DOI:
10.1039/C7QI00672A
(Research Article)
Inorg. Chem. Front., 2018,
5, 189-199
Structural diversities of squarate-based complexes: photocurrent responses and thermochromic behaviours enchanced by viologens†
Received
30th October 2017
, Accepted 14th November 2017
First published on 15th November 2017
Abstract
By using squarates as coordination building units, six new complexes formulated as [Mg(C4O4)(H2O)2]n (1), [Pr2(C4O4)2(OA)(H2O)4]n (2), [Eu2(C4O4)3(H2O)8]n (3) [Cd2(C4O4)2(bpdo)(H2O)4]n (4), [Cd(C4HO4)2(H2O)4]·(MV)2·(C4O4)2 (5), and [Cd(C4HO4)2(H2O)4]·(EV)·(C4HO4)2 (6), where (C4O4)2− = squarate, OA = oxalic acid, MV = methyl viologen, EV = ethyl viologen, and bpdo = 4,4′-dipyrdiyl-N,N′-dioxide, have been synthesized under hydro/solvothermal conditions. They present structural dimensions from 3-D networks (1, 2), 2-D layers (3, 4) to 0-D clusters (5, 6) based on the versatile bridged modes of squarates and the introduction of second ligands. In particular, 5 and 6 are the first examples incorporating squarates (electronic-rich species) with viologens (electronic-poor species) in one lattice. Consequently, they exhibit not only photocurrent responses but also reversible thermochromism, which will be beneficial for the design of multi-functional materials.
1. Introduction
As a well-known cyclic oxocarbon possessing D4h symmetry, the squarate ion (C4O4)2− is an electron rich species with a high degree of electronic delocalization.1 Therefore, squarate-based functional organic molecules have been extensively used in memory devices, molecular probes, solar cells and so on.2 In addition, the squarate dianion is a monocyclic carbon oxo anion with good aromaticity and strong ability to coordinate to metal ions, and therefore, in crystal engineering, it can be used as a poly-functional ligand for the construction of hybrid frameworks, including those built from supramolecular interactions such as hydrogen bonds or π–π interactions.3 These squarate-based hybrids have exhibited many interesting properties such as being heterogeneous catalysts, magnetic or luminescent.4 Until now, alkali/alkaline earth metals,1b,5a–c transition metals5d–h and rare earth metals5e,i–o have been used to coordinate with squarate to construct functional hybrids. The dianion of squarate in these complexes has illustrated versatile coordinated models, including 1,2-bidentate chelating, 1,2-bis(monodentate) and 1,3-bis(monodentate) bridging and even using all four oxygen atoms 1,2-bidentate chelate type.6 Most of these compounds are coordinated frameworks constructed from a single squarate ligand, but studies about the introduction of second ligands to tailor their structures and properties are relatively rare (such as the introduction of pyridine, 1,2-bis(4-pyridyl)ethane), besides this, the functions of the second ligands have not been fully considered.1a,4c,5j,k,l,7 As well-known good electron acceptors, viologens have been widely used in functional materials such as in solar energy storage, electrochromic display devices and organic electrical conductors.8 The incorporation of electron poor viologens with electron rich squarates seems significant, which might give rise to novel multi-functional materials. Therefore, in this work, by using squarates as coordination building units, six compounds exhibiting 0- to 3-D arrangements have been synthesized through hydrothermal and solvothermal methods, among which 5 and 6 are the first examples incorporating electronic poor viologens with electronic rich squarates in one lattice. They exhibit not only photocurrent responses but also reversible thermochromism, which will be beneficial for the design of multi-functional materials.
2. Experimental section
2.1 Materials and general methods
All chemicals except bpdo (bpdo = 4,4′-dipyrdiyl-N,N′-dioxide), MV·I2, and EV·I2 (methyl viologen and ethyl viologen, i.e. N′-dialkyl-4,4′-bipyridinium iodide) were of reagent grade quality obtained from commercial sources and used without further purification. Elemental analyses for C, H and N were performed on a Vario MICRO elemental analyzer. IR spectra were recorded on a PerkinElmer Spectrum-2000 FTIR spectrophotometer (4000–400 cm−1) on a powdered sample spread on a KBr plate. Optical diffuse reflectance spectra were measured on a PerkinElmer lambda 900 UV/VIS spectrophotometer equipped with an integrating sphere at 293 K, and BaSO4 plates were used as reference. Powder XRD patterns were obtained using a Philips X'Pert-MPD diffractometer with CuKα radiation (λ = 1.54056 A). The photocurrent experiments were performed on a CHI650 electrochemistry workstation with three-electrode systems. ESR signals were examined with a Bruker ESP 300E spectrometer. Fluorescence spectra were recorded using an Edinburgh FL-FS 920 TCSPC spectrometer.
Synthesis.
MV·I2 and EV·I2 were synthesized by a one-step N-alkylated reaction of 4,4′-bipyridine with iodomethane and iodoethane according to the literature method.9a MV·(NO3)2 and EV·(NO3)2 can be obtained by removing the iodine ion with an equal amount of AgNO3. bpdo was prepared by one-step oxidation of 4,4′-bipyridine using hydrogen peroxide at 80 °C.9b
[Mg(C4O4)(H2O)2]n (1).
1 was prepared by a solvothermal method. Squarate (0.1140 g, 1 mmol) was dissolved in 2.5 mL acetone solution containing 0.5 mmol MV·(NO3)2. In another beaker, MgCl2 (0.0476 g, 0.5 mmol) was dissolved 10 mL acetone. The above two solutions were mixed and stirred for 90 minutes. The suspension was transferred to a 25 mL Teflon-lined stainless steel vessel. The autoclave was heated to 160 °C and maintained at this temperature for 3 d. Then the autoclave was cooled to room temperature at a rate of 3 °C h−1. Block colorless crystals were obtained and washed with ether with a yield of 32.9% (0.0283 g, based on Mg). The desired outcome was an Mg/squarate anionic framework containing MV2+ cations. But out of our expectation, only an Mg/squarate neutral framework was obtained. Anal. calcd for C4H4MgO6 (172.38): C, 27.87; H, 2.34%. Found: C, 27.53; H, 2.56%. FT-IR (KBr, cm−1): 3320(s), 1723(w), 1510(s), 1099(m), 525(w).
[Pr2(C4O4)2(OA)(H2O)4]n (2).
Pr(NO3)3·6H2O (0.2175 g, 0.5 mmol), squarate (0.1140 g, 1.0 mmol) and oxalic acid (0.0900 g, 1 mmol) were dissolved in 10 mL H2O in a 25 mL Teflon-lined stainless steel vessel. The autoclave was heated to 180 °C at the rate of 15 °C h−1 and maintained at this temperature for 3 d, and then cooled slowly to room temperature at the rate of 5 °C h−1. Yellow block crystals can be obtained and washed with ether with a yield of 36.6% (0.0153 g, based on Pr). Anal. calcd for C5H4O8Pr (332.99): C, 18.03; H, 1.20%. Found: C, 18.22; H, 1.34%. FT-IR (KBr, cm−1): 3378(m), 3098(w), 1651(w), 1617(w), 1498(w), 1174(w), 1135(w), 1073(m).
