
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reversible ionically-crosslinked single chain nanoparticles as bioinspired and recyclable nanoreactors for N-heterocyclic carbene organocatalysis†
Sofiem
Garmendia
abcd,
Andrew P.
Dove
 d,
Daniel
Taton
ab and
Rachel K.
O'Reilly
*d
d,
Daniel
Taton
ab and
Rachel K.
O'Reilly
*d
aLaboratoire de Chimie des Polymères Organiques, Université de Bordeaux IPB-ENSCBP, F-33607 Pessac Cedex, France
bLaboratoire de Chimie des Polymères Organiques Centre National de la Recherche Scientifique, 16 Avenue Pey-Berland, F-33607 Pessac Cedex, France
cDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK
dSchool of Chemistry, The University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: r.oreilly@bham.ac.uk
First published on 16th October 2018
Abstract
The intrinsic advantages of poly(ionic liquid)s (PILs), based on their high chemical activity and flexible structure, have been harnessed by exploring their applicability as catalytic single chain nanoparticles (SCNPs). A non-covalent bioinspired approach has been established to ionically crosslink an imidazolium-based poly(ionic liquid) to form folded SCNPs. An amphiphilic styrenic-type coPIL was synthesized by reversible addition fragmentation chain transfer (RAFT) to include hydrophilic stabilizer units, hydrophobic spacers and two antagonist functionalities randomly distributed through the polymer backbone. The antagonist functionalities were then intramolecularly and ionically crosslinked using a simple anion metathesis reaction, which resulted in folding to form the SCNPs under mild conditions. The folding process enabled the protection of the N-heterocyclic carbene (NHC) functionality, through the benzoate–imidazolium interaction between antagonist monomer units. Upon the application of heat, free NHCs could be generated within the confined SCNPs, which could be further utilized in benzoin catalysis. Most importantly, the reversible nature of the crosslinking and reversible generation of the active functionality allowed for the utilization of the SCNPs as a recyclable catalytic support.
Introduction
Single-chain polymer nanoparticles (SCNPs) have recently attracted increasing interest due to their exceptional and unique properties in an array of applications in nanomedicine, catalysis and information storage.1–6 SCNPs are based on self-collapsed individual chains that mimic the structure of folded biomacromolecules, such as (poly)peptides or enzymes.7 Particularly in catalysis, these biomacromolecules serve as a source of inspiration for the construction of artificial nano-objects (i.e. nanoreactors) trying to emulate such precision pathways towards functionality, specificity and efficiency.7,8 The strategy followed is based on folding individual synthetic polymer chains through intramolecular crosslinking to obtain SCNPs containing confined catalytic entities.9–11 Whilst the majority of work has been particularly focused on the use of covalent intramolecular interactions for the synthesis of robust SCNPs, the number of approaches based on reversible intramolecular interactions for the development of stimuli-responsive SCNPs is increasing.6,12,13 Reversibly bonded SCNP synthesis was first achieved via non-covalent interactions through hydrogen-bonding.14 Subsequently, many other folding/collapsing routes have been investigated, based on hydrogen-bonding,15–19 host–guest interactions,20–22 dynamic covalent bonds,6 disulfide bridges,23 redox/pH/UV-responsive SCNPs24–26 or metal complexation.27–32 All of these SCNPs can be disassembled by applying different stimuli (e.g. pH/temperature/redox change, solvent polarity variation, and including host competitors). The formation of reversible SCNP linkages that rely on dynamic configuration processes represent probably the most promising pathway towards enzyme biomimicry since the nature of these non-covalent bonds could determine the function and performance of synthetic SCNPs as occurs in certain proteins.6 As a new type of non-covalent bonding in SCNP technology, ionic liquids (ILs or PILs in the polymerized version) display a solvent-independent ionization state and have found unusually broad potential applications such as catalysis, due to their negligible vapor pressure, high thermal stability and powerful solvation capability.33–35 Moreover, the freedom to choose the cation and anion and their tunable miscibility with numerous organic compounds/solvents make (P)ILs potential candidates as a new type of non-covalent bond based SCNP for catalysis. To this purpose, imidazolium-based (P)ILs have been found to serve as a latent source of catalytically active N-heterocyclic carbenes (NHCs), which are readily generated through a temperature-dependent acid–base equilibrium.36,37 These masked NHCs have been widely implemented in both molecular and supported macromolecular structures using a variety of different imidazolium precatalysts. Interestingly, supported imidazolium-based masked NHCs were previously decorated in our group with different basic counter anions in a wide array of different polymeric structures such as with HCO3− (in linear polymers),38,39 acetate (in both linear polymers and folded SCNPs),40,41 or sebacate (in gels),42 by simple anion exchange. Inspired by such a concept, supporting on the one hand the imidazolium cationic moieties (NHC source) and on the other hand the basic counter anions (thermally latent trigger) on the same single polymeric chain should enable the creation of a confined SCNP pocket, based on electrostatic interactions. This would facilitate the creation of thermally latent NHC based catalytic centers, which could then potentially be utilized as catalytic nanoreactors.In this context, a new methodology for the collapse of a linear imidazolium-based coPIL precursor, into an ionically-crosslinked SCNP, using simple anion exchange has been explored. The folding procedure has then been carried out under mild conditions, through deprotonation of the carboxylic acid functionality. The resulting carboxylate then has the dual role of physically crosslinking the SCNPs, whilst generating the masked NHC precatalyst. These SCNPs were further used as a thermally latent homogeneous catalyst for the benzoin condensation and could be easily recycled for a number of cycles without any significant loss of activity (Scheme 1). Functionalization of the SCNPs with carbon disulfide evidenced the generation of the active NHC and resulted in the unfolding of the ionically crosslinked SCNPs.
