
Dinuclear lanthanide–lithium complexes based on fluorinated β-diketonate with acetal group: magnetism and effect of crystal packing on mechanoluminescence†
Denis N.
Bazhin
 *ab,
Yulia S.
Kudyakova
a,
Artem S.
Bogomyakov
c,
Pavel A.
Slepukhin
ab,
Grigory A.
Kim
a,
Yanina V.
Burgart
ab and
Victor I.
Saloutin
ab
*ab,
Yulia S.
Kudyakova
a,
Artem S.
Bogomyakov
c,
Pavel A.
Slepukhin
ab,
Grigory A.
Kim
a,
Yanina V.
Burgart
ab and
Victor I.
Saloutin
ab
aPostovsky Institute of Organic Synthesis, the Ural Branch of the Russian Academy of Sciences, 620990 Ekaterinburg, Russian Federation. E-mail: dnbazhin@gmail.com
bUral Federal University named after the First President of Russia B.N. Eltsin, Mira Str. 19, 620002, Ekaterinburg, Russian Federation
cInternational Tomography Center, Siberian Branch of the Russian Academy of Sciences, 630090 Novosibirsk, Russian Federation
First published on 18th September 2018
Abstract
The synthesis of novel dinuclear lanthanide complexes with formulas [(LnL3)(LiL)(MeOH)] (1–3) and [(LnL3)(LiL)(H2O)] (4–6) (Ln = Eu(III), Tb(III), Dy(III)) based on functionalized lithium β-diketonate (LiL) is reported. The coordination environment of the Ln(III) ion is formed by oxygen atoms of both the 1,3-dicarbonyl moiety and the acetal group. Depending on the solvent used for the crystallization, methanol or water molecules participate in the coordination with Li(I) ions affecting the crystal structure of complexes. In contrast to [(LnL3)(LiL)(H2O)] 4–6, a series of complexes [(LnL3)(LiL)(MeOH)] 1–3 showed mechanoluminescence properties, thereby demonstrating the relationship between the observed optical properties and crystal packing architecture. The magnetic measurements of [(LnL3)(LiL)(MeOH)] 1–3 revealed SMM behavior of [(DyL3)(LiL)(MeOH)] 3 at low temperatures (Ueff is 37.7 cm−1).
Introduction
β-Diketones are among the most used tools in coordination chemistry providing a wide variety of metal–organic architectures such as homo- and heterometallic complexes, clusters of various nuclearities, MOFs, nanoparticles, and thin metal films.1 The applicability and efficiency of these ligands in the design and synthesis of advanced materials with optical, magnetic and catalytic properties are well documented.1e,f,2It is well known that β-diketones are coordinated with the lanthanide ion through the two oxygen atoms of the enolate anion to give lanthanide tris- and tetrakis-β-diketonates.1a–d Typically, the coordination sphere of the metal ion in this case consists of solvent or water molecules, which can be replaced by a variety of mono- or bidentate N,O,P-ligands, e.g. hydroxide, alcoholates, N-heterocycles, Schiff bases, ethers, carboxylates, and phosphines.3 Such an approach opens the door to a large family of coordination compounds. Lanthanide tetrakis-β-diketonate formation proceeds through the addition of a deprotonated enol form of β-diketone to the neutral Ln tris-diketonate.
Varying the substituents in diketones causes minor changes in the coordination environment of lanthanide ions influencing the complexes’ properties. The introduction of fluorine-containing substituents into the β-diketone fragment resulted in the increase of their thermal stability and solubility in organic solvents.1e,4 The replacement of the C–H bonds with lower-energy C–F oscillators plays a crucial role in the design of high performance luminescent molecular devices.5 In addition, electron-withdrawing perfluoroalkyl groups reduce the electron density on the oxygen atoms of β-diketones, thereby increasing the Lewis acidity of the metal center in the complex. In the reactions with other metal complexes or organic ligands, these compounds act as strong Lewis acids affording heterometallic or metal organic complexes and polymers.1e,9b,c,6 The weak intermolecular interaction caused by the presence of perfluoroalkyl groups determines the high volatility of such diketonates providing their further application in CVD processes.1e,9b,c,6 In some cases, the use of fluorinated diketonates instead of non-fluorinated analogues facilitates the crystallization of complexes due to less influence of intermolecular hydrogen bonds.7
Lanthanide complexes have attracted extensive attention in materials science due to their unique luminescence and magnetic properties.2,8,9 Showing extremely bright luminescence, Eu(III) and Tb(III) β-diketonates have been intensively studied in terms of their potential application in OLED devices.10 Furthermore, examples possessing triboluminescence (sub-division of mechanoluminescence) have been found among Ln diketonates.11 This property can be effectively used in detecting the deterioration of the principal components’ parts or their mechanical stress.
The chelation of β-diketone with Ln(III) salts has become a powerful tool for the construction of numerous examples of single-ion magnets.8,9,12,13 As has been recently shown, the structural variations of a β-diketone molecule within the Dy(III) heteroleptic cluster have a crucial effect on its magnetic properties.12g The mononuclear Ln systems provide ideal models to manipulate the magnetic properties by optimization of the ligand structure. One of the important factors in the realization of Ln SMM behavior is the obtainment of higher order local symmetry around the metal ion such as D4d (square antiprismatic structures), D∞i (linear 2-coordinated structures), D3h (tri coordinated structures), and large rotational symmetry.9a However, systematic studies of Ln tris- and tetrakis-diketonate series are relatively recent.8d,13 In 2010, Gao et al. reported the SMM behavior of Dy(acac)3·2H2O, which exhibited slow magnetic relaxation with an energy barrier of 64 K.13a Later, a significant SMM behavior improvement up to 187 K was achieved by the substitution of water molecules in Dy(acac)3·2H2O with the binuclear aza-coligand.13b,c In the series of tris-diketonates thus far studied the construction of the LnN2O6 core was based on readily available 1,3-diketones with a limited set of aliphatic and/or aromatic substituents.13 Another prospective and highly efficient strategy for improving the magnetic properties of polynuclear Ln systems is the incorporation of diamagnetic ions in direct coordination with the LnO8 core.9 The tuning of the Ln SMM behavior by doping with K(I), Li(I), Mg(II), Zn(II), Co(III) and Ti(IV) is reported.9h–o
The employment of non-symmetrical 1,3-diketones in coordination with lanthanides should also be stressed as a challenging approach for fine-tuning magnetic and luminescence properties. Moreover, the additional functional groups in the dicarbonyl fragment enable the metal ion coordination sphere to be filled without extra ligands (e.g. aza-coligands). We have therefore endeavored to investigate the potential of such β-diketonates for the synthesis of multifunctional lanthanide complexes.
Recently, we have elaborated the preparation of a novel lithium fluorinated β-diketonate bearing an acetal fragment (Fig. 1, LiL), which was successfully employed in the preparation of 3d metal complexes and heterocyclic derivatives.14 However, analyzing the literature data we have found only a few examples of heterometallic 3d or 4f metal complexes based on non-functional lithium β-diketonates.6d,12c,15 In all the cases thus far studied, the lithium ions were coordinated with metal-containing β-diketonate anions. In this work, we report the synthesis, crystal structures, and investigations of the optical and magnetic properties of novel Ln β-diketonates based on LiL. Among these complexes a new example of SMM was found (for Ln = Dy(III)) and crystal packing-controlled triboluminescence (TL) was observed (for Ln = Eu(III), Tb(III), Dy(III)).
