
Identification of the key factor promoting the enrichment of chiral polymorph A in zeolite beta and the synthesis of chiral polymorph A highly enriched zeolite beta†
Tingting
Lu‡
 a,
Liangkui
Zhu‡
a,
Xiaohe
Wang
a,
Wenfu
Yan
*a,
Wei
Shi
b and
Ruren
Xu
a
a,
Liangkui
Zhu‡
a,
Xiaohe
Wang
a,
Wenfu
Yan
*a,
Wei
Shi
b and
Ruren
Xu
a
aState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, Jilin University, Changchun 130012, P. R. China. E-mail: yanw@jlu.edu.cn
bKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Nankai University, Tianjin 300071, P. R. China
First published on 4th May 2018
Abstract
Chiral zeolites with intrinsically chiral frameworks are highly desired due to their potential applications in asymmetric catalysis and enantioseparation. Zeolite beta is an intergrowth of chiral polymorph A and achiral polymorph B in a ratio of ca. 44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :56. The synthesis of chiral polymorph A of zeolite beta or chiral polymorph A-enriched zeolite beta is one of the biggest challenges. By performing a series of control experiments, we identified the key factor promoting the enrichment of chiral polymorph A in pure-silica zeolite beta, i.e. the acidity of the initial mixture. With this discovery, we developed a generalized non-hydrofluoric acid-assisted synthesis route to synthesize polymorph A-enriched pure-silica zeolite beta. Zeolite beta crystals containing more than 70% chiral polymorph A were obtained. This acid assisted method could not only significantly enrich polymorph A in the crystals of pure-silica zeolite beta but also provide a way for avoiding the utilization of highly toxic concentrated hydrofluoric acid.
:56. The synthesis of chiral polymorph A of zeolite beta or chiral polymorph A-enriched zeolite beta is one of the biggest challenges. By performing a series of control experiments, we identified the key factor promoting the enrichment of chiral polymorph A in pure-silica zeolite beta, i.e. the acidity of the initial mixture. With this discovery, we developed a generalized non-hydrofluoric acid-assisted synthesis route to synthesize polymorph A-enriched pure-silica zeolite beta. Zeolite beta crystals containing more than 70% chiral polymorph A were obtained. This acid assisted method could not only significantly enrich polymorph A in the crystals of pure-silica zeolite beta but also provide a way for avoiding the utilization of highly toxic concentrated hydrofluoric acid.
Introduction
Of the 235 zeolitic framework types known so far, zeolite beta (*BEA-type) is one of the few examples that is of major importance in industrial applications.1–4 The unusual structural and chemical properties of zeolite beta make it an important material for petrochemical processes, such as adsorption and separation,5,6 isomerization,7 alkylation,8 and acylation.9 Initially, hydrothermal synthesis of zeolite beta was carried out in alkaline medium by Wadlinger et al.10 With the employment of fluoride as a mineralizing agent, the hydrothermal synthesis of zeolite beta was extended to nearly neutral aqueous conditions.11 From that point on, the synthesis in fluoride medium at near neutral pH has been applied successfully to the Lewis acid zeolites such as Ti-beta, Sn-beta, and Zr-beta.12–14The structure elucidation of zeolite beta has been very challenging. Until 1988, zeolite beta was determined as a highly faulted intergrowth of polymorph A and polymorph B in a ratio of ca. 44:56.15,16 A third hypothetical polymorph (polymorph C) was also proposed, but could only be experimentally synthesized 12 years later.17 From the point of view of structure, the three beta polymorphs are built from an identical centrosymmetric tetragonal building layer, but the layers are stacked in distinct sequences.18 Polymorph A (P4122 or P4322) was chiral whereas polymorph B (C2/c) and polymorph C (P42/mmc) were achiral.
To date, approximately 85% polymorph B-enriched zeolite beta and pure polymorph C zeolite beta have been synthesized from the initial mixture with specific chemical composition in the presence of specially designed organic structure directing agents (OSDAs).19,20 However, it has been still impossible to obtain the polymorph A form of zeolite beta. Chiral polymorph A is of particular interest due to its potential applications in asymmetric and enantioselective catalysis. Researchers have made extensive efforts to synthesise polymorph A-enriched zeolite beta in the last few years.11,21–24 However, the profiles of their experimental XRD patterns are very similar to those of the normal zeolite beta, suggesting that the proportion of polymorph A in their products was actually less than 50%. Until recently, the proportion of polymorph A has only been significantly improved (∼65%) by Tong et al., Guo et al., and Zhang et al. following an extremely concentrated fluoride route with achiral OSDAs,25–28 indicating that the chiral OSDA is not crucial for the enrichment of chiral polymorph A in zeolite beta.