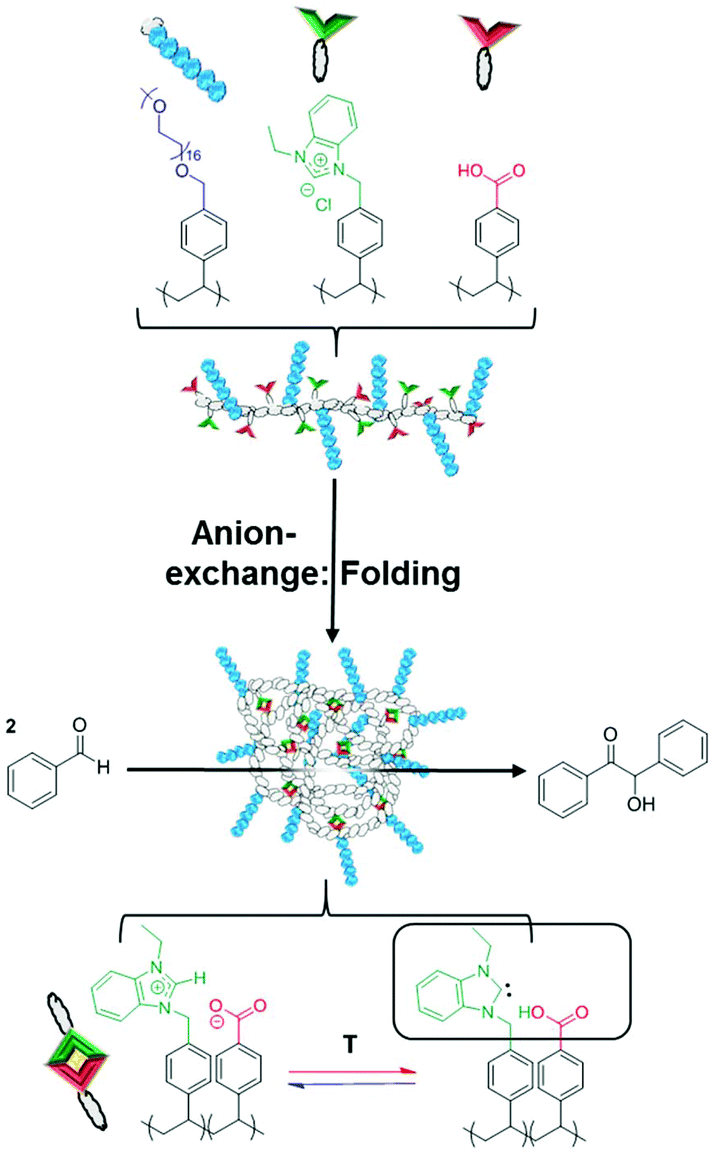 | ||
| Scheme 1 Design of catalytically active SCNPs for the benzoin condensation reaction. | ||
Experimental
Materials
Polymerization, catalyst loadings and catalysis experiments were carried out using syringe techniques under dry argon in baked glass tubes equipped with a two-way stopcock. Benzimidazole (>95%), 4-vinylbenzyl chloride (90%), ethyl bromide (99%) and 2-cyano-2-propyl benzodithioate (>97%) were obtained from Aldrich and used as received. Poly(ethylene oxide) monomethylester (CH3O-PEO16-OH, 750 g mol−1), triethylamine (TEA), carbon disulfide (CS2), 4-vinylbenzoic acid and 2-cyano-2-propyl benzodithioate (CTA) were obtained from Aldrich and used as received. Azobis(2-methylpropionitrile) (AIBN, 99%) was obtained from Aldrich and was purified by recrystallization from methanol. The synthesis of N-ethylbenzimidazole (1), vinyl ethyl benzimidazolium chloride (2) and PEG16-styrene (3) was performed as described in the literature.28 Styrene and benzaldehyde were dried over CaH2 and distilled prior to use. Tetrahydrofuran (THF) was distilled over Na/benzophenone. All synthesized polymers were purified by dialysis against methanol using “Standard Grade Regenerated Cellulose Dialysis Membranes (Spectra/Por6) Pre-wetted RC tubing” from Spectrum Lab with a molecular weight cut-off (MWCO) of 3.5 kDa and used after cleaning with pure water. All polymers were azeotropically dried using previously distilled THF before performing the catalysis.Characterization
1H NMR and 13C NMR spectra were recorded on a Bruker AC-400 spectrometer in appropriate deuterated solvents. All 13C measurements were performed at 298 K on a Bruker Avance III 400 spectrometer operating at 100.7 MHz and equipped with a 5 mm Bruker multinuclear direct cryoprobe. All DOSY (Diffusion Ordered Spectroscopy) measurements were performed at 298 K on a Bruker Avance III HD 400 spectrometer operating at 400.33 MHz and equipped with a 5 mm Bruker multinuclear z-gradient direct cryoprobe-head capable of producing gradients in the z direction with a strength of 53.5 G cm−1. For each sample, 3 mg was dissolved in 0.4 μL of DMSO-d6 for internal lock and spinning was used to minimize convection effects. DOSY spectra were acquired with the ledbpgp2s pulse program from the Bruker topspin software. The duration of the pulse gradients and the diffusion time were adjusted in order to obtain full attenuation of the signals at 95% of the maximum gradient strength. The values were 2.0 ms for the duration of the gradient pulses and 300 ms for the diffusion time. The gradient strength was linearly incremented in 16 steps from 5% to 95% of the maximum gradient strength. A delay of 3 s between echoes was used. The data were processed using 8192 points in the F2 dimension and 256 points in the F1 dimension with the Bruker topspin software. Field gradient calibration was accomplished at 25 °C using the self-diffusion coefficient of H2O + D2O at 19.0 × 10–10 m2 cm−1. Dimethylformamide soluble polymers were first solubilized in concentrations of 1 mg mL−1 and their masses were determined by size exclusion chromatography (SEC) in DMF at 50 °C using a refractive index (RI) detector (Varian). Analysis was performed using a three-column set of TSK gel TOSOH (G4000, G3000, and G2000 with pore sizes 20, 75, and 200 Å respectively, connected in series). A Bruker spectrometer was used for ATR-FTIR analysis. Hydrodynamic diameters (Dh) and size distributions of the SCNPs were determined by DLS on a Malvern Zetasizer Nano ZS system operating at 20 °C with a 4 mW He–Ne 633 nm laser module. Samples were filtered through a 0.2 μm PTFE filter prior to measurement and quartz