DOI:
10.1039/C7QM00492C
(Research Article)
Mater. Chem. Front., 2018,
2, 741-751
Effective removal of organic pollutants using GeO2/TiO2 nanoparticle composites under direct sunlight
Received
24th October 2017
, Accepted 19th January 2018
First published on 5th February 2018
Abstract
TiO2 and GeO2/TiO2 nanoparticle composites were synthesized from titanium tetraisopropoxide, germanium oxide, dry ethanol, and diethanolamine using a solution combustion method. The synthesized catalysts were characterized using powder X-ray diffraction, UV-Vis diffuse reflectance spectroscopy, Brunauer–Emmett–Teller surface area analysis, Fourier transform infrared spectroscopy, Fourier transform Raman spectroscopy, scanning electron microscopy, and transmission electron microscopy. The photocatalytic degradation of rhodamine B (RhB) dye in aqueous solution (2.06 × 10−5 M) was achieved using synthesized pristine TiO2 (P-T) and GeO2/TiO2 (GT) nanoparticle composite catalysts under direct sunlight. Among the synthesized catalysts, 1-GT afforded 100% RhB degradation in aqueous solution within 180 min. Furthermore, 85, 87, and 83% degradation was achieved using P-T, 2-GT, and 3-GT nanoparticles, respectively. RhB degradation was confirmed by chemical oxygen demand and total organic carbon analyses.
Introduction
Water pollution is a major problem resulting from increasing industrial activity to meet increasing human needs. The major cause of water pollution is the release of organic pollutants/waste from the chemical and dye industries. Most dye industry waste is composed of xanthenic, anthraquinonic, and heteropolyaromatic dyes. Among these dyes, rhodamine B (RhB) is notable for its high pigmentation and carcinogenic effects. The release of such waste directly to the environment is highly hazardous to flora, fauna, and humans. As RhB dye is harmful even at low concentrations, the abatement of such dye contaminants has become increasingly important.1–3 Although numerous methods have been adopted for abatement of polluted water/industrial waste, the disadvantages of these methods have highlighted the need for energy-efficient water treatment. Heterogeneous photocatalysis is an advanced oxidation process considered to be an effective method for the treatment of organic pollutants. In this process, various semiconductor nanomaterials have been widely utilized for the degradation of different organic pollutants under UV and visible light irradiation. Among the various types of nanomaterials, TiO2 is an effective photocatalyst owing to good properties, such as low toxicity, and chemical and thermal stability. TiO2 is also an effective catalyst for hydrogen generation (water splitting) via photocatalysis.4–6 Various TiO2 morphologies, such as nanoparticles, nanotubes, nanowires, nanorods, and nanospheres, have been investigated for the photocatalytic degradation of numerous organic pollutants/dyes.7–11 Among the different morphologies, TiO2 nanoparticles, including doped TiO2 and TiO2 nanocomposites, have been widely used for the photocatalytic degradation of organic compounds/pollutants under visible light (xenon lamp, simulated sunlight, and natural sunlight).12–14 As TiO2 is an UV-active catalyst, it has been effectively utilized for photocatalytic applications under UV irradiation. However, TiO2 can form visible-light active catalysts when doped with metal ions or coupled with narrow bandgap semiconductors.15–18 Synthetic methods used to prepare TiO2 nanocomposites include hydrothermal, solvothermal, sol–gel, solution and combustion methods. Methods other than solution combustion require more time for pristine/composite catalyst formation. To overcome these difficulties, solution combustion is an effective and rapid method for preparing nanomaterials/nanocomposites. The major advantage of this method is that high pressures are not required for material synthesis. In this study, pristine TiO2 (P-T) and GeO2/TiO2 (GT) nanoparticle composites with different GeO2 contents (1, 2, and 3 wt%; denoted as 1-GT, 2-GT, and 3-GT) were synthesized using a solution combustion method. The synthesized composite catalysts were applied to RhB photodegradation in aqueous solution under direct sunlight. The synthesized 1-GT catalyst showed better degradation efficiency than P-T, 2-GT and 3-GT catalysts under direct sunlight.
Experimental section
Materials
Titanium tetraisopropoxide (Ti(OCH(CH3)2)4) and germanium dioxide (GeO2) were purchased from Sigma-Aldrich India. Diethanol amine (DEA, HN(CH2CH2OH)2), dimethyl sulfoxide (DMSO), and 1,4-benzoquinone (BQ) were obtained from SD Fine Chemicals Pvt Ltd. The aforementioned chemicals were used as received. Ethanol was purchased from a local market and distilled before use. Deionized water was used to prepare the aqueous RhB dye solution.
Synthesis of pristine-TiO2 (P-T) and GeO2/TiO2 (GT) nanoparticle composites
To titanium tetraisopropoxide (TIP, 1 M) in dry ethanol (50 mL) in a 250 mL beaker was added GeO2 (1 wt%), followed by DEA (10 M), with constant stirring. The reaction mixture was heated to about 200 °C on a hot plate. After 10–15 min, the mixture autocombusted to form a black powder (GeO2/TiO2 nanocomposite). The as-obtained powder was crushed and calcined at 400 °C for 2 h using a ramp rate of 2 °C min−1. The calcined powder was denoted as 1 wt% GeO2/TiO2 (1-GT). Similarly, 2 wt% GeO2/TiO2 (2-GT) and 3 wt% GeO2/TiO2 (3-GT) were synthesized by adding 2 and 3 wt% GeO2, respectively, in the same procedure. Pristine TiO2 (P-T) was prepared using the same method in the absence of GeO2. Images of the GT nanoparticle composites (Fig. 1) show the colour change from black to off-white after calcination at 400 °C. This was mainly due to the removal of diethanolamine (fuel). The physical appearance and colour changes of P-T, 1-GT, 2-GT, and 3-GT catalysts were the same.
 |
| | Fig. 1 Schematic representation of the synthesis of GeO2/TiO2 nanocomposite (TIP, titanium tetraisopropoxide; C2H5OH, dry ethanol; GeO2, germanium dioxide; DEA, diethanolamine). | |
Characterization
Powder X-ray diffraction (PXRD) patterns of the synthesized P-T and GT photocatalysts were obtained using an X-ray diffractometer (XPERT-MPD model PW3050) with Cu Kα1 radiation in the 2θ range of 10–80°. The crystallite size was calculated from X-ray diffraction peak broadening of the (101) plane using the Scherrer equation (eqn (1)):| | D = kλ/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ cosθ | (1) |
where D is the crystallite size, k is the shape factor, λ is the wavelength of the X-ray source, β is the full-width at half maximum (FWHM) of the (101) diffraction peak, and θ is the Bragg angle. The bandgap was determined using UV-Vis diffuse reflectance spectroscopy (UV-Vis-DRS; Shimadzu UV-3101 PC spectrophotometer) equipped with an integrating sphere and BaSO4 as reference. The bandgaps of the synthesized catalysts were calculated using the following equation (eqn (2)):where Eg is the band gap energy (eV), h is Planck's constant (4.1357 × 10−15 eV s), c is the velocity of light (2.998 × 108 m s−1), and λ is the light wavelength (nm). The morphology of the synthesized catalysts was analyzed using scanning electron microscopy (SEM; JSM-7100F, field emission scanning electron microscope) and transmission electron microscopy (TEM; TEM-JEOL, JEM-2010 electron microscope). The surface areas, pore volumes, and pore diameters of the synthesized catalysts were determined by nitrogen adsorption studies at 423 K using a sorptometer (ASAP-2010, Micromeritics). The chemical properties of the synthesized catalysts were investigated by Fourier transform infrared spectroscopy (FTIR) analysis in the range 4000–400 cm−1. Fourier transform Raman (FT-Raman) spectroscopy was performed using an NXR FT-Raman analyzer.
Photocatalytic activity
The photocatalytic activity of the synthesized P-T and GT catalysts was evaluated by the degradation of RhB in aqueous solution (2.06 × 10−5 M) under direct sunlight (11.00 am to 3.00 pm). The intensity of incident sunlight was determined using a lux meter (Yopco) placed next to the round-bottomed flask containing the RhB solution and catalyst during experiments. Initially, P-T catalyst (50 mg) was accurately weighed into a round-bottomed flask and the RhB dye solution (100 mL, 2.06 × 10−5 M) was added. The mixture was sonicated for about 1 min and then kept in the dark for 30 min with constant stirring to reach an adsorption–desorption equilibrium. A sample of the reaction mixture (5 mL) was withdrawn to determine the percentage dye adsorption on the photocatalyst. The reaction mixture was then stirred under direct sunlight for up to 180 min, with 5 mL samples taken every 60 min. The RhB concentration (λmax = 554 nm) in the irradiated solution was determined by constructing a calibration curve of RhB concentration versus the absorbance of known concentrations using a Cary 500 UV-Vis spectrophotometer (Varian, Palo Alto, CA). The same experimental procedure was used for RhB degradation in the presence of 1-GT, 2-GT, 3-GT, and GeO2 catalysts. Room temperature fluorescence analysis was performed using a Fluorolog spectrophotometer (Horiba Jobinyvon) to analyse hydroxyl radicals (˙OH) using terephthalic acid (TPA) as a probe molecule with an excitation wavelength of 315 nm. Moreover, quenching experiments were performed using scavengers, such as DMSO and BQ, under the photocatalytic experimental conditions described above to determine the roles of ˙OH and ˙O2− radicals. The chemical oxygen demand (COD) and total organic carbon (TOC) content were measured for RhB degradation in aqueous solution using a Spectroquant NOVA 60 photometer and total organic carbon analyzer (Liqui Toc, Elementar, Germany), respectively.
