
Chiral metal–organic frameworks constructed from four-fold helical chain SBUs for enantioselective recognition of α-hydroxy/amino acids†
Ru-Xin
Yao
,
Huan-Huan
Fu
,
Bo
Yu
and
Xian-Ming
Zhang
 *
*
School of Chemistry & Materials Science, Shanxi Normal University, Linfen 041004, Shanxi, China. E-mail: zhangxm@dns.sxnu.edu.cn
First published on 24th November 2017
Abstract
The chiral recognition of racemic α-hydroxy/amino acids is an essential and challenging mission because enantiomers may profoundly differ in biological function, pharmacology and toxicity. Three stable, chiral 3D metal–carboxylate frameworks, namely [M2(bptc)(H2O)(MeOH)]·3H2O (bptc4− = 3,3′,5,5′-biphenyltetracarboxylate, M = Co, CoNi for 1–2, and [Ni2(bptc)(MeOH)2]·3H2O for 3) have been successfully obtained by spontaneous resolution with an achiral ligand H4bptc, and they crystallize in the chiral tetragonal space group I4122, and feature chiral four-fold helical metal chains as SBUs. In particular, the Co-MOF material could be used to rapidly and sensitively recognize racemic α-hydroxy/amino acids by the intensity change of circular dichroism signals. A large relative difference of 38.59% in circular dichroism signals for D/L-mandelic acid is achieved, which may be ascribed to the specific recognition sites (i.e., groove of helical chains) and different bonding energies of D/L-isomers in the chiral microenvironment of the crystal structure.
1. Introduction
The helix, one important origin of chirality, is an essential structural motif in biomolecules, such as the right-handed α-helix of proteins and the double helix of DNA,1 playing a crucial role in gene storage, protein–DNA binding and replication.2 Helical chiral self-assembly is a primary process in biological systems, which has gained much attention in biology and chemistry research,3 but it remains a huge challenge to control homochirality. Generally, enantiopure helical complexes can be designed and constructed in two ways. The first is by using chiral molecules to facilitate the synthesis of helical materials, where the inherent chirality of molecules transfers into the entire framework.4 Without any chiral source, the indirect method is based on the spatial arrangement of achiral precursors by symmetry breaking, which is a spontaneous process.5 However, the helix formed via spontaneous resolution tends to be racemic with an equivalent left/right-helix, due to the stochastic essence of crystallization.6 Only in very few situations can enantiopure or enantio-enriched crystals be obtained. For example, spontaneous chiral symmetry breaking can occur in the stirred crystallization of achiral NaClO3 (chiral autocatalysis), in which the secondary crystal nuclei may rapidly clone the parent structure, while the competitive crystallization of the opposite enantiomer is suppressed. Supramolecular interactions like H-bonding or π–π stacking may also induce symmetry breaking.7 This may promote chiral recognition and realize the origin of life in biological systems.As important chiral units in many natural products and drug molecules,8 α-hydroxy/amino acids have two enantiomers that exhibit identical physicochemical properties in an achiral environment. Among them, mandelic acids9 are used as antibacterial agents, cosmetics, as well as intermediates in the asymmetric synthesis of drug molecules with biological activities, such as optically pure amino acids and angiotensin converting enzyme inhibitors. Amino acids are the essential constituents of living organisms, and are present in body fluids, tissues and food.10 As essential building blocks of proteins, natural amino acids are found to have the L configuration, while the D configuration shows disease, aging, etc. negative symptoms, dramatically different in biology, pharmacology and toxicity. Hydroxy/amino acids are difficult to detect directly and recognize due to their active functional groups (carboxyl, hydroxyl or amino group), often requiring derivatization before detection.11 Therefore, a challenging and essential mission is the rapid, selective recognition of racemic α-hydroxy/amino acids in biological analysis, medical research and nutritional analysis.
As an ideal platform for host–guest chemistry, chiral MOFs have been exploited to stereoselectively recognise and separate racemic mixtures,12 owing to their stable chiral architectures, controllable open cavities, stereoselectivity and tunable intermolecular interactions. But the stereoselective recognition of α-hydroxy/amino acids remains very scarce. A chiral porous 1,1′-biphenol based MOF decorated with chiral dihydroxyl recognition sites is used to separate mandelic derivatives with up to 93.1% ee through chiral HPLC analyses.13 Homochiral quadruple-stranded helix cages with pyridyl-functionalized metallosalans exhibited enantioselective luminescence enhancement for amino acids in solution.14 A homochiral Zn-MOF based on pyrene-tetralactic acid, featuring concave shapes for guest inclusion, could selectively discriminate L-histidine by fluorescence quenching.15 It is worth noting that the chiral recognition capacity is related to not only high porosity, shape and size, but also the effective chiral recognition sites and the interaction (hydrogen bonding, π–π stacking, dipole–dipole etc.) of enantiomers and frameworks.16 Compared with chromatography, the fluorescence method possesses the merits of portability, high sensitivity and a short analysis time, without expensive columns and complicated derivatization processes, but requires strong luminescent materials.17 So it is desirable to explore a rapid, convenient and sensitive method for the recognition of α-hydroxy/amino acids. Circular dichroism (CD) spectroscopy is one of the most effective means in stereochemical analysis,18 superior to chromatography and fluorescence methods, but only used to simply judge the chirality of molecules, and rarely used to recognize enantiomers.
We have been conducting chiral MOF research by exploiting paramagnetic metal cluster SBUs19 and have efficiently separated pharmaceutically important (±)-ibuprofen racemate by HPLC with the MOF chiral stationary phase.20 Rod-shaped metal–carboxylate SBUs can improve the stability of MOFs and avoid the structure interpenetrated.21 A combination of rod-shaped SBUs with a groove of a helical structure is an effective strategy to obtain stable chiral recognition materials. As on-going research on chiral MOF materials, we present three stable, chiral 3D materials [M2(bptc)(H2O)(MeOH)]·3H2O (M = Co, CoNi for 1–2, and [Ni2(bptc)(MeOH)2]·3H2O for 3), which feature chiral quadruple-stranded helical metal–carboxylate chains as SBUs, exploiting an achiral ligand H4bptc via a spontaneous resolution process. Helical chiral Co-MOF can be used as a distinctive CD sensor for enantioselective recognition of racemic mandelic acids and amino acids.