cuvettes were used. Measurements were made at a detection angle of 173° (back scattering), and the data were analyzed using the Malvern DTS 6.20 software, using the multiple narrow modes setting. All measurements were made in triplicate, with 10 runs per measurement. TEM analyses were performed on a JEOL 2011 (LaB6) microscope operating at 200 keV, equipped with a GATAN UltraScan 1000 digital camera. The conventional bright field conditions were lacey carbon-coated copper grids (Agar Scientific, 400 mesh, S116-4) coated with a thin layer of graphene oxide. SCNP solutions were diluted to 2 mg mL−1 in MeOH before 4 μL of each sample were drop-deposited onto the graphene oxide coated grids, blotted immediately and allowed to air dry. Subsequent staining was applied using uranyl acetate to enhance the contrast. Images were analyzed using ImageJ software, and 50 particles were measured to produce a mean and standard deviation for the particle size (Dav). SAXS experiments were performed using a Xeuss 2.0 SAXS system equipped with two microfocus sources operating at 45 kV and 0.88 mA. The MicroMax-002 system based on a microfocus sealed tube S5 source module and an integrated X-ray generator unit producing Cu or Mo Kα transition photons at a wavelength of λ = 1.54 Å was used. Both the flight route and sample chamber in this equipment were under vacuum. The scattered X-rays were detected on a two-dimensional multiwire X-ray detector (Gabriel design, 2D-200X). The averaged scattered intensities were obtained as a function of momentum transfer q = 4πλ − 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sin(θ/2), where θ is the scattering angle. Ambient conditions were used for measurements with a sample to detector distance of 2 m. The prepared solutions (concentration: 1 mg mL−1, THF as solvent) were added into capillaries of 2 mm thickness and fixed perpendicularly to the beam. The data were corrected by subtracting the result of the blank capillary filled with the same THF employed for preparing the samples and applying the proper transmission corrections. All the SAXS measurements were carried out by Warwick Scientific Services.
sin(θ/2), where θ is the scattering angle. Ambient conditions were used for measurements with a sample to detector distance of 2 m. The prepared solutions (concentration: 1 mg mL−1, THF as solvent) were added into capillaries of 2 mm thickness and fixed perpendicularly to the beam. The data were corrected by subtracting the result of the blank capillary filled with the same THF employed for preparing the samples and applying the proper transmission corrections. All the SAXS measurements were carried out by Warwick Scientific Services.
Synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 8.3–6 (br, 34.3H, Ar–H), 5.9–5.5 (br, 2H, Ar–CH2–N), 4.7–4.1 (br, 4H, N–CH2–CH3, O–CH2–Ar), 4.1–3.1 (br, 67H, O–CH2–CH2, CH3–CH2–O), 2.3–0.9 (br, 13H, CH2–CH– (backbone), N–CH2–CH3) (Fig. S1A†); 13C NMR (DMSO-d6): 166.4, 164.9, 150.2, 145.5, 142.7, 141.3–126.2, 113.1, 72.1, 70.3, 68.7, 52.1, 50.9, 13.2 ppm (Fig. S2A†); IR: ν 3422.7 (OH), 1719.6 (CO), cm−1 (Fig. S3† in red).
N), 8.3–6.05 (br, 34.3, Ar–H), 6–5.55 (br, 2H, Ar–CH2–N), 4.83–4.05 (broad, 4H, N–CH2–CH3, Ar–CH2–O), 4.1–3.1 (br, 67H, O–CH2–CH2, CH3–CH2–O), 2.3–0.8 (br, 13H CH2–CH–(backbone), N–CH2–CH3) (Fig. S1B†); 13C NMR (DMSO-d6): 166.8, 150.4, 145.4, 132.4–127.3, 113.1, 72.1, 70.3, 51.9, 13.2 ppm (Fig. S2B†); IR: ν 1715.6 (CO), 1619.7, 1536.7 (COO−) cm−1 (Fig. S3† in blue).
N), 8.15–7.4 (m, 8H, Ar–H), 6.78–6.6 (dd, 1H, CH2–CH–Ar), 5.85 (d, 2H, CH2–CH–), 5.79 (s, 2H, Ar–CH2–N), 5.3 (d, 2H, CH2–CH–), 4.5 (m, 2H, N–CH2–CH3), 1.6 (s, 3H, −OOC–CH3), 1.5 (t, 3H, CH2–CH3) (Fig. S5†).