Results and discussion
PXRD analysis
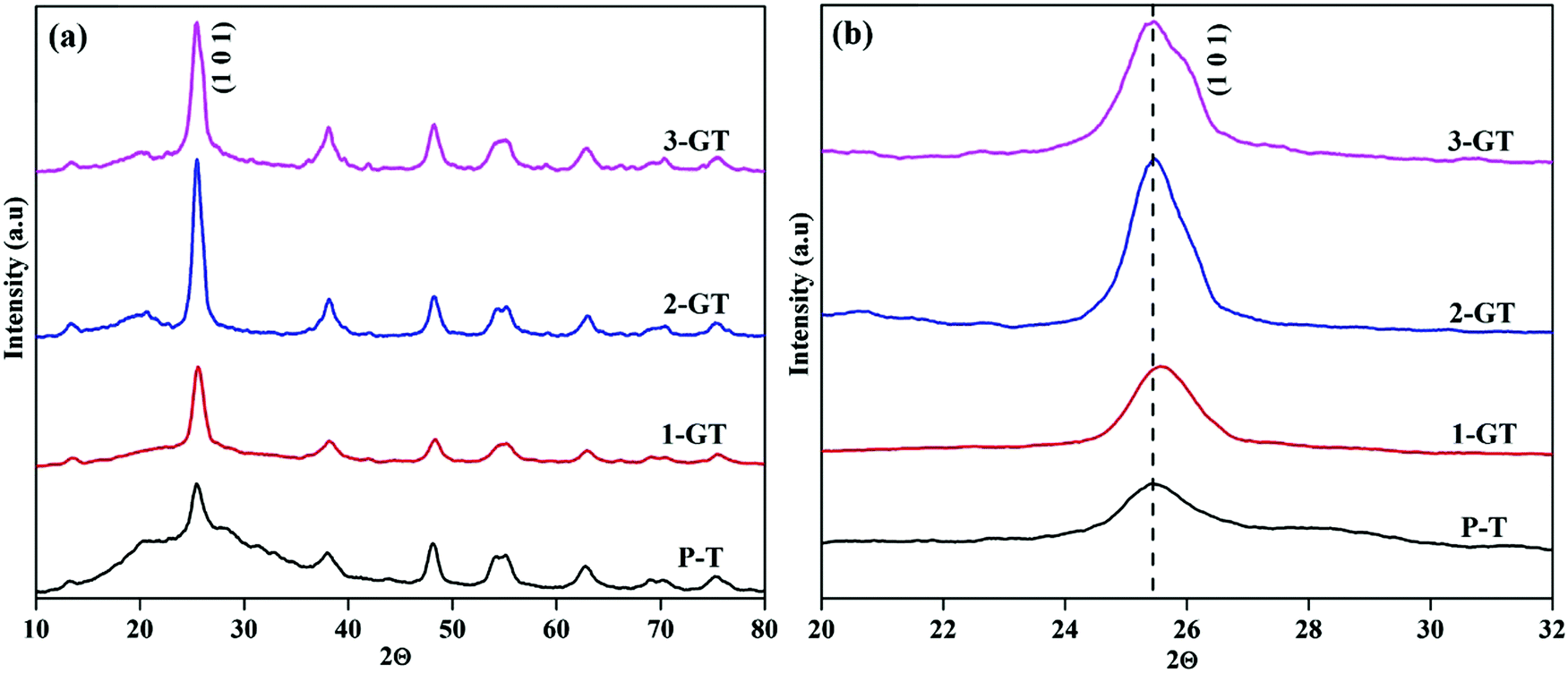
The PXRD patterns of the synthesized P-T and GT nanoparticle composites (Fig. 2) show the major peak (2θ = 25°) corresponding to the anatase phase. The intensity of the (101) plane increases with increasing in metal oxide (GeO2) content. No peaks of other impurities were observed in the synthesized GT nanocomposites. Furthermore, a peak shift was observed for the 1-GT nanocomposite catalyst, which suggested that the GeO2 dopant was well incorporated in the TiO2 lattice (Fig. 2b). This was attributed to Ge4+ having a lower ionic radius (0.054 nm) than Ti4+ (0.0605 nm), such that germanium oxide molecules can easily insert (isomorphism-based doping) in the TiO2 crystal lattice.19 However, the 2-GT and 3-GT nanocomposites showed no shift in the major peak (102), indicating that GeO2 clusters were deposited on the surface of TiO2 particles, but not inserted into the TiO2 lattice.20 The crystallite size was determined to be 3.19, 10.82, 5.96, and 4.42 nm for the synthesized P-T, 1-GT, 2-GT, and 3-GT catalysts, respectively. As shown in Table 1, a larger crystallite size was observed for 1-GT than for 2-GT. This was mainly attributed to the presence of excess GeO2 molecules adsorbed on TiO2 surface in 2-GT, which could suppress crystal growth of nanoparticles and lead to a decreased crystallite size.21–23 The increase in TiO2 crystallite size after GeO2 doping was also due to subsequent crystallite growth and collapse of the mesoporous structure.
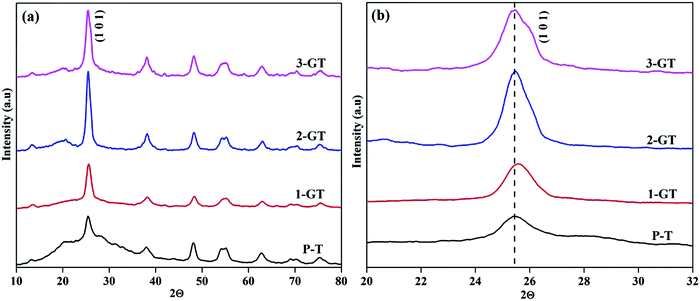 |
| | Fig. 2 (a) PXRD pattern and (b) enlarged pattern of the (101) plane of synthesized P-T and GT nanoparticles. | |
Table 1 Bandgap and band edge of synthesized P-T and GT catalysts
| Catalysts |
Crystallite size (nm) |
Bandgap (eV) |
Band edge (nm) |
S
BET (m2 g−1) |
D
pore (nm) |
V
total (cm3 g−1) |
| P-T |
3.19 |
3.28 |
377.7 |
124 |
4.97 |
0.22 |
| 1-GT |
10.82 |
3.2 |
386.4 |
107 |
4.62 |
0.18 |
| 2-GT |
5.96 |
3.24 |
381.6 |
115 |
4.65 |
0.19 |
| 3-GT |
4.42 |
3.27 |
379.2 |
113 |
4.97 |
0.16 |
UV-Vis DRS analysis
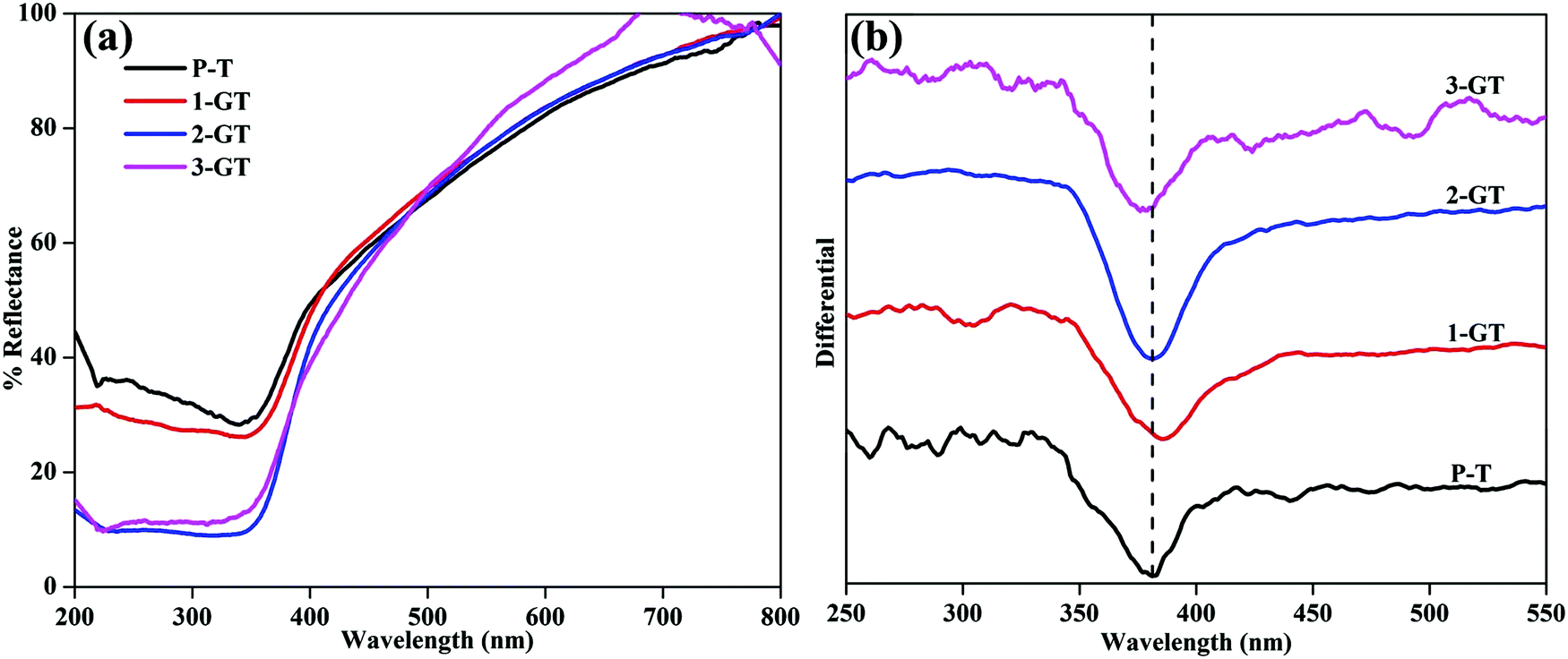
The photogenerated electrons and holes created by UV-Vis light excitation of the materials can be probed using UV-Vis DRS analysis. Fig. 3 shows UV-Vis DRS and differential spectra of the synthesized P-T and GT catalysts. All synthesised P-T and GT catalysts showed drops in the UV region (Fig. 3a). The band edge (Table 1) of 1-GT catalyst was redshifted slightly towards the visible region (Fig. 3b). Therefore, the absorbance of 1-GT was somewhat higher than the bare catalyst (P-T), which indicated the adhesion of GeO2 molecules over the TiO2 crystal lattice. For 2-GT and 3-GT, the band edge was shifted slightly back toward the UV region (Fig. 3b), which was mainly attributed to the excess amount of GeO2 not doped into the TiO2 lattice.24 This indicated that the synthesized 1-GT catalyst had the optimum amount of GeO2 ions to absorb visible light. Furthermore, the smaller bandgap of synthesized 1-GT indicated the localized nature of Ge species in the TiO2 crystal lattice. This contributed to the formation of GeO2 electronic energy levels and increase in grain/crystallite size. Furthermore, this is accompanied by the formation of surface oxygen vacancies (SOVs), which are another key factor in bandgap reduction. SOVs are formed by oxygen diffusion from the surface lattice to the