2. Experimental section
2.1 Materials and measurements
The reagents and solvents employed were commercially available and used without further purification. Elemental analyses were performed on a Vario EL-II elemental analyzer. The elemental composition of the samples was analyzed by energy-dispersive X-ray spectroscopy (EDX) attached to a SEM instrument JSM-7500F equipped with an EDAX CDU leap detector. The FTIR spectra were recorded using KBr pellets in the range of 4000–400 cm−1 on a Nicolet 5DX spectrometer. Powder X-ray diffraction (PXRD) data were collected using a Miniflex 600 diffractometer (Rigaku, Japan) with Cu Kα radiation at room temperature. Thermogravimetric analysis (TGA) was carried out under air using SETARAM LABSYS equipment at a heating rate of 10 °C min−1. CD experiments were performed on a Jasco J-815 circular dichroism spectrometer at room temperature. The magnetic measurement was made with Quantum Design SQUID MPMS XL-5 instruments. Data were corrected for the diamagnetic contribution calculated from Pascal constants and the sample container.2.2 Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Light red sheet crystals of 2 in a yield of 91.3% (based on H4bptc) were recovered. The relative molar ratio of Co/Ni in the heterometallic crystals of 2 determined by ICP analysis is 1:1, which roughly matches the EDS analysis values (Fig. S2†). Anal. calcd (%) for C17.5H14CoNiO13 (2): C, 38.22; H, 2.57. Found: C, 38.25; H, 2.52. IR data (KBr, cm−1): 3435(b), 2973(w), 2846(w), 2362(w), 1616(s), 1553(s), 1420(s), 1363(s), 1313(w), 1015(w), 913(w), 787(m), 717(m), 660(m), 590(w), 457(m).
:1. Light red sheet crystals of 2 in a yield of 91.3% (based on H4bptc) were recovered. The relative molar ratio of Co/Ni in the heterometallic crystals of 2 determined by ICP analysis is 1:1, which roughly matches the EDS analysis values (Fig. S2†). Anal. calcd (%) for C17.5H14CoNiO13 (2): C, 38.22; H, 2.57. Found: C, 38.25; H, 2.52. IR data (KBr, cm−1): 3435(b), 2973(w), 2846(w), 2362(w), 1616(s), 1553(s), 1420(s), 1363(s), 1313(w), 1015(w), 913(w), 787(m), 717(m), 660(m), 590(w), 457(m).
2.3 Crystallography studies
Single crystal X-ray diffraction measurements were carried out on an Agilent Technologies Gemini Eos diffractometer at 293.18 K using Cu Kα radiation (λ = 0.15418 Å) or Mo Kα radiation (λ = 0.71073 Å). The program SAINT was used for the integration of the diffraction profiles, and the program SADABS was used for absorption correction. The structures were solved with XS by direct methods and refined anisotropically by a full-matrix least-squares technique based on F2 using the Olex2 programs. All non-hydrogen atoms were refined with anisotropic thermal parameters. Hydrogen atoms of the organic ligand were generated theoretically onto the specific carbon and refined isotropically with fixed thermal factors, but the hydrogen atoms of the methanol molecules could not be determined. The water molecules of the crystal lattice are deduced according to the TGA data. Further details of structural analysis are summarized in Table S2,† and the corresponding bond lengths and bond angles are shown in Table S3.†3. Results and discussion
3.1 Description of structures
It is well known that the hydrolysis of DMF into dimethylamine and formic acid is apt to form metal formate frameworks.22 Addition of dilute HNO3 solution to a weakly acidic solution (pH = 6) may inhibit the hydrolysis of DMF and is in favor of bptc4− coordination. Single-crystal X-ray diffraction analyses reveal that complexes 1–3 are isostructural (Fig. S2–S4†), and thus only 1 is discussed herein in detail (Tables S1–S3†). Complex 1 crystallizes in a chiral tetragonal space group I4122 and consists of helical rod SBUs. There are one Co2+ ion, one water/methanol molecule (oxygen atom of water/methanol occupying the same site) and a quarter of bptc4− in the asymmetric unit (Fig. 1a). All of the atoms except for O1, O2, C2, C3 and C5 localize on the 2-fold axis with a site occupancy of 0.5. Each Co(1) center is coordinated by one μ2-H2O, one μ2-CH3OH and four oxygen atoms of bridging carboxyl groups from four different bptc4− ligands, which exhibits a slightly distorted octahedral geometry. While for 3, each Ni(1) center is coordinated by two μ2-CH3OH and four oxygen atoms of bridging carboxyl groups. Every bptc4− ligand links eight Co(II) centers, in which four of the carboxyl groups adopt syn, syn-μ2-η1:η1 mode (Fig. 1b). The Co–O bond distances are in the range of 2.057(8)–2.140(10) Å. The cis-O–Co–O bond angles are in the range of 83.8(4)–94.3(3)°, and trans-O–Co–O angles are in the range of 174.2 (5)–175.6(4)°. The corner-sharing CoO6 octahedron is connected by two bidentate carboxylate groups to form an infinite left-handed helical chain [Co2(H2O)(CH3OH)(CO2)4] (Co⋯Co distance is 3.599 Å) with a 4-fold screw axis and a pitch of 12.194(6) Å (Fig. 1c). Each helical chain as the secondary building unit is further bridged to four adjacent helical chains by the bptc4− ligand, and each ligand is linked to two helical chains (alternate left-handed 41 and 43 helices), thus producing a homochiral 3D framework with a one-dimensional helical channel of 15.35 × 15.35 Å size along the c-axis (Fig. 1d). PLATON calculations revealed the large solvent-accessible voids of 1376.8 Å3 to be 47.9% of per unit cell volume 2872.6 Å3. The chirality of 1 is a result of the 4-fold screw axis and the stereochemistry of the crosslinking ligand (bptc4−) in which two phenyl rings are not coplanar with a dihedral angle of 30.14° (Fig. S5†). Isostructural MOFs (Al, In, Mg, etc.) have been reported,23 which exhibited high adsorption capacities, but the chiral resolution ability has never been reported in chiral Co-MOF materials. The common helical rods include 3-fold, 4-fold and 6-fold helices, which can be linked by ditopic or polytopic organic ligands. For 4-fold helix SBUs, only a few compounds have been documented,3,24 which are observed in four nets (srs, ths, sra and rab) with tetragonal symmetry, and the etm net with orthorhombic symmetry. Only the srs net is a chiral net, in which all 4-fold helices have the same handedness. But compound 1 crystallizes in the chiral tetragonal space group I4122 with alternate 41 and 43 helices, to the best of our knowledge, which has been rarely reported.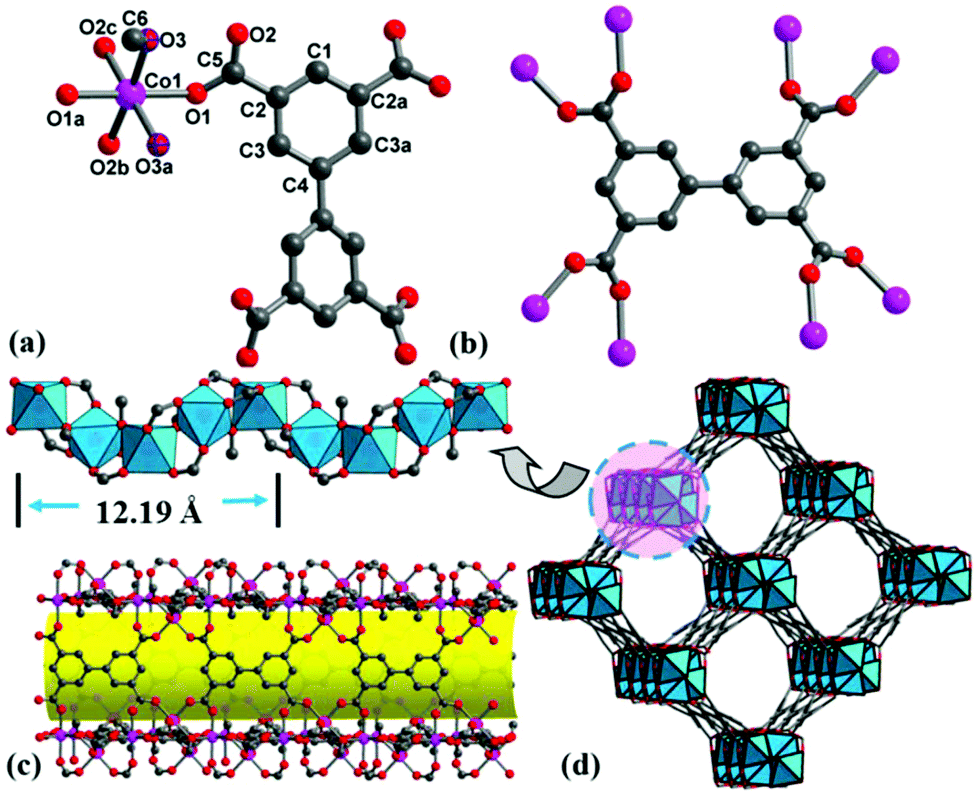 | ||
| Fig. 1 (a) Coordination environment of Co(II) in 1, (b) coordination mode of the bptc4− ligand, (c) the infinite Co-carboxylate 41 helical chain and four helical chains linked by ligands forming a helical channel, and (d) a 3D porous structure with chiral channels along the c-axis. | ||
To gain a better understanding of the morphology and crystallinity of MOFs, glass was selected as a substrate for the growth of Ni-MOF thin films, due to the smaller size of Ni-MOFs. As time goes on, the Ni-MOF film grows gradually from a few crystals to a large number of crystals (shown in Fig. 2). Finally, the supporting surface is fully covered with a homogeneous and compact MOF layer. Interestingly, the MOF film formed only on the pre-treated side of the glass substrate, leaving the other side almost intact or only some scattered single crystals are attached on the glass support. This is very important from both the synthesis and application points of view.25 The PXRD patterns of the synthesized Ni-MOF film on the glass substrate are consistent with those of the simulated 3 (Fig. S6†), which reveals a high crystallinity of crystals grown on the substrate.
 | ||