[Eu2(C4O4)3(H2O)8]n (3).
The synthesis conditions of 3 are similar to that of 2, except that Pr(NO3)3·6H2O is substituted with Eu(NO3)3·6H2O (0.2230 g, 0.5 mmol). Yellow block crystals with a yield of 37.2% (0.1840 g, based on Eu) can be obtained. Anal. calcd for C12H16Eu2O20 (784.17): C, 18.38, H 2.05%. Found: C, 18.56, H 1.93%. FT-IR (KBr, cm−1): 3425(m), 3210(m), 1815(w), 1603(w), 1502(s), 1094(m).
[Cd2(C4O4)2(bpdo)(H2O)4]n (4).
The squarate (0.0228 g, 0.2 mmol) and Cd(OH)2 (0.0183 g, 0.125 mmol) were dissolved in 3 mL of H2O, the pH of the solution was adjusted to 7.0 with 1 mol L−1 NaOH. In another beaker, bpdo (0.0376 g, 0.2 mmol) was dissolved in 7 mL H2O. The latter solution was added into the former and the mixture was transferred to a 25 mL Teflon-lined stainless steel vessel. The suspension was stirred for 90 minutes at room temperature. The autoclave was heated to 160 °C at the rate of 20 °C h−1 and maintained at this temperature for 3 d, and then cooled slowly to room temperature at the rate of 5 °C h−1. Yellow block crystals can be obtained and washed with ether with a yield of 20.5% (0.0091 g, based on Cd). Anal. calcd for C9H8CdNO7 (354.57): C, 30.48; H, 2.26; N, 3.95%. Found: C, 31.02; H, 2.14; N, 3.76%. FT-IR (KBr, cm−1): 3126(m), 1602(w), 1476(s), 1209(s), 1090(m), 771(m), 677(s), 531(m).
[Cd(C4HO4)2(H2O)4]·(MV)2·(C4O4)2 (5).
5 was prepared by hydrothermal reactions. Squarate (0.1140 g, 1.0 mmol) and Cd(OH)2 (0.0732 g, 0.5 mmol) were dissolved in 7.5 mL of H2O. Then 2.5 mL of water solution containing 0.5 mmol MV·(NO3)2 was added. The mixture was transferred to a 25 mL Teflon-lined stainless steel vessel and stirred for 90 minutes at room temperature. The autoclave was heated to 150 °C at the rate of 20 °C h−1 and maintained at this temperature for 5 d, and then cooled slowly to room temperature at the rate of 3 °C h−1. Yellow block crystals can be obtained and washed with ether with a yield of 60.8% (0.2500 g, based on Cd). Anal. calcd for C28H25CdN2O20 (821.91): C, 40.92; H, 3.07; N, 3.41%. Found: C, 41.22; H, 3.17; N, 3.56%. FT-IR (KBr, cm−1): 3246(w), 3140(w), 2994(w), 1815(m), 1656(m), 1562(m), 1476(m), 1376(m), 1070(m), 810(s), 551(w).
[Cd(C4HO4)2(H2O)4]·(EV)·(C4HO4)2 (6).
The synthesis process of 6 is similar to that of 5, except that MV·(NO3)2 was replaced with EV·(NO3)2. Yellow block crystals can be obtained with a yield of 79.3% (0.3376 g, based on Cd). In the synthesis of 4–6, only the products can only be obtained by using hydroxides as starting materials, which can be attributed to the part or full de-protonization of squarates in these compounds. Anal. calcd for C30H30CdN2O20 (850.96): C, 42.34; H, 3.55; N, 3.29%. Found: C, 42.21; H, 3.46%; N, 3.19%. FT-IR (KBr, cm−1): 3273(m), 3140(w), 2986(w), 1809(m), 1662(s), 1576(s), 1436(w), 1363(m), 1037(m), 831(s), 551(m).
2.2 X-ray data collection and structure determination
The intensity data of 1–6 were collected using a Bruker APEX II diffractometer using graphite-monochromated MoKα radiation (λ = 0.71073 Å) at room temperature. The empirical absorption corrections were based on equivalent reflections. Structures were solved by direct methods followed by successive difference Fourier methods. Computations were performed using SHELXTL and final full-matrix refinements were against F2.10 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms of C–H, N–H and O–H were generated geometrically. A summary of the crystal structure refinement data is provided in Table 1. The selected bond angles and distances are listed in Tables S1 and S2,† hydrogen bonds and π–π interaction details are given in Tables S3 and S4.†
Table 1 Summary of the crystal data and structure determination for 1–6
| Compound |
1
|
2
|
3
|
4
|
5
|
6
|
| Empirical formula |
C4H4MgO6 |
C5H4O8Pr |
C12H16Eu2O20 |
C9H8CdNO7 |
C28H25CdN2O20 |
C30H30CdN2O20 |
| Formula mass |
172.38 |
328.96 |
784.17 |
354.57 |
821.91 |
850.9 |
| Crystal system |
Cubic |
Monoclinic |
Monoclinic |
Orthorhombic |
Triclinic |
Monoclinic |
| Space group |
Pn![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) n n |
P21/c |
Pc
|
Pnnm
|
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
|
a [Å] |
16.2912(19) |
9.5960(19) |
11.956(2) |
12.1360(16) |
6.4103(10) |
6.3956(6) |
|
b [Å] |
16.2912(19) |
9.923(2) |
8.2251(16) |
8.2328(11) |
13.4345(18) |
19.0514(18) |
|
c [Å] |
16.2912(19) |
10.004(2) |
10.120(2) |
10.9375(15) |
19.156(3) |
27.414(2) |
|
α [°] |
90.00 |
90.00 |
90.00 |
90.00 |
72.349(2) |
90.00 |
|
β [°] |
90.00 |
118.60(3) |
96.08(3) |
90.00 |
80.678(2) |
100.310(2) |
|
γ [°] |
90.00 |
90.00 |
90.00 |
90.00 |
88.590(2) |
90.00 |
|
V [Å] |
4323.7(15) |
836.4(3) |
989.6(3) |
1092.8(3) |
1550.7(4) |
3286.3(5) |
|
Z
|
24 |
4 |
2 |
4 |
2 |
4 |
|
D
c [g cm−3] |
1.589 |
2.612 |
2.578 |
2.155 |
1.760 |
1.720 |
|
μ [mm−1] |
0.227 |
5.843 |
6.384 |
2.026 |
0.800 |
0.759 |
|
F(000) |
2112 |
612 |
716 |
692 |
830 |
1728 |
| Reflections, total |
33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 140 140 |
6120 |
7058 |
3714 |
8755 |
10808 |
| Reflections, unique |
837(Rint = 0.1044) |
1885 (Rint = 0.0473) |
3364 (Rint = 0.0383) |
1028 (Rint = 0.0186) |
4895 (Rint = 0.0195) |
5683 (Rint = 0.0246) |
| Reflections, observed |
617 |
1854 |
3321 |
862 |
4471 |
5076 |
| Goodness-of-fit on F2 |
1.135 |
1.068 |
1.075 |
1.085 |
1.071 |
1.172 |
| No. of parameters refined |
51 |
128 |
290 |
94 |
469 |
480 |
|
R[I > 2σ(I)] |
0.0756 |
0.0289 |
0.0292 |
0.0210 |
0.0554 |
0.0429 |
|
R
w [I > 2σ(I)] |
0.2319 |
0.0763 |
0.0771 |
0.0562 |
0.1564 |
0.1084 |
| Residual extremes (e Å−3) |
0.963, 1.045 |
1.555, −1.064 |
0.995, −1.683 |
0.818, −0.436 |
1.348, −1.156 |
0.473, −0.954 |
2.3 Preparation of the electrodes and photocurrent measurements
The films of electrodes were prepared by a solution coating method. 0.5 mg of the as-synthesized compounds 5 and 6 were dissolved in 0.5 mL DMF and the resulting solutions were coated on a precleaned FTO glass (0.6 × 0.6 cm). The solvents were removed by evaporation to produce the respective films. A high pressure xenon lamp (150 W, 15 cm far from the surface of the FTO electrode) was used as a light source. The photocurrent experiments were executed on a CHI650 electrochemistry workstation. In these measurements, a three electrode system (a sample coated 0.6 × 0.6 cm FTO glass plate as the working electrode, a Ag/AgCl as the counter and reference electrodes) was used, and Na2SO4 (50 mL, 0.2 mol L−1) was utilized as a supporting electrolyte solution. The lamp was kept on continuously, during which a manual shutter was utilized to block the light to the sample. The irradiation time of the samples was 120 s with an interval of 20 s. All photocurrent measurements were carried out under the same experimental conditions.