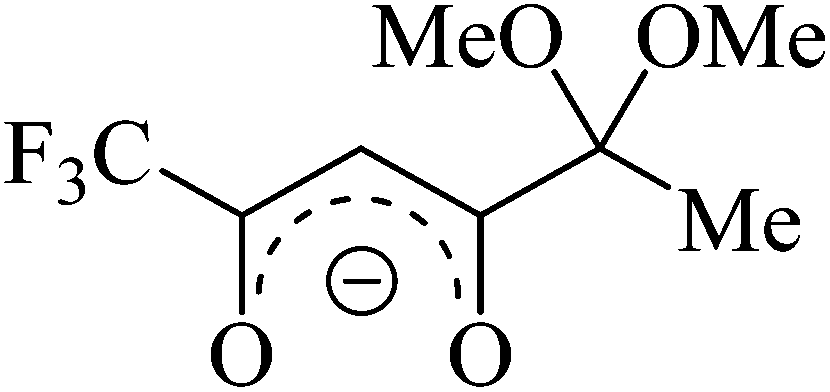 | ||
| Fig. 1 Structure of the starting β-diketonate ligand. | ||
Results and discussion
Syntheses and characterization of complexes
The ligand LiL is a non-symmetrical β-diketonate with a unique combination of substituents. On the one hand, we have a small and electron-withdrawing trifluoromethyl group. On the other hand, there is a bulky and strongly electron-donating substituent containing methyl group and two potentially coordinating methoxy functionalities. Enolate LiL provides a simple procedure for the synthesis of metal complexes as it does not require the use of any bases. In addition, LiL is readily soluble in many organic solvents and insoluble in water. We have chosen methanol as the mostly used solvent for the synthesis of Ln diketonates. NMR spectroscopy of a LiL solution in methanol-d4 showed that diketonate is mainly in an enolate form (see ESI†). In the 13C NMR spectrum, we observed differences in the chemical shift values of carbon atoms in the 1,3-dicarbonyl fragment, indicating unequal negative charge distribution in the molecule.The preparation reaction of [(LnL3)(LiL)(MeOH)] 1–3 was carried out according to the following scheme (eqn (1.1)):
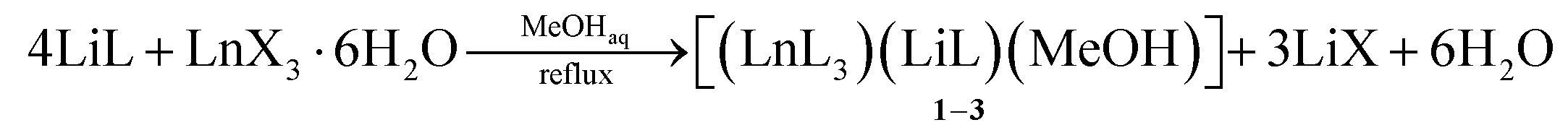 | (1.1) |
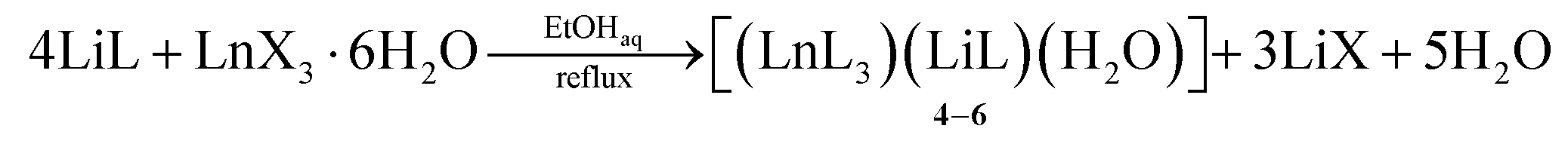 | (1.2) |
 | (1.3) |
The above reactions are simple and straightforward and do not need special conditions. Initially, assuming the formation of lanthanide tris-diketonates, we used the molar ratio of LiL to Ln(III) salt of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. However, in this case complexes 1–3 with formula [(LnL3)(LiL)(MeOH)] were obtained in good yields. In view of this result, we changed the molar ratio of LiL:Ln(III) salt to 4:1 and obtained the same complexes. This reaction is easily reproducible and scalable, allowing us to use up to 1 g of lithium diketonate at a single synthetic run. Complexes 1–3 suitable for XRD analysis and physicochemical property study can be obtained by direct crystallization from the reaction mixture under slow evaporation of the solvent.
:1. However, in this case complexes 1–3 with formula [(LnL3)(LiL)(MeOH)] were obtained in good yields. In view of this result, we changed the molar ratio of LiL:Ln(III) salt to 4:1 and obtained the same complexes. This reaction is easily reproducible and scalable, allowing us to use up to 1 g of lithium diketonate at a single synthetic run. Complexes 1–3 suitable for XRD analysis and physicochemical property study can be obtained by direct crystallization from the reaction mixture under slow evaporation of the solvent.
The complex formation occurs with different lanthanide salts (e.g., chlorides, acetates, and nitrates). We did not observe the influence of the nature of the Ln(III) salt anion on the yield or crystallization of 1–3. The presence of water molecules from the Ln(III) salt hydrates also did not prevent the complex formation. However, when ethanol was used as a solvent instead of methanol in the reaction of LiL with Ln(III) salts, the resulting complex structure was changed. In this case, the water molecule acts as a co-ligand of the lithium ion, forming the complex [(LnL3)(LiL)(H2O)] 4–6 (eqn (1.2)). We have found that these crystals can undergo reversible recrystallization: in methanol they give crystals of 1–3, heating of which at reflux in high-boiling ethanol (95%) leads again to 4–6 (eqn (1.3)). Clearly, the coordination of lithium ions with methanol, which is a stronger Lewis base, is preferred compared with the water molecule.
Furthermore, we attempted to obtain hetero-ligand complexes with a Ln tris-β-diketonate fragment by adding nitrogen-containing heterocycles as co-ligands. The reaction of lithium β-diketonate with Ln(III) salts was carried out in the presence of 2,2′-bipyridine and 4-pyridinecarboxylic acid in methanol. However, after crystallization from the reaction mixture, only the complexes of structures 1–3 were isolated and no other complexes were found.
The IR spectra of 1–3 and 4–6 are similar and display, typical of β-diketonates, strong absorption bands in the range 1635–1633 and 1519–1435 cm−1, which can be attributed to enolate C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CC vibrations, respectively.16 The carbonyl stretching frequency νs(CO) of LiL (1645 cm−1) shifts to 1635–1633 cm−1 in 1–6, thus indicating coordination of the carbonyl groups to the Ln(III) cation. Typical C–F vibrations are observed in the range 1198–1139 cm−1 (the most intensive peaks are at 1143–1139 cm−1), and weak C–H vibrations can be seen between 3002 and 2841 cm−1. The presence of coordinated water molecules in complexes 4–6 is determined by high-frequency absorption bands at 3537–3487 cm−1 while in IR spectra of complexes 1–3 the medium broad band is observed at 3395–3383 cm−1, corresponding to (O–H) vibrations of the solvate MeOH molecule.
O and CC vibrations, respectively.16 The carbonyl stretching frequency νs(CO) of LiL (1645 cm−1) shifts to 1635–1633 cm−1 in 1–6, thus indicating coordination of the carbonyl groups to the Ln(III) cation. Typical C–F vibrations are observed in the range 1198–1139 cm−1 (the most intensive peaks are at 1143–1139 cm−1), and weak C–H vibrations can be seen between 3002 and 2841 cm−1. The presence of coordinated water molecules in complexes 4–6 is determined by high-frequency absorption bands at 3537–3487 cm−1 while in IR spectra of complexes 1–3 the medium broad band is observed at 3395–3383 cm−1, corresponding to (O–H) vibrations of the solvate MeOH molecule.
X-Ray structural characterization
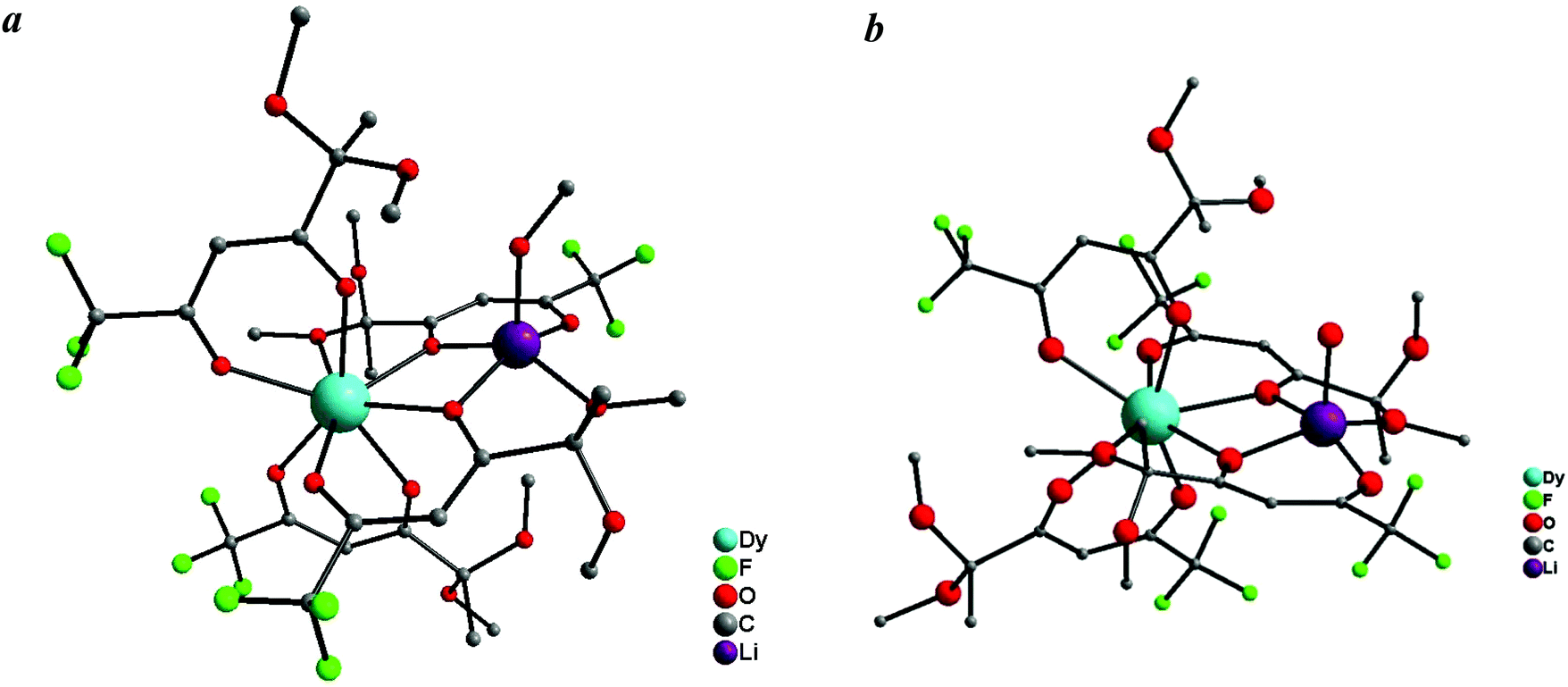 | ||
| Fig. 2 Molecular structures of Dy(III) complexes 3 (a) and 6 (b). Hydrogens are not shown for clarity. | ||
Therefore, the synthesized complexes 1–3 have similar compositions but different structures compared with the known Ln tetrakis-diketonates, which are polymeric and consist of the anionic Ln(diketonate)4 moiety and the bridging alkali metal cation. The discrete structure of heteronuclear complexes 1–3 results from the presence of sterically different substituents and donor methoxy groups in the starting diketonate as well as the small radius of the lithium ion.