In this work, we made a thorough investigation of the essential key factor promoting the enrichment of polymorph A in zeolite beta and experimentally confirmed that the acidity of the initial mixture greatly affected the enrichment of chiral polymorph A in zeolite beta. On the basis of this discovery, we developed a generalized non-hydrofluoric acid-assisted synthesis route to synthesize polymorph A-enriched pure-silica zeolite beta. This non-hydrofluoric acid-assisted method could not only significantly enrich polymorph A in the crystals of pure-silica zeolite beta but also avoid the utilization of highly toxic concentrated hydrofluoric acid.
Results and discussion
Scheme 1 shows the experimental protocol of this work. The molar composition of SiO2:0.5TEAOH:12H2O for the initial mixture was taken from that of SiO2:0.54TEAOH:0.54HF:7.25H2O reported by Camblor et al., to produce pure-silica zeolite beta that is free of defects in connectivity.11 The molar ratio of TEAOH/HF was set as 1/1 to keep the mixture neutral. Besides HF, NH4F was also used as a fluoride source in control experiments. Freeze-drying or heating at 80 °C was used to get rid of the water in the initial mixture for producing pure-silica zeolite beta under hydrothermal conditions. In experiment 1 of Scheme 1, freeze-drying was used to get rid of the water and HF was used as the fluoride source. The resulting mixture with the molar composition of SiO2:0.5TEAOH:0.5HF:H2O produces normal zeolite beta. The corresponding powder XRD pattern is shown in Fig. 1(a). However, when the identical initial mixture was heated at 80 °C to get rid of the extra water (experimental 2 in Scheme 1), chiral polymorph A-enriched zeolite beta was obtained, which agrees with the result reported by Tong et al.27 The proportion of polymorph A in the crystals of zeolite beta is as high as ca. 65%.27 It has been experimentally confirmed that the Hofmann degradation of tetraethylammonium hydroxide (TEAOH) occurred to form triethylamine (TEA) during the 80 °C heating dehydration process and the molar ratio of TEAOH/TEA in the final dehydrated mixture was approximately 2/3.27 However, dehydration by freeze-drying proved effective to avoid the decomposition of TEAOH.29 Compared to the final mixture in experiment 1, the final mixture in experiment 2 contains less TEAOH and more TEA.
 | ||
| Scheme 1 Experimental protocol for investigating the key factor promoting the enrichment of chiral polymorph A in zeolite beta (A = polymorph A enriched, N = normal). | ||
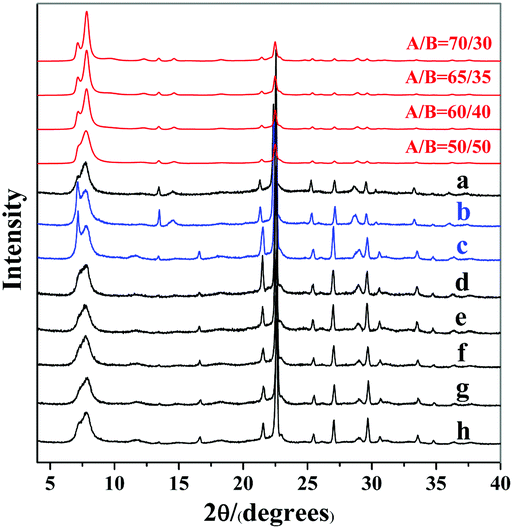 | ||
| Fig. 1 Experimental XRD patterns of zeolite beta mentioned in Scheme 1 (a: experiment 1; b: experiment 3; c: experiment 4; d: experiment 5; e: experiment 6; f, g, and h: experiment 7) and the simulated XRD patterns with different polymorph ratios of A/B. | ||
The above results show that the same initial mixture can produce the pure-silica zeolite beta with varied enrichment degree under different dehydration methods. Freeze-drying dehydration promotes the formation of normal pure-silica zeolite beta, while heating dehydration favours the enrichment of chiral polymorph A. The first thought that came to our mind as the reason for promoting the enrichment of chiral polymorph A in zeolite beta in experiment 2 of Scheme 1 was the co-template effect of TEA. However, the acidic nature of the final mixture is also a possible reason that cannot be pre-excluded because the quaternary ammonium hydroxide TEAOH has a stronger alkali than its decomposed product TEA. The final mixture in experiment 1 is neutral and that in experiment 2 is acidic. Thus, either the acidic nature of the initial mixture or the co-template effect of TEA promotes the enrichment of chiral polymorph A in the crystals of zeolite beta. To find out which factor is crucial for the enrichment of chiral polymorph A in zeolite beta, we conducted a series of control experiments.
