
Synthesis and properties of open-cage fullerene C60 derivatives: impact of the extended π-conjugation†
Yoshifumi
Hashikawa
 ,
Hidefumi
Yasui
,
Kei
Kurotobi
and
Yasujiro
Murata
*
,
Hidefumi
Yasui
,
Kei
Kurotobi
and
Yasujiro
Murata
*
Institute for Chemical Research, Kyoto University, Uji, Kyoto, 611-0011, Japan
First published on 10th November 2017
Abstract
We have developed a method for the synthesis of open-cage fullerene C60 derivatives with extended π-conjugation bearing thienyl groups. By applying this method to an asymmetric diketo derivative, the symmetric form can be obtained without changing the molecular formula. To investigate the structure–property relationship for the asymmetric and symmetric forms, we conducted electrochemical and photophysical measurements. The UV-vis absorption edge was shifted by 210 nm upon changing from the asymmetric to the symmetric form due to the narrower HOMO–LUMO gap, which was also demonstrated by electrochemical analyses. From theoretical calculations, the major contribution of the longest wavelength absorption for the symmetric form is assignable to unusual intramolecular charge transfer transitions whereas π–π* transitions are dominant for the asymmetric form.
Introduction
Fullerene C60 has been widely used as an excellent electron accepting material.1 So far, several fullerene-based electron acceptors have been developed by changing the shape and delocalization degree of the π-conjugated systems on the C60 cage, such as [6,6]-phenyl-C61-butyric acid methyl ester (PC61BM),2 methano-indene-fullerene (MIF)3 and bis(dimethylphenylsilylmethyl)fullerene (SIMEF).4 These methods for the σ-framework modification of C60 are based on the control of the degree of addition and pattern of addends. For further exploration of a variety of fullerene acceptors, it is required to create a new methodology and synthetic direction.We have proposed the potential utility of open-cage C60 derivatives as electron acceptors while they are known as precursors for endohedral fullerenes5 and heterofullerenes.6 In 2011, we demonstrated that open-cage C60 derivatives exhibited an improved open-circuit voltage which resulted in device performances comparable to that of PC61BM.7 In this context, we expected that open-cage C60 derivatives with effective π-conjugation should possess specific electronic and photophysical properties.8,9 The representative examples of π-extended open-cage C60 derivatives are shown in Fig. 1. These compounds can be obtained from C60via the Diels–Alder reactions and subsequent self-photooxygenation as key reactions. The unique π-extension reactions were also reported by Iwamatsu and co-workers, using diamino aryl compounds and open-cage C60 derivatives bearing carbonyl groups.10 However, except for a few reports,8 effects of the extended π-conjugation have not been studied in detail since these compounds have been synthesized as molecular containers.11 Thus, our current interest focuses on the development of a novel approach to synthesize π-extended open-cage C60 derivatives as well as elucidation of the relationship between the π-extended structure and fundamental properties. In this paper, we report the structural modification of open-cage C60 derivatives with extended π-conjugation bearing thienyl groups and discuss the electrochemical and photophysical properties in association with theoretical calculations.
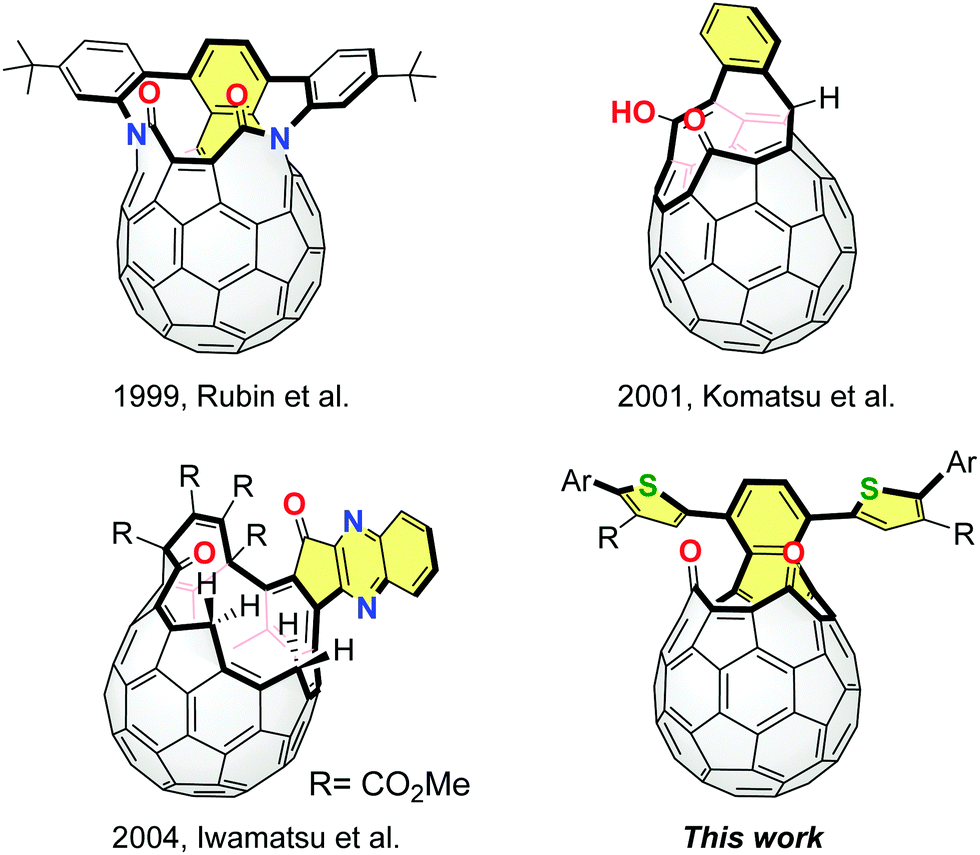 | ||
| Fig. 1 Representative examples of π-extended open-cage C60 derivatives and novel compounds studied herein (this work). | ||