external environment during high temperature annealing. The difference in the ionic radii and oxidation states of Ge (0.054 nm) and Ti ions (0.0605 nm) in the host matrix can favour or oppose SOV formation. These oxygen vacancies can lead to ohmic contact between GeO2 and TiO2, which altered the charge-transfer pathway (Fig. 10b) and maintained photocatalytic efficiency in 1-GT.25–27 Although the P-T catalyst had SOVs, the Ge impurities found in 1-GT increased the SOVs, leading to the smaller bandgap with respect to P-T. Therefore, integrated SOVs and the doped metal-ion/metal-oxide semiconductor (GeO2 in the present study) caused the reduction of the bandgap of 1-GT. Moreover, these SOVs can prolong the lifetime of charge carriers, which improves the separation efficiency of photogenerated e−/h+ pairs and leads to the enhanced photocatalytic efficiency of the 1-GT catalyst.28–30
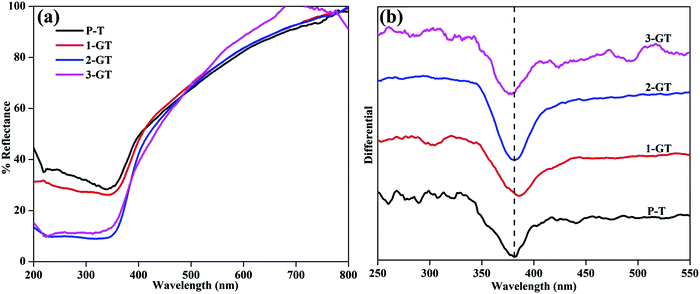 |
| | Fig. 3 (a) UV-DRS and (b) differential spectra of synthesized P-T and GT catalysts. | |
Surface area analysis
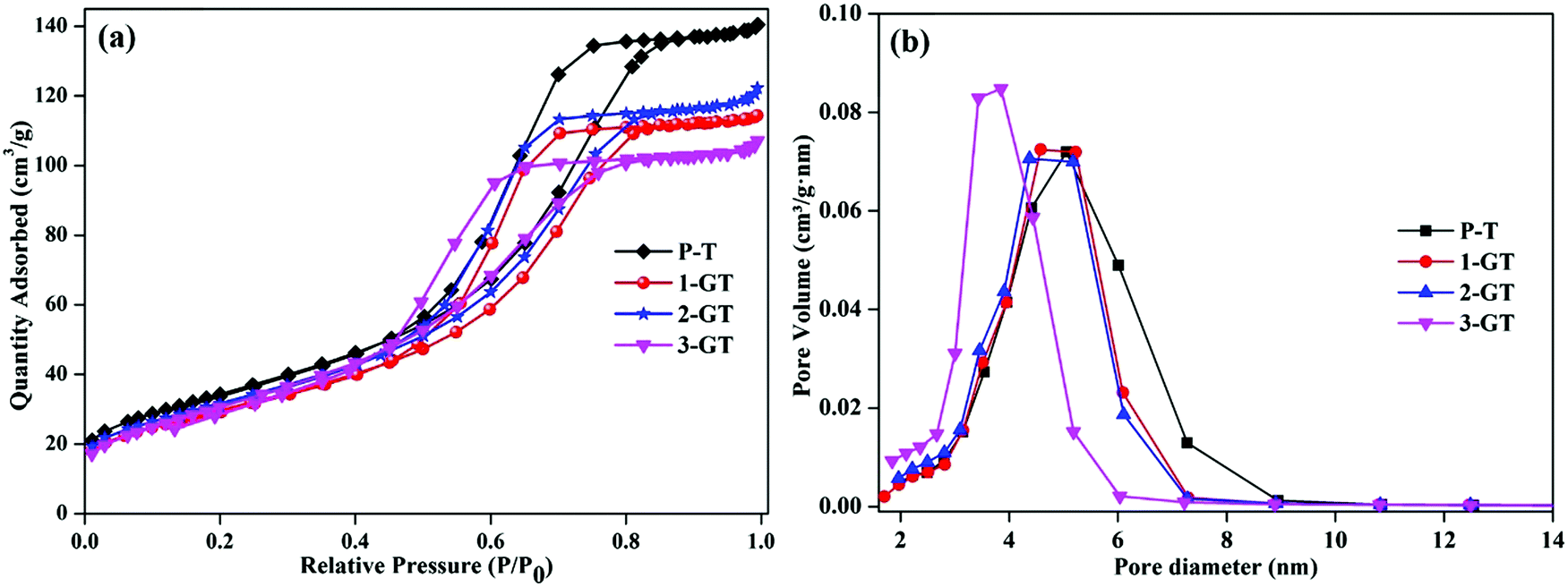
Fig. 4 shows N2 adsorption–desorption plot and pore size distribution curves of the synthesized P-T and GT catalysts. All synthesized catalysts showed adsorption isotherms with type-IV hysteresis, which is characteristic of mesoporous materials.31 This could indicate the porosity between the agglomerated nanoparticles in both P-T and GT catalysts. The surface area of the synthesized P-T catalyst was found to be 124 m2 g−1, which was higher than those of the GT catalysts. The lower surface areas of the GT catalysts might be due to the oxygen vacancies created by GeO2 interaction with the TiO2 crystal lattice. This enhanced the oxygen diffusion and particle size, and was observed by XRD analysis of the GT catalysts. Furthermore, the low melting point of GeO2 molecules relative to that of the TiO2 matrix assisted crystal rearrangement. This could explain the decrease in surface area of 1-GT compared with P-T.32 Furthermore, the surface areas of 2-GT and 3-GT were found to be larger than that of 1-GT (but not P-T) with a decrease in crystallite size. This was mainly attributed to the presence of excess GeO2 molecules. Meanwhile, the pore diameter of GT catalysts was found to be slightly lower than that of P-T, irrespective of the dopant quantity.
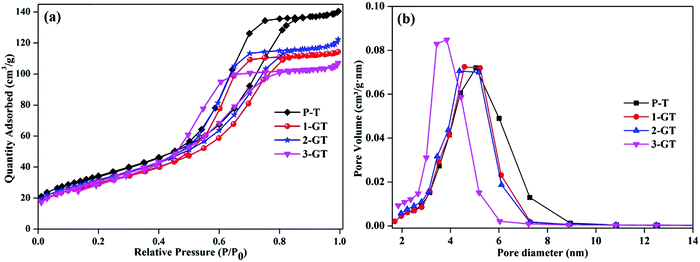 |
| | Fig. 4 (a) N2 adsorption–desorption isotherm plot; (b) pore size distribution plot of P-T and GT catalysts. | |
FTIR analysis
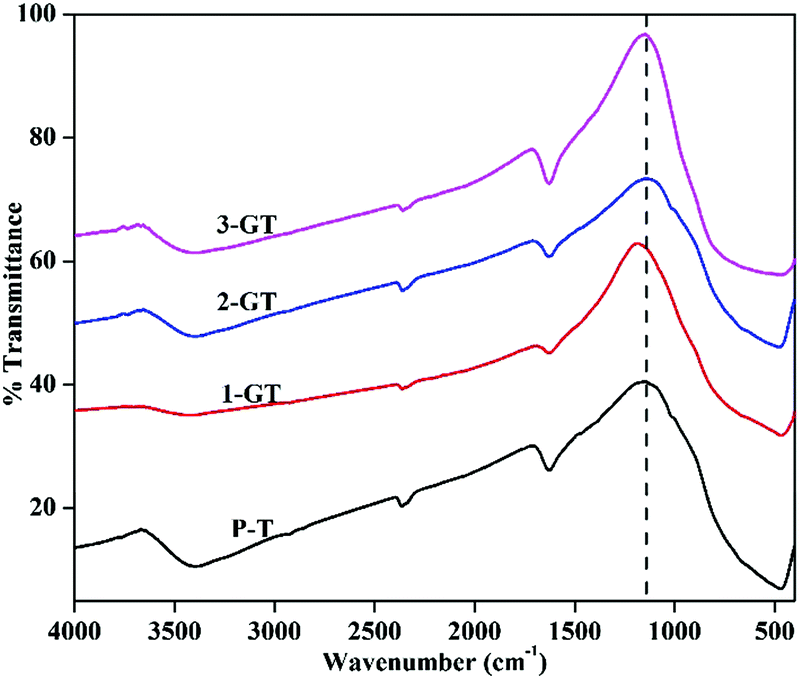
Fig. 5 shows FTIR spectra of the synthesized P-T, 1-GT, 2-GT, and 3-GT catalysts. For P-T catalyst, the band at around 450–900 cm−1 is mainly assigned to Ti–O–Ti lattice vibrations. Moreover, O–H bending and vibration bands were observed at around 1628 and 3398 cm−1, respectively.33
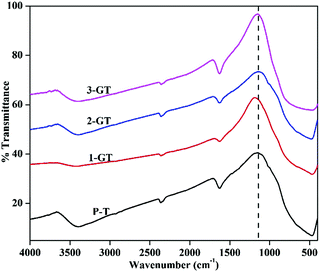 |
| | Fig. 5 FTIR analysis of synthesized P-T and GT catalysts. | |
These bands were mainly attributed to adsorbed water molecules and hydroxyl ions. The same type FTIR pattern was obtained for the GT nanoparticle composites. Furthermore, no peak for any impurity phase was observed even after GeO2 addition. Meanwhile, peak shifts and changes in the peak intensity were observed in the shoulder at around 1153 cm−1 (Fig. 5). This was attributed to TiO2 matrix disturbance due to the addition of GeO2, which led to crystal distortion in the GT composite catalysts.