N), 8.2–6.05 (br, Ar–H), 6–5.55 (br, 2H, Ar–CH2–N), 4.6–4.4 (br, 2H, N–CH2–CH3), 2.3–0.9 (br, 9H, −OOC–CH3, N–CH2–CH3, –CH2–CH– (backbone)) (Fig. S6†). SEC analysis “Đ” = 1.18; Mn = 14.8 kDa (Fig. S7†).
N), 8.3–6 (br, 34.3H, Ar–H), 5.9–5.5 (br, 2H, Ar–CH2–N), 4.7–4.1 (br, 4H, N–CH2–CH3, O–CH2–Ar), 4.1–3.1 (br, 67H, O–CH2–CH2, CH3–CH2–O), 2.3–0.9 (br, 13H, CH2–CH– (backbone), N–CH2–CH3) (Fig. S1A†); 13C NMR (DMSO-d6): 166.4, 164.9, 150.2, 145.5, 142.7, 141.3–126.2, 113.1, 72.1, 70.3, 68.7, 52.1, 50.9, 13.2 ppm (Fig. S2A†); IR: ν 3422.7 (OH), 1719.6 (CO), cm−1 (Fig. S3† in red).
N), 8.3–6.05 (br, 34.3, Ar–H), 6–5.55 (br, 2H, Ar–CH2–N), 4.83–4.05 (broad, 4H, N–CH2–CH3, Ar–CH2–O), 4.1–3.1 (br, 67H, O–CH2–CH2, CH3–CH2–O), 2.3–0.8 (br, 13H CH2–CH–(backbone), N–CH2–CH3) (Fig. S1B†); 13C NMR (DMSO-d6): 166.8, 150.4, 145.4, 132.4–127.3, 113.1, 72.1, 70.3, 51.9, 13.2 ppm (Fig. S2B†); IR: ν 1715.6 (CO), 1619.7, 1536.7 (COO−) cm−1 (Fig. S3† in blue).
N), 8.15–7.4 (m, 8H, Ar–H), 6.78–6.6 (dd, 1H, CH2–CH–Ar), 5.85 (d, 2H, CH2–CH–), 5.79 (s, 2H, Ar–CH2–N), 5.3 (d, 2H, CH2–CH–), 4.5 (m, 2H, N–CH2–CH3), 1.6 (s, 3H, −OOC–CH3), 1.5 (t, 3H, CH2–CH3) (Fig. S5†).
N), 8.2–6.05 (br, Ar–H), 6–5.55 (br, 2H, Ar–CH2–N), 4.6–4.4 (br, 2H, N–CH2–CH3), 2.3–0.9 (br, 9H, −OOC–CH3, N–CH2–CH3, –CH2–CH– (backbone)) (Fig. S6†). SEC analysis “Đ” = 1.18; Mn = 14.8 kDa (Fig. S7†).
General procedure for the benzoin condensation reaction
In a 10 mL glass tube, different catalysts were first azeotropically dried using distilled THF. Then, dry THF was added keeping a reference catalyst concentration of 20 mg mL−1. The solution was stirred, and previously distilled benzaldehyde (10 eq.) was added under argon. The resulting mixture was stirred under argon at 80 °C for 24 h. After cooling, an aliquot was immediately taken and analyzed by 1H NMR spectroscopy to determine the conversion by comparing the integral value of the aldehyde signal of the benzaldehyde starting material (s, 1H, 10 ppm) with that of the –CH– benzoin product signal (s, 1H, 6 ppm) (Fig. S8–10†).General catalyst recycling procedure
After the previous catalytic cycle was performed, the resulting mixture was cooled down, significantly diluted in methanol and purified by dialysis (3.5 kDa MWCO) against MeOH (1 L × 2 every 24 h) in order to remove unconverted benzaldehyde, formed benzoin and THF. The catalyst was dried under vacuum and azeotropically dried once again using THF before the next catalytic run.Results and discussion
Synthesis of catalytically active SCNPs