Results and discussion
For effective π-conjugation by the introduction of thiophene rings, we prepared 3,6-bis(4′-n-octylthiophen-2′-yl)pyridazine and 3,6-bis(3′-n-octyl-[2′,2′′-bithiophen]-5′-yl)pyridazine (see the ESI† for detailed synthetic procedures). We successfully obtained the single crystals of 3,6-bis(3′-n-octyl-[2′,2′′-bithiophen]-5′-yl)pyridazine from acetone solution. The single crystal X-ray analysis showed the packing structure co-crystallized with one acetone molecule (Fig. 2) as well as intermolecular hydrophobic interactions among the n-octyl groups (Fig. S26, ESI†).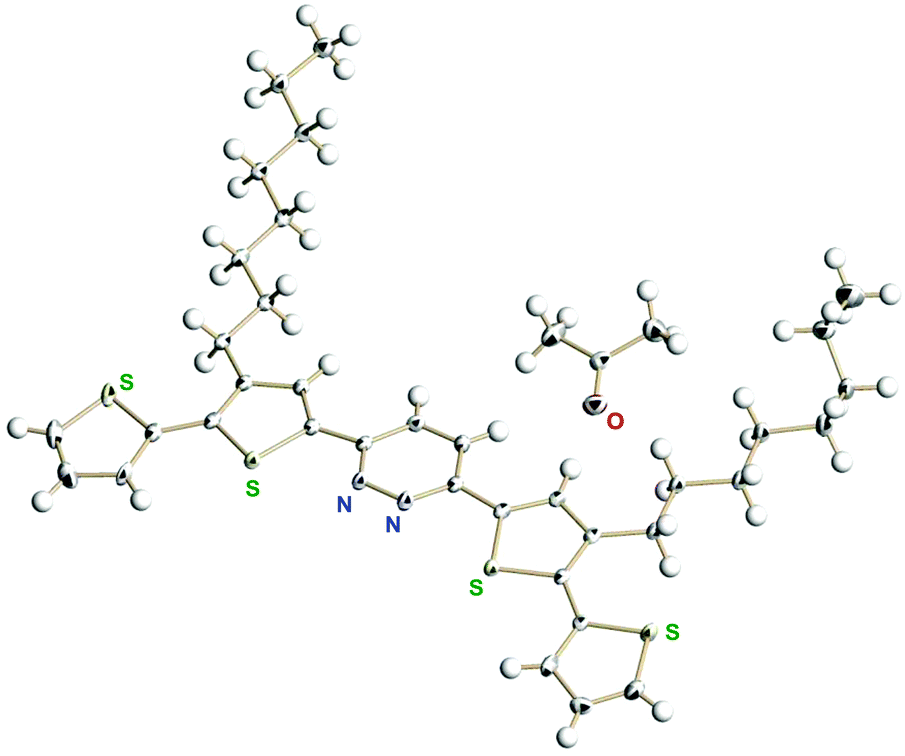 | ||
| Fig. 2 X-Ray structure of 3,6-bis(3′-n-octyl-[2′,2′′-bithiophen]-5′-yl)pyridazine. The disordered parts (two terminal thiophene rings and a part of the n-octyl group) are omitted for clarity. | ||
These pyridazine derivatives underwent the Diels–Alder reaction with C60 followed by extrusion of N2, intramolecular [4+4] cyclization and a retro [4+2] reaction to yield open-cage C60 derivatives 1 bearing thiophene rings (Scheme 1). Subsequent self-photooxygenation of 1 gave corresponding asymmetric diketo derivatives 2. As we have previously reported, diketo derivatives similar to 2 can be converted into structural analogues of 1 by the carbonyl coupling reaction using an excessive amount of phosphite or phosphine.5c,6b However, the stoichiometric reaction of 2 and triisopropyl phosphite resulted in the in-situ formation of epoxy derivatives 3. The treatment of 3a and 3b with sodium acetate in the presence of CuCl and subsequent hydrolysis afforded diol derivatives 4a and 4b, respectively. In this reaction, the cleavage of the epoxy moieties of 3 should be assisted by the nucleophilic attack of the acetoxy anions on the C4-atom where the LUMO (lowest unoccupied molecular orbital) is partially localized (Fig. S27, ESI†). Finally, diketo derivatives 5 were found to be obtained in the symmetric form by the photochemical oxidation of 4 in the presence of (diacetoxyiodo)benzene and iodine.12 This reaction was driven by the formation of the oxy radicals of 4 and re-aromatization to induce a (dithienyl)naphthalene substructure in 5 (coloured with yellow in Scheme 1). It should be noted that the thus obtained compounds 5a and 5b are regarded as structural isomers of 2a and 2b, respectively.
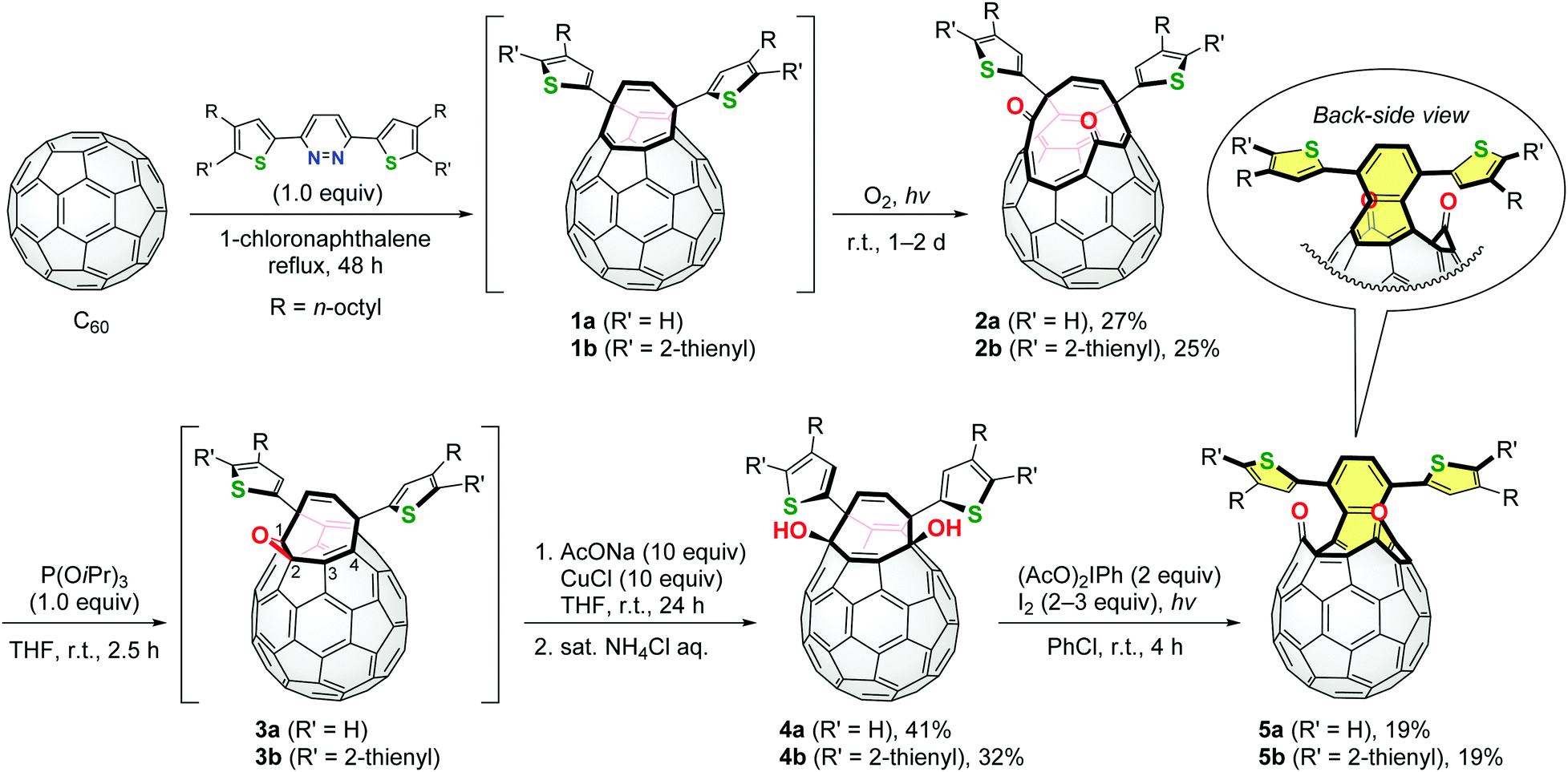 | ||
| Scheme 1 Synthesis of open-cage C60 derivatives 5a and 5b with extended π-conjugation. | ||
To verify the formation of the naphthalene substructures in 5, we measured 1H NMR spectra of 5a together with its structural isomer 2a. As shown in Fig. 3, the vinylene protons of asymmetric 2a appeared at δ 6.91 and 6.95 ppm as doublet signals with a coupling constant of 9.7 Hz in CD2Cl2 at a field strength of 500 MHz. These proton signals became one singlet in 5a reflecting the higher molecular symmetry and were downfield-shifted to δ 7.81 ppm by Δδ +0.88 ppm with respect to the vinylene protons of 2a. The similar down-field shift (Δδ +0.83 ppm) was also observed for asymmetric 2b (δ 6.97, 7.06 ppm, doublet, J = 9.8 Hz for vinylene, CDCl3) and symmetric 5b (δ 7.84 ppm for phenylene, CDCl3). These results indicated that the deshielding effect on these protons (coloured with blue in Fig. 3) should be ascribed to the construction of the naphthalene substructures in 5. These experimental results can be well-explained in terms of frontier orbitals (Fig. 6) and NICS(0) values (Fig. S34, ESI†).