3. Results and discussion
3.1 Structural description
[Mg(C4O4)(H2O)2]n (1).
1 crystallizes in the cubic system with a Pnn space group. The basic molecular unit contains one Mg(II) ion, one squarate ligand and two coordinated water molecules. The Mg center acquires regular octahedral geometry and is ligated by four oxygen atoms from four squarate ligands and two oxygen atoms from water molecules. The Mg–O bond lengths range among 2.046(3)–2.092(3) Å, and the bond angles of the MgO6 octahedron are in the range of 85.24(10)°–92.66(10)°, 179.06(16)° (for 1) (Table S1†), which deviate slightly from those of the ideal octahedron. The Mg(II)⋯Mg(II) distances bridged by the squarate (μ1,3-bridging) are 8.087 Å (Fig. 1). The C–C, C–O bond lengths of the squarate show a roughly D4h delocalized mode, and they have been averaged as observed in some analogous squarate-bridged complexes (Table S1†).11 Adjacent MgO6 octahedra are bridged by two crystallographic independent squarates (SA1, SA2) into 1-D chains along the b-axis through anti–anti μ1,3-O bridged squarate ions, adjacent 1-D chains are linked by SA2 into a two-dimensional (2D) rhombohedral grid-like layers along the bc-plane. These 2-D layers are actually composed of an infinite array of metallocycles formed by four Mg centres and four squarate ligands, with a dimension of 8.087 × 8.146 Å2 (Fig. 1). Finally, the 2-D layers extend into a 3-D network via bridged SA2 (Fig. 1a). Consequently, only one kind of coordinated model can be found in the squarate, in which the squarate coordinates to four Mg centers via its four oxygen atoms (tetramonodentate, Scheme 1e). In conclusion, 1 is a neutral three-dimensional framework (Fig. 1b). The void volumes of two compounds calculated by the PLATON analysis12 revealed a pore-accessible volume of 19.1% (823.8 out of 4323.7 Å3 per unit cell volume) with respect to the whole unit cell volume. Hydrogen bonds between the coordinated water molecules and squarates can be observed in two compounds (Table S3†). And furthermore, the intricate 3-D structural architecture of 1 is also stabilized by the π⋯π interaction with centroid-to-centroid distances of 3.644(3) and 3.711(3) Å (Table S4†).
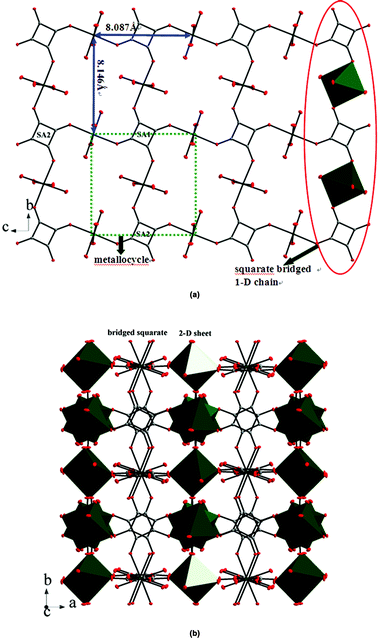 |
| | Fig. 1 (a) Illustration of the 2-D rhombohedral grid framework formed by the squarate bridged 1D chains and the metallocycle of 1. H atoms have been omitted for clarity; (b) 3-D network of 1 generated from bridged squarates. | |
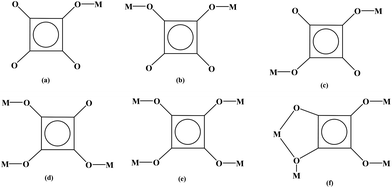 |
| | Scheme 1 The coordinated modes of squarate in this work. | |
[Pr2(C4O4)2(OA)(H2O)4]n (2).
2 crystallizes in space group P21/c, whose asymmetric unit contains one Pr3+ ion, one squarate, half of the oxalate ligand and two coordinated water molecules. The Pr center is nine-coordinated with five oxygen atoms from four squarate ligands, two oxygen atoms from one oxalate ligand and two oxygen atoms from the coordinated water molecules. Therefore, the coordinated geometry of Pr centers can be described as a distorted dicapped trigonal prism (Fig. 2a). The bond lengths and angles around the Pr center (Table S1†) are similar to the reported analogues of Ln-based compounds containing a similar set of ligands.4c,5o Two PrO9 dicapped trigonal prisms are connected into a Pr2O16 dimmer via an edge-sharing model (O(7)–O(7)#2, #2: −x + 1, −y, −z + 1, Fig. 2a), adjacent Pr2O16 dimmers are linked into a 1-D chain by using bridged squarate ligands (Fig. S1a†), furthermore, 1-D chains are extended into a 2-D layer via bridged oxalate ligands (Fig. S1b†), finally, these layers are linked into a 3-D network by a second squarate ligand (Fig. 2c). The deprotonation of the squarate ligands can be judged from their four averaged C–O bonds (Table S1†). Consequently, each squarate ligand acts as a tetradentate ligand to bind four lanthanide ions (μ4-1,2,3-tri(monodentate)-4-bisdentate, Scheme 1f), and oxalate adopts the common bisdentate-chelated mode (Fig. 2b).
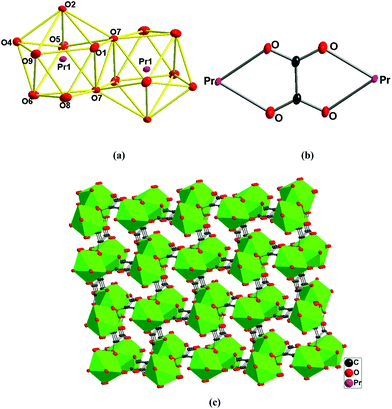 |
| | Fig. 2 (a) Pr2O16 dimer showing the PrO9 dicapped trigonal prism geometry; (b) the bridging mode of oxalate ligands; (c) 3-D network of 2 bridged by squarate and oxalate ligands. | |
[Eu2(C4O4)3(H2O)8]n (3).