In experiment 3 of Scheme 1, we first freeze-dried the initial mixture with a molar composition of SiO2:0.25TEAOH. Subsequently, calculated TEA and HF were added to keep the molar composition of the final mixture as SiO2:0.25TEAOH:0.25TEA:0.5HF:H2O.29 The powder XRD pattern of the resulting zeolite beta shown in Fig. 1(b) indicates that chiral polymorph A was enriched with a degree of ca. 65%.
Experiment 3 shows that we reproduced the results of experiment 2. However, we still cannot reach a conclusion as to which factor is crucial for the enrichment of chiral polymorph A, i.e. acidity of the initial mixture or the co-template effect of TEA. Experiment 4 is very similar to experiment 3 except that no extra TEA was used. The product of experiment 4 is polymorph A-enriched zeolite beta with an enrichment of ca. 65% and the corresponding powder XRD pattern is shown in Fig. 1(c). This experiment clearly excludes the co-template role of TEA in the enrichment of polymorph A in zeolite beta and suggests that the acidity of the mixture may be the key factor for the enrichment of polymorph A. Consequently, reducing the acidity of the initial mixture may suppress the enrichment of chiral polymorph A in the crystals of zeolite beta.
To decrease the acidity of the final mixture, we decreased the molar ratio of HF to SiO2 from 0.5 to 0.25 in experiment 5 and other conditions were maintained the same as those in experiment 4. The final mixture in experiment 5 will then be neutral. As expected, normal zeolite beta was obtained in experiment 5 (Fig. 1(d)). A further decrease in the molar ratio of HF to SiO2 from 0.25 to 0.15 leads to a basic mixture (experiment 6 in Scheme 1). It was not a surprise that normal zeolite beta was obtained, as confirmed by the corresponding XRD pattern shown in Fig. 1(e). Thus, it appears that the acidic nature of the final mixture favours the enrichment of chiral polymorph A in zeolite beta.
In experiments 1–6, HF was used as a mineralizer. The fluorine ion can balance the positive charge of TEA+. When the final mixture became acidic, both protons as well as fluorine ions were in excess. To distinguish the roles of protons and fluorine ions in promoting the enrichment of chiral polymorph A in zeolite beta, we performed experiment 7 of Scheme 1. In experiment 7, NH4F instead of HF was used with molar ratios to SiO2 of 0.15, 0.25, and 0.5. The powder XRD patterns of the corresponding products are shown in Fig. 1(f), (g), and (h), respectively, which confirmed that the products are normal zeolite beta.
The above results demonstrate that the acidity of the concentrated mixture containing SiO2–TEAOH–HF–H2O is a crucial factor promoting the enrichment of chiral polymorph A in the crystals of zeolite beta. Because concentrated HF solution is very toxic and if the above conclusion is correct, a combination of NH4F and other type of acid should also result in the chiral polymorph A-enriched zeolite beta, which can avoid the utilization of highly toxic concentrated HF. With this deduction, we performed experiment 8 of Scheme 1. In experiment 8, the initial mixture with the molar composition of SiO2:0.25TEAOH was first freeze-dried; subsequently, 0.25NH4F (to SiO2) and an appropriate (optimized) amount of the other types of acids such as 0.12 citric acid monohydrate (C6H8O7·H2O), 0.12 phosphorus pentoxide (P2O5), 0.10 acetic acid (CH3COOH, 98 wt%), 0.15 oxalic acid dihydrate (H2C2O4·2H2O), or 0.06 phosphoric acid (H3PO4, 85% aqueous solution) were added. The powder XRD patterns and scanning electron microscopy (SEM) images, thermogravimetric analysis (TGA) curves, and nitrogen adsorption–desorption isotherms of the products are shown in Fig. 2, S1, and S2,† respectively.