To evaluate the geometry of the DyO8 coordination polyhedron, we used the SHAPE 2.1 software.17,18 On the basis of the analysis performed, the geometry of the octa-coordinated Dy(III) ion can be best described as a distorted triangular dodecahedron (TDD-8) with the D2d symmetry (Fig. 3a), and the penta-coordinated Li(I) ion – as a distorted square pyramid (SPY-5) with the C4v symmetry (Fig. 3b). In addition, we calculated the trigonality index τ519 for the LiO5 coordination polyhedron. Its value (0.155) confirms a slightly distorted square pyramidal geometry and, therefore, is in agreement with SHAPE measurements. The complete results of geometric analysis for complexes 1–3 are given in the ESI (Tables S2 and S3, ESI†).
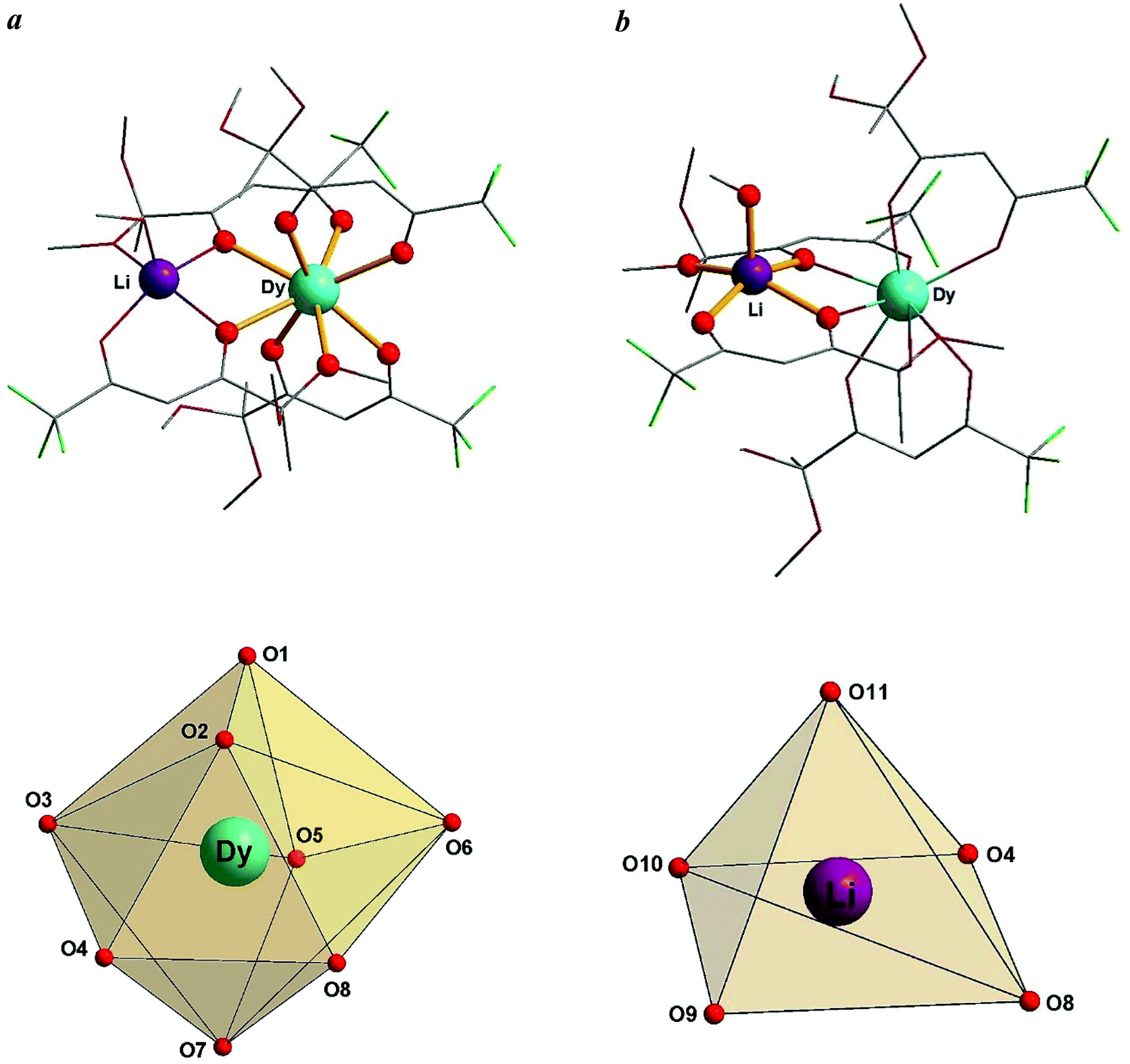 | ||
| Fig. 3 Polyhedra representing the geometries of the coordination environments of the metal ions: (a) DyO8 coordination polyhedron; (b) LiO5 coordination polyhedron. | ||
Bond-length distribution in the four six-membered chelate cycles indicates strong π-electron delocalization. Therefore, this structural ligand fragment represents a depronated enol form of 1,3-diketone. The five-membered cycles formed by the oxygen atom of the acetal fragment are obviously less stable because the Ln–O bond length in this case is relatively longer (about 2.5 Å) compared with the other Ln–O bonds (2.3–2.4 Å). Because of lanthanide contraction, a systematic decrease of Ln–O bond lengths from complex 1 to 3 is observed (Table S6, ESI†). Taking into consideration the Li–Ln distance in all the investigated complexes (>3.5 Å), the metal–metal interaction is unlikely and electron exchange through the bridging O atoms of the ligands takes place. The shortest Ln⋯Ln distances between the dinuclear molecules are 10.527, 10.552, and 10.615 Å for 1, 2 and 3, respectively, thus indicating well-isolated dinuclear units. The distinctive feature of heterometallic crystal structures 1–3 is the absence of inversion centers in the space group symmetry (Fig. 4).
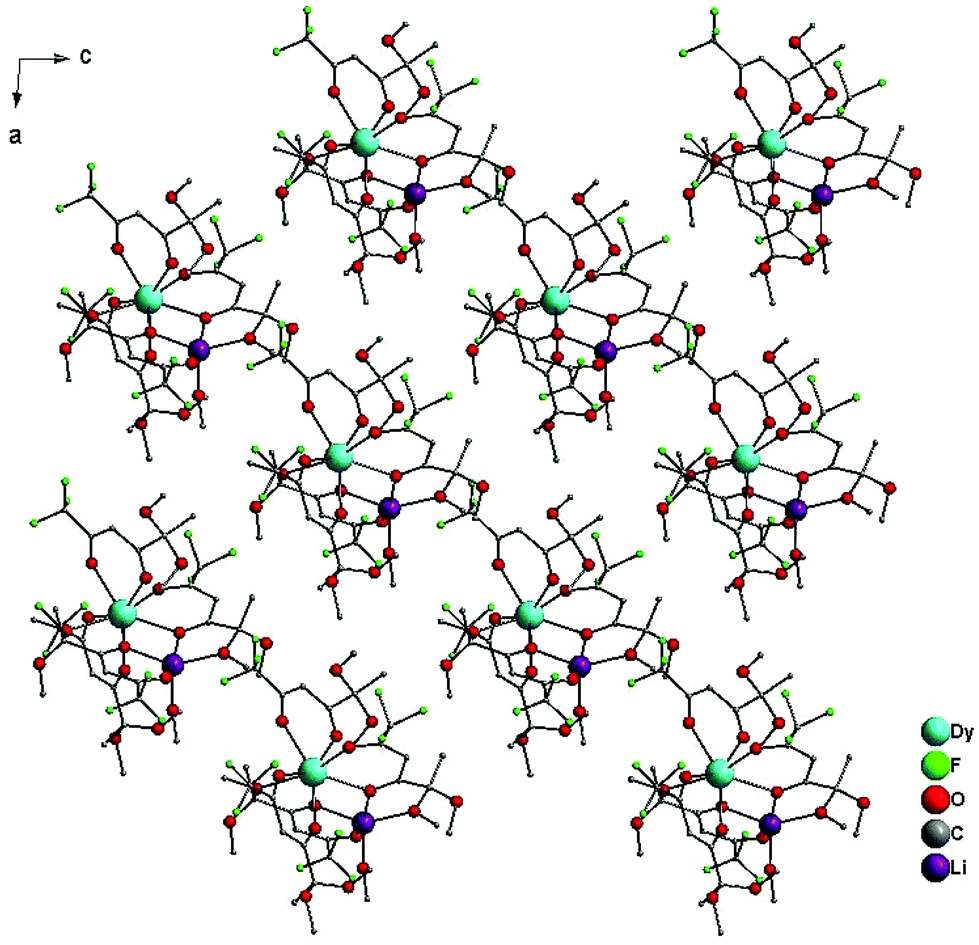 | ||
| Fig. 4 Crystal packing of 3 along the b axis. Hydrogens are not shown for clarity. | ||
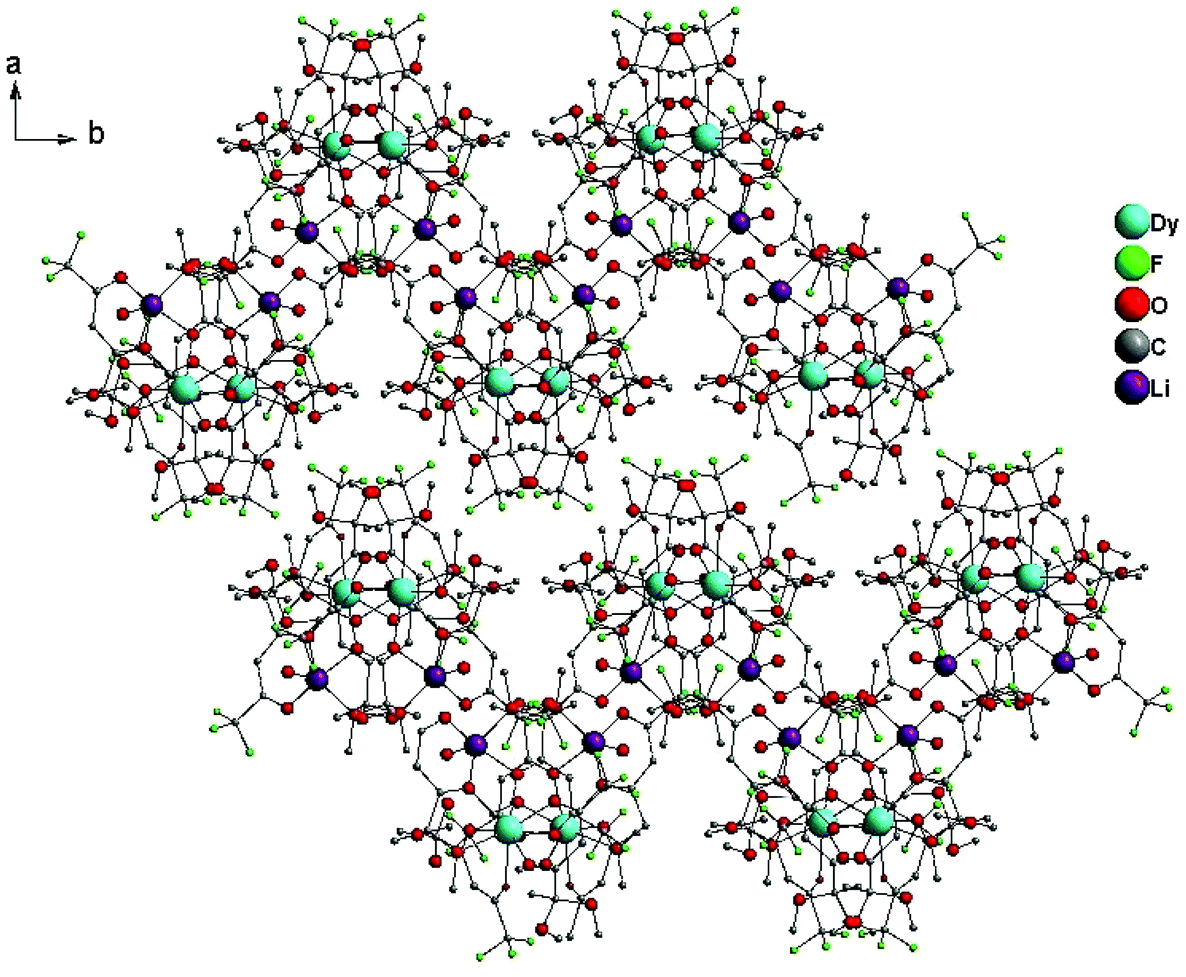 | ||
| Fig. 5 Crystal packing of [(DyL3)(LiL)H2O] 6 along the c axis. Hydrogens are not shown for clarity. | ||
Similar to 1–3, the Ln–O bond lengths gradually decrease from 4 to 6 because of the lanthanide contraction effect (Table S7, ESI†). In addition, with a decrease in the ionic radius of Ln(III), the Ln⋯Ln distance gradually increases.
Luminescence of complexes
All complexes were illuminated by a standard laboratory UV lamp at 254 nm and showed visible luminescence. The solid-state luminescence properties of complexes 1–6 were measured at room temperature. Upon excitation at 340 nm, complexes showed the characteristic emission bands of the corresponding Ln(III) ion originating from the following transitions: 5D0 → 7FJ for 1, 4; 5D4 → 7FJ for 2, 5; and 4F9/2 → 6HJ for 3, 6 (Table S9, ESI†). The dysprosium-based complexes showed less intensive luminescence compared with europium- and terbium-containing ones. For Dy(III) diketonates 3 and 6, near-infrared transitions are identified as a result of the overlapping of 4F9/2 → 6F11/2 and 4F9/2 → 6H9/2 transitions at ∼750 nm. Based on the literature data, we can assume that the sensitization mechanism for luminescent complexes involves the usual triplet pathway, in which the transfer of energy absorbed by the