Single-crystal X-ray analysis revealed that 3 is a neutral 2-D bis-layer. In this neutral bis-layer, each europium atom is coordinated to four oxygen atoms from four squarate ligands and four oxygen atoms from water molecules in a severely distorted bicapped trigonal prismatic geometry (Fig. 3a). The Eu–O distances are in the range of 2.376(6)–2.458(6) Å (Table S1†), which can be comparable with other Eu-squarate compounds.13 Two kinds of bridged modes in the squarate ligand can be found: μ3-trimonodentate and μ2-1,2-bis(monodentate) trimonodentate mode (Scheme 1b and d). Adjacent EuO8 bicapped trigonal prisms are bridged by squarate ligands along the b-axis to give a 1-D chain (Fig. S2a†), these 1-D chains are connected into a 2-D mono-layer along the ab-plane via the third O atom of the squarate ligands (Fig. S2b†), in other words, the 2-D mono-layer is constructed from the μ3-connected coordinated mode of the squarate ligands. Finally, two mono-layers are linked by bridged squarate ligands to generate a bis-layer via the μ2-connected mode (Fig. 3b). We also could not find the H atoms on coordinated water molecules, so no hydrogen-bonding interactions can be shown. In addition, there is no π⋯π stacking interactions in the lattice.
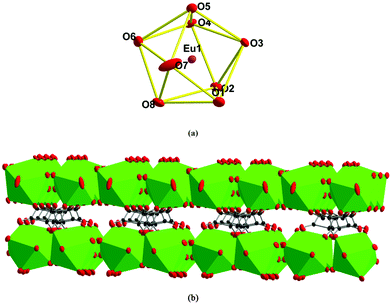 |
| | Fig. 3 (a) EuO8 bicapped trigonal prism; (b) neutral 2-D bis-layer of 3. | |
[Cd2(C4O4)2(bpdo)(H2O)4]n (4).
By using a bridged ligand bpdo (4,4′-dipyrdiyl-N,N′-dioxide), 4 presents a 2-D layer arrangement. Compared with the [Cd(C4HO4)2(H2O)4] cluster in 5 and 6, two coordinated water molecules of the CdO6 octahedron in 4 are replaced by oxygen atoms from bpdo ligands. The coordinated model of the O atom in bpdo is μ2-bridged, consequently, two CdO6 octahedra are linked into a Cd2O10 dimmer (Fig. 4a). These Cd2O10 dimers are linked into a 1-D double-chain along the b-axis by bis-deprotonated squarates (μ2-1,3-bis(monodentate) mode, Scheme 1c). The bis-deprotonated squarate can be deduced from their four averaged C–O bonds (Table S2†). And furthermore, these double-chains are extended into a 2-D layer along the ab-plane via bridged bpdo ligands. The adjacent layers are connected into a psedo-3-D network via strong hydrogen bonds between the coordinated water molecules (as donors) and squarate/bpdo (as acceptors, Fig. 4b).
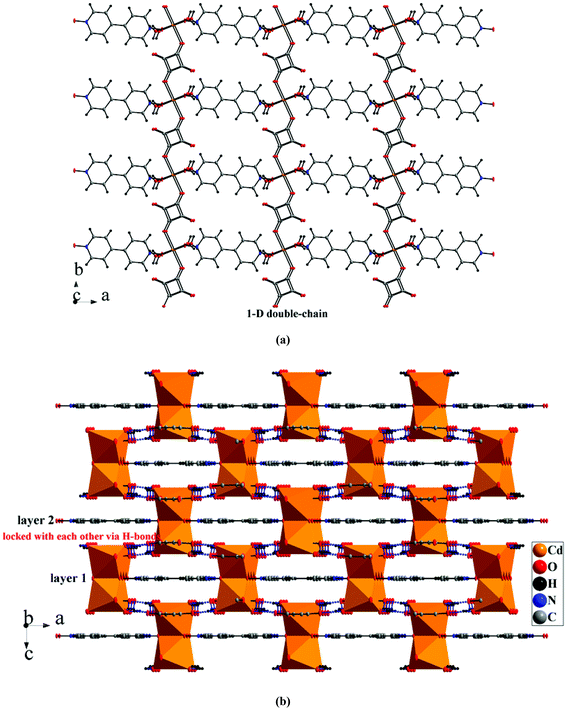 |
| | Fig. 4 (a) 2-D layer of 4via bridged bpdo ligands; (b) 3-D network based on hydrogen bonds. | |
[Cd(C4HO4)2(H2O)4]·(MV)2·(C4O4)2 (5) and [Cd(C4HO4)2(H2O)4]·(EV)·(C4HO4)2 (6).
5 and 6 consist of neutral [Cd(C4HO4)2(H2O)4] clusters, MV2+/EV2+ counteractions and two isolated deprotonated squarate ligands ((C4O4)2− for 5 and (C4HO4)− for 6). Due to the incorporation of larger countercations MV2+/EV2+, the infinite metal–organic moieties were split to be 0-D clusters. Versatile hydrogen bonds contribute to the formation of 2-D supramolecular arrangements. The Cd centers appear in distorted octahedral geometries coordinated by two oxygen atoms from squarate ligands and four water molecules (Fig. 5a). So the coordinated mode of the squarate is μ1-monodentate (Scheme 1a). The Cd–O distances are in the range of 2.221(5)–2.336(5) Å (for 5)/2.263(3)–2.322(3) Å (for 6). The angles of the CdO6 octahedron are 86.0(3)–91.7(3)°, 176.7(2)° (for 5) and 82.21(9)–96.64(10)°, 177.39(9)° (for 6), deviating obviously from 90° and 180° for an ideal octahedron (Table S2†). The mono-deprotonation of squarate ligands in [Cd(C2HO4)2(H2O)4] clusters in two compounds can be deduced from their three averaged and one longer C–O bonds (Fig. 5a, Fig. S3a and Table S2†). This situation can also be found in two isolated squarate ligands (C2HO4)− of 6 (1.222(5)–1.259(5) Å vs. 1.302(5)/1.310(5) Å, Table S2†). But in 5, the isolated squarates are bis-deprotonated with four averaged C–O bonds (Table S2†). The bond lengths in MV2+ and EV2+ are normal. There are also versatile hydrogen bonds in two compounds. Intra-molecular hydrogen bonds in the [Cd(C4HO4)2(H2O)4] clusters can be observed in 5 and 6. But in 5, inter-molecular ones between the [Cd(C4HO4)2(H2O)4] clusters, isolated squarate ligands and MV2+ cations can be found, in 6, only inter-molecular ones between the [Cd(C4HO4)2(H2O)4] clusters and isolated squarate ligands were observed (Table S3†). Consequently, adjacent [Cd(C2HO4)2(H2O)4] clusters are linked into a 1-D chain along the a-axis via inter-molecular hydrogen bonds with three coordinated water molecules as donors and isolated squarate ligands as acceptors, and furthermore, the adjacent chains are extended into a 2-D layer via inter-molecular hydrogen bonds among the coordinated and isolated squarate ligands (Fig. 5b and Fig. S3b†). In addition, π–π stacking interactions with a centroid-to-centroid distance of 3.556(6)–3.675(3) Å between free and coordinated squarate ligands in two compounds stabilize the structures (Table S4†). Finally, in 5, the hydrogen bonds between MV2+ and the {[Cd(C4HO4)2(H2O)4]·(C4O4)2}4− layer lead to the formation of a 3-D network (Fig. 5c). But in 6, no such interactions can be detected, EV2+ cations accommodate among {[Cd(C2HO4)2(H2O)4]·2(C2HO4)}2− layers only through electrostatic interactions (Fig. 5d). In particular, in 5, the isolated squarates were bis-deprotonated accompanied by two MV2+ countercations, but in 6, only one proton was left in isolated squarates with one EV2+ counteraction. The deprotonated degree of squarate in this work could be controlled by the lengths of substituted groups of viologens. The packing stability will decrease if two EV2+ with a larger ethyl group were incorporated into the lattice, and as a result, the squarate was presented as (C2HO4)− to decrease the number of counteractions.