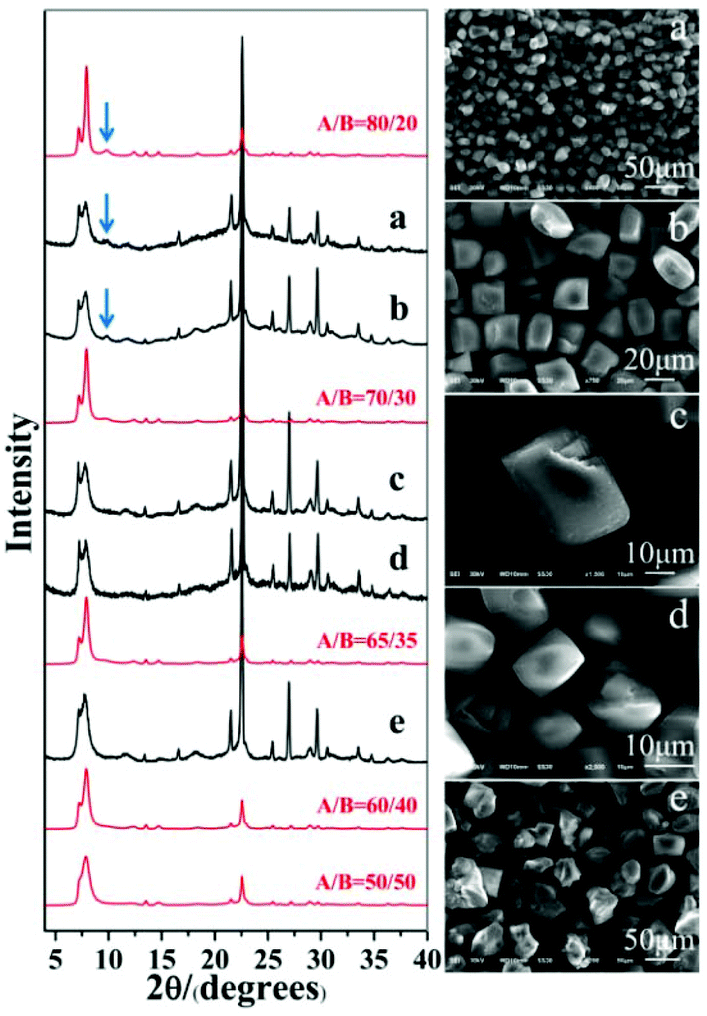 | ||
| Fig. 2 XRD patterns (left) and SEM images (right) of C6H8O7-beta-0.12 (a), P2O5-beta-0.12 (b), CH3COOH-beta-0.1 (c), H2C2O4-beta-0.15 (d), and H3PO4-beta-0.06 (e). The appearance of the characteristic peak at 2θ = 9.7° marked with arrows indicates that the enrichment of chiral polymorph A was greater than 70%. | ||
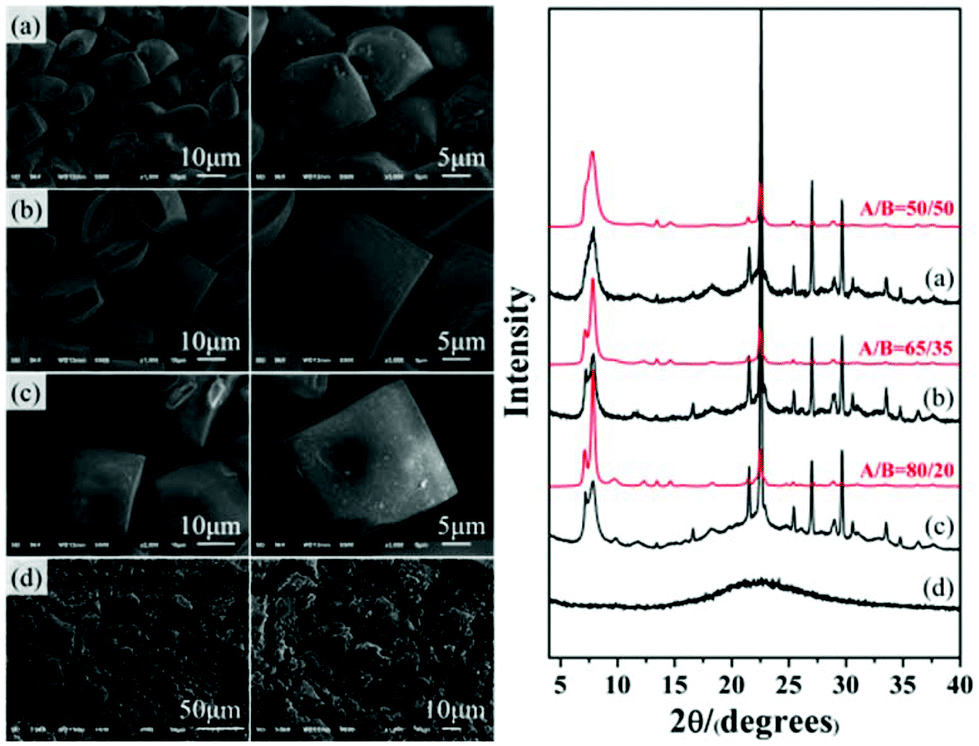
As expected, polymorph A-enriched zeolite beta crystals with varied proportions of chiral polymorph A are obtained. The proportion of polymorph A in zeolite beta is usually estimated by comparing an experimental XRD pattern with the simulated XRD patterns with different A/B polymorph ratios, especially the low angle fingerprint region 5°–10°. Fig. 2 displays the experimental XRD patterns of the products synthesized in the presence of the above five acid or acidic additives and simulated XRD patterns with polymorph ratios A/B of 50/50, 60/40, 65/35, 70/30, and 80/20, indicating that the proportions of polymorph A in these products are all greater than 60%. The scanning electron microscopy (SEM) images (Fig. 2) of the polymorph A-enriched zeolite beta show that the samples were free of impurities, although some particles seem to be broken or congregated. As shown in Fig. 2 (right), the chiral polymorph A-enriched zeolite beta crystals synthesized with different types of acid additives have a similar irregular cuboid morphology with a diameter of around 20 μm, which is totally different from the traditional truncated bipyramidal shapes. Thermogravimetric analysis (TGA) curves of the as-synthesized products shown in Fig. S1† all display a weight loss at about 20%, which is related to the removal of water, the decomposition of TEAOH molecules and