Amphiphilic copolymer 4 was synthesized by reversible addition fragmentation chain transfer (RAFT) polymerization in methanol at 80 °C, in the presence of 2-cyano-2-propyl benzodithioate as a chain transfer agent and AIBN as an initiator. Copolymers were prepared by randomly copolymerizing the commercially available monomers 4-vinyl benzoic acid and styrene as well as the synthesized vinyl ethyl benzimidazolium chloride (2) and PEG16-styrene (3). Note that monomer 2 was polymerized in its chloride form, which enabled the use of methanol as the copolymerization solvent (Scheme 2). To obtain SCNPs with comparable size and similar amphiphilic character to those reported in the literature,28,41 the total theoretical degree of copolymerization of different styrenic monomers was kept around 80. The copolymerization was not carried out to full conversion (instead to ca. 40%) to avoid side reactions which might have led to a loss of a key functionality or to an increase in dispersity. Consequently, the molar mass distribution (ĐM) was narrow (1.2) for all of the copolymerizations. The conversion of the monomers was quantified via1H NMR spectroscopy by both the disappearance of the vinyl peaks and the broadening of the C2-proton during copolymerization. The almost identical consumption of the four different styrenic monomers, together with the moderate conversion, indicated that a statistical copolymer distribution was achieved. The composition of benzoic acid, styrene, 2 and 3 in copolymer 4 was then found to be 0.15/0.55/0.15/0.15, respectively, as identified by 1H NMR spectroscopy (Fig. S1A†).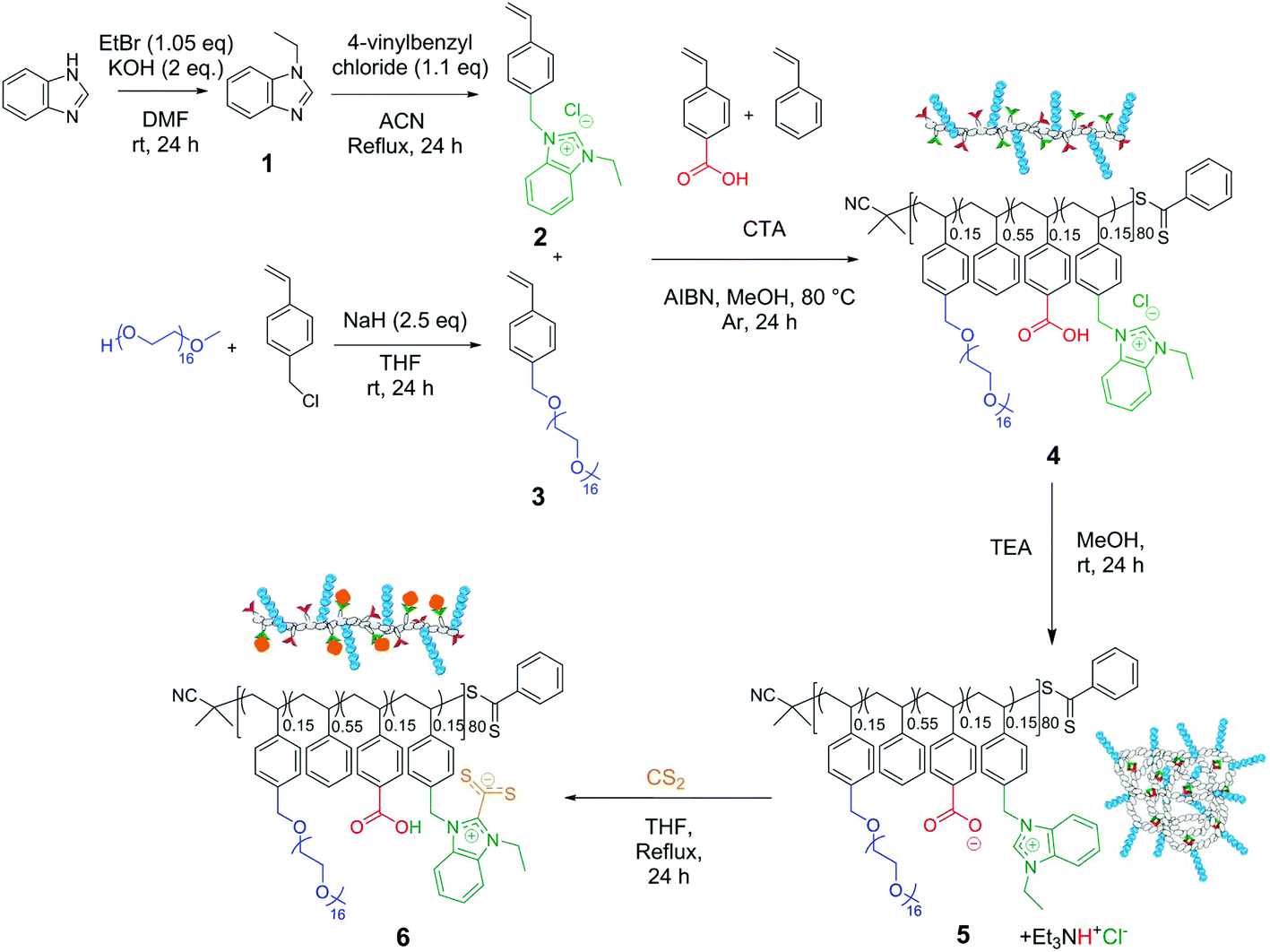 | ||
| Scheme 2 Synthetic route to the parent linear copolymer 4 by RAFT copolymerization of 2, 3, 4-vinylbenzoic acid and styrene, followed by a folding step caused by anion metathesis leading to the formation of SCNP 5. Unfolded copolymer 6 could be obtained by irreversibly post-functionalizing SCNP 5 with CS2. | ||
In the previous work from our group, SCNP-supported NHCs were generated upon heating to a temperature of 80 °C involving a post-functionalization approach to confer the thermally latent behavior to NHC precatalyst moieties.40,41 As a more facile and scalable alternative, we apply here an anion-exchange method enabling the triggering of the self-folding of the RAFT-derived copolymer precursor. This provides, on the one hand, a three-dimensional robust SCNP structure and, on the other hand, a basic counter anion for the benzimidazolium moieties, hence enabling their utility as a latent NHC source upon thermal triggering. Indeed, the ionic nature of the crosslinking requires only mild conditions for the triggering of SCNP folding and also confers reversibility in this process. In detail, linear copolymer precursor 4 was subjected to an anion metathesis reaction under diluted conditions (1 mg mL−1), replacing the chloride anions (present in benzimidazolium moieties) by carboxylate ones (present in benzoic acid moieties) in the presence of triethylamine (TEA) to afford SCNP 5.