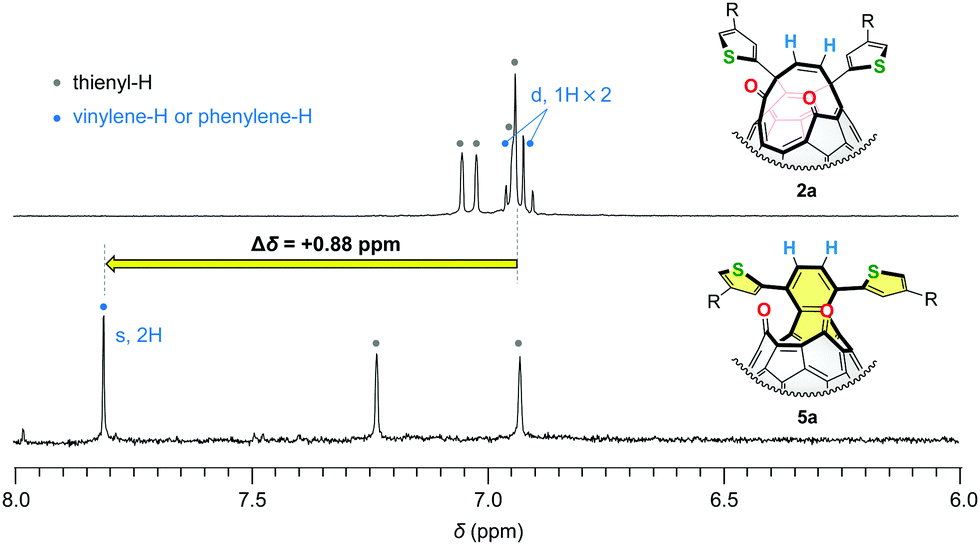 | ||
| Fig. 3 1H NMR spectra of 2a and 5a (500 MHz, CD2Cl2). | ||
The electrochemical properties of C60, 2a, 5a and 5b were also studied by cyclic voltammetry (CV). The measurements were conducted using 1.0 mM samples with 0.10 M n-Bu4N·BF4 as a supporting electrolyte in benzonitrile at a scan rate of 20 mV s−1 (Fig. 4). Whereas the asymmetric diketo derivative 2a has an irreversible oxidation potential comparable to C60 (Epa +1.28 V for each compound), it exhibited a reversible reduction wave at E1/2 −0.81 V which is anodically shifted by ΔE +0.09 V with reference to that of C60. This originates from the introduction of two carbonyl groups in 2a. Surprisingly, the oxidation and reduction potentials of structural isomer 5a were significantly shifted to Epa +0.84 V (ΔE −0.44 V vs. C60) and E1/2 −0.58 V (ΔE +0.32 V vs. C60), respectively. This consequently led to the narrower HOMO–LUMO gap of 5a compared to that of 2a. The additional extension of the conjugated π-systems in 5b bearing two thienylthiophene groups resulted in further cathodic shift of the oxidation potential (Epa +0.67 V) as well as anodic shift of the reduction potential (E1/2 −0.54 V). This structural modification retaining the original chemical formula, i.e., the transformation of asymmetric 2 into symmetric 5, has a drastic effect on their electronic properties.
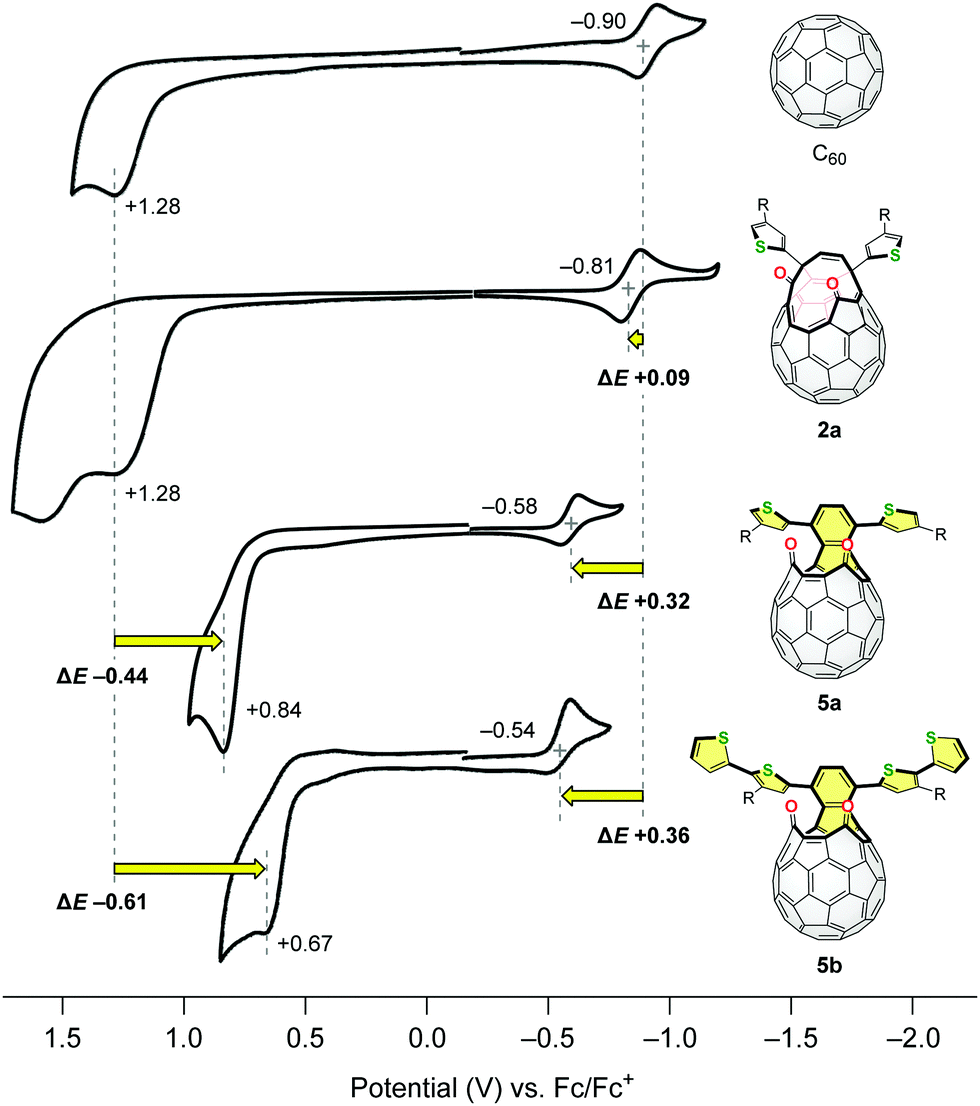 | ||
| Fig. 4 Cyclic voltammograms of C60, 2a, 5a and 5b using 1.0 mM samples with 0.10 M n-Bu4N·BF4 in benzonitrile at a scan rate of 20 mV s−1. | ||
The UV-vis absorption spectra of C60, 2a, 5a and 5b were recorded using 100 μM solutions in chloroform (Fig. 5a). Compared with C60, the absorption edge of asymmetric 2a was shifted by Δλedge +60 nm. In the case of symmetric 5a, a remarkable bathochromic shift (Δλedge +270 nm vs. C60) was observed with increasing absorption coefficients. π-Extended 5b exhibited an intense absorption over the measured range with further bathochromic shift (Δλedge +350 nm vs. C60). These considerable shifts of long wavelength absorption with enhanced absorption coefficients imply that the narrower HOMO–LUMO gaps as well as the π-conjugation extended to the thienyl groups through the naphthalene moieties in 5. These results are in good agreement with CV measurements. We subsequently examined the solvent effect on the UV-vis absorption behaviour of 5a. As depicted in Fig. 5b, the absorption band at around 740 nm in benzonitrile was appreciably broadened and the edge reached 1220 nm although the absorption spectra in toluene and chloroform showed a negligible change. These results suggest that the excited states of 5a are notably stabilized in polar solvents.