the partial fluorine in the framework. After calcination at 550 °C for 8 h, all products show the type-I adsorption isotherm (see Fig. S2†), which is typical for pure-silica zeolite beta. The inconspicuous hysteresis loop could be ascribed to the grain gathered accumulation during crystallization by a dry-gel method.
Notably, the appearance of a characteristic peak at 2θ = 9.7° marked with arrows in Fig. 2 indicates that the enrichment of chiral polymorph A in C6H8O7-beta-0.12 and P2O5-beta-0.12 remarkably improved to greater than 70%. Fig. 3 shows the careful comparison of the experimental XRD patterns of C6H8O7-beta-0.12 and P2O5-beta-0.12 with the simulated XRD patterns in the fingerprint area. By comparing the degrees (2θ) of the first low-angle peaks with those of the simulated XRD patterns, it can be concluded that the proportions of chiral polymorph A in C6H8O7-beta-0.12 and P2O5-beta-0.12 were greater than 70% and approximately 80%, respectively. To the best of our knowledge, this is among the highest proportion reported so far. Fig. 4 shows the high resolution transmission electron microscopy (HRTEM) images of C6H8O7-beta-0.12 and P2O5-beta-0.12 viewed along the [100] direction. The white fold lines are added in the images to mark the stacking sequences of the 12-ring channels. The HRTEM images actually present the projection of zeolite beta in the [100] direction. As shown in Fig. 4, both polymorph A type (ABAB…) and polymorph B type (ABCABC…) stacking sequences are found in these areas, and no polymorph C type which should exhibit an AAA… stacking sequence is observed. The alternation of areas observed indicates the intergrowths inside the bulk crystals and the polymorph A type was dominant in C6H8O7-beta-0.12 and P2O5-beta-0.12.