This folding process, in which a carboxylate counter anion ionically interacts with the benzimidazolium cation, was evidenced using a range of analysis methods, including 1H and 13C NMR spectroscopy, diffusion ordered spectroscopy (DOSY), Fourier transform infrared spectroscopy (FTIR), size exclusion chromatography (SEC), dynamic light scattering (DLS), transmission electron microscopy (TEM), and small-angle X-ray scattering (SAXS). In particular, 1H NMR spectroscopy revealed the disappearance of the acidic –OH proton signal, as well as the shift of the benzimidazolium C2-proton signal from 10.5 to 11.4 ppm (Fig. S1A†). FTIR confirmed the disappearance of the acidic –OH vibration signal around 3300 cm−1, and the presence of the acetate symmetric and asymmetric vibrations in the 1500–1600 cm−1 (specifically at 1590 cm−1) region (Fig. S3†). It is known that PILs supporting polycations and polyanions in the same backbone are difficult to characterize by SEC analysis due to electrostatic interactions with the stationary phase.43,44 Thus, after testing a number of different solvents, the use of DMF containing 10 mM ammonium tetrafluoroborate at 50 °C was found to be optimal. An increase in the retention time for SCNP 5 compared to linear precursor 4 was observed, which was attributed to the decrease in hydrodynamic volume, confirming successful folding of the co(PILs) (Fig. 1A).
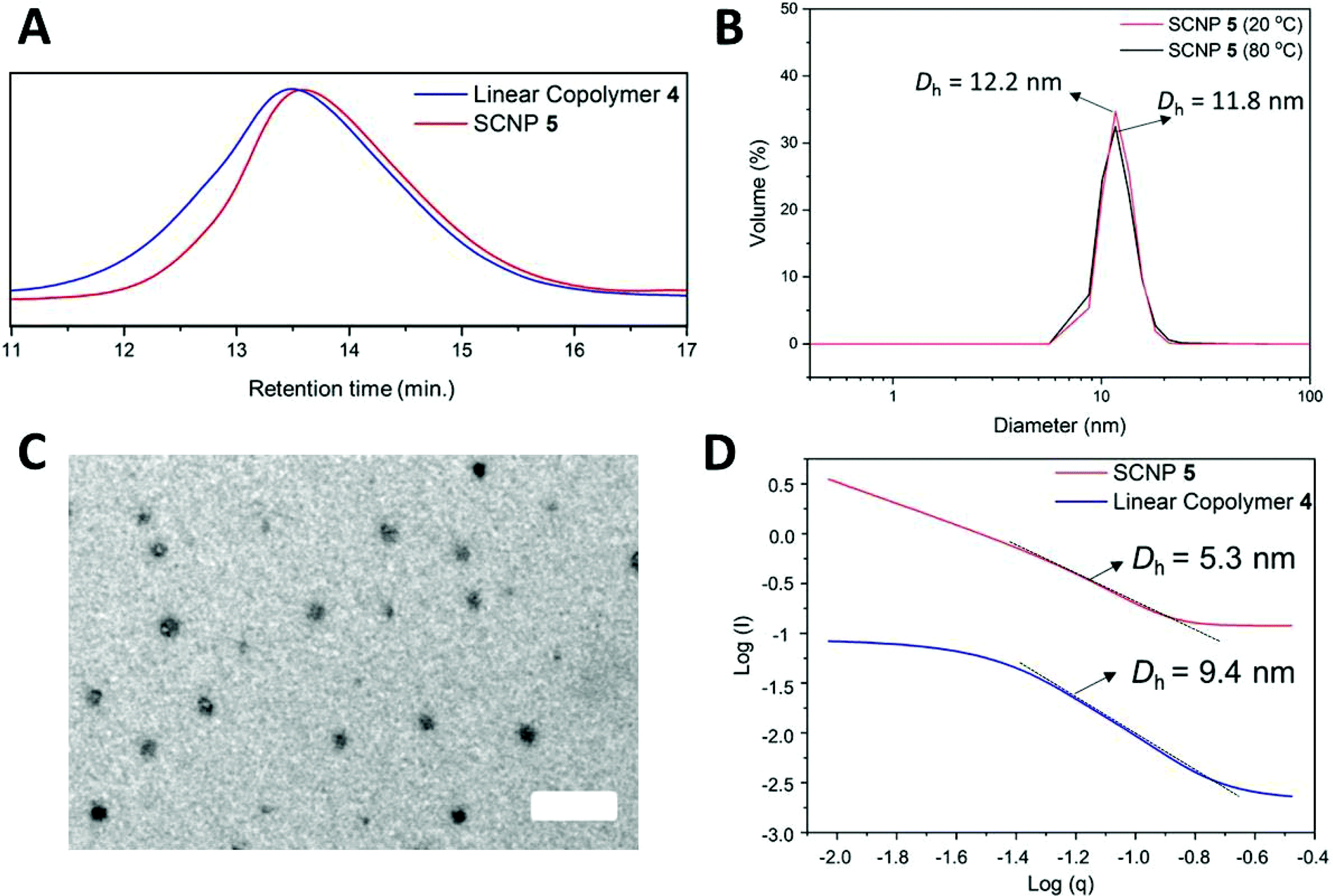 | ||
| Fig. 1 (A) SEC responses (in DMF in the presence of 10 mM NH4BF4; RI detector) of SCNP 5 (red line) obtained via cross-linking of copolymer precursor 4 (blue line) by anion exchange. (B) DLS analysis of SCNP 5 at rt (solid red line) and at catalysis temperature (solid black line) distributed by volume in THF (conc. = 5 mg mL−1). (C) TEM images of SCNP 5 (conc. = 1 mg mL−1 in methanol) stained with uranyl acetate; scale bar = 50 nm. (D) Kratky log–log plot from the SAXS analysis of precursor 4 (solid blue line) and folded 5 (solid red line) in THF (conc. = 1 mg mL−1; fitted to a compact particle model). | ||
Dynamic light scattering (DLS) experiments were performed to confirm the single chain folding of the prepared copolymers. The folded SCNP 5 was analyzed at a concentration of 5 mg mL−1 and showed a single peak around 12 nm for the hydrodynamic diameter (distribution in volume) at room temperature. However, we were also interested in the particle size at catalysis temperature (i.e. 80 °C in THF) and hence variable temperature DLS analysis was performed, as summarized in Fig. 1B (see Fig. S11† for 20–80 °C range). This analysis confirmed a relatively homogeneous diameter of the SCNPs from room temperature (rt) to catalytic conditions. Dry state TEM, stained with uranyl acetate, revealed particle diameters of 7–10 nm, confirming the results obtained by DLS analysis (Fig. 1C†).