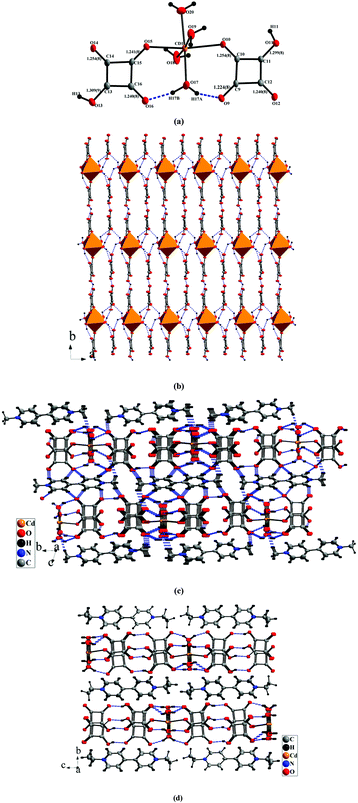 |
| | Fig. 5 (a) Structure of the [Cd(C2HO4)2(H2O)4] cluster showing intra-molecular hydrogen bonds; (b) structure of the {[Cd(C4HO4)2(H2O)4]·(C4O4)2}4− layer constructed from intermolecular hydrogen bonds and π–π stacking interactions; (c) crystal packing showing hydrogen bonds between the {[Cd(C4HO4)2(H2O)4]·(C4O4)2}4− layer and MV2+ cations in 5; (d) packing diagram showing the position of EV2+ in 6. | |
In this work, six squarate-containing compounds exhibit 3-D networks (1, 2), 2-D layers (3, 4) and 0-D clusters (5, 6) based on the versatile coordinated modes of squarate ligands and the introduction of second ligands. The tetramonodentate (μ4-connected) mode was observed in 1, μ2-1,3-bis(monodentate) (for 4) and μ1-monodentate (for 5, 6) were engaged. These modes were commonly observed in squarate-containing compounds.4 But the μ4-1,2,3-tri(monodentate)-4-bisdentate in 2 and μ3-trimonodentate and μ2-1,2-bis(monodentate) in 3 are seldom reported.5b,14 The coordinated modes of the squarate in this work are summarized in Scheme 1. Interestingly, the introduction of larger counteractions may lead to the formation of 0-D clusters (5, 6). This might be helpful for the design of new functional materials.
3.2 Optical diffuse-reflection spectra
The phase purities of bulk compounds 1–6 have been verified by powder X-ray diffraction (PXRD). It is obvious that the peaks of the experimental patterns are consistent with the corresponding simulated ones, suggesting their good phase purities (Fig. S4a–f†). The solid-state optical diffuse-reflection spectra of 1–6 were recorded from powder samples at room temperature (Fig. 6). In the ultra-violet regions, the intense bands around 200–320 nm can be assigned to the π–π* transitions of squarate, bpdo (5) and viologen ligands (5, 6), which are similar to literature compounds.5l,12 Judging from Fig. 6, for 1–3, there is no adsorption in the visible region, but for 4–6, strong adsorption bands in the visible region (among 400–550 nm) can be observed. The presence of the visible region might be assigned to the absorption of charge transfer (CT), partial electron transfer between the (C4HO4)−/(C4O4)2− anions and MV2+/EV2+ cations or the much stronger contacts such as hydrogen bond interactions.12a
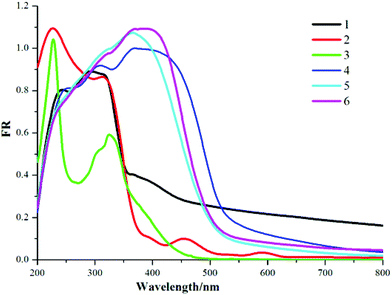 |
| | Fig. 6 Solid state electronic spectra of 1–6. | |
3.3 Fluorescence spectra
We have conducted photoluminescence measurements on all compounds at room temperature, but only the Eu-containing one exhibits photoluminescence behaviour. It is universally accepted that lanthanide-containing complexes show promising luminescent device characteristics. When excited at 337 nm, 3 produces emissions in visible regions (Fig. 7), in which the strong emission peak at 614 nm is presented, and weaker shoulder peaks at 590, 593, 618, 627, 678, 690, 719 nm are detected. Comparing the adsorption spectrum with the excited spectrum of 3 (Fig. 7, left and middle), there are some overlapped wavelength ranges between 285 and 445 nm, suggesting that the emissions stem mainly from the energy absorbed from metal centers. Therefore, the defined emissions are assigned to 5D0–7FJ (J = 0, 1, 2, 3, 4) in the Eu3+ center, but the weaker wide emission bands below 580 nm might stem from the squarate ligands.15 The universally accepted emission mechanism of lanthanide/organic is as follows: the ligands are excited with the energy being transferred from the singlet state S1 to the triplet state T1, then the energy transfers from the lowest excited triplet state to the excited states of the central lanthanide centers, finally, the central lanthanides produce fluorescence via radiative transitions from the excited states to the ground states.16 As shown in Fig. 6, the organic emissions are much weaker, suggesting that the triplet state of the squarate ligand is not matched well to Eu3+ ion's emitting resonance levels 5D0.
