by heating to 350 °C for 15 min. Finally, to anneal the sample, the temperature of zone 2 was increased stepwise to the final temperature.STM measurements
STM measurements were performed with a constant current mode using a commercial instrument (PicoSPM; Keysight Technologies Inc., formerly Molecular Imaging) under an Ar atmosphere at room temperature. All STM images were acquired at a tip bias of 0.2 V with a constant current of 5–20 pA. An electrochemically etched Pt–Ir (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20) wire was used as the STM tip.
:20) wire was used as the STM tip.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by JSPS KAKENHI Grant Numbers JP17H05154, JP17H02724, JP16H00967, JP16K13608, JP16K17893, and JP17K14471 and the “Zero-Emission Energy Research” of IAE, Kyoto University. The Super Computer System, Institute for Chemical Research, Kyoto University, was used for the calculations. Z. X. acknowledges the scholarship of the China Scholarship Council, affiliated with the Ministry of Education of the P. R. China.Notes and references
- L. Chen, Y. Hernandez, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2012, 51, 7640–7654 CrossRef CAS PubMed.
- M. Ezawa, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 045432 CrossRef.
- Y.-W. Son, M. L. Cohen and S. G. Louie, Phys. Rev. Lett., 2006, 97, 216803 CrossRef PubMed.
- H. Huang, D. Wei, J. Sun, S. L. Wong, Y. P. Feng, A. H. C. Neto and A. T. S. Wee, Sci. Rep., 2012, 2, 983 CrossRef PubMed.
- D. Prezzi, D. Varsano, A. Ruini, A. Marini and E. Molinari, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 041404 CrossRef.
- S. Okada, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 041408 CrossRef.
- T. Wassmann, A. P. Seitsonen, A. M. Saitta, M. Lazzeri and F. Mauri, Phys. Rev. Lett., 2008, 101, 096402 CrossRef PubMed.
- P. Wagner, C. P. Ewels, J.-J. Adjizian, L. Magaud, P. Pochet, S. Roche, A. Lopez-Bezanilla, V. V. Ivanovskaya, A. Yaya, M. Rayson, P. Briddon and B. Humbert, J. Phys. Chem. C, 2013, 117, 26790–26796 CAS.
- G. D. Nguyen, H.-Z. Tsai, A. A. Omrani, T. Marangoni, M. Wu, D. J. Rizzo, G. F. Rodgers, R. R. Cloke, R. A. Durr, Y. Sakai, F. Liou, A. S. Aikawa, J. R. Chelikowsky, S. G. Louie, F. R. Fischer and M. F. Crommie, Nat. Nanotechnol., 2017, 12, 1077–1082 CrossRef CAS PubMed.
- E. Carbonell-Sanromà, J. Hieulle, M. Vilas-Varela, P. Brandimarte, M. Iraola, A. Barragán, J. Li, M. Abadia, M. Corso, D. Sánchez-Portal, D. Peña and J. I. Pascual, ACS Nano, 2017, 11, 7355–7361 CrossRef PubMed.
- C. Bronner, S. Stremlau, M. Gille, F. Brauße, A. Haase, S. Hecht and P. Tegeder, Angew. Chem., Int. Ed., 2013, 52, 4422–4425 CrossRef CAS PubMed.
- R. R. Cloke, T. Marangoni, G. D. Nguyen, T. Joshi, D. J. Rizzo, C. Bronner, T. Cao, S. G. Louie, M. F. Crommie and F. R. Fischer, J. Am. Chem. Soc., 2015, 137, 8872–8875 CrossRef CAS PubMed.
- S. Kawai, S. Saito, S. Osumi, S. Yamaguchi, A. S. Foster, P. Spijker and E. Meyer, Nat. Commun., 2015, 6, 8098 CrossRef CAS PubMed.
- J. Cai, C. A. Pignedoli, L. Talirz, P. Ruffieux, H. Söde, L. Liang, V. Meunier, R. Berger, R. Li, X. Feng, K. Müllen and R. Fasel, Nat. Nanotechnol., 2014, 9, 896–900 CrossRef CAS PubMed.
- G. D. Nguyen, F. M. Toma, T. Cao, Z. Pedramrazi, C. Chen, D. J. Rizzo, T. Joshi, C. Bronner, Y.-C. Chen, M. Favaro, S. G. Louie, F. R. Fischer and M. F. Crommie, J. Phys. Chem. C, 2016, 120, 2684–2687 CAS.
- Z. Chen, H. I. Wang, J. Teyssandier, K. S. Mali, T. Dumslaff, I. Ivanov, W. Zhang, P. Ruffieux, R. Fasel, H. J. Räder, D. Turchinovich, S. De Feyter, X. Feng, M. Kläui, A. Narita, M. Bonn and K. Müllen, J. Am. Chem. Soc., 2017, 139, 3635–3638 CrossRef CAS PubMed.