| Fig. 2 Photos of Ni-MOF films on the glass substrate with different growth times: (a) one day, (b) two days, (c) three days, and (d) four days. | ||
3.2 Powder X-ray diffraction and stability
The phase purity of 1–3 was tested by comparison of the simulated and experimental PXRD patterns, which are well consistent with each other (Fig. S7†), indicating that they have good phase purity. As shown in Fig. S8,† isostructural 1–3 have similar thermal stabilities. For 1, the initial weight loss of about 10.51% (calcd 9.68%) in the temperature range of 25–160 °C corresponds to the removal of free water molecules. The second weight loss of 64.42% in the temperature range of 160–450 °C is in agreement with the removal of bptc4− ligands, accompanied by the decomposition of the framework. The final residua of 31.23% (calcd 29.13%) for 1 and 27.82% (calcd 26.79%) for 3 are close to the percentage of Co3O4 and NiO, respectively. For 2, the final residua are the mixture of CoO and NiO. To further investigate the stability of 3, it was immersed in aqueous solutions of different pH values. The PXRD patterns indicated that the structure is retained after soaking in pH = 2–12 aqueous solutions (Fig. S9†), confirming the strong chemical stability, which is ascribed to helical chain [Ni2(CH3OH)2(CO2)4] based SBUs.3.3 Enantioselective recognition
To confirm the optical activity of 1–3, CD spectra were recorded in 5 × 10−4 mol L−1 aqueous solution (Fig. S10†). The CD spectra of 1 exhibit a positive Cotton effect at λmax = 251 nm along with a negative Cotton effect around 254 nm, indicating that 1 possesses optical activity in aqueous solution. With Co ions being gradually replaced by Ni ions, the CD spectra of 2 and 3 exhibit a small difference from that of 1. For 2, there are a strong positive Cotton effect at 254 nm and a negative Cotton effect at 276 nm, which show a wider range related to 1. For 3, the spectra show a positive Cotton effect at 240 nm and a negative Cotton effect at 256 nm, which show a blue-shift relative to 1. The solid-state CD spectra of 1 were obtained on bulk crystals from four batches, using pressed KBr pellets. As shown in Fig. S11,† the CD spectra exhibit similar signals, a positive Cotton effect at about 250 nm and a negative Cotton effect at 260 nm, further confirming the optical activity of bulk materials. The characteristic CD signals of 1–3 may be attributed to the four-fold helical chains, aromatic ring, carboxylate chromophores (π–π* and n–π* transitions), and the ligand to metal charge transition (LMCT).The helical environments of 1 motivated us to explore its potential for chiral resolution and separation. Considering the 4-fold helical channel of the structure, it can be used to recognize racemic organic molecules with an active carboxyl group. The same amounts of 1 were soaked in an aqueous solution of three pairs (Fig. 3) of enantiomers with the same concentration (5 × 10−5 mol L−1) at room temperature. After ultrasonic processing for 30 min, the solutions were detected by CD spectroscopy under the identical conditions. It was observed that the extent of CD signal reduction (calculated by the maximum wavelength) varies with the amount of 1 to different racemic molecules (Fig. S12–14†), which indicates that 1 has a different discerning ability from racemic organic molecules.15,26 As shown in Fig. 4a, the order of recognition efficiency (η) is L-mandelic acid (98.82%) > L-histidine (88.97%) > L-tryptophan (47.81%); the recognition efficiency is defined as: 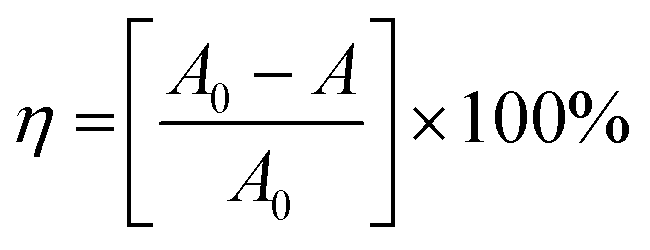 , where A0 and A are the initial and final CD signal intensity, respectively. The relative different changes of CD signals for D/L-mandelic acid, D/L-histidine and D/L-tryptophan are 38.59%, 28.32% and 14.86%, respectively, indicating that 1 can effectively, enantioselectively