ligand to the Ln(III) ion takes place from the ligand-centered triplet excited state. Here we would like to put the accent on the unique structure of 1–6 providing their bright luminescence: despite the presence of solvent molecules, which often quench the luminescence of metal complexes,20,21 in our case they are not in the direct coordination environment of the lanthanide ion. Also the high luminescence performances of the obtained complexes are in good agreement with the low symmetry coordination environment of lanthanide ions. There is a significant difference in the phosphorescence lifetimes between Eu(III) complexes 1 and 4 (90 μs) and Tb(III) complexes 2 and 5 (128 μs), while for the less luminescent Dy(III) complexes 3 and 6 this difference is negligible (7 μs) (Table S9, ESI†). We assume that the decrease of phosphorescence lifetimes in the case of complexes 4–6 is the result of quenching effects based on the nonradiative channels to the ground state. As we have shown above, the incorporation of a water molecule into the structure of 4–6 changes their crystal packing. Furthermore, the decrease of the Ln⋯Ln distance in complexes 4–6 by ∼1 Å compared with that of 1–3 (Tables S6 and S7, ESI†) can bring about intersystem crossing and reduce their phosphorescence lifetimes.Complexes 1 and 2 exhibited strong triboluminescence upon crushing, which can be seen with the naked eye as bright red (for 1) and green (for 2) flashes at daylight or in the dark (see Fig. S3–S6 and video in ESI†). The intensity of yellow TL for 3 is much lower and can be observed only in the dark (Fig. S5, ESI†). The TL spectra showed the same emission lines as in the corresponding PL spectra of complexes 1–3 (Fig. S7–S9, ESI†). The differences between the PL and TL emission spectra are mainly caused by the differences in resolution and spectral sensitivity of the spectrophotometer used.
The interpretation of the triboluminescence mechanism in lanthanide complexes is still under discussion.11j To gain deeper insight into the structural aspects of mechanoluminescence properties, more detailed criteria should be postulated. Recently it was proposed that weak intermolecular interactions within the layered crystal architectures are preferred for TL compared with π-stacking or hydrogen bonding in a wavy chain network.11a,i,j Here, considering a series of two different types of crystals, we proved the validity of this idea. The crucial point is that complexes 1–3 and 4–6 are formed from the same ligand and have similar dinuclear structures. However, by changing the co-ligand of the Li(I) ion, different packing modes can be obtained, allowing us to obtain either the 2D parallel-layered (layers follow the ab diagonal of the cell) structure in 1–3 (Fig. S10a, ESI†) or the wavy chain frameworks in the case of 4–6 (Fig. S10b, ESI†). The crystals of 1–3 have no symmetry centers and, therefore, their crushing along the separate layers of Ln–Li complex molecules (so-called cleavage planes over which the crystal fracturing can easily take place) is possible (Fig. S11, ESI†). The weak intermolecular interactions of face-to-face oriented trifluoromethyl groups in 1–3 are also favorable for the sliding effect between the layers.6k Otherwise, the crystal packing of 4–6 is built up via the intermolecular H-bonding caused by the presence of water molecules, and the contribution of intermolecular fluoroalkyl interactions in this case is negligible (Fig. S12, ESI†).