To further confirm the change in size upon folding, the linear and folded co(PILs) were characterized by DOSY NMR spectroscopy in DMSO-d6. This characterization technique confirmed the faster diffusion coefficient for the SCNP 5 (D = 3.14 × 10−11 m s−2) compared to the linear precursor 4 (D = 2.39 × 10−11 m2 s−1) and also allowed for the calculation of the hydrodynamic diameter of the SCNPs (of ∼7 nm) using the Stokes–Einstein equation (Fig. S12†). Furthermore, despite the suggested presence of some larger aggregates, SAXS measurements (fitted to a compact particle model) unambiguously revealed a compaction from 9.4 nm to 5.3 nm calculated from the Porod region in the Kratky plot (Fig. 1D). All of these characterization methods afforded similar values for SCNP diameter (with variations attributable to the different measurement conditions, solvents, etc.) and confirmed the successful folding of the linear coPIL 4 to form the SCNP 5.
Benzoin condensation catalysed by SCNP 5
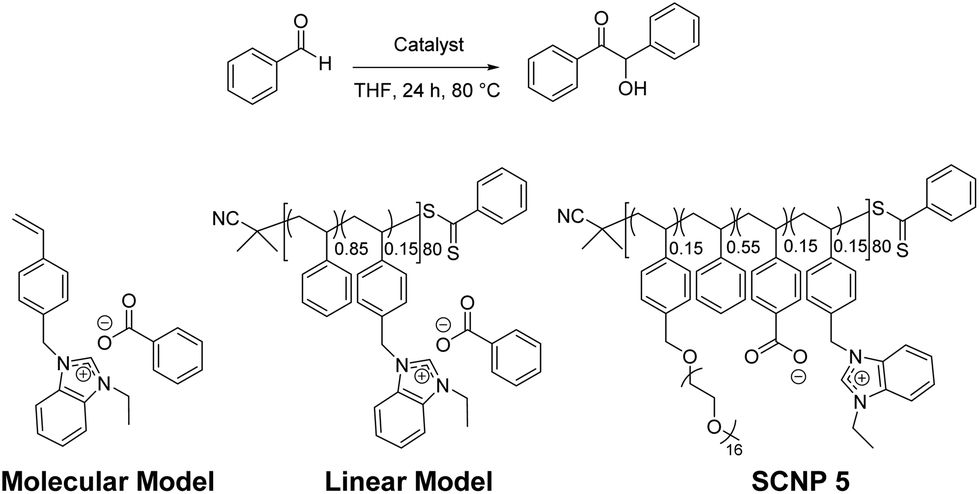
This particular folding technique was of interest as it provided a method for the generation of a thermally latent NHC precatalyst, which could be activated upon thermal cleavage of the intrachain crosslinks. To prove that an active NHC species was generated upon heating SCNP 5, an NHC-CS2 adduct forming reaction was carried out (copolymer 6; Scheme 2).40 Upon the addition of CS2 irreversible and selective “trapping” of the generated active NHCs in the SCNPs was expected. This was confirmed by identifying the characteristic signals of the NHC-CS2 betaine (223.9 ppm, corresponding to the dithiolate group in the 13C NMR spectrum; Fig. S4B†).45 Masked carbenes in their active form are known for their versatility and capability of acting as (organo)catalysts in a wide array of (macro)molecular reactions.36,37,46,47 Since the folding step afforded a 3D conformational masked NHC precatalyst, no further post-functionalization was required prior to catalysis. We hypothesized that the active NHC species could be generated upon heating as previously proven by NHC-CS2 selective formation, resulting in the generation of a thermally latent catalyst, which could be utilized in an organic transformation.Benzoin condensation was chosen as model reaction because it is almost exclusively catalysed by carbenes,48 and hence would permit the evaluation of the catalytic performance of our system. Therefore, this reaction was first implemented in THF using benzaldehyde as the substrate and 10 mol% of SCNP 5 (reference concentration: 20 mg mL−1) relative to benzimidazolium moieties. In the first catalyst run using these conditions, 71% of benzaldehyde units were converted into benzoin after 24 h (Run 1; Table 1) and no higher conversions were observed when prolonging the experiment time up to 48 h (Run 2). Interestingly, owing to both the incorporated side oligo-PEG in the polymeric structure and thermo-latent behavior of SCNPs, the increased solubility allowed polymer-supported catalyst recycling after catalyst deactivation, namely, by dialysis against methanol (see the Experimental section). Conversions remained around 70% after 24 h of reaction, and after three consecutive catalytic cycles when SCNP 5 was applied. This highlights the robust and reversible nature of our latently crosslinking approach (Runs 3 and 4).