recognise them. The lower relative difference of 14.86% for D/L-tryptophan may be related to the large steric hindrance of the tryptophan molecule. To further understand the recognition efficiency of 1 for mandelic acid, the plots of ΔCD signal vs. crystal amount have been studied, which present a linear relationship (Fig. 5). The equation is ΔCD (mdeg) = 1.907X (mg) + 6.530 (for L-Man), and ΔCD = 1.167X − 7.255 (for D-Man). As shown in Fig. 5, the slope of L-Man is obviously larger than that of D-Man with the same amount of 1, which may be due to a stronger interaction between 1 and L-Man than that between 1 and D-Man. For tryptophan, ΔCD = 0.2933X + 0.0933 (for L-Trp), and ΔCD = 0.2065X − 0.4934 (for D-Trp). Evidently, the D/L isomers of α-hydroxy/amino acids are discerned by the Co-MOF. The PXRD patterns of 1 immersed in the aqueous solution of three pairs of enantiomers are similar to those of the prepared crystals (Fig. 4b), except that some peaks of Trp-containing MOFs are slightly different from other PXRD patterns, indicating that 1 maintained its crystallinity and basic skeleton.
, where A0 and A are the initial and final CD signal intensity, respectively. The relative different changes of CD signals for D/L-mandelic acid, D/L-histidine and D/L-tryptophan are 38.59%, 28.32% and 14.86%, respectively, indicating that 1 can effectively, enantioselectively recognise them. The lower relative difference of 14.86% for D/L-tryptophan may be related to the large steric hindrance of the tryptophan molecule. To further understand the recognition efficiency of 1 for mandelic acid, the plots of ΔCD signal vs. crystal amount have been studied, which present a linear relationship (Fig. 5). The equation is ΔCD (mdeg) = 1.907X (mg) + 6.530 (for L-Man), and ΔCD = 1.167X − 7.255 (for D-Man). As shown in Fig. 5, the slope of L-Man is obviously larger than that of D-Man with the same amount of 1, which may be due to a stronger interaction between 1 and L-Man than that between 1 and D-Man. For tryptophan, ΔCD = 0.2933X + 0.0933 (for L-Trp), and ΔCD = 0.2065X − 0.4934 (for D-Trp). Evidently, the D/L isomers of α-hydroxy/amino acids are discerned by the Co-MOF. The PXRD patterns of 1 immersed in the aqueous solution of three pairs of enantiomers are similar to those of the prepared crystals (Fig. 4b), except that some peaks of Trp-containing MOFs are slightly different from other PXRD patterns, indicating that 1 maintained its crystallinity and basic skeleton.
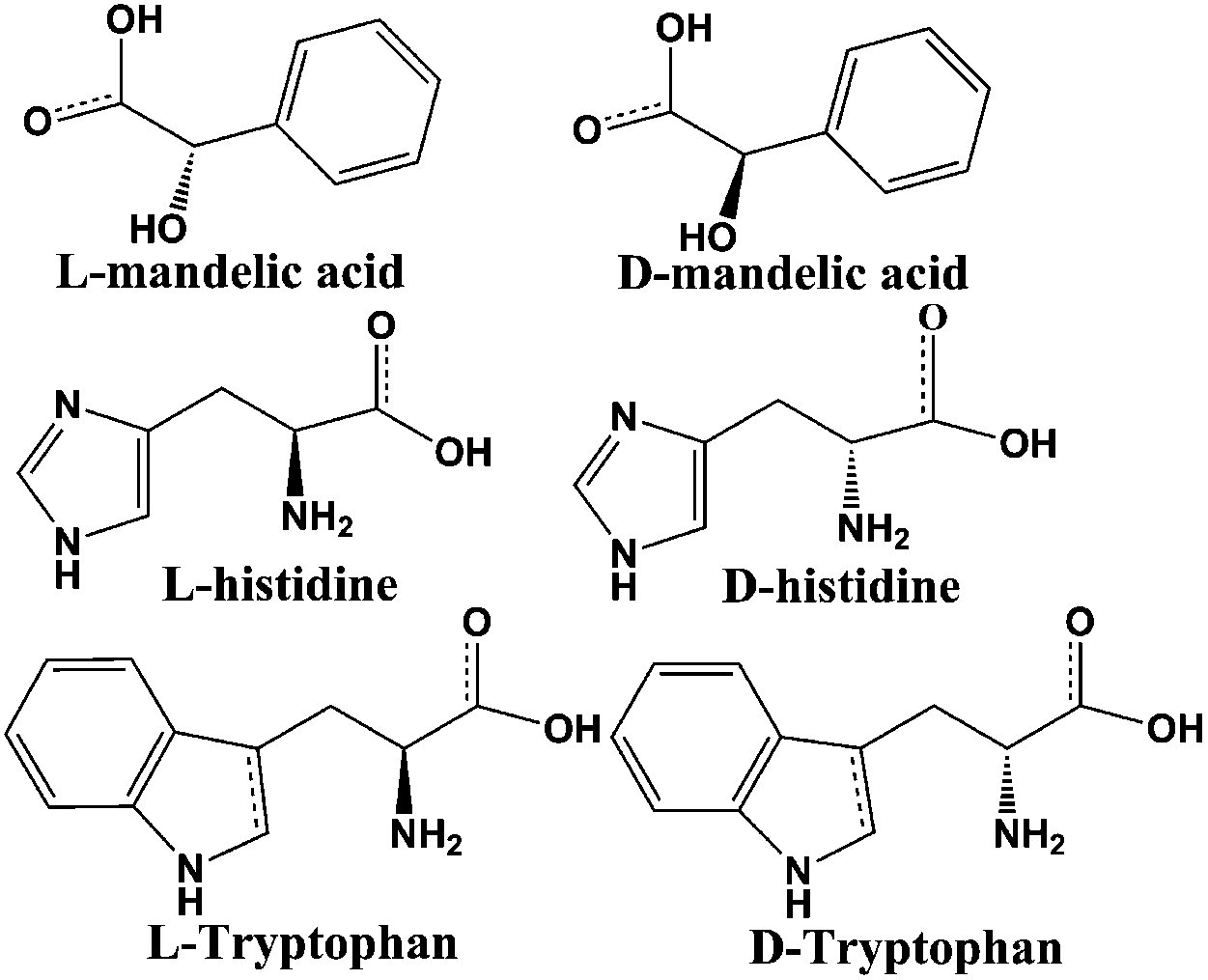 | ||
| Fig. 3 Three pairs of detected enantiomers. | ||
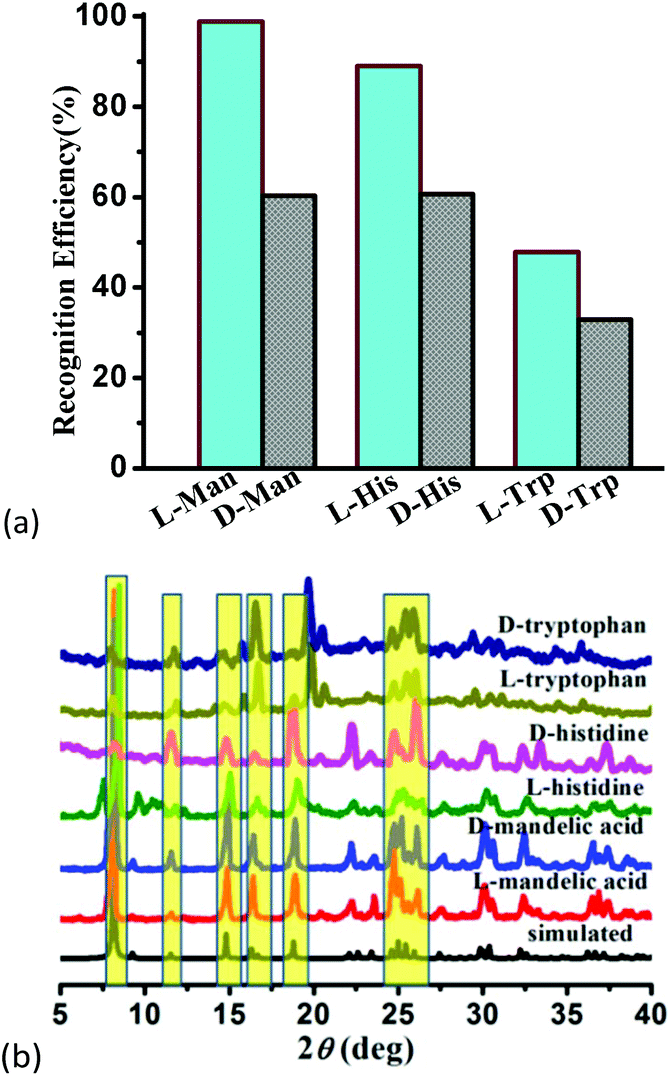 | ||
| Fig. 4 (a) Recognition efficiency of different enantiomers upon the addition of MOF-1, and (b) the PXRD patterns of 1 soaked in the solution of mandelic acid and amino acids. | ||
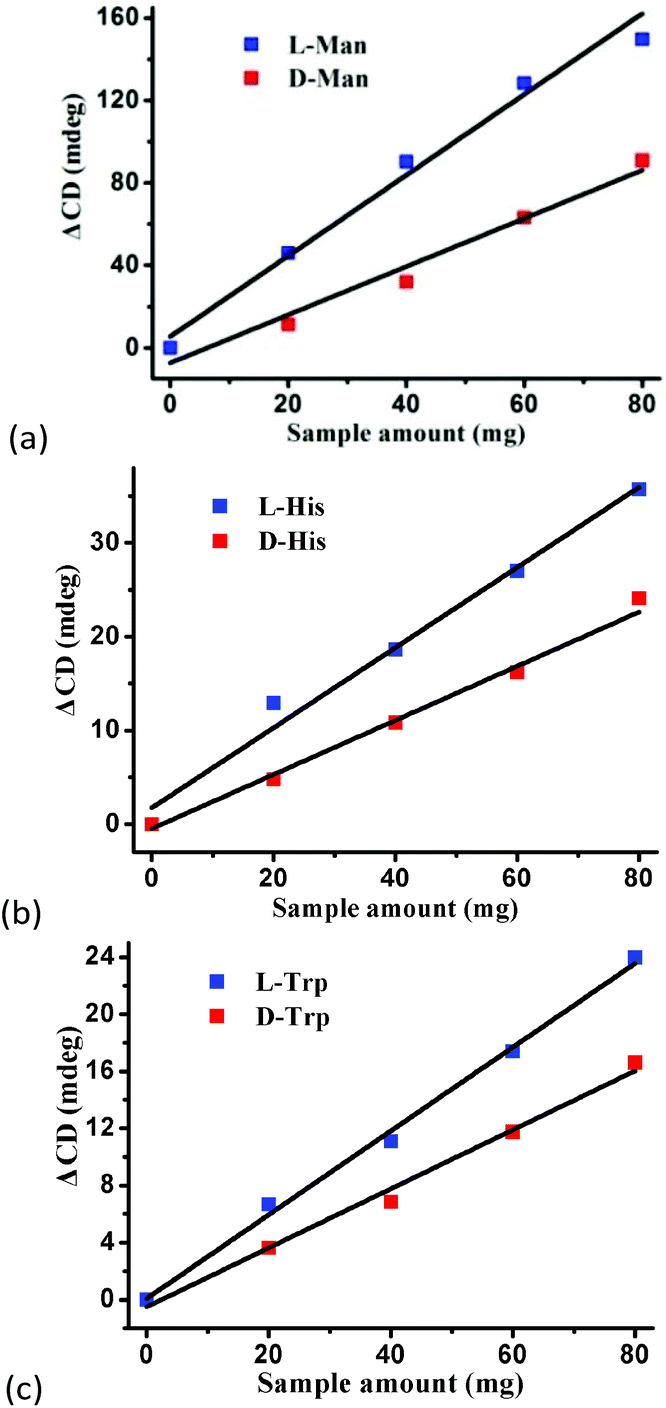 | ||
| Fig. 5 The plots of ΔCD signal vs. crystal amount for (a) mandelic acid, (b) histidine, and (c) tryptophan in aqueous solutions. | ||
To provide microscopic insight into the mechanism of the enantioselective recognition of mandelic acid and amino acids, molecular simulations were conducted by using Materials Studio software. The geometry of mandelic acid was optimized by density-functional theory in DMol3 (section S1†).27Fig. 6 illustrates the bonding sites of 1 with mandelic acid, which are preferentially located in the groove formed by 4-fold helical chains. However, the orientation of the L-isomer is distinctly different from that of the D-isomer, which is crucial for the optical discrimination. Furthermore, the specific bonding energies are −5.11 and −2.77 kcal mol−1 for L and D-enantiomers, respectively, which indicates that L-Man has a stronger interaction with 1 than the D-counterpart, causing enantioselective resolution.