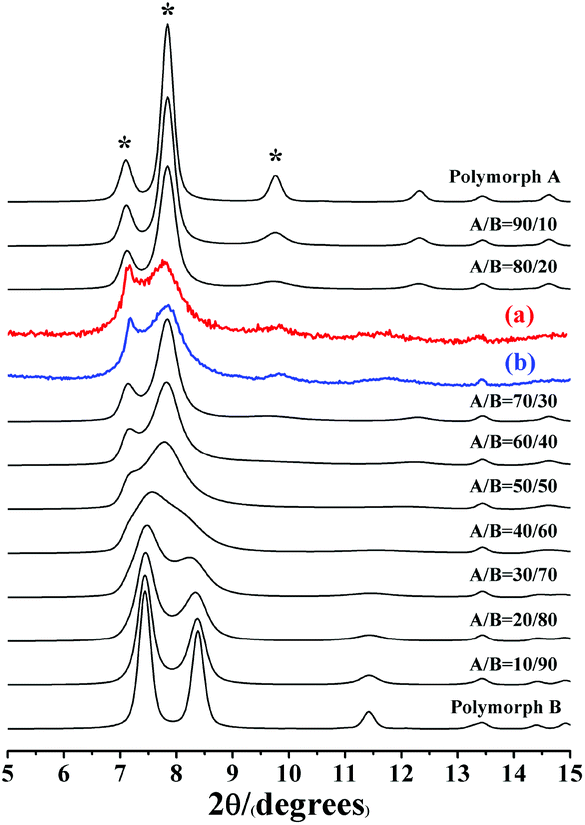 | ||
| Fig. 3 Experimental XRD patterns of C6H8O7-beta-0.12 (a), P2O5-beta-0.12 (b), and the simulated XRD patterns in the fingerprint area with different polymorph ratios of A/B. | ||
 | ||
| Fig. 4 HRTEM images of C6H8O7-beta-0.12 (a) and P2O5-beta-0.12 (b) aligned along the [100] direction. | ||
Fig. S3† displays the simulated XRD patterns of three pure zeolite beta polymorphs (polymorphs A, B, and C). The characteristic peaks of polymorph A at 2θ = 7.08° and 9.76° are very close to those of polymorph C (2θ = 7.0° and 9.72°). Therefore, the possibility of the existence of polymorph C in the investigated products cannot be excluded solely on the basis of powder XRD analyses. Because only polymorph C contains the double four membered ring (D4R) cage, which exhibits high affinity for fluorine ions, we performed 19F MAS NMR analyses to detect the chemical environments of the fluorine ions. For comparison, the 19F MAS NMR spectra of normal zeolite beta synthesized in neutral medium (HF-beta-0.25 (experiment 5 in Scheme 1)) and polymorph A-enriched zeolite beta synthesized in acidic medium (HF-beta-0.5 (experiment 4 in Scheme 1), C6H8O7-beta-0.12, and P2O5-beta-0.12) are shown in Fig. 5. In these four 19F MAS NMR spectra, three distinct signals at around −37, −59, and −71 ppm are observed. The first signal is from the fluorine ions occluded in the [46] (D4R) cage, whereas the latter two signals are attributed to the fluorine ions occluded in the [4354] cages of zeolite beta.30 Since neither polymorph A nor polymorph B contains D4R cages, the detected D4R cages in these samples are most likely associated with polymorph C. Notably, no characteristic peak of polymorph C in the XRD pattern of the normal zeolite beta (HF-beta-0.25, experiment 5 in Scheme 1, Fig. 1(d)) was observed, indicating that the long-range order of D4R was not constructed. Therefore, polymorph C is not XRD detectable if it does exist in the normal zeolite beta crystals, which rules out the possibility of the presence of a considerable amount of polymorph C. In addition, the intensities of the 19F signal at −37 ppm in normal zeolite beta (Fig. 5(a)) and polymorph A-enriched zeolite beta (Fig. 5(b), (c), and (d)) were basically the same, suggesting that the proportion of polymorph C in the chiral polymorph A-enriched zeolite beta (HF-beta-0.5, C6H8O7-beta-0.12, and P2O5-beta-0.12) is no more than that in normal beta. Thus, it appears that the existence of a considerable amount of polymorph C could be neglected in the polymorph A-enriched zeolite beta synthesized in acidic medium.