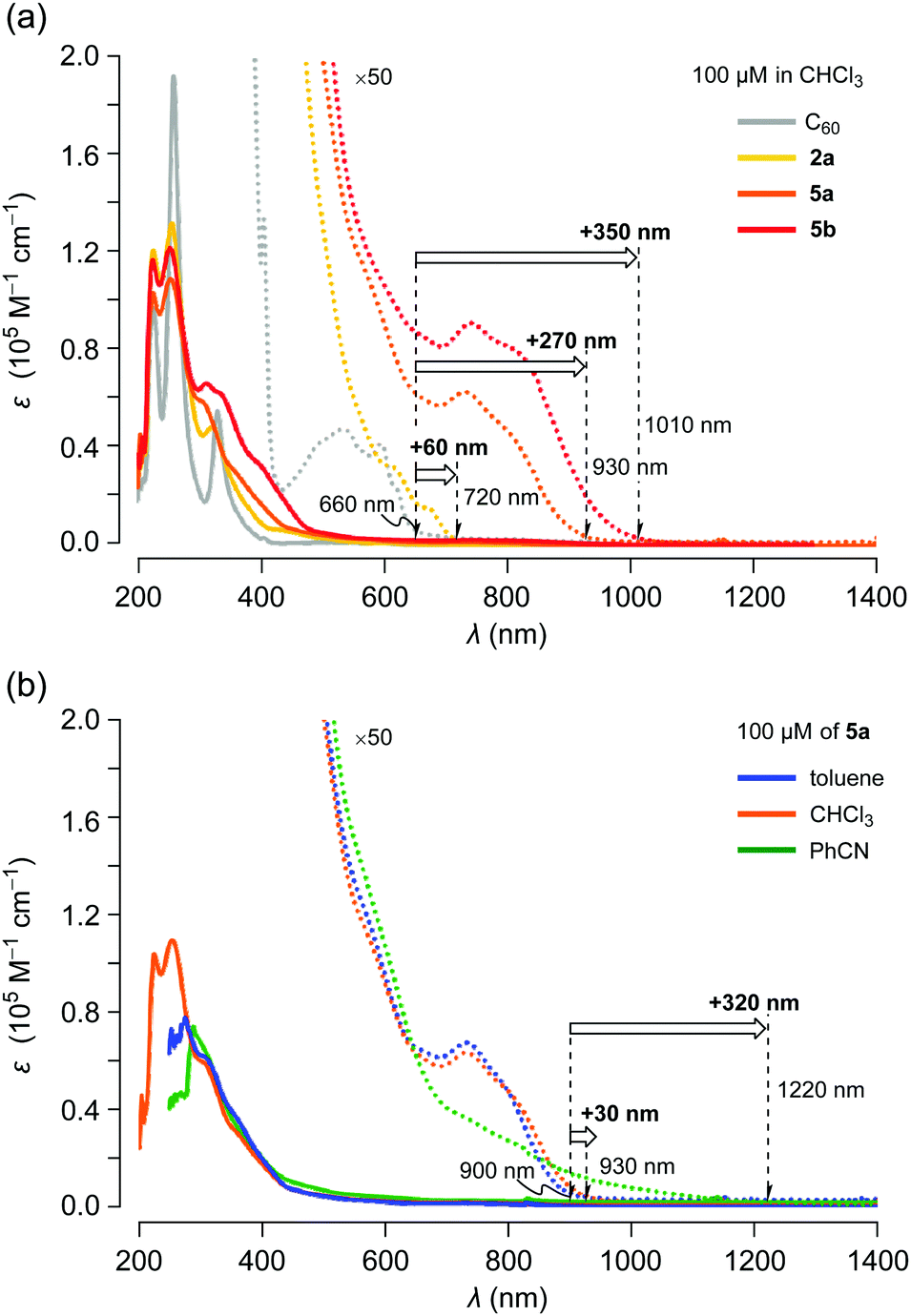 | ||
| Fig. 5 UV-vis absorption spectra for 100 μM solutions of (a) C60, 2a, 5a and 5b in chloroform and (b) 5a in toluene, chloroform and benzonitrile. | ||
For further understanding of the electrochemical and photophysical properties, we performed TD-DFT calculations (Fig. 6).13 The n-octyl groups in 2a, 5a and 5b were replaced with the methyl groups for the calculations to be simplified. The structures of 2a′, 5a′ and 5b′ were optimized at the B3LYP/6-31G(d) level of theory. Using these optimized structures, we calculated the optical transitions with oscillator strengths at the TD CAM-B3LYP/6-31G(d) level of theory. The transition energies were calibrated by using a factor of 0.75. Although the HOMO level of asymmetric 2a′ (−5.93 eV) is comparable to that of C60 (−5.99 eV),14 symmetric 5a′ and 5b′ have elevated HOMO levels of −5.36 and −5.10 eV, respectively. According to the computational results, the HOMO of 2a′ is distributed on the C60 skeleton. However, frontier orbitals including the HOMO and HOMO−4 for 5a′ are localized on the dithienylnaphthalene moiety, which originates from the HOMO and HOMO−1 of 1,4-bis(4′-methylthiophen-2′-yl)naphthalene, respectively (Fig. S31, ESI†). Likewise, the HOMO and HOMO−1 of 5b′ are related to those for 1,4-bis(4′-methyl-5′-thienylthiophen-2′-yl)naphthalene (Fig. S32, ESI†). In contrast to the HOMOs, the LUMOs of 2a′, 5a′ and 5b′ are distributed on the whole of the C60 skeleton covering the two carbonyl groups and their energy levels become deeper in the order of C60 (−3.23 eV), 2a′ (−3.38 eV), 5a′ (−3.53 eV) and 5b′ (−3.55 eV). These results are well-reproducible for the experimental data obtained from cyclic voltammograms.