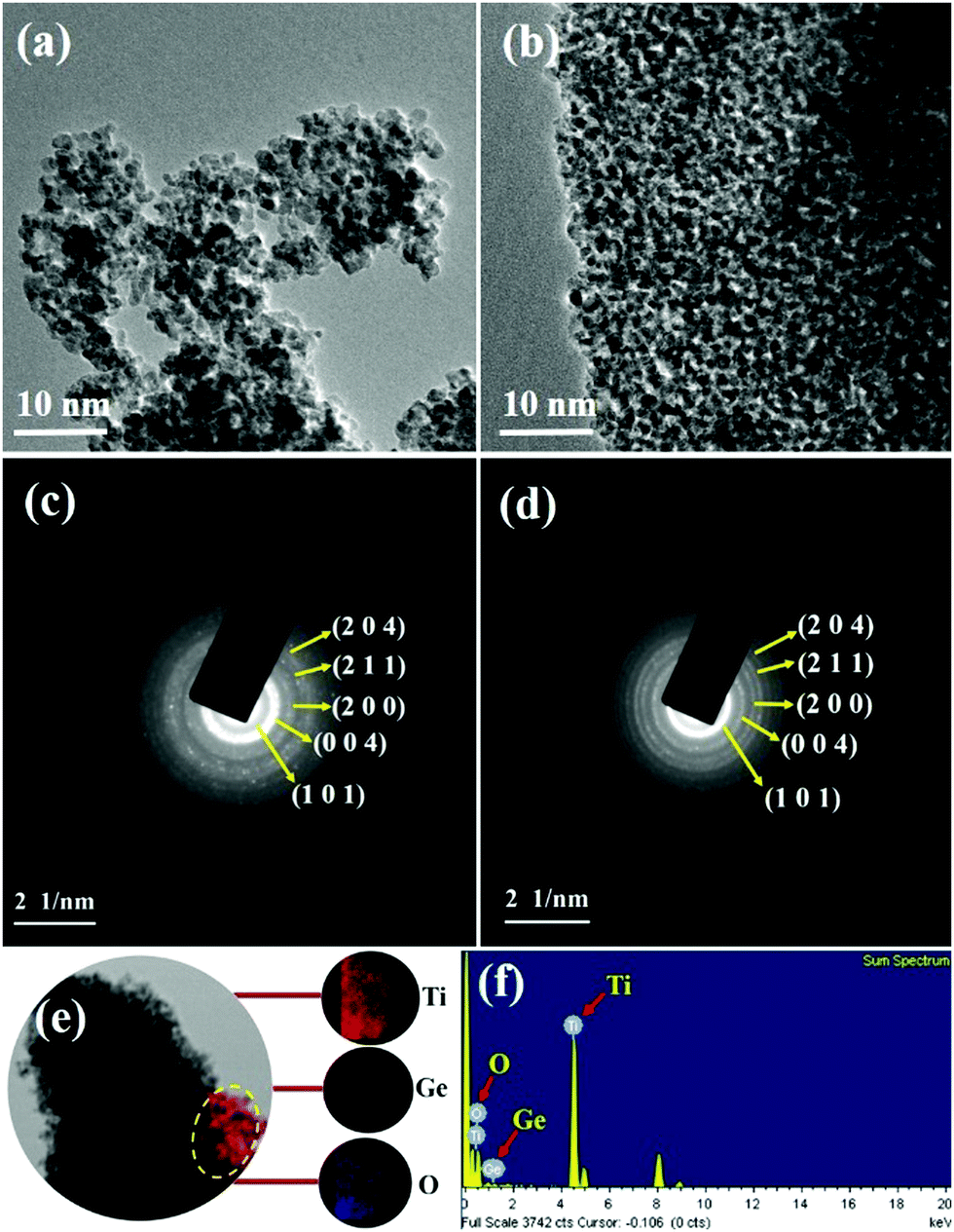
SEM and TEM analysis
Fig. 6 shows SEM images of the synthesized P-T and 1-GT catalysts, which show the particle nature of both catalysts. Tightly agglomerated nanoparticles were observed for both P-T (Fig. 6a and b) and 1-GT (Fig. 6c and d) catalysts. The size of the TiO2 particles formed during synthesis was restricted to the size of liquid droplets before combustion.34
 |
| | Fig. 6 SEM images of (a and b) P-T and (c and d) 1-GT catalysts. | |
Consequently, the formed nanoparticles were obtained in the nanometre range, which is common for GT nanoparticle composites. Significant agglomeration in the synthesized PT nanoparticles could be due to the combination of single and primary particles, which was also the case for GT nanoparticle composites.35 The particle nature of the synthesized P-T and 1-GT catalysts was also observed using TEM analysis (Fig. 7a and b). The SAED pattern (Fig. 7c and d) of P-T and 1-GT catalysts showed similar ring patterns, which suggested that the presence of GeO2 did not affect the TiO2 crystal structure. The presence of germanium, titanium, and oxygen molecules was also confirmed using TEM mapping analysis of 1-GT catalyst (Fig. 7e and f). The agglomerated nature of the nanoparticles observed in P-T was retained in the 1-GT catalyst.
 |
| | Fig. 7 (a and b) TEM images and (c and d) SAED patterns of P-T and 1-GT catalysts; (e) TEM mapping image and (f) EDS of 1-GT catalyst. | |
EDS spectra (Fig. 7f) of the synthesized 1-GT catalyst confirmed the presence of germanium, titanium, and oxygen, while a minor peak at around 8 keV corresponded to the copper grid. Using EDS analysis, the elemental composition of P-T and GT (Table 2) catalysts was specified in terms of the weights and atomic ratios of their respective elements.
Table 2 Elemental composition of P-T and 1-GT catalysts
| Catalysts |
Weight % of elements |
Atomic % of elements |
| Ti |
O |
Ge |
Ti |
O |
Ge |
| P-T |
82.4 |
17.6 |
— |
62.1 |
37.9 |
— |
| 1-GT |
63.5 |
35.6 |
0.9 |
36.7 |
62.9 |
0.4 |
| 2-GT |
61.9 |
36.3 |
1.8 |
33.9 |
64.8 |
1.3 |
| 3-GT |
57.2 |
39.6 |
3.2 |
29.4 |
67.9 |
2.7 |
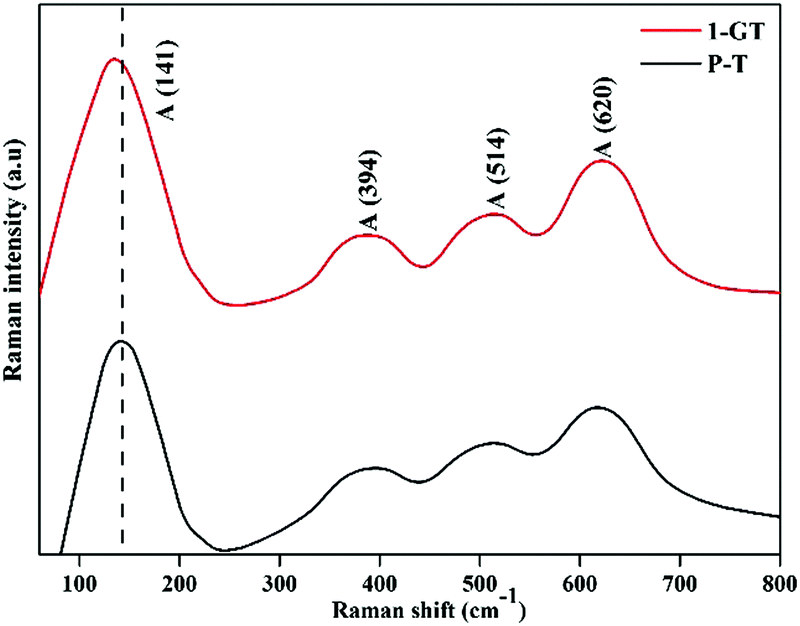
FT-Raman analysis
Fig. 8 shows Raman spectra of the synthesized P-T and 1-GT catalysts. For P-T catalyst, the major peaks at around 141, 394, 514, and 620 cm−1 represented the Raman-active vibrational modes. These bands were well matched with that of crystalline anatase phase and consistent reported values.36,37 Moreover, Raman spectra of the synthesized 1-GT catalyst showed the same band position in accordance with the P-T catalyst. A small shift was observed in the major peak at around 141 cm−1, which was mainly attributed to GeO2 loading on the host TiO2. Moreover, no peaks corresponding to other impurities were observed. This implied that the addition of GeO2 to the host TiO2 matrix did not affect the local crystal structure.