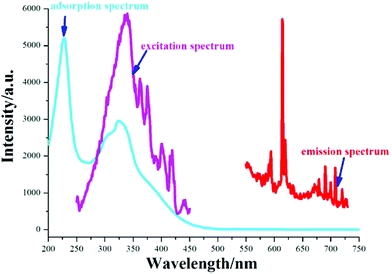 |
| | Fig. 7 UV-Vis absorption, excitation and emission spectra of 3. | |
3.4 Photocurrent response properties
Recently, considerable attention has been paid to the photocurrent response materials, which can be explained as the effective electron transfer between the electron donors and the electron acceptors.17 According to the typical method, three electrode photo/electrochemical cells consisting of the sample modified FTO electrode were used in this system (a more detailed description is given in the Experimental section), and the results can be seen from Fig. 8a. Interestingly, upon repetitive irradiation with xenon light on and off (interval 20 s), effective and repeatable photocurrent responses can be observed on two compounds. For 5, the photocurrent decreased to some extent firstly and then stabilized after four cycles. In particular, the cathodic photocurrents (square curves) can be achieved quickly without delay, though their photocurrent intensities are weaker compared with other complexes (the photocurrent densities are 0.4 μA for 5 and 0.3 μA for 6).17 Generally, the following factors (metal centers, coordination models of ligands, the structural dimensions, and the interactions between cations and anions) can affect the photocurrent responses. According to the structural and adsorption spectra information, in 5 and 6, the layer-like packing modes with the existence of many interactions such as electrostatic interactions, hydrogen bond interactions and face-to-face π–π stacking interactions between squarate and viologen moieties have been observed, and the near infrared CT bands can be detected. So a conclusion could be drawn that there exist strong charge transfers and even partial electron transfer from the squarate moiety to the viologen cations in these ion pair compounds, which play an important role in the enhancement of the photocurrent.17d The photocurrent response mechanisms can be explained by the following electron-transfer routes for the [Cd(C4HO4)2(H2O)4]·(MV)2·(C4O4)2/ITO and [Cd(C4HO4)2(H2O)4]·(EV)·(C4HO4)2/ITO electrodes in solution (Fig. 8b). Firstly, after irradiation, the photosensitive MV2+/EV2+ ions are excited to generate the MV˙+/EV˙+ radicals, at the same time, electron transfers can also occur from the (C4O4)2−/(C4HO4)− donors to the MV2+/EV2+ cations. Then the MV˙+/EV˙+ radicals transfer their electrons to the ITO electrodes with the re-generation of MV2+/EV2+ cations. The squarate moieties lose one electron to form the squarate˙+ radicals to produce the electron conductive pair of squarate˙+–MV˙+/EV˙+. Consequently, the effective electron flow in these cycles can be maintained. According to this mechanism, more counteractions will lead to higher mobility, so the photocurrent density of 5 is higher than that of 6.
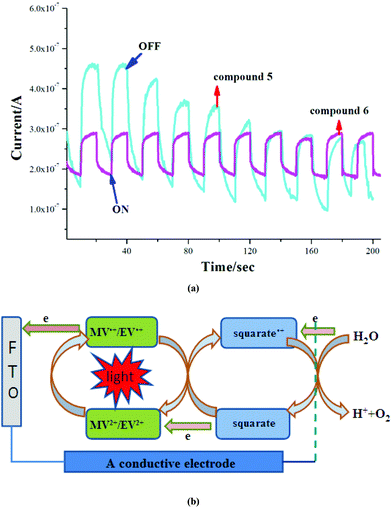 |
| | Fig. 8 (a) Photocurrent response behaviours; (b) proposed photocurrent response mechanism in 5 and 6. | |
3.5 Thermochromic properties
The thermochromic properties of the all compounds have been studied. However, only viologen-containing ones (5 and 6) exhibit interesting reversible thermochromic behaviours (Fig. 9 and 10). Their colours can change clearly upon heating in air. In detail, the pale yellow crystalline samples of 5 and 6 start to become pink when heated to 110 and 130 °C, and they finally change to orange at 130 and 150 °C and remain unchanged. In particular, these processes are reversible while they are cooled to room temperature (Fig. 9(a) and 10(a)). Meanwhile, the cyclical colour changes can be repeated up to at least 10 times by heating, which imply their typical thermochromic behaviours. Until now, many thermosensitive mechanisms have been proposed for explaining the thermochromic properties, for example, the conformation change of the crystal structure and electron transfer.18 In order to disclose the thermochromic behaviours, electron spin resonance (ESR) measurements were carried out to make sure of the presence of electron transfers upon heating. The results show that two compounds are ESR silent before heating, but after heating, they show single-line signals at g = 2.0010 (for 5) and g = 2.0008 (for 6), and furthermore, after cooling down, the ESR singles return to be zero (Fig. 9(b) and 10(b)). The cadmium atom can be excluded to be a paramagnetic center, because CdI and CdIII atoms are unstable in this case. However, the g values are consistent with MV2+ or other viologen compounds.19 Besides this, we also attempt to determine the crystal structures after heating. Here we set compound 6 as an example, whose structure after thermochromism has been determined by the X-ray diffraction single crystal method. Upon comparison of the structures before and after heating, we can find that the bond lengths of the [Cd(C4HO4)2(H2O)4] cluster and free (C4HO4) change little, but the bond lengths and angles around the N atoms of pyridine (N(1), N(2)) and C–C bonds linking two pyridine rings (C(21)–C(22)) change significantly when EV2+ is converted to EV˙+ (Fig. 10c and d). Therefore, the thermochromism of 5 and 6 can be mainly assigned to the reversible interconversion between the monocation radical [V]+˙ and the dication [V]2+, which are in agreement with the cases observed in reported viologen-based compounds.19d
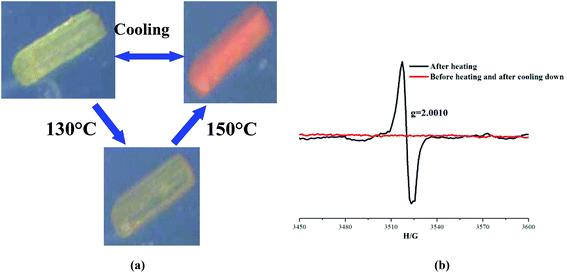 |
| | Fig. 9 (a) Thermochromic behaviour of 5; (b) ESR spectrum of before and after heating and after cooling down in 5. | |
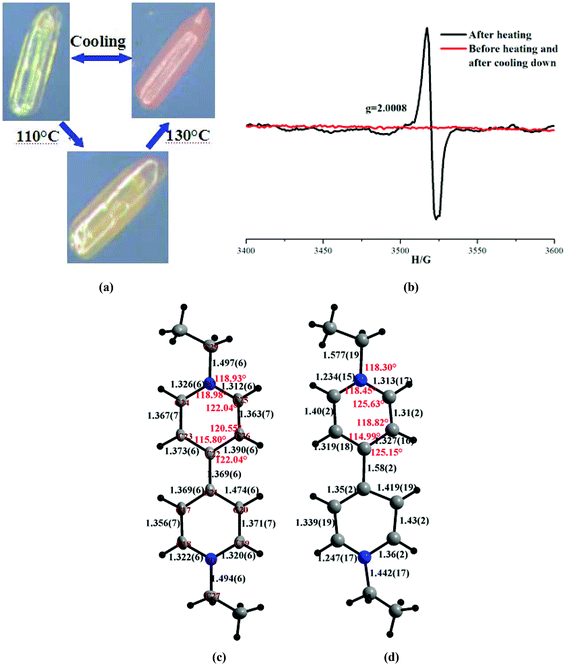 |
| | Fig. 10 (a) Thermochromic behaviour of 6; (b) ESR spectrum of before and after heating and after cooling down in 6; structure of EV2+ before (c) and after heating (d). | |
4. Conclusions
In summary, six new squarate-based complexes have been synthesized and structurally determined. They present structural dimensions from 3-D networks, 2-D layers to 0-D clusters based on the versatile bridged modes of squarates and the introduction of second ligands. In particular, 5 and 6 are the first examples incorporating squarates (electronic-rich species) with viologens (electronic-poor species) in one lattice. Consequently, electrodes with films of 5 and 6 are prepared to investigate their photocurrent response behaviors, and they exhibit effective and repeatable photocurrent responses, besides this, they show reversible thermochromism, whose mechanisms were discussed. This work will be beneficial for the design of multi-functional materials.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
We acknowledge the support for this research by the National Natural Science Foundation of China (No.: 21271043, 21771038) and the Qishan Scholars Program of Fuzhou University (No.: XQS-1403).