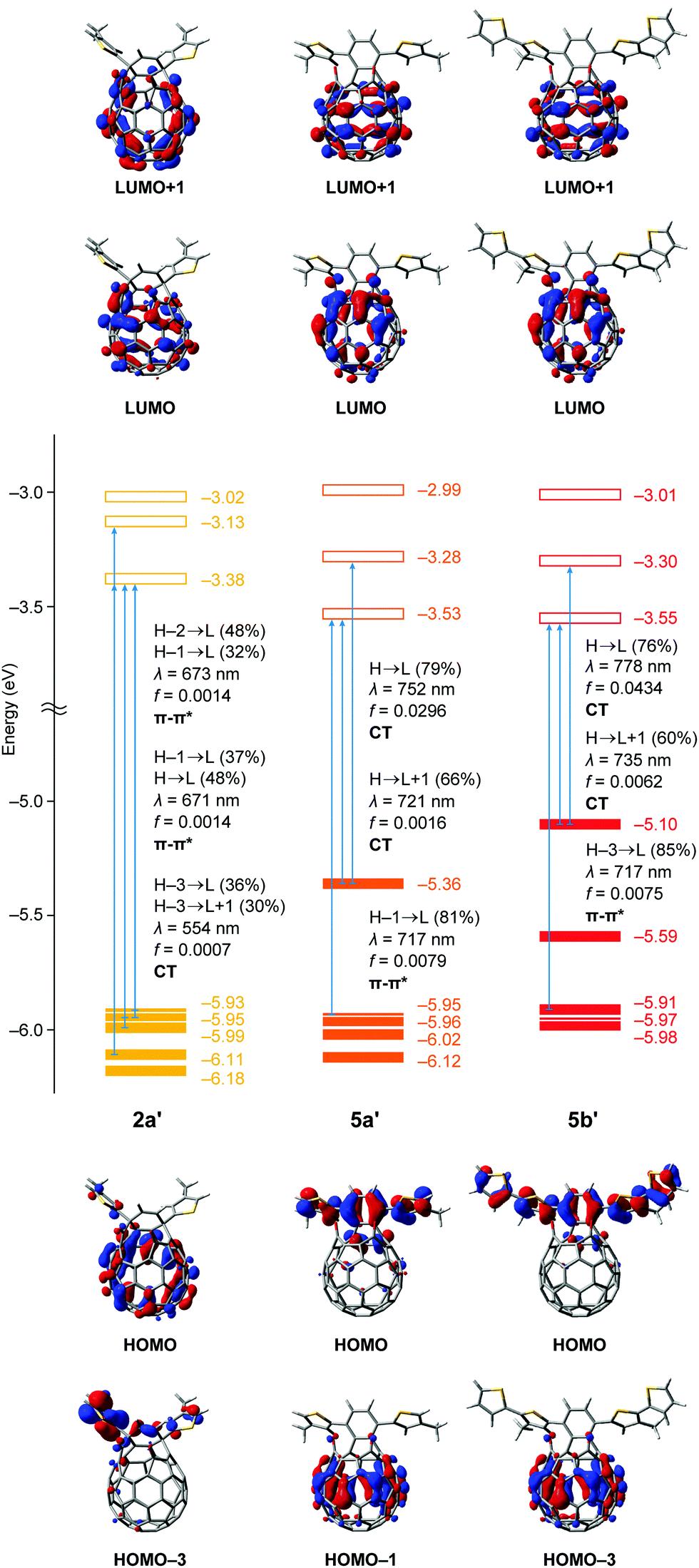 | ||
| Fig. 6 Pictorial representation of frontier orbitals with a plot of the Kohn–Sham HOMO and LUMO levels (B3LYP/6-31G(d)) and optical transitions with oscillator strengths (TD CAM-B3LYP/6-31G(d)//B3LYP/6-31G(d)) of 2a′, 5a′ and 5b′. The transition energies were calibrated by using a factor of 0.75. The n-octyl groups in 2a, 5a and 5b were replaced with the methyl groups in 2a′, 5a′ and 5b′. | ||
For the absorption behaviour of 2a′, the longest wavelength absorption bands at 673 and 671 nm are attributed to the optical transitions including the HOMO−2 to LUMO, HOMO−1 to LUMO and HOMO to LUMO with oscillator strengths of 0.0014. In terms of the frontier orbitals of 2a′, these transitions are regarded as π–π* transitions whereas the intramolecular charge transfer (ICT) transitions corresponding to the HOMO−3 to LUMO (contribution: 36%) and HOMO−3 to LUMO+1 (30%) have a smaller oscillator strength of 0.0007 at 554 nm. Interestingly, symmetric 5 exhibits a unique optical transition character different from asymmetric 2. The longest wavelength absorptions of 5a′ (λ = 752 and 721 nm) and 5b′ (λ = 778 and 735 nm) are mainly based on the ICT transitions corresponding to the HOMO to LUMO and HOMO to LUMO+1 in each case. The oscillator strength of the HOMO to LUMO transition for symmetric 5a′ is 0.0296 which is considerably larger than that for asymmetric 2a′ (f = 0.0007). By introduction of the additional thiophene rings, the oscillator strength further increases up to 0.0434 for 5b′. These results are in good accordance with the UV-vis measurements (Fig. 5) and the two observed longest wavelength absorption bands (741 and 807 nm for 5a and 752 and 846 nm for 5b) should be ascribed to ICT transitions of the HOMO to LUMO+1 and HOMO to LUMO, respectively. This stands in contrast to 2a with the π–π* transition character which can rationally explain the longest wavelength absorption bands at 625 and 683 nm observed in the UV-vis absorption spectrum (Fig. 5). From these experimental and computational results, we concluded that the extended π-conjugation in 5 should effectively enhance the contribution of the characteristic ICT transitions toward the bathochromically shifted absorption with intense coefficients.
Conclusions
In summary, we have developed a method for the synthesis of open-cage C60 derivatives possessing extended π-systems. The electrochemical and photophysical properties were drastically influenced just by changing the molecular symmetry retaining the molecular formula. The CV measurements demonstrated that symmetric 5a has a narrower HOMO–LUMO gap than 2a, which resulted in a longer wavelength absorption in the UV-vis spectra. Interestingly, TD-DFT calculations suggested that the longest wavelength absorption is attributed to π–π* transitions for 2a′ but ICT transitions for 5a′ and 5b′ with larger oscillator strengths. Thus, the effective π-conjugation can be considered to impart a new function to open-cage C60 derivatives as n-type materials with intense absorption coefficients covering a much longer wavelength region.Experimental
Instrumentation and materials
Melting points were determined on a Yanaco MP-500D apparatus. The 1H and 13C NMR measurements were carried out at room temperature using Varian MERCURY 300 and JEOL JNM ECA500 instruments. The NMR chemical shifts were reported in ppm with reference to residual protons and carbons of CDCl3 (δ 7.26 ppm in 1H NMR, δ 77.00 ppm in 13C NMR), CD2Cl2 (δ 5.32 ppm in 1H NMR), and C6D6 (δ 7.15 ppm in 1H NMR, δ 128.00 ppm in 13C NMR). APCI mass spectra were measured on a JEOL MStation JMS-700 and a Bruker micrOTOF-Q II. UV-vis absorption spectra were measured using a Shimadzu UV-3150 spectrometer. Cyclic voltammetry was conducted on a BAS Electrochemical Analyzer ALS620C using a three-electrode cell with a glassy carbon working electrode, a platinum wire counter electrode, and an Ag/AgNO3 reference electrode. The measurements were carried out under a N2 atmosphere using benzonitrile solutions of 1.0 mM samples and 0.10 M tetrabutylammonium tetrafluoroborate (n-Bu4N·BF4) as a supporting electrolyte. The redox potentials were calibrated with ferrocene used as an internal standard which was added after each measurement. High-performance liquid chromatography (HPLC) was performed with the use of a Cosmosil Buckyprep column (250 mm in length, 4.6 mm in inner diameter) for analytical purposes and the same columns (two directly connected columns; 250 mm in length, 20 mm in inner diameter) for preparative purposes. Thin layer chromatography (TLC) was performed on glass plates coated with 0.25 mm thick silica gel 60F-254 (Merck). Column chromatography was performed using PSQ 60B or PSQ 100B (Fuji Silysia).All reactions were carried out under an Ar atmosphere. Unless otherwise noted, materials purchased from commercial suppliers were used without further purification.
General procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)) afforded 3,6-bis(4′-n-octylthiophen-2′-yl)pyridazine as a yellow powder (3.03 g, 6.46 mmol, 52% based on 3-octylthiophene).
:2)) gave 3,6-bis(5′-bromo-4′-n-octylthiophen-2′-yl)pyridazine as a white powder (1.79 g, 2.86 mmol, 89%).
:2)) gave 3,6-bis(3′-n-octyl-[2′,2′′-bithiophen]-5′-yl)pyridazine as a yellow powder (6.22 g, 9.83 mmol, 62% based on 2-bromothiophene).
:1)) gave 2a as dark brown solids (241 mg, 0.208 mmol, 28%).
:1)) afforded 3,6-bis(4′-n-octylthiophen-2′-yl)pyridazine as a yellow powder (3.03 g, 6.46 mmol, 52% based on 3-octylthiophene).
:2)) gave 3,6-bis(5′-bromo-4′-n-octylthiophen-2′-yl)pyridazine as a white powder (1.79 g, 2.86 mmol, 89%).
:2)) gave 3,6-bis(3′-n-octyl-[2′,2′′-bithiophen]-5′-yl)pyridazine as a yellow powder (6.22 g, 9.83 mmol, 62% based on 2-bromothiophene).
:1)) gave 2a as dark brown solids (241 mg, 0.208 mmol, 28%).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 1H NMR (300 MHz, CDCl3) δ 7.07 (s, 1H, thienyl), 7.00 (d, J = 9.9 Hz, 1H, vinyl), 6.97 (s, 1H, thienyl), 6.91 (d, J = 9.9 Hz, 1H, vinyl), 6.90 (s, 2H, thienyl), 2.59–2.53 (m, 4H, CH2), 1.60–1.51 (m, 4H, CH2), 1.28–1.26 (m, 20H, CH3), 0.89–0.84 (m, 6H, CH3); 13C NMR (75 MHz, CDCl3) δ 199.20, 191.56, 153.40, 153.32, 150.26, 148.98, 148.46, 148.13, 148.03, 147.61, 147.16, 146.61, 146.43, 146.28, 146.15, 146.05, 145.97, 145.85, 145.58, 145.31, 145.08, 145.04, 144.87, 144.62, 144.30, 144.12, 143.65, 143.22, 142.88, 142.81, 142.56, 142.31, 142.25, 141.84, 141.32, 140.97, 140.59, 140.14, 139.92, 139.71, 139.11, 138.68, 137.77, 137.51, 136.56, 136.35, 135.71, 134.44, 133.58, 132.59, 132.18, 131.97, 131.50, 130.92, 130.70, 129.46, 127.22, 120.94, 120.39, 55.78, 49.41, 32.08, 30.86, 30.72, 30.40, 29.63, 29.55, 29.49, 22.90, 14.36; HRMS (FAB, positive ion mode) calcd for C88H40O2S2 (M + H+) 1193.2550, found 1193.2546.
:1)) gave 2b as dark brown solids (948 mg, 0.715 mmol, 26%).
O); 1H NMR (300 MHz, CDCl3) δ 7.30–7.27 (m, 2H, thienyl), 7.10–7.09 (m, 3H, thienyl), 7.06 (d, J = 9.9 Hz, 1H, vinyl), 7.04–7.01 (m, 2H, thienyl), 6.97 (d, J = 9.8 Hz, 1H, vinyl), 6.97 (s, 1H, thienyl), 2.75–2.67 (m, 4H, CH2), 1.62–1.55 (m, 4H, CH2), 1.25–1.22 (m, 20H, CH3), 0.90–0.83 (m, 6H, CH3); 13C NMR (75 MHz, CDCl3) δ 198.87, 191.55, 152.96, 151.48, 150.24, 148.96, 148.12, 148.00, 147.85, 147.59, 147.13, 146.61, 146.48, 146.36, 146.28, 146.11, 146.03, 145.99, 145.93, 145.85, 145.57, 145.32, 145.03, 144.87, 144.62, 144.26, 144.11, 143.19, 142.86, 142.82, 142.57, 142.34, 142.28, 141.84, 141.36, 140.92, 140.56, 140.31, 140.20, 140.16, 139.88, 139.83, 139.75, 139.35, 139.25, 138.73, 137.68, 137.50, 136.59, 136.29, 135.77, 135.72, 134.16, 133.58, 132.47, 132.04, 131.89, 131.81, 131.46, 130.95, 130.83, 128.69, 127.60, 126.55, 126.40, 125.90, 125.78, 55.66, 49.35, 32.09, 32.06, 30.78, 30.71, 29.80, 29.70, 29.62, 29.52, 29.50, 22.9, 14.38; HRMS (APCI, negative ion mode) calcd for C96H44O2S4 (M˙−) 1356.2230, found 1356.2210.
:1)) δ 7.28 (d, J = 2.1 Hz, 2H, thienyl), 6.99 (d, J = 1.5 Hz, 2H, thienyl), 6.94 (d, J = 5.4 Hz, 2H, thienyl), 6.78 (dd, J = 2.1, 5.4 Hz, 2H, thienyl) 6.68 (s, 2H, vinyl) 3.90 (s, 2H, alcohol) 2.64 (t, J = 7.4 Hz, 4H, CH2), 1.54–1.49 (m, 4H, CH2), 1.19 (m, 20H, CH2), 0.87–0.83 (t, J = 6.5 Hz, 6H, CH3); 13C NMR (150 MHz, CS2/C6D6 (1:1)) δ 149.90, 148.76, 148.09, 147.21, 146.54, 146.36, 146.20, 146.02, 145.75, 145.68, 145.63, 145.26, 145.10, 145.03, 144.79, 144.59, 144.23, 143.79, 143.47 142.65, 142.21, 141.37, 141.12, 139.93, 139.52, 139.37, 139.24, 138.66, 137.33, 136.52, 135.38, 133.54, 133.25, 132.67,130.70, 126.66, 126.11, 88.90, 50.27, 32.85, 31.63, 30.56, 30.43, 30.36, 30.16, 23.77, 15.06; HRMS (FAB, positive ion mode) calcd for C96H46O2S4 (M˙+) 1358.2381, found 1358.2340.
:1)) gave 5a as dark brown solids (6.6 mg, 0.0055 mmol, 19%).