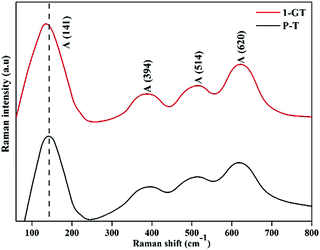 |
| | Fig. 8 FT-Raman spectra of the synthesized P-T and GT catalysts. | |
Photocatalytic activity of P-T and GT catalysts
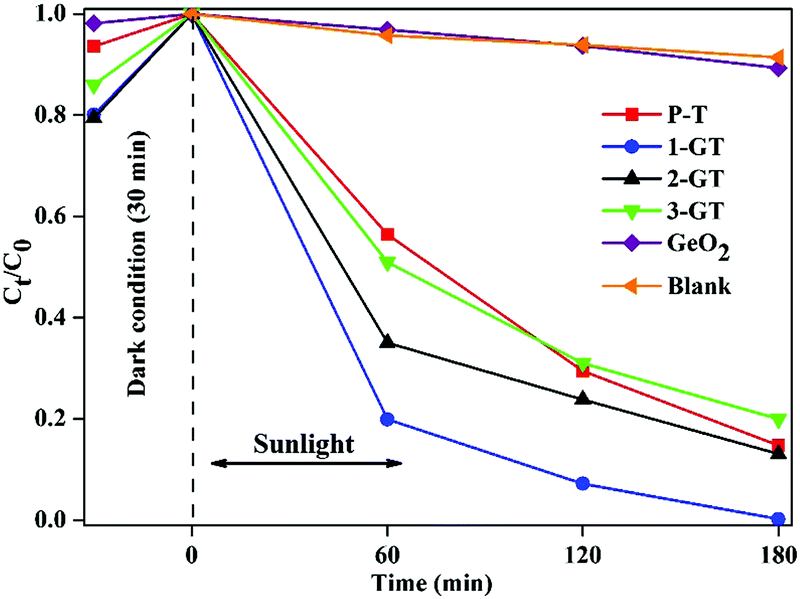
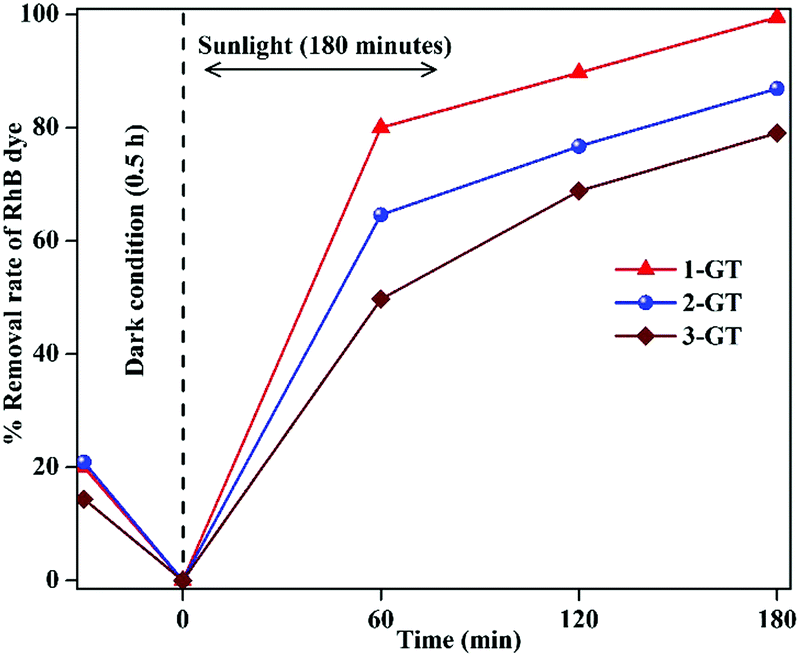
The photocatalytic degradation of RhB dye in aqueous solution (1.96 × 10−5 M) was studied using the synthesized P-T and GT catalysts under direct sunlight. Complete degradation of RhB was observed using 1-GT (Fig. 9), while 85, 87, 83, and 11% degradation were achieved using P-T, 2-GT, 3-GT, and GeO2 catalysts, respectively, after 180 min. Without any catalyst, the self-photodegradation of RhB under sunlight was performed, with the result showing that only about 8.5% degradation was achieved after 180 min. The higher degradation rate achieved by 1-GT (Fig. 9) was mainly attributed to its GeO2 content, which has a stronger ability to reduce electron–hole pair recombination. Furthermore, the larger particle size of 1-GT nanoparticle composites compared with PT produces more effective degradation. Meanwhile, the lower degradation rates achieved by 2-GT and 3-GT were mainly due to the presence of excess GeO2. Although the crystallite size of 1-GT was larger and the surface area smaller, it still showed better photocatalytic activity than P-T. This can be attributed to the GeO2 loading on TiO2, which acts as an electron trap and prevents electron–hole recombination, which is an important factor38 in determining photocatalytic activity, as shown in Fig. 10. The higher activity of the 1-GT catalyst was also attributed to the transformation of e−/h+ pairs, which resulted in a narrower bandgap compared with P-T.39
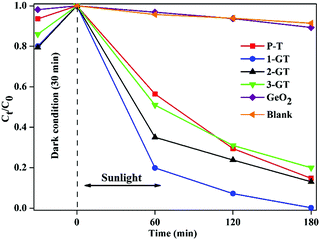 |
| | Fig. 9 Percentage degradation rate of RhB dye in GeO2, blank, and synthesized P-T and GT catalysts under direct sunlight. | |
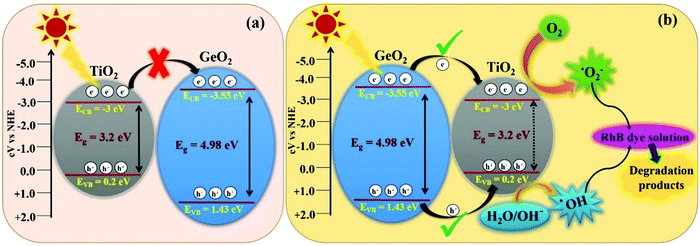 |
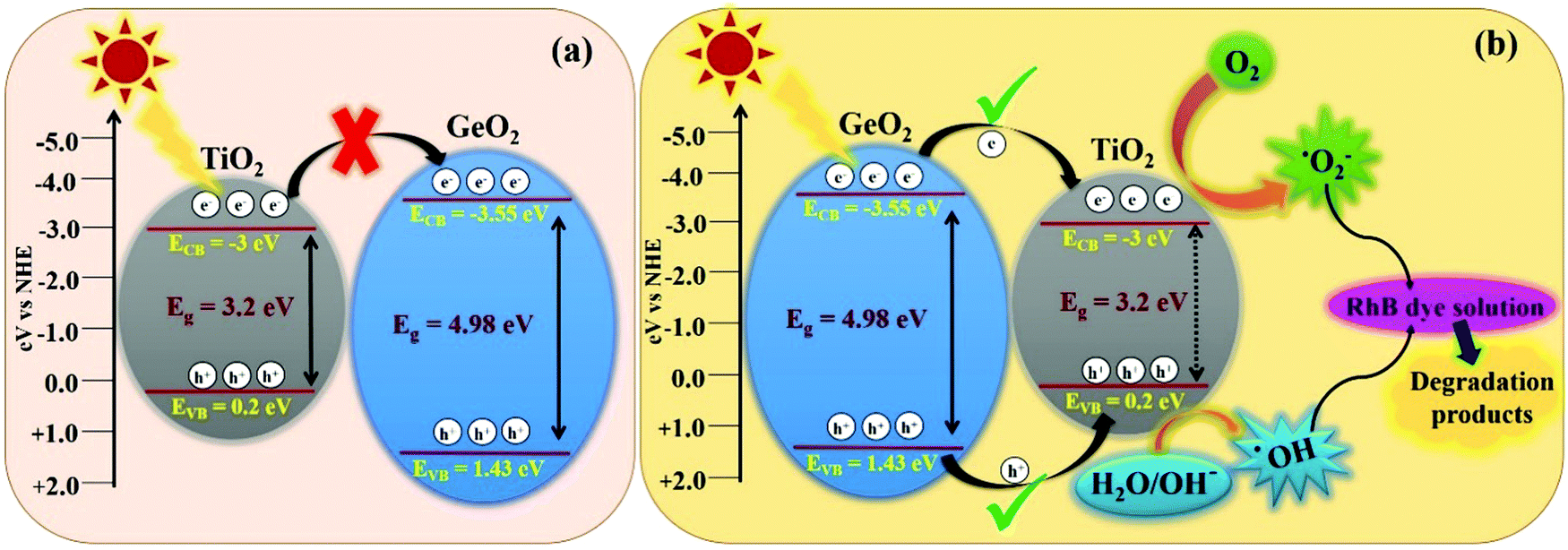
| | Fig. 10 Schematic representation of the band structure of 1-GT catalyst and its subsequent electron charge transfer mechanism showing (a) the usual pathway and (b) an unusual feasible pathway via a type-I mechanism. | |
Furthermore, the higher crystallinity of 1-GT compared with P-T facilitated the efficient transfer of photoelectrons from the bulk to the surface and inhibited recombination, which led to its enhanced photocatalytic activity.40 When doping with metal-ions/metal-oxides (GeO2 in the present study), some Ti4+ ions may be converted into Ti3+ ions, which can act as active sites for trapping photogenerated electron–hole pairs. Meanwhile, recombination centres can form if Ti3+ accumulates on the surface, leading to a decrease in the photocatalytic removal of RhB dye/organic pollutant.41 A plausible electron–hole pair excitation mechanism for 1-GT catalyst under sunlight irradiation and its charge transfer followed by the formation of superoxide and hydroxyl radicals is shown in Fig. 10. The adhered GeO2 molecules can effectively trap the photogenerated electrons, which reduces recombination with holes42 and leads to effective degradation of RhB within 180 min. COD and TOC analysis have also been performed to confirm complete degradation using the 1-GT catalyst in the present study. In COD and TOC, 95 and 85% reductions in RhB were determined, respectively, for 1-GT catalyst.
Bandgap structure and feasible mechanism for photocatalytic degradation of RhB dye using 1-GT catalyst
In the present study, 1-GT showed a higher photocatalytic removal efficiency than P-T, 2-GT, 3-GT, and GeO2 catalysts. Using the valence band (VB) and conduction band (CB) edge potentials for GeO2 and TiO2 in 1-GT, the degradation mechanism can be explained from the bandgap structure. VB and CB edge potentials were calculated using the following empirical equations:where EVB is the VB edge potential, ECB is the CB edge potential, X is the electronegativity of the semiconductor, Ee is the energy of free electrons on the hydrogen scale (≈4.5 eV), and Eg is the bandgap energy of the semiconductor. The values of X for GeO2 and TiO2 were 3.44 and 3.1, respectively.
In the present study, the smaller bandgap of TiO2 should receive incident photons from sunlight, and is thought to transfer excited e−/h+ pairs to GeO2. However, the transfer of photogenerated electrons from the CB of TiO2 to CB of GeO2 was restricted, mainly due to the higher negative CB edge potential of GeO2 (−3.55 eV) compared with that of TiO2 (−3 eV). Furthermore, if the transfer had proceeded (Fig. 10a), the formation of ROS (˙OH and ˙O2−) required for RhB degradation was not feasible. However, the degradation pathway might be favoured via a type-I mechanism (Fig. 10b), which is unusual.43 Using this route, the photogenerated electrons are transferred from the CB of GeO2 to the CB of TiO2. Consequently, the recombination of e−/h+ pairs can take place simultaneously in both TiO2 and GeO2, resulting in lower photocatalytic efficiency. However, photoinduced e−/h+ pairs accumulated in TiO2 can easily convert O2 to ˙O2− in the CB and H2O/OH− to ˙OH radicals in the VB.44,45 Although the CB of GeO2 is more negative than that of TiO2, the electrons could easily migrate to the CB of TiO2, where the ROS, namely ˙O2− radicals, were formed effectively. Furthermore, the formation of ˙OH radicals in the VB of TiO2 also proceeded well.46
Accordingly, effective photocatalytic degradation by 1-GT was favoured, which only occurs with a low GeO2 loading. Furthermore, the low GeO2 loading (1 wt% in 1-GT) gives effective degradation of RhB due to GeO2 being well dispersed with good Ti-O-Ge ion linkages. However, the recombination of e−/h+ pairs that is dominant in 1-GT is overwhelmed by the attractive force interaction between oxygen vacancies and photo-induced charges, which might explain the enhancement in photocatalytic efficiency.47 The type-I mechanism of e−/h+ pair generation and transformation believed to occur in the 1-GT photocatalytic system is consistent with the reported mechanism.48 Therefore, we concluded that the enhanced photocatalytic efficiency of as-prepared 1-GT was mainly attributed to the appropriate weight ratio and synergetic effects of GeO2, despite having a larger bandgap than TiO2. This resulted in the effective trapping of e−/h+ pairs and fast charge carrier transport across the heterojunction, which suppressed recombination by GeO2 and caused the complete degradation of RhB dye under direct sunlight.