Magnetic properties
The crystal structure of Ln complexes 1–3 based on a non-symmetrical β-diketonate prompted us to evaluate their SMM behavior as highly anisotropic properties can be expected. The μeff(T) dependences for polycrystalline samples of complexes 1–3 are shown in Fig. 6. For complex 1 the μeff is 3.34μB at 300 K and decreases with lowering the temperature to 0.32μB at 2 K. The ground state of Eu(III) is singlet (7F0), but there are closely spaced levels with different J-states, whose population decreases dramatically with a decrease in the temperature. The equation for the magnetic susceptibility of Eu(III) describes well the experimental μeff(T) dependence (Fig. 6, red line). The best fit value of the spin–orbital coupling parameter λ is 340 cm−1.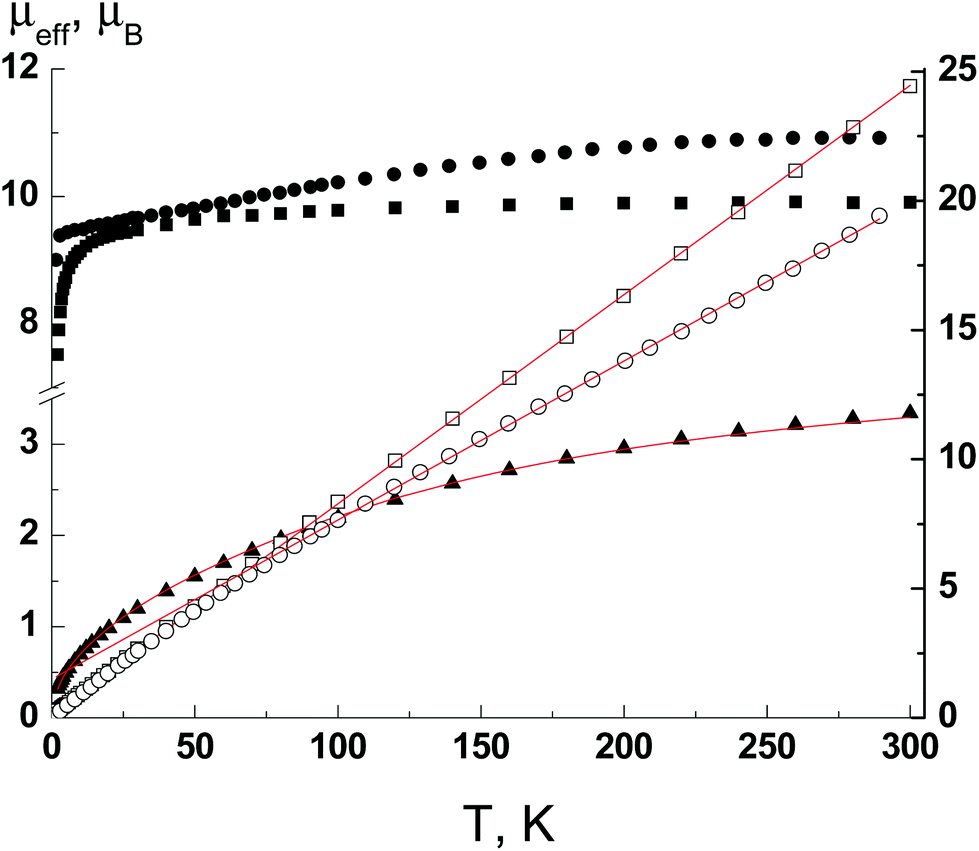 | ||
| Fig. 6 The μeff(T) and 1/χ(T) dependencies for complexes 1 (▲), 2 (■, □) and 3 (●, ○). Solid lines are theoretical curves. | ||
The high temperature values of μeff are 9.91 and 10.92μB for complexes 2 and 3 and agree with the theoretical values of 9.72 and 10.63μB for Tb(III) (7F6 ground state with gJ = 3/2) and Dy(III) (6H15/2 ground state with gJ = 4/3) ions, respectively. The 1/χ(T) dependencies are linear in the temperature range 300–2 K for 2 and 300–80 K for 3 and obey the Curie–Weiss law with the best fit parameters C and Θ of 12.35 K cm3 mol−1 and −2.2 K for complex 2 and 16.26 K cm3 mol−1 and −24.4 K for complex 3. For complex 2 the Curie constant is close to the theoretical value of 11.81 K cm3 mol−1 for the Tb(III) ion. In the case of complex 3, the Curie constant is substantially larger than the theoretical value of 14.17 K cm3 mol−1. In conjunction with a large Weiss constant, it indicates the presence of significant anisotropy. The decrease of μeff with lowering the temperature is also a consequence of the mixture of anisotropy and possible intermolecular interactions. So, only complex 3 displays SMM behavior at low temperatures. The temperature dependencies of the in-phase (χ′) and out-of-phase (χ′′) parts of magnetic susceptibility for complex 3 are frequency dependent under a dc field of 1000 Oe (Fig. 7).
 | ||
| Fig. 7 The temperature dependencies of the in-phase (χ′) (a) and out-of-phase (χ′′) (b) parts of magnetic susceptibility for complex 3 at different frequencies. | ||
Complex 3 displays SMM behavior at low temperatures. To identify the quantum tunneling process and obtain an effective energy barrier, the temperature dependencies of the in-phase (χ′) and out-of-phase (χ′′) parts of magnetic susceptibility are frequency dependent under a dc field of 1000 Oe (Fig. 7).
To extract the relaxation times and their distribution, the frequency dependencies of the in-phase (χ′) and out-of-phase (χ′′) parts of magnetic susceptibility were fitted simultaneously with the generalized Debye model (Fig. S13, ESI†) in the temperature range 2–10.8 K. The best fit values of χT, χS, τ and α are listed in Table S10 in ESI.† At lower temperatures, the distribution broadens drastically, with α increasing to 0.324 at 2 K. Taken together with the shapes of the in-phase magnetic susceptibilities at low temperature, this large increase of the distribution width suggests more than one relaxation process. Logarithmic variation of the relaxation time τ (τ = (2πυ)−1) for 3 as a function of 1/T under 1000 Oe dc field is nonlinear (Fig. 8), suggesting different relaxation processes. The equation for the thermally activated (τ0exp(−Ueff/kT)) slow magnetic relaxation and Raman (CTn) processes describes well the ln(τ) versus T−1 dependency. The best fit values of C, n, τ0 and Ueff are 0.0691, 4.361, 9.857 × 10−6 s and 37.7 cm−1.
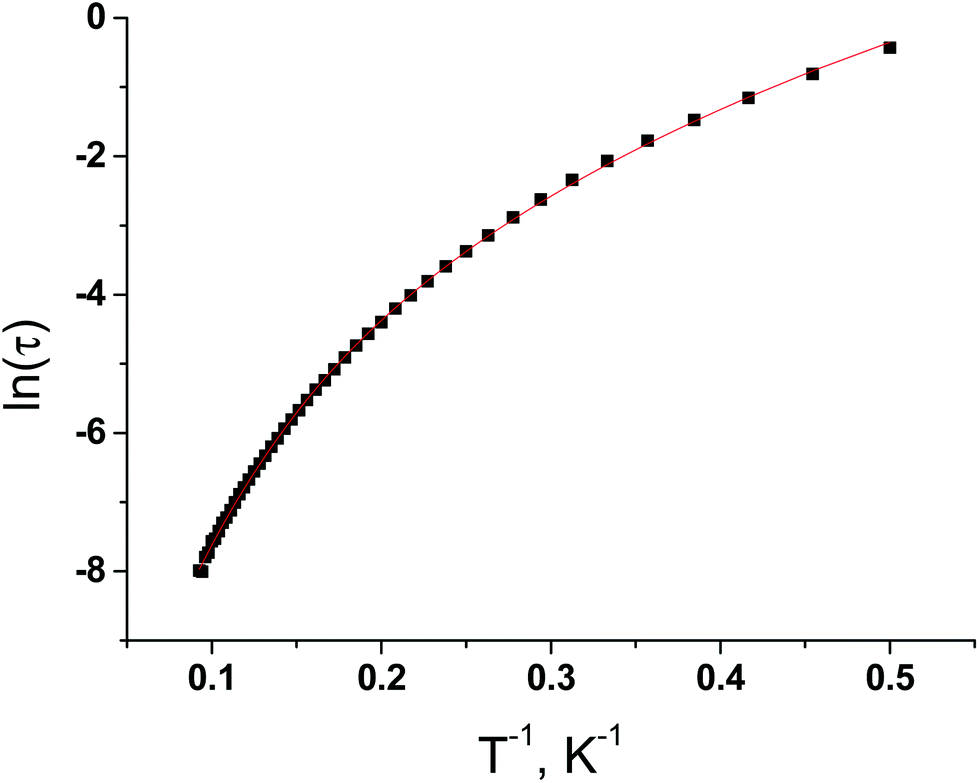 | ||
| Fig. 8 The logarithmic variation of the relaxation time τ as a function of 1/T under 1000 Oe dc field for 3. (Solid line – a theoretical curve.) | ||
The symmetry of the LnO8 coordination sphere in complexes 1–3 is a key point influencing their magnetic properties. The impressive SMM examples of Ln tris- and tetrakis-diketonates have highly symmetrical structures with the square-antiprismatic (SAP) coordination environment and approximate D4d axial symmetry. The increase of the coordination environment symmetry is known to result in the decrease of the QTM process effect at low temperatures. However, a slight change in the coordination environment, the ligand field strength and the complicated inter-molecular interactions can regulate the anisotropy and tune the magnetic dynamics of the mononuclear Ln SMM systems. For example, notwithstanding the TDD geometry of the LnO8 coordination polyhedron in complex 3 its energy barrier value (Ueff = 53 K) is twice higher than that of [K{Dy(hfac)4}]12a having a SAP geometry (Ueff = 23 K). To thoroughly investigate the magnetic properties and elaborate the “structure–property” relationships, the study should be broadened by looking at further examples of Ln β-diketonates with an LnO8 coordination polyhedron. From our point of view, this research can be continued by varying the nature of the alkali metal in the series of Ln complexes based on the described diketonate. In this case there is a more specific and obvious task for magnetism, since we can expect more significant changes in the structure of complexes.
Conclusions
Herein, a novel lithium β-diketonate was used for the construction of a series of lanthanide-based complexes. As in the case of tetrakis-diketonates, we have obtained structures including four β-diketonate-anions. However, the described complexes [(LnL3)(LiL)(MeOH)] 1–3 and [(LnL3)(LiL)(H2O)] 4–6 are strongly different from the known M[Ln(β-diketonate)4] (M is an alkali metal). Unusual assembly of Ln–Li dinuclear complexes becomes possible due to the presence of methoxy-groups in the structure of the starting β-diketonate participating in the coordination with metal ions. In this context, the employment of functionalized β-diketonates is a straightforward and efficient approach to the novel metal organic complexes.We investigated the influence of crystal packing on the mechanoluminescence properties of complexes 1–6. The intermolecular H-bonds between the layers in the structures of [(LnL3)(LiL)H2O] 4–6 prevent their mechanical destruction and, therefore, suppress triboluminescence. In this case, the manipulation of crystal structure with different co-ligands is of crucial importance along with the construction of a complex framework by placing the metal ion in the coordination shell of the organic ligand.