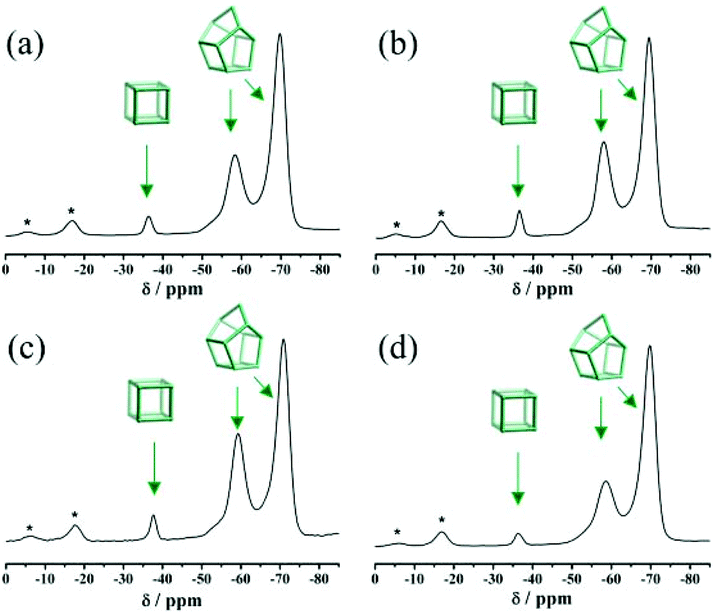 | ||
| Fig. 5 19F MAS NMR spectra of the as-synthesized samples of HF-beta-0.25 (a), HF-beta-0.5 (b), C6H8O7-beta-0.12 (c), and P2O5-beta-0.12 (d). The asterisks indicate spinning side bands. | ||
Thus, by stepwise verification, the acidity of the initial mixture was confirmed to be the crucial factor promoting the enrichment of chiral polymorph A in zeolite beta during the crystallization process. In general, increasing the amount of acid additive did result in a better enrichment of chiral polymorph A. However, we also found that the crystallization time needs to be prolonged to 10 d to reach the crystallinity similar to that synthesized under neutral conditions for 2 d. Further increasing the acidity will lead to the amorphous silica instead of crystalline zeolite beta. For those acidic initial mixtures that can crystallize polymorph A-enriched zeolite beta, we also found that a more acidic condition results in a larger crystal size and more well-defined crystal morphology. For example, Fig. 6 shows the SEM images with different magnifications and the corresponding XRD patterns of the products crystallized from the mixture with different amounts of P2O5. With the increased amount of P2O5, more amorphous product is formed, as confirmed by the XRD pattern shown in Fig. 6, however, the resulting crystals have a larger size and more well-defined morphology, as illustrated by the SEM images shown in Fig. 6. When the P2O5 is in excess, only amorphous silica was obtained (Fig. 6(d)). These results suggest that the number of nuclei in acidic medium is less than that in neutral medium, i.e. the nucleation of zeolite beta was inhibited in acidic medium, which usually causes larger crystal size and the formation of more amorphous silica. Combined with previous results that Al can increase the ratio of the nucleation rate and the crystal growth rate31 and our previous result that introduction of Al species results in a decrease in the degree of enrichment of chiral polymorph A in zeolite beta,32 inhibition of nucleation seems to be the essential factor for the enrichment of chiral polymorph A in zeolite beta. Although zeolite beta has no particular energetic preference for either polymorph A or B,15,33 restraining nucleation might promote growth of polymorph A in a more orderly manner, which provides us a new perspective to synthesize the chiral polymorph A-enriched zeolite beta.
 | ||
| Fig. 6 SEM images of P2O5-beta-0.02 (a), P2O5-beta-0.085 (b), P2O5-beta-0.12 (c), and P2O5-beta-0.18 (d) with different magnifications and the corresponding XRD patterns. | ||
Conclusions
In summary, we have developed a generalized non-hydrofluoric acid-assisted route to synthesize polymorph A-enriched zeolite beta. The control experiments showed that the acidity of the initial mixture certainly contributes to the enrichment of polymorph A. With this non-hydrofluoric acid-assisted strategy, the proportion of polymorph A can be significantly promoted to greater than 70% and approximately 80% under optimized conditions and the utilization of toxic hydrofluoric acid can be avoided by using more benign NH4F as the safe fluoride source. The results suggested that the slowed nucleation rate might be an essential reason for the enrichment of chiral polymorph A in zeolite beta. This acid-assisted method also provided us an opportunity to understand the enrichment mechanism of polymorph A during crystallization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (21571075, 21320102001, and 21621001), the National Key Research and Development Program of China (Grant no. 2016YFB0701100), the 111 Project (B17020 and B12015), and the Program for JLU Science and Technology Innovative Research Team.Notes and references
- http://www.iza-structure.org/databases/ .
- R. R. Xu, W. Q. Pang, J. H. Yu, Q. S. Huo and J. S. Chen, Chemistry of Zeolites and Related Porous Materials: Synthesis and Structure, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010 Search PubMed.
- J. Čejka, A. Corma and S. Zones, Zeolites and Catalysis: Synthesis, Reactions and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010 Search PubMed.
- S. Kulprathipanja, Zeolites in Industrial Separation and Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010 Search PubMed.
- P. Li and F. T. Handan, Microporous Mesoporous Mater., 2007, 98, 94 CrossRef.
- S. K. Brand, J. E. Schmidt, M. W. Deem, F. Daeyaert, Y. Ma, O. Terasaki, M. Orazov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5101 CrossRef PubMed.
- M. G. Kuba, R. Prins and G. D. Pirngruber, Appl. Catal., 2007, 333, 78 CrossRef.