Effect of GeO2 loading on photocatalytic activity
The photocatalytic activity of GT nanoparticle composites was enhanced by the addition of GeO2. In addition to 1-GT (1 wt% GeO2/TiO2) and 2-GT (2 wt% GeO2/TiO2), 3-GT nanoparticle composite (3 wt% GeO2/TiO2) was utilized for the photocatalytic removal of RhB dye under direct sunlight using the same experimental conditions.
It was observed that the removal rate of RhB dye decreased with increasing GeO2 loading (Fig. 11). This was mainly attributed to the higher recombination probability of trapped electrons with holes. Moreover, the excess amount of GeO2 (in both 2-GT and 3-GT) over the active sites of host TiO2 led to the possibility of a higher degree of light scattering.38,49 Therefore, the effective trapping of excited e−/h+ pairs was reduced due to the presence of excess GeO2 molecules on the TiO2 matrix surface. This eventually congested the optical system, resulting in the lower photocatalytic efficiency of 2-GT and 3-GT compared with that of 1-GT.
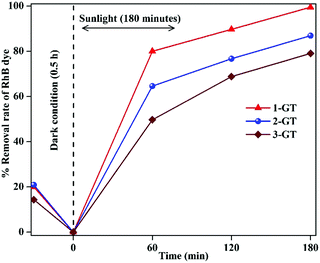 |
| | Fig. 11 Effect of GeO2 on the photocatalytic activity of GT nanoparticle composites. | |
Fluorescence analysis
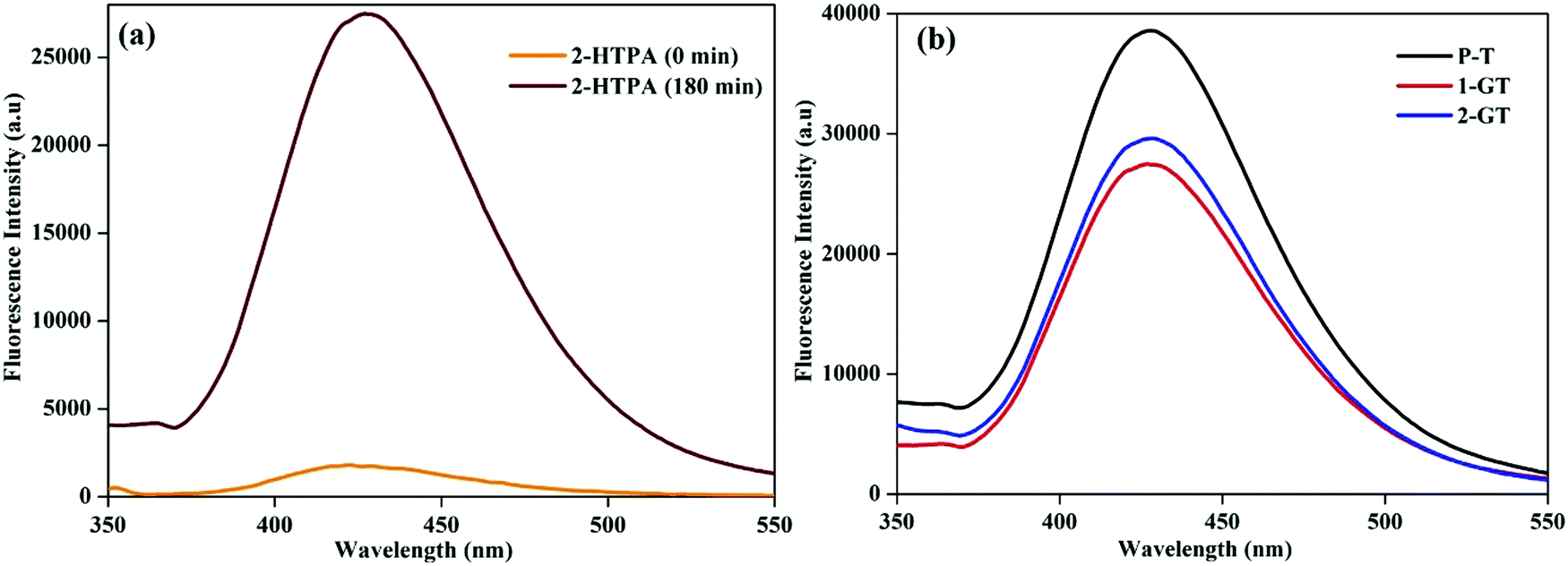
The formation of ˙OH radicals from photogenerated holes on the catalyst (1-GT) surface was analysed by fluorescence using terephthalic acid (TPA) as the probe molecule. During photocatalytic processing, 2-hydroxyterephthalic acid (2-HTPA) is the fluorescent product formed due to the reaction of TPA with ˙OH radicals.50Fig. 12a shows the fluorescence intensity of 2-HTPA at around 427 nm, which increased up to 180 min. This was due to the intensity of 2-HTPA being proportional to the amount of ˙OH radicals.51
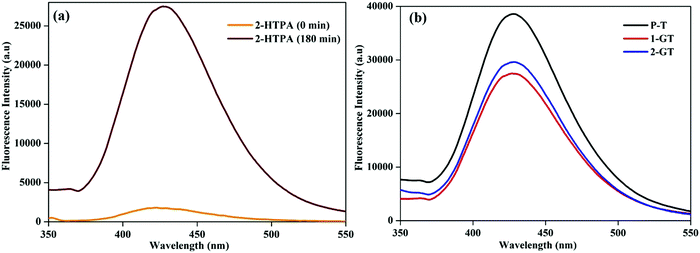 |
| | Fig. 12 Fluorescence spectra of (a) 2-HTPA before and after sunlight illumination in the presence of 1-GT, and (b) P-T, 1-GT and 2-GT catalysts. | |
This increasing 2-HTPA intensity (Fig. 12a) confirmed the formation of ˙OH radicals, which strictly favoured a higher degradation efficiency. Fig. 12b shows fluorescence spectra of the synthesized P-T, 1-GT, and 2-GT catalysts utilized for the photocatalytic degradation of RhB. The peak intensities of 1-GT and 2-GT catalysts were lower than that of P-T catalyst. This was mainly attributed to the faster recombination of e−/h+ pairs in P-T catalyst than in the composite catalysts (1-GT and 2-GT). Furthermore, the lower intensity of 1-GT implied its higher photonic efficiency, which results in less e−/h+ pair recombination.52,53 Therefore, in 1-GT, GeO2 acted as a separation centre for charge carriers, but, in 2-GT, acted as recombination centres. This ultimately led to the more effective degradation of RhB using 1-GT than for 2-GT and P-T (bare TiO2) catalyst under direct sunlight. As 1-GT has a lower luminescence intensity than 2-GT (Fig. 12b), the 1 wt% GeO2 could be an optimum loading54 on the host TiO2, leading to the effective removal of RhB dye.
Quenching analysis of the role of oxidizing species
In the photocatalytic reactions concerned, the formation of active e−/h+ pairs after illumination is the key process, followed by ROS formation. ROS, namely, ˙OH, H2O2, 1O2, ˙HO2, and ˙O2−, are the main species responsible for the photo-oxidation of organic and biological pollutants under UV/Visible irradiation. As 1-GT was the most effective photocatalyst in the present study, DMSO55 and BQ56 were used as scavengers of hydroxyl (˙OH) and superoxide (˙O2−) radicals, respectively, in its photocatalytic reactions.
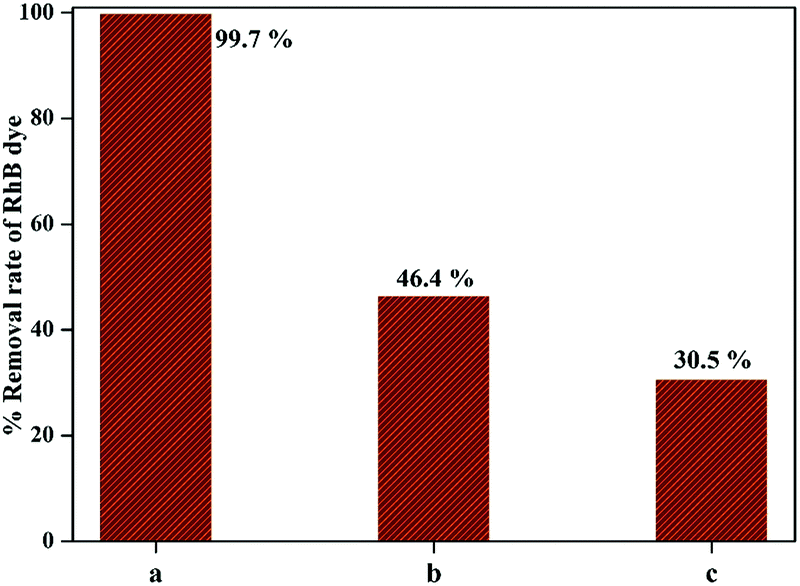
The photocatalytic efficiency of 1-GT catalyst in the presence of DMSO and BG as scavengers under the same experimental conditions is shown in Fig. 13. The RhB removal rate was decreased significantly in the presence of scavengers. Using DMSO afforded a lower photocatalytic efficiency (46.4% RhB removal), indicating that ˙OH radicals were trapped after light illumination (up to 180 min). Meanwhile, 30.5% RhB removal was achieved in the presence of BQ scavenger, indicating that ˙O2− radicals were trapped over the same reaction duration. Therefore, the scavengers appeared to affect the RhB removal rate to different extents (Fig. 13), which affected the outcome of the photocatalytic reaction. In the present study, BQ inhibited RhB removal more than DMSO. This suggested that ˙OH radicals contributed more to the photocatalytic removal of RhB than ˙O2− radicals while using 1-GT under direct sunlight.