- J. P. Llinas, A. Fairbrother, G. B. Barin, W. Shi, K. Lee, S. Wu, B. Y. Choi, R. Braganza, J. Lear, N. Kau, W. Choi, C. Chen, Z. Pedramrazi, T. Dumslaff, A. Narita, X. Feng, K. Müllen, F. Fischer, A. Zettl, P. Ruffieux, E. Yablonovitch, M. Crommie, R. Fasel and J. Bokor, Nat. Commun., 2017, 8, 633 CrossRef PubMed.
- T. J. Sisto, Y. Zhong, B. Zhang, M. T. Trinh, K. Miyata, X. Zhong, X.-Y. Zhu, M. L. Steigerwald, F. Ng and C. Nuckolls, J. Am. Chem. Soc., 2017, 139, 5648–5651 CrossRef PubMed.
- Z. Chen, W. Zhang, C. A. Palma, A. Lodi Rizzini, B. Liu, A. Abbas, N. Richter, L. Martini, X. Y. Wang, N. Cavani, H. Lu, N. Mishra, C. Coletti, R. Berger, F. Klappenberger, M. Kläui, A. Candini, M. Affronte, C. Zhou, V. De Renzi, U. del Pennino, J. V. Barth, H. J. Räder, A. Narita, X. Feng and K. Müllen, J. Am. Chem. Soc., 2016, 138, 15488–15496 CrossRef CAS PubMed.
- L. Talirz, P. Ruffieux and R. Fasel, Adv. Mater., 2016, 28, 6222–6231 CrossRef CAS PubMed.
- Y. Segawa, H. Ito and K. Itami, Nat. Rev. Mater., 2016, 1, 15002 CrossRef CAS.
- H. Sakaguchi, Y. Kawagoe, Y. Hirano, T. Iruka, M. Yano and T. Nakae, Adv. Mater., 2014, 26, 4134–4138 CrossRef CAS PubMed.
- T. Kojima, Y. Bao, C. Zhang, S. Liu, H. Xu, T. Nakae, K. P. Loh and H. Sakaguchi, Langmuir, 2017, 33, 10439–10445 CrossRef CAS PubMed.
- H. Sakaguchi, S. Song, T. Kojima and T. Nakae, Nat. Chem., 2017, 9, 57–63 CAS.
- S. Song, G. Huang, T. Kojima, T. Nakae, H. Uno and H. Sakaguchi, Chem. Lett., 2017, 46, 1525–1527 CrossRef CAS.
- S. Song, T. Kojima, T. Nakae and H. Sakaguchi, Chem. Commun., 2017, 53, 7034–7036 RSC.
- J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. Feng, K. Müllen and R. Fasel, Nature, 2010, 466, 470–473 CrossRef CAS PubMed.
- P. Han, K. Akagi, F. Federici Canova, H. Mutoh, S. Shiraki, K. Iwaya, P. S. Weiss, N. Asao and T. Hitosugi, ACS Nano, 2014, 8, 9181–9187 CrossRef CAS PubMed.
- P. Han, K. Akagi, F. Federici Canova, R. Shimizu, H. Oguchi, S. Shiraki, P. S. Weiss, N. Asao and T. Hitosugi, ACS Nano, 2015, 9, 12035–12044 CrossRef CAS PubMed.
- D. G. de Oteyza, A. Garcia-Lekue, M. Vilas-Varela, N. Merino-Díez, E. Carbonell-Sanromà, M. Corso, G. Vasseur, C. Rogero, E. Guitián, J. I. Pascual, J. E. Ortega, Y. Wakayama and D. Peña, ACS Nano, 2016, 10, 9000–9008 CrossRef CAS PubMed.
- P. H. Jacobse, A. van den Hoogenband, M. E. Moret, R. J. Klein Gebbink and I. Swart, Angew. Chem., Int. Ed., 2016, 128, 13052–13055 CrossRef PubMed.
- F. Schulz, P. H. Jacobse, F. Federici Canova, J. van der Lit, D. Z. Gao, A. van den Hoogenband, P. Han, R. J. M. Klein Gebbink, M.-E. Moret, P. M. Joensuu, I. Swart and P. Liljeroth, J. Phys. Chem. C, 2017, 121, 2896–2904 CAS.
- K. A. Simonov, N. A. Vinogradov, A. S. Vinogradov, A. V. Generalov, E. M. Zagrebina, G. I. Svirskiy, A. A. Cafolla, T. Carpy, J. P. Cunniffe, T. Taketsugu, A. Lyalin, N. Mårtensso and A. B. Preobrajenski, ACS Nano, 2015, 9, 8997–9011 CrossRef CAS PubMed.
- J. I. Urgel, H. Hayashi, M. Di Giovannantonio, C. A. Pignedoli, S. Mishra, O. Deniz, M. Yamashita, T. Dienel, P. Ruffieux and H. Yamada, J. Am. Chem. Soc., 2017, 139, 11658–11661 CrossRef CAS PubMed.
- Q. Sun, C. Zhang, H. Kong, Q. Tan and W. Xu, Chem. Commun., 2014, 50, 11825–11828 RSC.
- C. Zhang, Q. Sun, H. Chen, Q. Tan and W. Xu, Chem. Commun., 2015, 51, 495–498 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qm00577f |
| This journal is © the Partner Organisations 2018 |