The magnetic properties of [(LnL3)(LiL)(MeOH)] 1–3 were studied. The dysprosium-based complex [(DyL3)(LiL)(MeOH)] 3, in which the DyO8 coordination polyhedron is close to the triangular dodecahedron (TDD-8) in shape, revealed SMM behavior at low temperatures with Ueff being 37.7 cm−1. We foresee the further development of these types of complexes in the field of advanced multifunctional materials.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Financial support of this work (the synthesis part and structure characterization) by the Russian Foundation for Basic Research (grant no. mol_a_dk 16-33-60048) is gratefully acknowledged. ASB is thankful to the FASO (0033-2017-0001). VIS is thankful to the Ural Branch of the Russian Academy of Sciences (AAAA-A18-118020290158-5). The authors would like to thank Dr Djamila Guettas for assistance in the optical and magnetic property measurements. The authors acknowledge Prof. Dominique Luneau, Prof. Olivier Maury, Prof. Boris Le Guennic, Dr Francois Riobe, and Dr Guillaume Pilet for their interest in this work and fruitful discussions. We also thank Prof. Michael Kiskin for his assistance in the SHAPE measurements. XRD experiments and physicochemical studies were carried out using the equipment of the Center for Joint Use “Spectroscopy and Analysis of Organic Compounds” at the Postovsky Institute of Organic Synthesis UB RAS. We also thank both reviewers for helpful suggestions, improving the visibility of the presented results.Notes and references
- (a) G. G. Condorelli, G. Malandrino and I. L. Fragala, Coord. Chem. Rev., 2007, 251, 1931–1950 CrossRef CAS; (b) G. Aromí, P. Gamez and J. Reedijk, Coord. Chem. Rev., 2008, 252, 964–989 CrossRef; (c) B. I. Kharisov, P. E. Martínez, V. M. Jiménez-Pérez, O. V. Kharissova, B. N. Martínez and N. Pérez, J. Coord. Chem., 2009, 63, 1–25 CrossRef; (d) P. A. Vigato, V. Peruzzo and S. Tamburini, Coord. Chem. Rev., 2009, 253, 1099–1201 CrossRef CAS; (e) S. Mishra and S. Daniele, Chem. Rev., 2015, 115, 8379–8448 CrossRef CAS PubMed; (f) P. Z. Chen, L. Y. Niu, Y. Z. Chen and Q. Z. Yang, Coord. Chem. Rev., 2017, 350, 196–216 CrossRef CAS.
- (a) J.-C. G. Bünzli, J. Lumin., 2016, 170, 866–878 CrossRef; (b) J. Kljun and I. Turel, Eur. J. Inorg. Chem., 2017, 1655–1666 CrossRef CAS; (c) K. Binnemans, Chem. Rev., 2009, 109, 4283–4374 CrossRef CAS PubMed; (d) A. T. Wagner and P. W. Roesky, Eur. J. Inorg. Chem., 2016, 782–791 CrossRef CAS.
- (a) S.-Y. Wang, W.-M. Wang, H.-X. Zhang, H.-Y. Shen, L. Jiang, J.-Z. Cui and H.-L. Gao, Dalton Trans., 2016, 45, 3362–3371 RSC; (b) L.-F. Wang, J.-Z. Qiu, J.-Y. Hong, Y.-C. Chen, Q.-W. Li, J.-H. Jia, J. Jover, E. Ruiz, J.-L. Liu and M.-L. Tong, Inorg. Chem., 2017, 56, 8829–8836 CrossRef CAS; (c) W. Huang, Z. Zheng and D. Wu, Inorg. Chem. Commun., 2017, 84, 40–44 CrossRef CAS; (d) H. Sun, L. Wu, W. Yuan, J. Zhao and Y. Liu, Inorg. Chem. Commun., 2016, 70, 164–167 CrossRef CAS; (e) C. Shi, R. Nie, X. Yao, S. Fan, G. An, Y. Dong and G. Li, RSC Adv., 2017, 7, 49701–49709 RSC; (f) G. Huang, X. Yi, J. Jung, O. Guillou, O. Cador, F. Pointillart, B. Le Guennic and K. Bernot, Eur. J. Inorg. Chem., 2018, 326–332 CrossRef CAS; (g) A. N. Swinburne, L. M. H. Paden, T. L. Chan, S. Randall, F. Ortu, A. M. Kenwright and L. S. Natrajan, Inorganics, 2016, 4, 27–44 CrossRef; (h) Z. Wang, N. Liu, H. Li, P. Chen and P. Yan, Eur. J. Inorg. Chem., 2017, 2211–2219 CrossRef CAS; (i) L.-R. Lin, H.-H. Tang, Y.-G. Wang, X. Wang, X.-M. Fang and L.-H. Ma, Inorg. Chem., 2017, 56, 3889–3900 CrossRef CAS; (j) Y. Dong, P. Yan, X. Zou, X. Yao, G. Hou and G. Li, Dalton Trans., 2016, 45, 9148–9157 RSC; (k) S. Biju, D. B. A. Raj, M. L. P. Reddy and B. M. Kariuki, Inorg. Chem., 2006, 45, 10651–10660 CrossRef CAS; (l) S. Petit, F. Baril-Robert, G. Pilet, C. Reber and D. Luneau, Dalton Trans., 2009, 6809–6815 RSC; (m) D. Guettas, V. Montigaud, G. F. Garcia, P. Larini, O. Cador, B. Le Guennic and G. Pilet, Eur. J. Inorg. Chem., 2018, 3–4, 333–339 CrossRef; (n) B. Yao, Y. Cong and B. Zhang, New J. Chem., 2016, 40, 3664–3670 RSC; (o) S. Dasari, S. Singh, S. Sivakumar and A. K. Patra, Chem. – Eur. J., 2016, 22, 17387–17396 CrossRef CAS PubMed; (p) M. R. Felício, T. G. Nunes, P. M. Vaz, A. M. P. Botas, P. Ribeiro-Claro, R. A. S. Ferreira, R. O. Freire, P. D. Vaz, L. D. Carlos, C. D. Nunes and M. M. Nolasco, J. Mater. Chem. C, 2014, 2, 9701–9711 RSC; (q) C. Freund, W. Porzio, U. Giovanella, F. Vignali, M. Pasini and S. Destri, Inorg. Chem., 2011, 50, 5417–5429 CrossRef CAS; (r) S. Zhang, H. Ke, Q. Shi, J. Zhang, Q. Yang, Q. Wei, G. Xie, W. Wang, D. Yang and S. Chen, Dalton Trans., 2016, 45, 5310–5320 RSC; (s) K. Singh, R. Boddula and S. Vaidyanathan, Inorg. Chem., 2017, 56, 9376–9390 CrossRef CAS; (t) Yu. S. Kudyakova, D. N. Bazhin, Y. V. Burgart and V. I. Saloutin, Mendeleev Commun., 2016, 26, 54–56 CrossRef CAS; (u) Z. Ahmed and K. Iftikhar, Polyhedron, 2015, 85, 570–592 CrossRef CAS.
- (a) C. Aronica, G. Chastanet, G. Pilet, B. Le Guennic, V. Robert, W. Wernsdorfer and D. Luneau, Inorg. Chem., 2007, 46, 6108–6119 CrossRef CAS; (b) V. V. Krisyuk, S. V. Tkachev, I. A. Baidina, I. V. Korolkov, A. E. Turgambaeva and I. K. Igumenov, J. Coord. Chem., 2015, 68, 1890–1902 CrossRef CAS; (c) V. V. Krisyuk, I. A. Baidina, N. A. Kryuchkova, V. A. Logvinenko, P. E. Plyusnin, I. V. Korolkov, G. I. Zharkova, A. E. Turgambaeva and I. K. Igumenov, Dalton Trans., 2017, 46, 12245–12256 RSC; (d) K. Assim, M. Melzer, M. Korb, T. Rüffer, A. Jakob, J. Noll, C. Georgi, S. E. Schulz and H. Lang, RSC Adv., 2016, 6, 102557–102569 RSC.
- (a) M. L. P. Reddy, V. Divya and R. Pavithran, Dalton Trans., 2013, 42, 15249–15262 RSC; (b) E. A. Varaksina, I. V. Taydakov, S. A. Ambrozevich, A. S. Selyukov, K. A. Lyssenko, L. T. Jesus and R. O. Freire, J. Lumin., 2018, 196, 161–168 CrossRef CAS; (c) L. N. Puntus, K. J. Schenk and J.-C. G. Bünzli, Eur. J. Inorg. Chem., 2005, 2005(23), 4739–4744 CrossRef; (d) S. Biju, Y. K. Eom, J.-C. G. Bünzli and H. K. Kim, J. Mater. Chem. C, 2013, 1, 6935–6944 RSC; (e) Q. Zhong, H. Wang, G. Qian, Z. Wang, J. Zhang, J. Qiu and M. Wang, Inorg. Chem., 2006, 45, 4537–4543 CrossRef CAS.