- F. J. Maldonado, T. Bécue, J. M. Silva, M. F. Ribeiro, P. Massiani and M. Kermarec, J. Catal., 2000, 195, 342 CrossRef.
- M. Bejblová, D. Procházková and J. Čejka, ChemSusChem, 2009, 2, 486 CrossRef PubMed.
- R. L. Wadlinger, G. T. Kerr and E. J. Rosinski, US4579994, 1967 Search PubMed.
- M. A. Camblor, A. Corma and S. Valencia, Chem. Commun., 1996, 20, 2365 RSC.
- T. Blasco, M. A. Camblor, A. Corma, P. Esteve, A. Martinez, C. Prieto and S. Valencia, Chem. Commun., 1996, 20, 2367 RSC.
- A. Corma, L. T. Nemeth, M. Renz and S. Valencia, Nature, 2001, 412, 423 CrossRef PubMed.
- Y. Zhu, G. Chuah and S. Jaenicke, Chem. Commun., 2003, 21, 2734 RSC.
- J. M. Newsam, M. M. J. Treacy, W. T. Koetsier and C. B. D. Gruyter, Proc. R. Soc. London, Ser. A, 1988, 420, 375 CrossRef.
- J. B. Higgins, R. B. LaPierre, J. L. Schlenker, A. C. Rohrman, J. D. Wood, G. T. Kerr and W. J. Rohrbaugh, Zeolites, 1988, 8, 446 CrossRef.
- A. Corma, M. T. Navarro, F. Rey, J. Rius and S. Valencia, Angew. Chem., Int. Ed., 2001, 40, 2277 CrossRef PubMed.
- M. Tong, D. Zhang, L. Zhu, J. Xu, F. Deng, R. Xu and W. Yan, CrystEngComm, 2016, 18, 1782 RSC.
- Á. Cantín, A. Corma, M. J. Díaz-Cabañas, J. L. Jordá, M. Moliner and F. Rey, Angew. Chem., Int. Ed., 2006, 45, 8013 CrossRef PubMed.
- A. Corma, M. Moliner, Á. Cantín, M. J. Díaz-Cabañas, J. L. Jordá, D. Zhang, J. Sun, K. Jansson, S. Hovmöller and X. Zou, Chem. Mater., 2008, 20, 3218 CrossRef.
- M. E. Davis and R. F. Lobo, Chem. Mater., 1992, 4, 756 CrossRef.
- Q. H. Xia, S. C. Shen, J. Song, S. Kawi and K. Hidajat, J. Catal., 2003, 219, 74 CrossRef.
- Y. Takagi, T. Komatsu and Y. Kitabata, Microporous Mesoporous Mater., 2008, 109, 567 CrossRef.
- F. Taborda, T. Willhammar, Z. Wang, C. Montes and X. Zou, Microporous Mesoporous Mater., 2011, 143, 196 CrossRef.
- M. Tong, W. Yan, J. H. Yu and R. R. Xu, Chem. J. Chin. Univ., 2013, 34, 494 Search PubMed.
- W. Guo, W. Yan, R. R. Xu, Y. Wang and X. H. Mu, Chem. J. Chin. Univ., 2014, 35, 1363 Search PubMed.
- M. Tong, D. Zhang, W. Fan, J. Xu, L. Zhu, W. Guo, W. Yan, J. Yu, S. Qiu, J. Wang, F. Deng and R. Xu, Sci. Rep., 2015, 5, 11521 CrossRef PubMed.
- G. Zhang, B. Wang, W. Zhang, M. Li and Z. Tian, Dalton Trans., 2016, 45, 6634 RSC.
- T. Lu, R. Xu and W. Yan, Microporous Mesoporous Mater., 2016, 226, 19 CrossRef.
- M. A. Camblor, P. A. Barrett, M. a.-J. Díaz-Cabañas, L. A. Villaescusa, M. Puche, T. Boix, E. Pérez and H. Koller, Microporous Mesoporous Mater., 2001, 48, 11 CrossRef.
- M. A. Camblor, A. Corma and S. Valencia, J. Mater. Chem., 1998, 8, 2137 RSC.
- T. Lu, P. Gao, J. Xu, Y. Wang, W. Yan, J. Yu, F. Deng, X. Mu and R. Xu, Chin. J. Catal., 2015, 36, 889 CrossRef.
- S. M. Tomlinson, R. A. Jackson and C. R. A. Catlow, J. Chem. Soc., Chem. Commun., 1990, 11, 813 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and additional characterization of products. See DOI: 10.1039/c8qi00265g |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2018 |