O); 1H NMR (300 MHz, CDCl3) δ 7.84 (s, 2H, phenyl), 7.22 (s, 2H, thienyl), 6.89 (s, 2H, thienyl), 2.53 (t, J = 7.4 Hz, 4H, CH2), 1.50–1.49 (m, 4H, CH2), 1.25–1.19 (m, 20H, CH2), 0.84 (t, J = 6.5 Hz, 6H, CH3); 13C NMR (75 MHz, CDCl3) δ 184.83, 149.42, 149.19, 148.61, 148.49, 147.55, 146.46, 145.53, 145.30, 145.09, 145.02, 144.72, 144.38, 144.35, 144.09, 143.65, 143.53, 143.11, 142.90, 141.51, 140.56, 140.42, 139.78, 139.46, 138.41, 137.41, 136.25, 135.90, 135.84, 134.95, 131.96, 130.62, 129.62, 121.16, 32.13, 30.68, 30.51, 29.69, 29.52, 29.36, 22.92, 14.39; HRMS (FAB, positive ion mode) calcd for C88H40O2S2 (M + H+) 1193.2550, found 1193.2533.
:1)) and then HPLC equipped with a Buckyprep column (toluene) gave 5b as dark brown solids (1.8 mg, 0.0013 mmol, 19%).
O); 1H NMR (300 MHz, CD2Cl2) δ 7.84 (s, 2H, phenyl), 7.31 (d, J = 5.0 Hz, 2H, thienyl), 7.20 (s, 2H, thienyl), 7.14 (d, J = 3.0 Hz, 2H, thienyl), 7.06 (dd, J = 3.0, 5.0 Hz, 4H, thienyl), 2.75–2.65 (m, 4H, CH2), 1.50 (m, 4H, CH2), 1.20 (m, 20H, CH2), 0.84 (t, J = 6.4 Hz, 6H, CH3); 13C NMR (150 MHz, CD2Cl2) δ 184.62, 149.24, 149.02, 148.43, 148.30, 147.36, 146.47, 145.36, 145.10, 144.92, 144.85, 144.55, 144.20, 143.92, 143.50, 143.37, 142.71, 141.91, 141.35, 140.39, 140.28, 139.68, 139.30, 139.13, 138.27, 137.33, 136.12, 136.04, 135.53, 135.33, 134.86, 131.67, 131.57, 130.99, 130.25, 127.33, 125.81, 125.20, 77.23, 77.02, 76.81, 31.94, 30.41, 29.51, 29.34, 29.29, 29.23, 22.72, 14.19; HRMS (APCI, negative ion mode) calcd for C96H44O2S4 (M˙−) 1356.2230, found 1356.2180.
O); 1H NMR (300 MHz, CDCl3) δ 7.07 (s, 1H, thienyl), 7.00 (d, J = 9.9 Hz, 1H, vinyl), 6.97 (s, 1H, thienyl), 6.91 (d, J = 9.9 Hz, 1H, vinyl), 6.90 (s, 2H, thienyl), 2.59–2.53 (m, 4H, CH2), 1.60–1.51 (m, 4H, CH2), 1.28–1.26 (m, 20H, CH3), 0.89–0.84 (m, 6H, CH3); 13C NMR (75 MHz, CDCl3) δ 199.20, 191.56, 153.40, 153.32, 150.26, 148.98, 148.46, 148.13, 148.03, 147.61, 147.16, 146.61, 146.43, 146.28, 146.15, 146.05, 145.97, 145.85, 145.58, 145.31, 145.08, 145.04, 144.87, 144.62, 144.30, 144.12, 143.65, 143.22, 142.88, 142.81, 142.56, 142.31, 142.25, 141.84, 141.32, 140.97, 140.59, 140.14, 139.92, 139.71, 139.11, 138.68, 137.77, 137.51, 136.56, 136.35, 135.71, 134.44, 133.58, 132.59, 132.18, 131.97, 131.50, 130.92, 130.70, 129.46, 127.22, 120.94, 120.39, 55.78, 49.41, 32.08, 30.86, 30.72, 30.40, 29.63, 29.55, 29.49, 22.90, 14.36; HRMS (FAB, positive ion mode) calcd for C88H40O2S2 (M + H+) 1193.2550, found 1193.2546.
:1)) gave 2b as dark brown solids (948 mg, 0.715 mmol, 26%).
O); 1H NMR (300 MHz, CDCl3) δ 7.30–7.27 (m, 2H, thienyl), 7.10–7.09 (m, 3H, thienyl), 7.06 (d, J = 9.9 Hz, 1H, vinyl), 7.04–7.01 (m, 2H, thienyl), 6.97 (d, J = 9.8 Hz, 1H, vinyl), 6.97 (s, 1H, thienyl), 2.75–2.67 (m, 4H, CH2), 1.62–1.55 (m, 4H, CH2), 1.25–1.22 (m, 20H, CH3), 0.90–0.83 (m, 6H, CH3); 13C NMR (75 MHz, CDCl3) δ 198.87, 191.55, 152.96, 151.48, 150.24, 148.96, 148.12, 148.00, 147.85, 147.59, 147.13, 146.61, 146.48, 146.36, 146.28, 146.11, 146.03, 145.99, 145.93, 145.85, 145.57, 145.32, 145.03, 144.87, 144.62, 144.26, 144.11, 143.19, 142.86, 142.82, 142.57, 142.34, 142.28, 141.84, 141.36, 140.92, 140.56, 140.31, 140.20, 140.16, 139.88, 139.83, 139.75, 139.35, 139.25, 138.73, 137.68, 137.50, 136.59, 136.29, 135.77, 135.72, 134.16, 133.58, 132.47, 132.04, 131.89, 131.81, 131.46, 130.95, 130.83, 128.69, 127.60, 126.55, 126.40, 125.90, 125.78, 55.66, 49.35, 32.09, 32.06, 30.78, 30.71, 29.80, 29.70, 29.62, 29.52, 29.50, 22.9, 14.38; HRMS (APCI, negative ion mode) calcd for C96H44O2S4 (M˙−) 1356.2230, found 1356.2210.
:1)) δ 7.28 (d, J = 2.1 Hz, 2H, thienyl), 6.99 (d, J = 1.5 Hz, 2H, thienyl), 6.94 (d, J = 5.4 Hz, 2H, thienyl), 6.78 (dd, J = 2.1, 5.4 Hz, 2H, thienyl) 6.68 (s, 2H, vinyl) 3.90 (s, 2H, alcohol) 2.64 (t, J = 7.4 Hz, 4H, CH2), 1.54–1.49 (m, 4H, CH2), 1.19 (m, 20H, CH2), 0.87–0.83 (t, J = 6.5 Hz, 6H, CH3); 13C NMR (150 MHz, CS2/C6D6 (1:1)) δ 149.90, 148.76, 148.09, 147.21, 146.54, 146.36, 146.20, 146.02, 145.75, 145.68, 145.63, 145.26, 145.10, 145.03, 144.79, 144.59, 144.23, 143.79, 143.47 142.65, 142.21, 141.37, 141.12, 139.93, 139.52, 139.37, 139.24, 138.66, 137.33, 136.52, 135.38, 133.54, 133.25, 132.67,130.70, 126.66, 126.11, 88.90, 50.27, 32.85, 31.63, 30.56, 30.43, 30.36, 30.16, 23.77, 15.06; HRMS (FAB, positive ion mode) calcd for C96H46O2S4 (M˙+) 1358.2381, found 1358.2340.