 | ||
| Fig. 6 Possible binding sites of (a) L-Man and (b) D-Man with 1. | ||
3.4 Magnetic studies
The variable-temperature magnetic susceptibility of 1–3 was analyzed in the temperature range of 300–2 K under 1000 Oe. For 1, the χmT value per Co2 unit is 4.31 cm3 K mol−1 at 300 K, which is higher than the value of two isolated high-spin Co(II) ions (S = 3/2, 3.75 cm3 K mol−1), owing to the orbital contribution of Co(II) ions.28 Upon cooling, the χmT value decreases to 2.57 cm3 K mol−1 at 16.6 K, exhibits a small shoulder peak with 2.61 cm3 K mol−1 at 11.8 K, and finally decreases to 0.83 cm3 K mol−1 at 2 K, which may be due to the onset of magnetic order, the spin–orbit coupling or zero-field splitting of the Co(II) ion itself. The reciprocal magnetic susceptibility (χm−1) above 10 K follows the Curie–Weiss law with C = 4.592 (2) cm3 K mol−1 and θ = −20.40(7) K (Fig. 7a). The C value corresponds to g = 2.21, being normal for Co2+ compounds. The negative Weiss constant suggests strong antiferromagnetic (AF) interactions within the vertex-sharing Co2 cluster. The M–H curve at 2 K presents a drastic rise at low field, and gradually reached 2.91Nβ per Co2 unit at 5 T, far below the expected value 6.0Nβ (Fig. 7b). Moreover, field-cooled (FC) and zero-field-cooled (ZFC) curves have a clear bifurcation at 15 K, indicating long-range magnetic ordering below the critical temperature (Fig. S15a†). A narrow hysteresis loop of 1 is observed in the low field at 2 K (Fig. S15b†). Neglecting the zero field splitting and spin–orbit coupling of Co(II) ions, the magnetic data were fitted with the isotropic Fisher model for a linear chain of N spins.29 This model gives a very satisfactory matching of the magnetic properties in the high temperature range with the following parameters: g = 2.219(4), J1 = −5.785(6) and R2 = 0.9940 (Fig. S16†). The negative J value also confirms the AF interaction between Co2 units. | (1) |
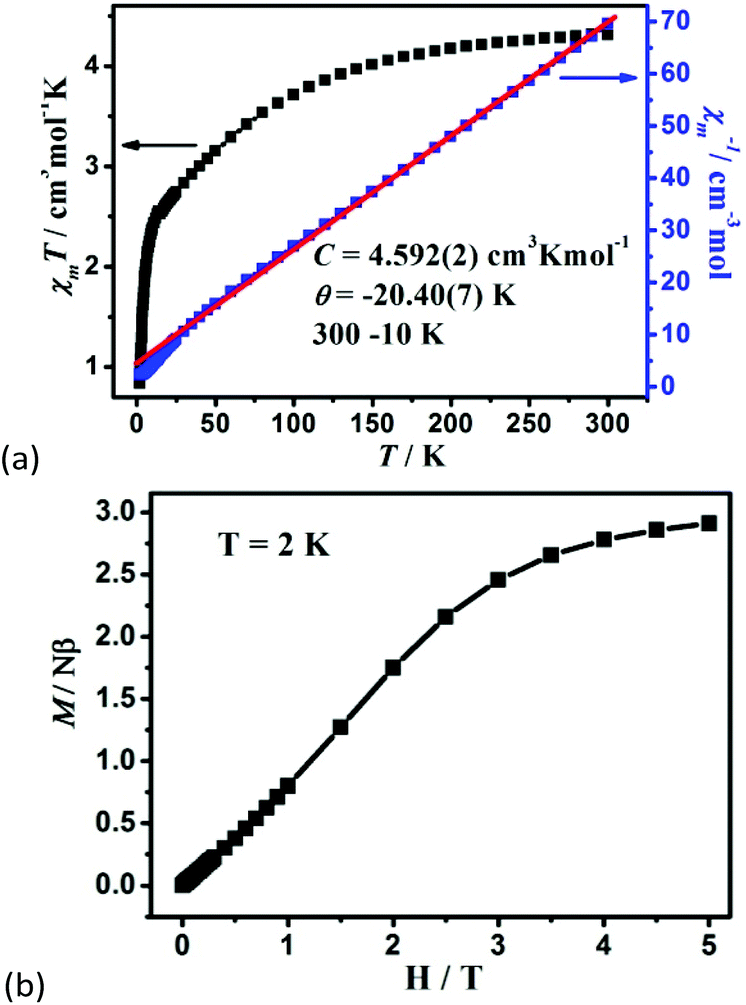 | ||
| Fig. 7 (a) The χmT vs. T plots and the Curie–Weiss fitting of χm−1vs. T curves for 1 in the range of 10–300 K per Co2 unit, and (b) the field dependence of magnetization for 1 at 2 K. | ||
For 3, the χmT value per Ni2 unit is 1.95 cm3 K mol−1 at 300 K, which is close to the expected value 2.00 cm3 K mol−1 (S = 1, g = 2.00). As the temperature drops, the χmT value decreases continuously and rapidly reaches 1.26 cm3 K mol−1 at 2 K, which is characteristic of AF coupling, supported by the field dependence of the magnetization (Fig. S17†). The magnetic data in the range of 10–300 K obey the Curie–Weiss law with C = 1.95(2) cm3 K mol−1 and θ = −8.08(1) K. Similar to 3, light red crystal 2 with a dopant of Co/Ni also presents AF behavior (Fig. S18†).