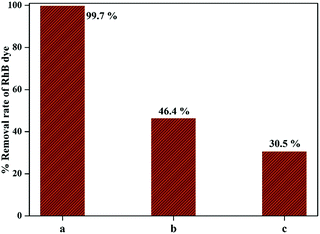 |
| | Fig. 13 Comparison of RhB removal rate (a) without quencher, (b) with DMSO, and (c) with BQ. | |
Kinetic analysis
Kinetic analysis is important for the photocatalytic degradation of any organic compound. In the present study, kinetic analysis was performed on the degradation of RhB solution under sunlight using P-T, GT, and GeO2 catalysts.
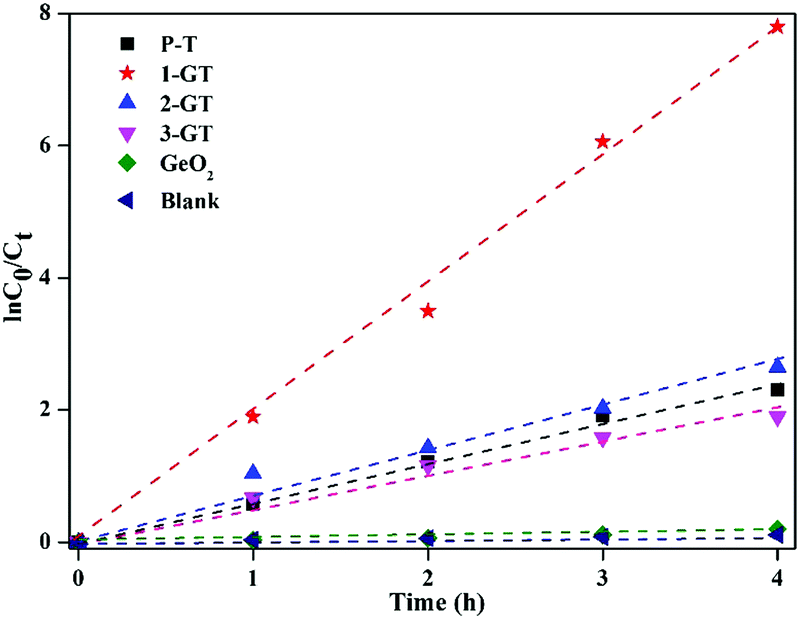
The results imply that all photocatalytic degradation reactions followed pseudo-first-order kinetics ([ln(C0/Ct) = kappt]), where, C0 is the initial dye concentration, and Ct is the concentration at time t, and kapp is the apparent first-order rate constant. kapp was calculated by linear regression of the slope of the plot of ln(C0/Ct) vs. time (Fig. 14). RhB degradation followed first-order kinetics (approximately fits a straight line) for all synthesized catalysts under sunlight. Among all catalysts utilized in the present study, 1-GT showed the maximum activity, as confirmed by its maximum intensity for ln(C0/Ct) vs. time (Fig. 14). This provided additional evidence of effective photocatalytic degradation of RhB in aqueous solution using 1-GT under sunlight.
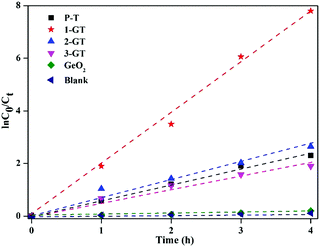 |
| | Fig. 14 Kinetic plot of RhB degradation using P-T, GT, and GeO2 catalysts. | |
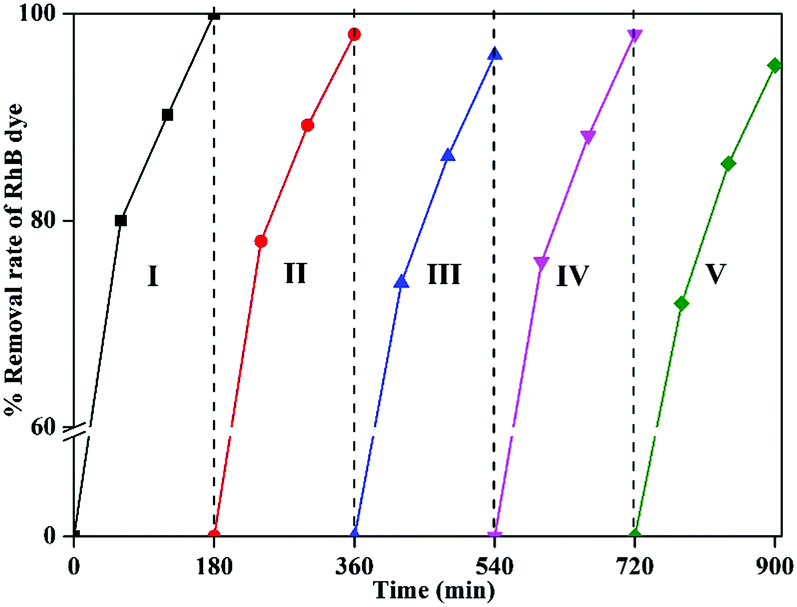
Recyclability study of 1-GT catalyst for RhB degradation
Recyclability is an important parameter for any catalytic reaction using nanomaterials. In this study, RhB degradation was achieved effectively while using 1-GT catalyst. This catalyst was recycled up to five times in the degradation reaction. After the first cycle, the catalyst was recovered by centrifugation, washed twice with water, and dried at 60 °C for 2–3 h.
The dried catalyst was then used in the second cycle reaction under sunlight. Using the same isolation method, third, fourth, and fifth reaction cycles were performed, each for 180 min. The results showed a 3–5% reduction in the degradation rate by the end of fifth cycle, which might be attributed to RhB dye molecules adsorbed on the surface of used 1-GT from previous cycles (Fig. 15). This partial suppression of the degradation rate at the end of fifth cycle demonstrated the stability of 1-GT catalyst under sunlight for RhB degradation.
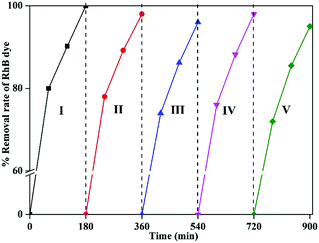 |
| | Fig. 15 Recyclability study of synthesized 1-GT catalyst for RhB removal in aqueous solution under direct sunlight. | |
Comparison of RhB dye photodegradation with previous reports
The utilization of direct sunlight for the effective removal of RhB dye from aqueous solution in the present study can be compared with similar work reported previously. Table 3 shows a comparison of recent reported literature on RhB degradation with the present study. Although five of these studies achieved higher photocatalytic efficiency over a shorter period than in the present study, the catalyst synthesis method used herein is a more rapid approach than those previously reported. Therefore, 1-GT nanoparticle composites prepared by solution combustion could be an effective alternative to previously reported approaches for the removal of RhB dye from aqueous solution under direct sunlight/simulated sunlight.
Table 3 Comparison of present study with previous reported literature using other catalysts
| Catalyst |
Synthesis method |
Dye (ppm) |
Light source |
Degradation (time) |
Ref. |
| BiVO4/Bi2S3/MoS2 heterojunction |
In situ hydrothermal |
RhB (10) |
Natural sunlight |
97% (300 min) |
57
|
| Perylene-3,4,9,10-tetracarboxylic dianhydride–ZnO |
Hydrothermal |
RhB dye (30) |
Natural sunlight |
100% (80 min) |
58
|
| Bi2WO6/RGO composite |
Hydrothermal |
RhB (60) |
Natural sunlight |
100% (360 min) |
59
|
| Titanosilicate/g-C3N4 nanocomposite |
Hydrothermal |
RhB (5) |
Natural sunlight |
99% (40 min) |
60
|
| Fe doped CuS |
Chemical coprecipitation |
RhB (10) |
Simulated sunlight irradiation (AM 1.5 G filter and 150 W Xe lamp) |
98.5% (80 min) |
61
|
| 3D flower-like Bi2O3 |
Hydrothermal |
RhB (10) |
Simulated sunlight irradiation (300 W) |
86% (240 min) |
62
|
| Iodine doped TiO2 nanoparticles |
Sol–gel |
RhB (10) |
Natural sunlight |
98% (240 min) |
63
|
| TiO2 (binary system) |
— |
RhB (5) |
Simulated sunlight (ULTRA VITALUX 300 W 230 V E27) |
100% (120 min) |
64
|
| ZnO–Ag nanocomposite |
Solid-state thermal decomposition |
RhB (5) |
Natural sunlight |
100% (60 min) |
65
|
| Tin(II) sulfide |
Template-free chemical route |
RhB (10) |
Natural sunlight |
88.4% (240 min) |
66
|
| GeO2/TiO2 (1-GT) nanoparticle composite |
Solution Combustion |
RhB (10) |
Natural sunlight |
100% (180 min) |
Present study |
Conclusion
Pristine TiO2 (P-T) and GeO2/TiO2 (GT) nanoparticle composites were synthesized by a solution combustion method and applied to the degradation of RhB dye in aqueous solution (2.06 × 10−5 M) under direct sunlight. Among the synthesized catalysts, 1-GT was more effective than the synthesized P-T and 2-GT catalysts, affording 100% degradation efficiency in 180 min. Using 1-GT, 95% and 85% RhB degradation were confirmed by COD and TOC analyses, which were higher than those of PT, 2-GT, and 3-GT catalysts. Kinetic studies showed that RhB degradation followed first-order kinetics for 1-GT catalyst. The recyclability of 1-GT catalyst was also studied for up to five cycles, which resulted in only a 3–5% reduction in the RhB degradation rate. Overall, this study shows that the synthesized 1-GT catalyst could be a more effective visible-light-active photocatalyst than pristine TiO2 for RhB dye removal under direct sunlight.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge CSMCRI for allowing publication of this work (Grant number: CSIR-CSMCRI-147/2017). We also wish to thank Mr Jayesh Chaudry, Mr Gopal Ram, and Mr Vakani Viral of the Analytical Division & Centralized Instrument Facility (ADCIF) of the institute for kind support.
References
- M. Sundararajan, V. Sailaja, L. John Kennedy and J. Judith Vijaya, Ceram. Int., 2017, 43, 540–548 CrossRef CAS.
- M. A. Alvi, A. A. Al-Ghamdi and M. ShaheerAkhtar, Mater. Lett., 2017, 204, 12–15 CrossRef CAS.