:1)) gave 5a as dark brown solids (6.6 mg, 0.0055 mmol, 19%).
O); 1H NMR (300 MHz, CDCl3) δ 7.84 (s, 2H, phenyl), 7.22 (s, 2H, thienyl), 6.89 (s, 2H, thienyl), 2.53 (t, J = 7.4 Hz, 4H, CH2), 1.50–1.49 (m, 4H, CH2), 1.25–1.19 (m, 20H, CH2), 0.84 (t, J = 6.5 Hz, 6H, CH3); 13C NMR (75 MHz, CDCl3) δ 184.83, 149.42, 149.19, 148.61, 148.49, 147.55, 146.46, 145.53, 145.30, 145.09, 145.02, 144.72, 144.38, 144.35, 144.09, 143.65, 143.53, 143.11, 142.90, 141.51, 140.56, 140.42, 139.78, 139.46, 138.41, 137.41, 136.25, 135.90, 135.84, 134.95, 131.96, 130.62, 129.62, 121.16, 32.13, 30.68, 30.51, 29.69, 29.52, 29.36, 22.92, 14.39; HRMS (FAB, positive ion mode) calcd for C88H40O2S2 (M + H+) 1193.2550, found 1193.2533.
:1)) and then HPLC equipped with a Buckyprep column (toluene) gave 5b as dark brown solids (1.8 mg, 0.0013 mmol, 19%).
O); 1H NMR (300 MHz, CD2Cl2) δ 7.84 (s, 2H, phenyl), 7.31 (d, J = 5.0 Hz, 2H, thienyl), 7.20 (s, 2H, thienyl), 7.14 (d, J = 3.0 Hz, 2H, thienyl), 7.06 (dd, J = 3.0, 5.0 Hz, 4H, thienyl), 2.75–2.65 (m, 4H, CH2), 1.50 (m, 4H, CH2), 1.20 (m, 20H, CH2), 0.84 (t, J = 6.4 Hz, 6H, CH3); 13C NMR (150 MHz, CD2Cl2) δ 184.62, 149.24, 149.02, 148.43, 148.30, 147.36, 146.47, 145.36, 145.10, 144.92, 144.85, 144.55, 144.20, 143.92, 143.50, 143.37, 142.71, 141.91, 141.35, 140.39, 140.28, 139.68, 139.30, 139.13, 138.27, 137.33, 136.12, 136.04, 135.53, 135.33, 134.86, 131.67, 131.57, 130.99, 130.25, 127.33, 125.81, 125.20, 77.23, 77.02, 76.81, 31.94, 30.41, 29.51, 29.34, 29.29, 29.23, 22.72, 14.19; HRMS (APCI, negative ion mode) calcd for C96H44O2S4 (M˙−) 1356.2230, found 1356.2180.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was partially provided by the JSPS KAKENHI Grant Number JP15H00993, JP17H05152, JP17H06119 and JP17H06789.References
- T. Torres and G. Bottari, Organic Nanomaterials: Synthesis, Characterization, and Device Applications, John Wiley & Sons, West Sussex, UK, 2013 Search PubMed.
- J. C. Hummelen, B. W. Knight, F. LePeq, F. Wudl, J. Yao and C. L. Wilkins, J. Org. Chem., 1995, 60, 532 CrossRef CAS.
- Y. Matsuo, J. Kawai, H. Inada, T. Nakagawa, H. Ota, S. Otsubo and E. Nakamura, Adv. Mater., 2013, 25, 6266 CrossRef CAS PubMed.
- (a) Y. Matsuo, A. Iwashita, Y. Abe, C.-Z. Li, K. Matsuo, M. Hashiguchi and E. Nakamura, J. Am. Chem. Soc., 2008, 130, 15429 CrossRef CAS PubMed; (b) Y. Matsuo, Y. Sato, T. Niinomi, I. Soga, H. Tanaka and E. Nakamura, J. Am. Chem. Soc., 2009, 131, 16048 CrossRef CAS PubMed.
- (a) K. Komatsu, M. Murata and Y. Murata, Science, 2005, 307, 238 CrossRef CAS PubMed; (b) Y. Morinaka, F. Tanabe, M. Murata, Y. Murata and K. Komatsu, Chem. Commun., 2010, 46, 4532 RSC; (c) K. Kurotobi and Y. Murata, Science, 2011, 333, 613 CrossRef CAS PubMed; (d) A. Krachmalnicoff, R. Bounds, S. Mamone, S. Alom, M. Concistrè, B. Meier, K. Kouřil, M. E. Light, M. R. Johnson, S. Rols, A. J. Horsewill, A. Shugai, U. Nagel, T. Rõõm, M. Carravetta, M. H. Levitt and R. J. Whitby, Nat. Chem., 2016, 8, 953 CrossRef CAS PubMed.
- (a) J. C. Hummelen, B. Knight, J. Pavlovich, R. González and F. Wudl, Science, 1995, 269, 1554 CAS; (b) Y. Hashikawa, M. Murata, A. Wakamiya and Y. Murata, Org. Lett., 2014, 16, 2970 CrossRef CAS PubMed; (c) Y. Hashikawa, M. Murata, A. Wakamiya and Y. Murata, J. Am. Chem. Soc., 2016, 138, 4096 CrossRef CAS PubMed.
- M. Murata, Y. Morinaka, Y. Murata, O. Yoshikawa, T. Sagawa and S. Yoshikawa, Chem. Commun., 2011, 47, 7335 RSC.
- (a) Z. Xiao, G. Ye, Y. Liu, S. Chen, Q. Peng, Q. Zuo and L. Ding, Angew. Chem., Int. Ed., 2012, 51, 9038 CrossRef CAS PubMed; (b) Y. Yu, L. Xu, X. Huang and L. Gan, J. Org. Chem., 2014, 79, 2156 CrossRef CAS PubMed.
- (a) G. Schick, T. Jarrosson and Y. Rubin, Angew. Chem., Int. Ed., 1999, 38, 2360 CrossRef CAS; (b) Y. Murata and K. Komatsu, Chem. Lett., 2001, 896 CrossRef CAS.
- (a) S.-i. Iwamatsu, T. Uozaki, K. Kobayashi, S. Re, S. Nagase and S. Murata, J. Am. Chem. Soc., 2004, 126, 2668 CrossRef CAS PubMed; (b) S.-I. Iwamatsu and S. Murata, Tetrahedron Lett., 2004, 45, 6391 CrossRef CAS.
- (a) K. Komatsu and Y. Murata, Chem. Lett., 2005, 34, 886 CrossRef CAS; (b) M. Murata, Y. Murata and K. Komatsu, Chem. Commun., 2008, 6083 RSC; (c) G. C. Vougioukalakis, M. M. Roubelakis and M. Orfanopoulos, Chem. Soc. Rev., 2010, 39, 817 RSC.
- A. Boto, C. Betancor, T. Prangé and E. Suárez, J. Org. Chem., 1994, 59, 4393 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- Y. Hashikawa, M. Murata, A. Wakamiya and Y. Murata, Org. Lett., 2016, 18, 6348 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data, detailed experimental procedures and computational data. CCDC 1577019. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qm00449d |
| This journal is © the Partner Organisations 2018 |