Notes and references
-
(a) O. Z. Yeşilel, H. Erer and O. Büyükgüngör, Inorg. Chem. Commun., 2009, 12, 724–727 CrossRef
 ;
(b) C. M. Wang and K. H. Lii, Inorg. Chem., 2009, 48, 6335–6337 CrossRef CAS PubMed ;
(c) S. Horike, Y. Kamitsubo, M. Inukai, T. Fukushima, D. Umeyama, T. Itakura and S. Kitagawa, J. Am. Chem. Soc., 2013, 135, 4612–4615 CrossRef CAS PubMed ;
(d) C. C. Wang, C. H. Yang, G. H. Lee and H. L. Tsai, Eur. J. Inorg. Chem., 2005, 1334–1342 CrossRef CAS .
;
(b) C. M. Wang and K. H. Lii, Inorg. Chem., 2009, 48, 6335–6337 CrossRef CAS PubMed ;
(c) S. Horike, Y. Kamitsubo, M. Inukai, T. Fukushima, D. Umeyama, T. Itakura and S. Kitagawa, J. Am. Chem. Soc., 2013, 135, 4612–4615 CrossRef CAS PubMed ;
(d) C. C. Wang, C. H. Yang, G. H. Lee and H. L. Tsai, Eur. J. Inorg. Chem., 2005, 1334–1342 CrossRef CAS .
-
(a) J. V. Ros-Lis, R. Martínez-Máñez, K. Rurack, F. Sancenón, J. Soto and M. Spieles, Inorg. Chem., 2004, 43, 5183–5185 CrossRef CAS PubMed ;
(b) M. C. Basheer, S. Alex, K. G. Thomas, C. H. Suresh and S. A. Das, Tetrahedron, 2006, 62, 605–610 CrossRef CAS ;
(c) C. Chen, R. Wang, L. Guo, N. Y. Fu, H. H. Dong and Y. F. Yuan, Org. Lett., 2011, 13, 1162–1165 CrossRef CAS PubMed ;
(d) S. Alex, U. Santhosh and S. Das, J. Photochem. Photobiol., A, 2005, 172, 63–71 CrossRef CAS ;
(e) J. H. Yum, P. Walter, S. Huber, D. Rentsch, T. Geiger, F. Nüesch, F. De Angelis, M. Grätzel and M. K. Nazeeruddin, J. Am. Chem. Soc., 2007, 129, 10320–10321 CrossRef CAS PubMed .
-
(a) J. A. C. Van Ooijen, J. Reedijk and A. L. Spek, Inorg. Chem., 1979, 18, 1184–1189 CrossRef CAS ;
(b) K. J. Lin and K. H. Lii, Angew. Chem., Int. Ed. Engl., 1997, 36, 2076–2077 CrossRef CAS ;
(c) S. O. H. Gutschke, M. Molinier, A. K. Powell and P. T. Wood, Angew. Chem., Int. Ed. Engl., 1997, 36, 991–992 CrossRef CAS .
-
(a) F. Gandara, B. Gomez-Lor, M. Iglesias, N. Snejko, E. Gutie rrez-Puebla and A. Monge, Chem. Commun., 2009, 2393–2395 RSC ;
(b) S. Biswas, H. S. Jena, A. Adhikary and S. Konar, Inorg. Chem., 2014, 53, 3926–3928 CrossRef CAS PubMed ;
(c) C. De Mello Donega, S. J. L. Ribeiro, R. R. Goncalves and G. Blasse, J. Phys. Chem. Solids, 1996, 57, 1727–1734 CrossRef CAS .
-
(a) A. Bouhali, C. Trifa, S. Bouacida, C. Boudaren and T. Bataille, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, m1130–m1131 CAS ;
(b) N. Mahe and T. Bataille, Inorg. Chem., 2004, 43, 8379–8386 CrossRef CAS PubMed ;
(c) N. Petrova, B. Shivachev, T. Kolev and R. Petrova, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, m1359–m1361 CAS ;
(d) I. Castro, J. Sletten, L. K. Glaerum, J. Cano, F. Lloret, J. Fausa and M. Julve, J. Chem. Soc., Dalton Trans., 1995, 3207–3213 RSC ;
(e) P. M. T. Piggot, S. Seenarine and L. A. Hall, Inorg. Chem., 2007, 46, 5243–5251 CrossRef CAS PubMed ;
(f) E. Krupicka, A. Griinert and A. Lentz, Z. Kristallogr. - New Cryst. Struct., 2000, 215, 573–574 CAS ;
(g) R. Kirchmaier, E. Altin and A. Lentz, Z. Kristallogr. - New Cryst. Struct., 2003, 218, 541–542 CAS ;
(h) S. Neeraj, M. L. Noya, C. N. R. Rao and A. K. Cheethama, Solid State Sci., 2002, 4, 1231–1236 CrossRef CAS ;
(i) P. M. T. Piggott, L. A. Hall;, A. J. P. White and D. J. Williams, Inorg. Chem., 2003, 42, 8344–8352 CrossRef PubMed ;
(j) G. Ferey, S. C. Mellot-Drazniek, C. Serre and F. Millange, Acc. Chem. Res., 2005, 38, 217–225 CrossRef CAS PubMed ;
(k) B. Zheng and J. F. Bai, CrystEngComm, 2009, 11, 271–273 RSC ;
(l) T. Kajiwara, M. Nakano, Y. Kaneko, S. Takaishi, T. Ito, M. Yamashita, A. Igashira-Kamiyama, H. Nojiri, Y. Ono and N. Kojima, J. Am. Chem. Soc., 2005, 127, 10150–10151 CrossRef CAS PubMed ;
(m) B. Zheng, H. Dong, J. F. Bai, Y. Z. Li, S. H. Li and M. Scheer, J. Am. Chem. Soc., 2008, 130, 7778–7779 CrossRef CAS PubMed ;
(n) H. Akkari, S. Bouacida, P. Bénard-Rocherullé, H. Merazig and T. Roisnel, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m1566–m1567 CAS ;
(o) L. Wang, W. Gu, X. J. Deng, L. F. Zheng, S. Y. Liao, M. Zhang, L. Y. Yang and X. Liu, Aust. J. Chem., 2011, 64, 1373–1382 CrossRef CAS .