- (a) C. M. Lieberman, Z. Wei, A. S. Filatov and E. V. Dikarev, Inorg. Chem., 2016, 55, 3946–3951 CrossRef CAS; (b) C. M. Lieberman, A. S. Filatov, Z. Wei, A. Y. Rogachev, A. M. Abakumov and E. V. Dikarev, Chem. Sci., 2015, 6, 2835–2842 RSC; (c) M. C. Barry, Z. Wei, T. He, A. S. Filatov and E. V. Dikarev, J. Am. Chem. Soc., 2016, 138, 8883–8887 CrossRef CAS; (d) Z. Wei, H. Han, A. S. Filatov and E. V. Dikarev, Chem. Sci., 2014, 5, 813–818 RSC.
- T. T. Cunha, J. Jung, M.-E. Boulon, G. Campo, F. Pointillart, C. L. M. Pereira, B. Le Guennic, O. Cador, K. Bernot, F. Pineider, S. Golhen and L. Ouahab, J. Am. Chem. Soc., 2013, 135, 16332–16335 CrossRef.
- (a) P. Zhang, Y.-N. Guo and J. Tang, Coord. Chem. Rev., 2013, 257, 1728–1763 CrossRef CAS; (b) I. Cimatti, X. Yi, R. Sessoli, M. Puget, B. Le Guennic, J. Jung, T. Guizouarn, A. Magnani, K. Bernot and M. Mannini, Appl. Surf. Sci., 2018, 432, 7–14 CrossRef CAS; (c) F. Pointillart, B. Le Guennic, O. Cador, O. Maury and L. Ouahab, Acc. Chem. Res., 2015, 48, 2834–2842 CrossRef CAS PubMed; (d) R. Sessoli and A. K. Powell, Coord. Chem. Rev., 2009, 253, 2328–2341 CrossRef CAS; (e) Y. Dong, P. Yan, X. Zou and G. Li, Inorg. Chem. Front., 2015, 2, 827–836 RSC; (f) D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS; (g) J. Long, Y. Guari, R. A. S. Ferreira, L. D. Carlos and J. Larionova, Coord. Chem. Rev., 2018, 363, 57–70 CrossRef CAS; (h) J.-H. Jia, Q.-W. Li, Y.-C. Chen, J.-L. Liu and M.-L. Tong, Coord. Chem. Rev., 2018 DOI:10.1016/j.ccr.2017.11.012; (i) S. G. McAdams, A.-M. Ariciu, A. K. Kostopoulos, J. P. S. Walsh and F. Tuna, Coord. Chem. Rev., 2017, 346, 216–239 CrossRef CAS; (j) F. Pointillart, O. Cador, B. Le Guennic and L. Ouahab, Coord. Chem. Rev., 2017, 346, 150–175 CrossRef CAS; (k) K. L. M. Harriman and M. Murugesu, Acc. Chem. Res., 2016, 49, 1158–1167 CrossRef CAS; (l) Y. Zheng, C. Zhang and Q. Wang, Sens. Actuators, B, 2017, 245, 622–640 CrossRef CAS; (m) K. Lunstroot, K. Driesen, P. Nockemann, K. Van Hecke, L. Van Meervelt, C. Görller-Walrand, K. Binnemans, S. Bellayer, L. Viau, J. Le Bideau and A. Vioux, Dalton Trans., 2009, 298–306 RSC; (n) S. V. Eliseeva and J.-C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC; (o) L. Sorace, C. Benelli and D. Gatteschi, Chem. Soc. Rev., 2011, 40, 3092–3104 RSC; (p) W.-Y. Zhang, Y.-Q. Zhang, S.-D. Jiang, W.-B. Sun, H.-F. Li, B.-W. Wang, P. Chen, P.-F. Yan and S. Gao, Inorg. Chem. Front., 2018, 5, 1575–1586 RSC; (q) H.-R. Tu, W.-B. Sun, H.-F. Li, P. Chen, Y.-M. Tian, W.-Y. Zhang, Yi.-Q. Zhang and P.-F. Yan, Inorg. Chem. Front., 2017, 4, 499–508 RSC; (r) H.-M. Chen, W.-M. Wang, X.-Q. Li, X.-Y. Chu, Y.-Y. Nie, Z. Liu, S.-X. Huang, H.-Y. Shen, J.-Z. Cui and H.-L. Gao, Inorg. Chem. Front., 2018, 5, 394–402 RSC; (s) J. Li, R.-M. Wei, T.-C. Pu, F. Cao, L. Yang, Y. Han, Y.-Q. Zhang, J.-L. Zuo and Y. Song, Inorg. Chem. Front., 2017, 4, 114–122 RSC.
- (a) J. Tang and P. Zhang, Lanthanide single molecular magnets, Springer, Heidelberg, NY, 2015 Search PubMed; (b) S. Biswas, S. Das, J. Acharya, V. Kumar, J. van Leusen, P. Kögerler, J. M. Herrera, E. Colacio and V. Chandrasekhar, Chem. – Eur. J., 2017, 23, 5154–5170 CrossRef CAS; (c) J. Acharya, S. Biswas, J. van Leusen, P. Kumar, V. Kumar, R. S. Narayanan, P. Kögerler and V. Chandrasekhar, Cryst. Growth Des., 2018, 18, 4004–4016 CrossRef CAS; (d) S. Biswas, K. S. Bejoymohandas, S. Das, P. Kalita, M. L. P. Reddy, I. Oyarzabal, E. Colacio and V. Chandrasekhar, Inorg. Chem., 2017, 56, 7985–7997 CrossRef CAS; (e) J.-R. Jiménez, I. F. Díaz-Ortega, E. Ruiz, D. Aravena, S. J. A. Pope, E. Colacio and J. M. Herrera, Chem. – Eur. J., 2016, 22, 14548–14559 CrossRef; (f) C. Das, A. Upadhyay and M. Shanmugam, Inorg. Chem., 2018, 57, 9002–9011 CrossRef CAS; (g) A. Upadhyay, K. R. Vignesh, C. Das, S. K. Singh, G. Rajaraman and M. Shanmugam, Inorg. Chem., 2017, 56, 14260–14276 CrossRef CAS; (h) S. K. Singh, B. Pandey, G. Velmurugan and G. Rajaraman, Dalton Trans., 2017, 46, 11913–11924 RSC; (i) J. Goura, J. Brambleby, P. Goddard and V. Chandrasekhar, Chem. – Eur. J., 2015, 21, 4926–4930 CrossRef CAS PubMed; (j) J. P. Costes, S. Titos-Padilla, I. Oyarzabal, T. Gupta, C. Duhayon, G. Rajaraman and E. Colacio, Inorg. Chem., 2016, 55, 4428–4440 CrossRef CAS PubMed; (k) K. R. Vignesh, S. K. Langley, K. S. Murray and G. Rajaraman, Inorg. Chem., 2017, 56, 2518–2532 CrossRef CAS PubMed; (l) A. Upadhyay, C. Das, S. Vaidya, S. K. Singh, T. Gupta, R. Mondol, S. K. Langley, K. S. Murray, G. Rajaraman and M. Shanmugam, Chem. – Eur. J., 2017, 23, 4903–4916 CrossRef CAS PubMed; (m) A. Upadhyay, C. Das, S. K. Langley, K. S. Murray, A. K. Srivastavad and M. Shanmugam, Dalton Trans., 2016, 45, 3616–3626 RSC; (n) S. Das, K. S. Bejoymohandas, A. Dey, S. Biswas, M. L. Reddy, R. Morales, E. Ruiz, S. Titos-Padilla, E. Colacio and V. Chandrasekhar, Chem. – Eur. J., 2015, 21, 6449–6464 CrossRef CAS PubMed; (o) F. Pointillart, K. Bernot, G. Poneti and R. Sessoli, Inorg. Chem., 2012, 51, 12218–12229 CrossRef CAS.
- (a) D. Kovacs and K. E. Borbas, Coord. Chem. Rev., 2018, 364, 1–9 CrossRef CAS; (b) H. Xu, Q. Sun, Z. An, Y. Wei and X. Liu, Coord. Chem. Rev., 2015, 293–294, 228–249 CrossRef CAS; (c) J.-C. G. Bünzli, Coord. Chem. Rev., 2015, 293–294, 19–47 CrossRef.
- (a) B. V. Bukvetskii, A. G. Mirochnik and A. S. Shishov, J. Lumin., 2018, 195, 44–48 CrossRef CAS; (b) H.-Y. Wong, W.-S. Lo, W. T. K. Chan and G.-L. Law, Inorg. Chem., 2017, 56, 5135–5140 CrossRef CAS; (c) P. C. R. Soares-Santos, H. I. S. Nogueira, F. A. A. Paz, R. A. Sá Ferreira, L. D. Carlos, J. Klinowski and T. Trindade, Eur. J. Inorg. Chem., 2003, 3609–3617 CrossRef CAS; (d) A. Mech, Polyhedron, 2008, 27, 393–405 CrossRef CAS; (e) S. Akerboom, M. S. Meijer, M. A. Siegler, W. T. Fu and E. Bouwman, J. Lumin., 2014, 145, 278–282 CrossRef CAS; (f) E. E. S. Teotonio, F. A. Silva, D. K. S. Pereira, L. M. Santo, H. F. Brito, W. M. Faustino, M. C. F. C. Felinto, R. H. Santos, R. Moreno-Fuquen, A. R. Kennedy and D. Gilmore, Inorg. Chem. Commun., 2010, 13, 1391–1395 CrossRef CAS; (g) S. V. Eliseeva, D. N. Pleshkov, K. A. Lyssenko, L. S. Lepnev, J.-C. G. Bünzli and N. P. Kuzmina, Inorg. Chem., 2010, 49, 9300–9311 CrossRef CAS PubMed; (h) S. Kariaka, V. A. Trush, S. S. Smola, Ye. M. Fadieiev, V. V. Dyakonenko, S. V. Shishkina, T. Y. Sliva and V. M. Amirkhanov, J. Lumin., 2018, 194, 108–115 CrossRef; (i) Y. Hirai, T. Nakanishi, Y. Kitagawa, K. Fushimi, T. Seki, H. Ito and Y. Hasegawa, Angew. Chem., Int. Ed., 2017, 56, 7171 CrossRef CAS PubMed; (j) J.-C. G. Bünzli and K.-L. Wong, J. Rare Earths, 2018, 36, 1–41 CrossRef.