- A. I. Borhan, P. Samoila, V. Hulea, A. R. Iordan and M. N. Palamaru, J. Photochem. Photobiol., A, 2014, 279, 17–23 CrossRef CAS.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- S. Kment, F. Riboni, S. Pausova, L. Wang, L. Wang, H. Han, Z. Hubicka, J. Krysa, P. Schmuki and R. Zboril, Chem. Soc. Rev., 2017, 46, 3716–3769 RSC.
- M. Ge, J. Cai, J. Iocozzia, C. Cao, J. Huang, X. Zhang, J. Shen, S. Wang, S. Zhang, K. Q. Zhang, Y. Lai and Z. Lin, Int. J. Hydrogen Energy, 2017, 42, 8418–8449 CrossRef CAS.
- W. Fang, M. Xing and J. Zhang, J. Photochem. Photobiol., C, 2017, 32, 21–39 CrossRef CAS.
- R. J. Tayade and D. L. Key, Mater. Sci. Forum, 2010, 657, 62–74 CrossRef CAS.
- W. K. Jo and R. J. Tayade, J. Mater. Eng. Perform., 2016, 25, 83–90 CrossRef CAS.
- K. Natarajan, R. I. Kureshy, H. C. Bajaj and R. J. Tayade, Mater. Sci. Forum, 2016, 855, 45–57 CrossRef.
- T. S. Natarajan, K. Natarajan, H. C. Bajaj and R. J. Tayade, Ind. Eng. Chem. Res., 2011, 50, 7753–7762 CrossRef CAS.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- M. Dahl, Y. Liu and Y. Yin, Chem. Rev., 2014, 114, 9853–9889 CrossRef CAS PubMed.
- M. Kapilashrami, Y. Zhang, Y. S. Liu, A. Hagfeldt and J. Guo, Chem. Rev., 2014, 114, 9662–9707 CrossRef CAS PubMed.
- S. Girish Kumar and L. Gomathi Devi, J. Phys. Chem. A, 2011, 115, 13211–13241 CrossRef PubMed.
- S. Yadav and G. Jaiswar, J. Chin. Chem. Soc., 2017, 64, 103–116 CrossRef CAS.
- H. Liu, L. Yu, W. Chen and Y. Li, J. Nanomater., 2012, 235879 Search PubMed.
- T. S. Natarajan, K. Natarajan, H. C. Bajaj and R. J. Tayade, J. Nanopart. Res., 2013, 15(1669), 1–18 Search PubMed.
- V. Stengl, T. M. Grygar, F. Oplustil and T. Nemec, J. Hazard. Mater., 2012, 227-228, 62–67 CrossRef CAS PubMed.
- M. Lei, N. Wang, L. Zhu, Q. Zhou, G. Nie and H. Tang, Appl. Catal., B, 2016, 182, 414–423 CrossRef CAS.
- M. Hamadanian, A. Reisi-Vanani and A. Majedi, J. Iran. Chem. Soc., 2010, 7, S52–S58 CrossRef CAS.
- R. J. Tayade, H. C. Bajaj and R. V. Jasra, Desalination, 2011, 275, 160–165 CrossRef CAS.
- R. J. Tayade, R. G. Kulkarni and R. V. Jasra, Ind. Eng. Chem. Res., 2006, 45, 5231–5238 CrossRef CAS.
- M. Zhang, J. Wu, D. D. Lu and J. Yang, Int. J. Photoenergy, 2013, 471674 Search PubMed.
- S. G. Kumar and K. S. R. K. Rao, Appl. Surf. Sci., 2016, 391, 124–148 CrossRef.
- L. G. Devi, S. G. Kumar, B. N. Murthy and N. Kottam, Catal. Commun., 2009, 10, 794–798 CrossRef CAS.
- L. Pan, J. Zhang, X. Jia, Y. H. Ma, X. Zhang, L. Wang and J. J. Zou, Chin. J. Catal., 2017, 38, 253–259 CrossRef CAS.
-
H. R. Zhang, H. W. Zheng, Y. Z. Gu, Y. Liang, Y. R. Cao and K. Q. Tan, The 7th National Conference on Functional Materials and Applications, Scientific Research Publishing, USA, 2010, 978-1-935068-41-9.
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
- M. Jahurul Islam, D. Amaranatha Reddy, J. Choi and T. K. Kim, RSC Adv., 2016, 6, 19341–19350 RSC.
- E. C. Kohlrausch, M. J. M. Zapata, R. V. Gonçalves, S. Khan, M. O. Vaz, J. Dupont, S. R. Teixeira and M. J. L. Santos, RSC Adv., 2015, 5, 101276–101286 RSC.
- J. Reszczynska, D. A. Esteban, M. Gazda and A. Zaleska, Physicochem. Probl. Miner. Process., 2014, 50, 515–524 CAS.
- A. R. Ocwelwang and L. Tichagwa, Int. J. Adv. Res. Chem. Sci., 2014, 1, 28–37 Search PubMed.
- O. Jongprateep, R. Puranasamriddhi and J. Palomas, Ceram. Int., 2015, 41, S169–S173 CrossRef CAS.
- S. L. Chung and C. M. Wang, J. Mater. Sci. Technol., 2012, 28, 713–722 Search PubMed.
- U. Balachandran and N. G. Eror, J. Solid State Chem., 1982, 42, 276–282 CrossRef CAS.
- A. L. Bassi, D. Cattaneo, V. Russo, C. E. Bottani, E. Barborini, T. Mazza, P. Piseri, P. Milani, F. O. Ernst, K. Wegner and S. E. Pratsinis, J. Appl. Phys., 2005, 98, 1–9 CrossRef.
- K. Natarajan, H. C. Bajaj and R. J. Tayade, Sol. Energy, 2017, 148, 87–97 CrossRef CAS.
- R. Nainani, P. Thakur. and M. Chaskar, J. Mater. Sci. Eng. B, 2012, 2, 52–58 CAS.
- G. Yang, Z. Jiang, H. Shi, T. Xiao and Z. Yan, J. Mater. Chem., 2010, 20, 5301–5309 RSC.
- D. Chen, Q. Zhu, Z. Lv, X. Deng, F. Zhou and Y. Deng, Mater. Res. Bull., 2012, 47, 3129–3134 CrossRef CAS.
- J. X. Low, B. Cheng and J. Yu, Appl. Surf. Sci., 2017, 392, 658–686 CrossRef CAS.
- M. Janczarek and E. Kowalska, Catalysts, 2017, 7, 1–26 CrossRef.
- W. Zhang, X. Xiao, L. Zheng and C. Wan, Appl. Surf. Sci., 2015, 358, 468–478 CrossRef CAS.
-
S. V. P. Vattikuti and C. Byon, Nanoscaled Films and Layers, 2017, ch. 9, pp. 240–254 Search PubMed.
- T. Yu, L. Liu and F. Yang, Chin. J. Catal., 2017, 38, 270–277 CrossRef CAS.
- T. Xie, L. Xu, C. Liu, J. Yang and M. Wang, Dalton Trans., 2014, 43, 2211–2220 RSC.
- G. Li, N. M. Dimitrijevic, L. Chen, T. Rajh and K. A. Gray, J. Phys. Chem. C, 2008, 112, 19040–19044 CAS.
- G. Kaur, O. P. Pandey and K. Singh, Phys. Status Solidi A, 2012, 209, 1231–1238 CrossRef CAS.
- Q. Xiao, Z. Si, J. Zhang, C. Xiao and X. Tan, J. Hazard. Mater., 2008, 150, 62–67 CrossRef CAS PubMed.
- K. Ishibashi, A. Fujishima, T. Watanabe and K. Hashimoto, Electrochem. Commun., 2000, 2, 207–210 CrossRef CAS.
- C. W. Lai and S. Sreekantan, Electrochim. Acta, 2013, 87, 294–302 CrossRef CAS.
- I. A. Castro, W. Avansi and C. Ribeiro, CrystEngComm, 2014, 16, 1514–1524 RSC.
- A. A. Ismail, I. Abdelfattah, A. Helal, S. A. Al-Sayari, L. Robben and D. W. Bahnemann, J. Hazard. Mater., 2016, 307, 43–54 CrossRef CAS PubMed.
- L. Luo, T. Li, X. Ran, P. Wang and L. Guo, J. Nanomater., 2014, 947289 Search PubMed.
- R. Ma, X. Wang, J. Huang, J. Song, J. Zhang and X. Wang, Vacuum, 2017, 141, 157–165 CrossRef CAS.
- J. Wang, J. Jin, X. Wang, S. Yang, Y. Zhao, Y. Wu, S. Dong, J. Sun and J. Sun, J. Colloid Interface Sci., 2017, 505, 805–815 CrossRef CAS PubMed.
- S. Radhika and J. Thomas, J. Environ. Chem. Eng., 2017, 5, 4239–4250 CrossRef CAS.
- S. Dong, X. Ding, T. Guo, X. Yue, X. Han and J. Sun, Chem. Eng. J., 2017, 316, 778–789 CrossRef CAS.
- A. K. Adepu, R. Anumula and V. Narayanan, Microporous Mesoporous Mater., 2017, 247, 86–94 CrossRef CAS.
- N. Sreelekha, K. Subramanyam, D. A. Reddy, G. Murali, K. R. Varma and R. P. Vijayalakshmi, Solid State Sci., 2016, 62, 71–81 CrossRef CAS.
- Y. Bao, T. T. Lim, Z. Zhong, R. Wang and X. Hu, J. Colloid Interface Sci., 2017, 505, 489–499 CrossRef CAS PubMed.
- R. P. Barkul, M. K. Patil, S. M. Patil, V. B. Shevale and S. D. Delekar, J. Photochem. Photobiol., A, 2017, 349, 138–147 CrossRef CAS.
- D. Ariyanti, M. Maillot and W. Gao, J. Environ. Chem. Eng., 2018, 6, 539–548 CrossRef CAS.
- K. Rokesh, S. ChandraMohan, S. Karuppuchamy and K. Jothivenkatachalam, J. Environ. Chem. Eng. DOI:10.1016/j.jece.2017.01.023.
- S. Kabouch, B. Bellal, Y. Louafi and M. Trari, Mater. Chem. Phys., 2017, 195, 229–235 CrossRef.
|
| This journal is © the Partner Organisations 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ab and
Rajesh J.
Tayade
ab and
Rajesh J.
Tayade