4. Conclusions
In summary, we have presented three chiral 3D metal–carboxylate frameworks, which feature chiral four-fold helical metal chains as SBUs. Particularly, the Co-MOF material may conveniently, sensitively, and enantioselectively recognize α-hydroxy/amino acids by the intensity change of CD signals, averting the time-consuming or sophisticated process of chromatography. A large relative difference of 38.59% in CD signals for D/L-mandelic acid is achieved, which may be attributed to the specific recognition sites (i.e., groove of helical chains) of the framework. This work will provide a promising approach and practical application of MOFs in enantioselective recognition and separation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from 10000 Talents Plan, the Ministry of Education of China (Grant IRT1156), and NSFC (Grant 21401119 & 20925101) is greatly appreciated. We also gratefully acknowledge the assistance in the fitting of magnetic properties provided by Jun-Liang Liu.References
- (a) M. Deng, L. Zhang, Y. Jiang and M. Liu, Angew. Chem., Int. Ed., 2016, 55, 15062–15066 CrossRef CAS PubMed; (b) Z.-G. Gu, C. Zhan, J. Zhang and X. Bu, Chem. Soc. Rev., 2016, 45, 3122–3144 RSC.
- (a) X. Zhang, B. Li and J. Zhang, Inorg. Chem., 2016, 55, 3378–3383 CrossRef CAS PubMed; (b) S. Ido, K. Kimura, N. Oyabu, K. Kobayashi, M. Tsukada, K. Matsushige and H. Yamada, ACS Nano, 2013, 7, 1817–1822 CrossRef CAS PubMed; (c) J. Rodriguez, J. Mosquera, R. Garcia-Fandino, M. E. Vazquez and J. L. Mascarenas, Chem. Sci., 2016, 7, 3298–3303 RSC.
- Z.-L. Wu, J. Dong, W.-Y. Ni, B.-W. Zhang, J.-Z. Cui and B. Zhao, Inorg. Chem., 2015, 54, 5266–5272 CrossRef CAS PubMed.
- (a) B.-Q. Song, D.-Q. Chen, Z. Ji, J. Tang, X.-L. Wang, H.-Y. Zang and Z.-M. Su, Chem. Commun., 2017, 53, 1892–1895 RSC; (b) B. Ma, Y. Wu, S. Zhang, S. Wang, J. Qiu, L. Zhao, D. Guo, J. Duan, Y. Sang, L. Li, H. Jiang and H. Liu, ACS Nano, 2017, 11, 1973–1981 CrossRef CAS PubMed.
- (a) Z.-M. Zhang, X. Duan, S. Yao, Z. Wang, Z. Lin, Y.-G. Li, L.-S. Long, E.-B. Wang and W. Lin, Chem. Sci., 2016, 7, 4220–4229 RSC; (b) P. C. Rao, S. P. Chaudhary, D. Kuznetsov and S. Mandal, Inorg. Chem., 2016, 55, 12669–12674 CrossRef CAS PubMed.
- (a) Y.-G. Huang, B. Mu, P. M. Schoenecker, C. G. Carson, J. R. Karra, Y. Cai and K. S. Walton, Angew. Chem., Int. Ed., 2011, 50, 436–440 CrossRef CAS PubMed; (b) J. Zhang, S. Chen, R. A. Nieto, T. Wu, P. Feng and X. Bu, Angew. Chem., Int. Ed., 2010, 49, 1267–1270 CrossRef CAS PubMed; (c) X. Jing, C. He, D. Dong, L. Yang and C. Duan, Angew. Chem., Int. Ed., 2012, 51, 10127–10131 CrossRef CAS PubMed.
- (a) D. K. Kondepudi, R. J. Kaufman and N. Singh, Chiral Symmetry Breaking in Sodium Chlorate Crystallizaton, Science, 1990, 250, 975–976 CAS; (b) Q. X. Yang, Z. J. Chen, J. S. Hu, Y. Hao, Y. Z. Li, Q. Y. Lu and H. G. Zheng, Chem. Commun., 2013, 49, 3585–3587 RSC; (c) K.-T. Chihiro, I. Megumi, S. Toshiaki, K. Chikahide, H. Takunori and K. Reiko, Inorg. Chem., 2012, 51, 7502–7507 CrossRef PubMed; (d) P. Cui, L. J. Ren, Z. Chen, H. C. Hu, B. Zhao, W. Shi and P. Cheng, Inorg. Chem., 2012, 51, 2303–2310 CrossRef CAS PubMed.
- (a) L. A. Joyce, M. S. Maynor, J. M. Dragna, G. M. da Cruz, V. M. Lynch, J. W. Canary and E. V. Anslyn, J. Am. Chem. Soc., 2011, 133, 13746–13752 CrossRef CAS PubMed; (b) H.-L. Liu, Q. Peng, Y.-D. Wu, D. Chen, X.-L. Hou, M. Sabat and L. Pu, Angew. Chem., Int. Ed., 2010, 49, 602–606 CrossRef CAS PubMed; (c) S. Superchi, R. Bisaccia, D. Casarini, A. Laurita and C. Rosini, J. Am. Chem. Soc., 2006, 128, 6893–6902 CrossRef CAS PubMed.
- (a) R. K. Hylton, G. J. Tizzard, T. L. Threlfall, A. L. Ellis, S. J. Coles, C. C. Seaton, E. Schulze, H. Lorenz, A. Seidel-Morgenstern, M. Stein and S. L. Price, J. Am. Chem. Soc., 2015, 137, 11095–11104 CrossRef CAS PubMed; (b) K.-J. Xiao, L. Chu and J.-Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 2856–2860 CrossRef CAS PubMed.
- (a) C. Han and H. Li, Small, 2008, 4, 1344–1350 CrossRef CAS PubMed; (b) Y. Zhou and J. Yoon, Chem. Soc. Rev., 2012, 41, 52–67 RSC; (c) L. Dutot, K. Wright, A. Gaucher, M. Wakselman, J.-P. Mazaleyrat, M. D. Zotti, C. Peggion, F. Formaggio and C. Toniolo, J. Am. Chem. Soc., 2008, 130, 5986–5992 CrossRef CAS PubMed.
- (a) H. Lorenz and A. Seidel-Morgenstern, Angew. Chem., Int. Ed., 2014, 53, 1218–1250 CrossRef CAS PubMed; (b) X. Zhang, J. Yin and J. Yoon, Chem. Rev., 2014, 114, 4918–4959 CrossRef CAS PubMed.