- (a) D. Zeng, M. Ren, S.-S. Bao and L.-M. Zheng, Inorg. Chem., 2014, 53, 795–801 CrossRef CAS; (b) L. A. Galán, B. L. Reid, S. Stagni, A. N. Sobolev, B. W. Skelton, M. Cocchi, J. M. Malicka, E. Zysman-Colman, E. G. Moore, M. I. Ogden and M. Massi, Inorg. Chem., 2017, 56, 8975–8985 CrossRef PubMed; (c) W. G. Quirino, C. Legnani, R. M. B. Santos, K. C. Teixeira, M. Cremona, M. A. Guedes and H. F. Brito, Thin Solid Films, 2008, 517, 1096–1100 CrossRef CAS; (d) X.-L. Li, M. Hu, Z. Yin, C. Zhu, C.-M. Liu, H.-P. Xiao and S. Fang, Chem. Commun., 2017, 53, 3998–4001 RSC; (e) B. Monteiro, M. Outis, H. Cruz, J. P. Leal, C. A. T. Laia and C. C. L. Pereira, Chem. Commun., 2017, 53, 850–853 RSC; (f) D. T. Thielemann, M. Klinger, T. J. A. Wolf, Y. Lan, W. Wernsdorfer, M. Busse, P. W. Roesky, A.-N. Unterreiner, A. K. Powell, P. C. Junk and G. B. Deacon, Inorg. Chem., 2011, 50, 11990–12000 CrossRef CAS PubMed; (g) K. Zhang, V. Montigaud, O. Cador, G.-P. Li, B. Le Guennic, J.-K. Tang and Y.-Y. Wang, Inorg. Chem., 2018, 57, 8550–8557 CrossRef CAS PubMed.
- (a) S. D. Jiang, B. W. Wang, G. Su, Z. M. Wang and S. Gao, Angew. Chem., Int. Ed., 2010, 49, 7448–7451 CrossRef CAS PubMed; (b) G. J. Chen, C. Y. Gao, J. L. Tian, J. Tang, W. Gu, X. Liu, S. P. Yan, D. Z. Liao and P. Cheng, Dalton Trans., 2011, 40, 5579–5583 RSC; (c) G. J. Chen, Y. N. Guo, J. L. Tian, J. K. Tang, W. Gu, X. Liu, S. P. Yan, P. Cheng and D. Z. Liao, Chem. – Eur. J., 2012, 18, 2484–2487 CrossRef CAS PubMed; (d) Y. Bi, Y. N. Guo, L. Zhao, Y. Guo, S. Y. Lin, S. D. Jiang, J. K. Tang, B. W. Wang and S. Gao, Chem. – Eur. J., 2011, 17, 12476–12481 CrossRef CAS PubMed; (e) G. J. Chen, Y. Zhou, G. X. Jin and Y. B. Dong, Dalton Trans., 2014, 43, 16659–16665 RSC; (f) C. M. Liu, D. Q. Zhang and D. B. Zhu, Inorg. Chem., 2013, 52, 8933–8940 CrossRef CAS PubMed; (g) D. P. Li, T. W. Wang, C. H. Li, D. S. Liu, Y. Z. Li and X. Z. You, Chem. Commun., 2010, 46, 2929–2931 RSC; (h) Y. Z. Tong, C. Gao, Q. L. Wang, B. W. Wang, S. Gao, P. Cheng and D. Z. Liao, Dalton Trans., 2015, 44, 9020–9026 RSC; (i) J. Zhu, C. Z. Wang, F. Luan, T. Q. Liu, P. F. Yan and G. M. Li, Inorg. Chem., 2014, 53, 8895–8901 CrossRef CAS PubMed; (j) Z. G. Wang, J. Lu, C. Y. Gao, C. Wang, J. L. Tian, W. Gu, X. Liu and S. P. Yan, Inorg. Chem. Commun., 2013, 27, 127–130 CrossRef CAS; (k) D. P. Li, X. P. Zhang, T. W. Wang, B. B. Ma, C.-H. Li, Y. Z. Li and X. Z. You, Chem. Commun., 2011, 47, 6867–6869 RSC.
- (a) D. N. Bazhin, D. L. Chizhov, G.-V. Röschenthaler, Yu. S. Kudyakova, Ya. V. Burgart, P. A. Slepukhin, V. I. Saloutin and V. N. Charushin, Tetrahedron Lett., 2014, 55, 5714–5717 CrossRef CAS; (b) D. N. Bazhin, Yu. S. Kudyakova, G.-V. Röschenthaler, Y. V. Burgart, P. A. Slepukhin, M. L. Isenov, V. I. Saloutin and V. N. Charushin, Eur. J. Org. Chem., 2015, 5236–5245 CrossRef CAS; (c) D. N. Bazhin, Y. S. Kudyakova, A. Y. Onoprienko, P. A. Slepukhin, Y. V. Burgart and V. I. Saloutin, Chem. Heterocycl. Compd., 2017, 53, 1324–1329 CrossRef CAS; (d) D. N. Bazhin, Yu. S. Kudyakova, P. A. Slepukhin, Ya. V. Burgart, N. M. Malysheva, A. N. Kozitsina, A. V. Ivanova and V. I. Saloutin, Mendeleev Commun., 2018, 28, 202–204 CrossRef CAS; (e) D. N. Bazhin, Y. S. Kudyakova, Y. V. Burgart and V. I. Saloutin, Russ. Chem. Bull. Int. Ed., 2018, 67, 497–499 CrossRef CAS; (f) V. V. Krisyuk, S. U. Kyzy, T. V. Rybalova, I. A. Baidina, I. V. Korolkov, D. L. Chizhov, D. N. Bazhin and Y. S. Kudyakova, J. Coord. Chem., 2018 DOI:10.1080/00958972.2018.1479746.
- (a) A. Navulla, L. Huynh, Z. Wei, A. S. Filatov and E. V. Dikarev, J. Am. Chem. Soc., 2012, 134, 5762–5765 CrossRef CAS PubMed; (b) J. Priya, N. K. Gondia, A. K. Kunti and S. K. Sharma, ECS J. Solid State Sci. Technol., 2006, 5, R166–R171 CrossRef; (c) K. C. Teixeira, G. F. Moreira, W. G. Quirino, C. Legnani, R. A. Silva, M. Cremona, H. F. Brito and C. A. Achete, J. Therm. Anal. Calorim., 2011, 106, 587–593 CrossRef CAS; (d) M. C. Barry, C. M. Lieberman, Z. Wei, R. Clérac, A. S. Filatov and E. V. Dikarev, Inorg. Chem., 2018, 57, 2308–2313 CrossRef CAS PubMed; (e) H. Han, Z. Wei, M. C. Barry, J. C. Carozza, M. Alkan, A. Y. Rogachev, A. S. Filatov, A. M. Abakumov and E. V. Dikarev, Chem. Sci., 2018, 9, 4736–4745 RSC.
- (a) K. Nakamoto, in Infrared and Raman Spectra of Inorganic and Coordination Compounds. Part B. Organometallic and Coordination compounds, Wiley, Hoboken, NJ, 2009, ch. 1, pp. 96–105 Search PubMed; (b) K. Binnemans, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneidner Jr., J.-C. G. Bunzli and V. K. Pecharsky, Elsevier, Amsterdam, 2004, ch. 225, vol. 35, pp. 107–272 Search PubMed.
- D. Casanova, M. Llunell, P. Alemany and S. Alvarez, Chem. – Eur. J., 2005, 11, 1479–1494 CrossRef CAS PubMed.
- M. Llunell, D. Casanova, J. Cirera, P. Alemany and S. Alvarez, SHAPE v 2.1, Barcelona, 2013 Search PubMed.
- A. W. Addison, N. T. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 7, 1349–1356 RSC.
- J.-C. G. Bünzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed.
- (a) Y. Xie and Z. Li, Chem., 2018, 4, 943–971 CrossRef CAS; (b) D. O. Olawale, T. Dickens, W. G. Sullivan, O. I. Okoli, J. O. Sobanjo and B. Wang, J. Lumin., 2011, 131, 1407–1418 CrossRef CAS; (c) J.-S. Kang, Y.-K. Jeong, Y. S. Shim, S. Rout, K. T. Leung, Y. Sohn and J.-G. Kang, J. Lumin., 2016, 178, 368–374 CrossRef CAS; (d) N. Terasaki, H. Zhang, Y. Imai, H. Yamada and C.-N. Xu, Thin Solid Films, 2009, 518, 473–476 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1855391–1855396. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8qi00772a |
| This journal is © the Partner Organisations 2019 |