-
(a) K. Sumida, M. Hu, S. Furukawa and S. Kitagawa, Inorg. Chem., 2016, 55, 3700–3705 CrossRef CAS PubMed ;
(b) A. K. Ghosh, A. D. Jana, D. Ghoshal, G. Mostafa and N. R. Chaudhuri, Cryst. Growth Des., 2006, 6, 701–707 CrossRef CAS ;
(c) M. I. Khan, Y. D. Chang, Q. Chen, J. Salta, Y. S. Lee, C. J. O'Connor and J. Zubieta, Inorg. Chem., 1994, 33, 6340–6350 CrossRef CAS ;
(d) I. Castro, M. L. Calatayad, J. Sletten, F. Llore and M. Julve, J. Chem. Soc., Dalton Trans., 1997, 811–818 RSC .
-
(a) T. F. Liu, D. Fu, S. Gao, Y. Z. Zhang, H. L. Sun, G. Su and Y. J. Liu, J. Am. Chem. Soc., 2003, 125, 13976–13977 CrossRef CAS PubMed ;
(b) X. J. Liu and R. Warmuth, J. Am. Chem. Soc., 2006, 128, 14120–14127 CrossRef CAS PubMed ;
(c) C. C. Correa, R. Diniz, L. H. Chagas, B. L. Rodrigues, M. I. Yoshida, W. M. Teles, F. C. Machado, L. Fernando and C. deOliveira, Polyhedron, 2007, 26, 989–995 CrossRef CAS .
-
(a) J. T. Sampanthar, K. G. Neoh, S. W. Ng, E. T. Kang and K. L. Tan, Adv. Mater., 2000, 12, 1536–1539 CrossRef CAS ;
(b) D. S. Guo, L. H. Wang and Y. Liu, J. Org. Chem., 2007, 72, 7775–7778 CrossRef CAS PubMed .
-
(a) L. P. Gao, G. J. Ding, Y. C. Wang and Y. L. Yang, Appl. Surf. Sci., 2011, 258, 1184–1191 CrossRef CAS ;
(b) J. H. Jia, A. J. Blake, N. R. Champness, P. Hubberstey, C. Wilson and M. Schroder, Inorg. Chem., 2008, 47, 8652–8664 CrossRef CAS PubMed .
-
G. M. Sheldrick, SHELXTL-97, Program for Crystal Structure Solution and Refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed .
-
(a) R. Soules, F. Dahan, J. P. Laurent and P. Castan, J. Chem. Soc., Dalton Trans., 1988, 587–590 RSC ;
(b) J. A. C. Ooijen, J. Reedijk and A. L. Spek, Inorg. Chem., 1979, 18, 1184–1189 CrossRef ;
(c) T. K. Maji, G. Mostafa, S. Sain, J. S. Prasad and N. R. Chaudhuri, CrystEngComm, 2001, 3, 155–158 RSC .
-
(a) M. T. Reetx, S. Hoger and K. Harms, Angew. Chem., Int. Ed. Engl., 1994, 33, 181–183 CrossRef ;
(b) J. Szklarzewicz, A. Samotus, B. Nowicka, J. Burgess, J. Fawcettand and D. R. Russell, Transition Met. Chem., 1999, 24, 177–182 CrossRef CAS .
- B. D. Alleyne, A. R. Williams, A. Lincoln and L. A. Hall, Inorg. Chem., 2001, 40, 1045–1051 CrossRef CAS .
- J. C. Trombe, J. F. Petit and A. Gleizes, Inorg. Chim. Acta, 1990, 167, 69–81 CrossRef CAS .
- J. B. Yu, L. Zhou, H. J. Zhang, Y. X. Zheng, H. R. Li, R. P. Deng, Z. P. Peng and Z. F. Li, Inorg. Chem., 2005, 44, 1611–1618 CrossRef CAS PubMed .
-
(a) Y. X. Zheng, L. S. Fu, Y. H. Zhou, J. B. Yu, Y. N. Yu, S. B. Wang and H. J. Zhang, J. Mater. Chem., 2002, 12, 919–923 RSC ;
(b) J. Kido and Y. Okamot, Chem. Rev., 2002, 102, 2357–2368 CrossRef CAS PubMed .
-
(a) J. P. Meng, Y. Gong and J. H. Lin, RSC Adv., 2016, 6, 73869–73878 RSC ;
(b) Y. D. Huang, P. Huo, M. Y. Shao, J. X. Yin, W. C. Shen, Q. Y. Zhu and J. Dai, Inorg. Chem., 2014, 53, 3480–3487 CrossRef CAS PubMed ;
(c) M. Q. Yang, B. Weng and X. J. Xu, J. Mater. Chem. A, 2014, 2, 1710–1720 RSC ;
(d) Y. G. Sun, S. F. Ji, P. Huo, J. X. Yin, Y. D. Huang, Q. Y. Zhu and J. Dai, Inorg. Chem., 2014, 53, 3078–3087 CrossRef CAS PubMed .
-
(a) Z. Tong, S. Pu, Q. Xiao, G. Liu and S. Cui, Tetrahedron Lett., 2013, 54, 474–477 CrossRef CAS ;
(b) Z. J. Zhang, S. C. Xiang, G. C. Guo, G. Xu, M. S. Wang, J. P. Zou, S. P. Guo and J. S. Huang, Angew. Chem., Int. Ed., 2008, 47, 4149–4152 CrossRef CAS PubMed ;
(c) J. J. Shen, X. X. Li, T. L. Yu, F. Wang, P. F. Hao and Y. L. Fu, Inorg. Chem., 2016, 55, 8271–8273 CrossRef CAS PubMed .
-
(a) G. Xu, G. C. Guo, M. S. Wang, Z. J. Zhang, W. T. Chen and J. S. Huang, Angew. Chem., Int. Ed., 2007, 46, 3249–3251 CrossRef CAS PubMed ;
(b) R. G. Lin, G. Xu, G. Lu, M. S. Wang, P. X. Li and G. C. Guo, Inorg. Chem., 2014, 53, 5538–5545 CrossRef CAS PubMed ;
(c) M. S. Wang, C. Yang, G. E. Wang, G. Xu, X. Y. Lv, Z. N. Xu, R. G. Lin, L. Z. Cai and G. C. Guo, Angew. Chem., Int. Ed., 2012, 51, 3432–3435 CrossRef CAS PubMed ;
(d) H. Yoshikawa, S. Nishikiori, T. Watanabe, T. Ishida, G. Watanabe, M. Murakami, K. Suwinska, R. Luboradzki and J. Lipkowski, J. Chem. Soc., Dalton Trans., 2002, 1907–1917 RSC ;
(e) J. B. Wu, C. Y. Tao, Y. Li, J. Y. Li and J. H. Yu, Chem. Sci., 2015, 6, 2922–2927 RSC ;
(f) C. Y. Tao, J. B. Wu, Y. Yan, C. Shi and J. Y. Li, Inorg. Chem. Front., 2016, 3, 541–546 RSC .
Footnote |
| † Electronic supplementary information (ESI) available: Selected bond lengths and angles, hydrogen bond details, π–π stacking interactions, additional structural figures, and PXRD patterns. CCDC 802888 (3), 803940 (2), 1433796 (5), 1447563 (4), 1449157 (1), 1449156 (6). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qi00672a |
|
| This journal is © the Partner Organisations 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 * and
Zhi-Rong
Chen
*
* and
Zhi-Rong
Chen
*