- (a) L. Lin, R. Yu, X.-Y. Wu, W.-B. Yang, J. Zhang, X.-G. Guo, Z.-J. Lin and C.-Z. Lu, Inorg. Chem., 2014, 53, 4794–4796 CrossRef CAS PubMed; (b) Y. Peng, T. Gong, K. Zhang, X. Lin, Y. Liu, J. Jiang and Y. Cui, Nat. Commun., 2014, 5, 4406 CAS; (c) C. Zhu, G. Yuan, X. Chen, Z. Yang and Y. Cui, J. Am. Chem. Soc., 2012, 134, 8058–8061 CrossRef CAS PubMed.
- Y. Peng, T. Gong and Y. Cui, Chem. Commun., 2013, 49, 8253–8255 RSC.
- W. Xuan, M. Zhang, Y. Liu, Z. Chen and Y. Cui, J. Am. Chem. Soc., 2012, 134, 6904–6907 CrossRef CAS PubMed.
- P. Chandrasekhar, A. Mukhopadhyay, G. Savitha and J. N. Moorthy, Chem. Sci., 2016, 7, 3085–3091 RSC.
- (a) L. A. Barrios, J. Salinas-Uber, O. Roubeau, S. J. Teat and G. Aromi, Chem. Commun., 2015, 51, 4631–4634 RSC; (b) A. Berthod, Anal. Chem., 2006, 78, 2093–2099 CrossRef PubMed.
- (a) P. Li, Y. He, J. Guang, L. Weng, J. Zhao, S. Xiang and B. Chen, J. Am. Chem. Soc., 2014, 136, 547–549 CrossRef CAS PubMed; (b) J. Zhang, Z. Li, W. Gong, X. Han, Y. Liu and Y. Cui, Inorg. Chem., 2016, 55, 7229–7232 CrossRef CAS PubMed; (c) A. Akdeniz, L. Mosca, T. Minami and P. Anzenbacher, Chem. Commun., 2015, 51, 5770–5773 RSC; (d) L. Pu, Acc. Chem. Res., 2012, 45, 150–163 CrossRef CAS PubMed.
- (a) C. Wolf and K. W. Bentley, Chem. Soc. Rev., 2013, 42, 5408–5424 RSC; (b) F. Zhu, X. Li, Y. Li, M. Yan and S. Liu, Anal. Chem., 2015, 87, 357–361 CrossRef CAS PubMed; (c) R. Schreiber, N. Luong, Z. Fan, A. Kuzyk, P. C. Nickels, T. Zhang, D. M. Smith, B. Yurke, W. Kuang, A. O. Govorov and T. Liedl, Nat. Commun., 2013, 4, 2948 Search PubMed; (d) N. Berova, L. D. Bari and G. Pescitelli, Chem. Soc. Rev., 2007, 36, 914–931 RSC.
- (a) R.-X. Yao, X. Cui, J. Wang and X.-M. Zhang, Chem. Commun., 2015, 51, 5108–5111 RSC; (b) C.-R. Li, S.-L. Li and X.-M. Zhang, Cryst. Growth Des., 2009, 9, 1702–1707 CrossRef CAS.
- R. Hailili, L. Wang, J. Qv, R.-X. Yao, X.-M. Zhang and H. Liu, Inorg. Chem., 2015, 54, 3713–3715 CrossRef CAS PubMed.
- (a) A. Schoedel, M. Li, D. Li, M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2016, 116, 12466–12535 CrossRef CAS PubMed; (b) N. L. Rosi, J. Kim, M. Eddaoudi, B. Chen, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 1504–1518 CrossRef CAS PubMed.
- (a) R.-X. Yao, X. Xu and X.-M. Zhang, Chem. Mater., 2011, 24, 303–310 CrossRef; (b) M. E. Medina, Y. Dumont, J.-M. Grenèche and F. Millange, Chem. Commun., 2010, 46, 7987–7989 RSC.
- (a) M. Savage, I. da Silva, M. Johnson, J. H. Carter, R. Newby, M. Suyetin, E. Besley, P. Manuel, S. Rudić, A. N. Fitch, C. Murray, W. I. F. David, S. Yang and M. Schröder, J. Am. Chem. Soc., 2016, 138, 9119–9127 CrossRef CAS PubMed; (b) R. A. Peralta, A. Campos-Reales-Pineda, H. Pfeiffer, J. R. Álvarez, J. A. Zárate, J. Balmaseda, E. Gonzalez-Zamora, A. Martinez, D. Martinez-Otero, V. Jancik and I. A. Ibarra, Chem. Commun., 2016, 52, 10273–10276 RSC; (c) S. Yang, A. J. Ramirez-Cuesta, R. Newby, V. Garcia-Sakai, P. Manuel, S. K. Callear, S. I. Campbell, C. C. Tang and M. Schröder, Nat. Chem., 2015, 7, 121–129 CrossRef CAS PubMed; (d) Q. P. Lin, T. Wu, S.-T. Zheng, X. H. Bu and P.-Y. Feng, Chem. Commun., 2011, 47, 11852–11854 RSC.
- Q.-Y. Liu, W.-L. Xiong, C.-M. Liu, Y.-L. Wang, J.-J. Wei, Z.-J. Xiahou and L.-H. Xiong, Inorg. Chem., 2013, 52, 6773–6775 CrossRef CAS PubMed.
- E. Shamsaei, X. Lin, L. Wan, Y. Tong and H. Wang, Chem. Commun., 2016, 52, 13764–13767 RSC.
- (a) F. Wang, Y.-H. Tang and J. Zhang, Inorg. Chem., 2015, 54, 11064–11066 CrossRef CAS PubMed; (b) T. Senthilkumar and S. K. Asha, Chem. Commun., 2015, 51, 8931–8934 RSC; (c) K. W. Bentley and C. Wolf, J. Am. Chem. Soc., 2013, 135, 12200–12203 CrossRef CAS PubMed.
- Materials Studio, 6.0 V, Accelrys, Inc., San Diego, CA, 2010 Search PubMed.
- J. S. Miller, Chem. Soc. Rev., 2011, 40, 3266–3296 RSC.
- M. E. Fisher, Am. J. Phys., 1964, 32, 343–346 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Calculation details, IR spectra, PXRD pattern, TGA plots, crystallographic data, and CD spectra. CCDC 1552232–1552234 for compounds 1–3. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qi00615b |
| This journal is © the Partner Organisations 2018 |