|
|
|||||
|---|---|---|---|---|---|
| Run | Catalyst | Cat. (mol%) | Cycle | Time (h) | Conv.a (%) |
| a Conversions were calculated by 1H NMR spectroscopy (Fig. S8). b Active monomer based molecular model. c Linear model containing randomly ordered spacers and active monomer units. d Entry 19: the catalysis reaction was run in water. The reference catalyst concentration used was 20 mg mL−1 and temperature was set at 80 °C. In all catalytic experiments, no enantiomeric selectivity was observed. | |||||
| 1 | SCNP 5 | 10 | 1st | 24 | 71 |
| 2 | SCNP 5 | 10 | 1st | 48 | 71 |
| 3 | SCNP 5 | 10 | 2nd | 24 | 69 |
| 4 | SCNP 5 | 10 | 3rd | 24 | 68 |
| 5 | SCNP 5 | 5 | 1st | 24 | 76 |
| 6 | SCNP 5 | 1 | 1st | 24 | 85 |
| 7 | SCNP 5 | 0.1 | 1st | 24 | 80 |
| 8 | Molecular model | 10 | 1st | 24 | 74 |
| 10 | Molecular model | 5 | 1st | 24 | 62 |
| 11 | Molecular model | 1 | 1st | 24 | 61 |
| 12 | Molecular model | 0.1 | 1st | 24 | 45 |
| 13 | Linear model | 10 | 1st | 48 | 61 |
| 14 | Linear model | 5 | 1st | 24 | 61 |
| 15 | Linear model | 1 | 1st | 24 | 50 |
| 16 | Linear model | 0.1 | 1st | 24 | 50 |
| 17 | 2 | 10 | 1st | 24 | 0 |
| 18 | 4 | 10 | 1st | 24 | 0 |
| 19d | SCNP 5 | 10 | 1st | 24 | 0 |
| 20 | — | — | 1st | 24 | 0 |
The role of confined space, where the reagents are able to efficiently interact with the thermally latent NHC, was then explored. The amount of SCNP 5 catalyst was thus decreased from 10 mol% to 1 mol%. Interestingly, conversions increased from 71% to 85%, reducing the mol% of the SCNP 5 ten-fold (Runs 5 and 6). However, when the catalyst amount was dropped further to 0.1 mol% (Run 7), the conversion did not increase, suggesting a product inhibition effect in the catalytic pocket of SCNP 5, which has been observed before in similar confined systems.49 More specifically, we hypothesized that this inhibition effect could come from the generated –OH moieties in benzoin molecules that would inhibit the NHC catalyst as similarly occurred in other masked NHC catalytic systems.38–40,50
To examine the relationship between the confinement effect and inhibition provoked by the catalysed benzoin product, SCNP 5 was compared with a linear polymeric model, i.e. only bearing the NHC precatalyst and spacer moieties, and a molecular model based on an active benzimidazolium monomer. Both molecular and macromolecular models were assessed for the benzoin condensation reaction under the same catalysis conditions as SCNP 5. No appreciable confinement effect was found when the molecular model was used, showing a progressive decrease in conversion from 74% to 45% when decreasing the catalytic amount from 10 to 0.1 mol% (Runs 8–12). The lower catalytic performance of the molecular model compared to SCNP 5 could be due to the poor solubility of benzimidazolium benzoate and subsequent aggregation in the solvent used (i.e. THF). Conversely, the linear copolymeric model based on styrenic benzimidazolium benzoate moieties showed higher solubility compared to the molecular model, but led to a lower catalytic activity than both the molecular model and, more importantly, SCNP 5. In fact, only 61% conversion was obtained when using 10 mol% of the linear copolymer, which dropped up to 50% when the catalyst loading was decreased down to 0.1 mol% (Runs 13–16). The observed plateau of 50% conversion when using either 1 or 0.1 mol% catalyst (Runs 15 and 16, respectively) suggested that the inhibition effect was probably more apparent in the linear model, since the catalytic sites were more exposed to the obtained product (i.e. benzoin), and hence, more prone to inhibition than SCNP 5. This is proposed to be due to the confinement effect on SCNP 5, leading to an energy-free driven catalytic nanoreactor system based on the osmotic gradient between the SCNP pocket and the external solution.49,51–53
Otherwise, for comparison, the chlorine-containing monomer 2 and the linear parent coPIL 4 were also tested under the same conditions (Runs 17 and 18). However, no benzoin product was obtained, providing further evidence for the role of benzoate counter-anions to generate in situ catalytic SCNP-supported NHCs. Finally, control experiments were performed in order to successfully discard any catalytic activity of SCNP 5 in H2O (Run 19) or spontaneously generated benzoin without a catalyst under previously used conditions (Run 20). It is also worth mentioning that no benzoin conversion was observed when the experiments were run at room temperature, in good agreement with previously reported studies of our group reporting similar systems.40–42
This non-covalently crosslinked SCNP approach illustrates the importance of having the normally ionic catalyst tethered to the SCNP scaffold for efficient catalysis of generated soluble products which may entail inhibition in relatively apolar solvent conditions.
Conclusions
In summary, a novel type of non-covalently bonded SCNP obtained by RAFT copolymerization is reported. Exploiting the PIL nature of the parent linear precursor, amphiphilic SCNP folding was triggered by a mild anion metathesis intramolecular reaction. The collapse was confirmed by different characterization methods (1H/13C NMR spectroscopy, SEC, DLS, SAXS, TEM and FTIR). These SCNPs contain latent NHC-type active species which could be thermally activated without the need for an exogenous reagent. The relatively high conversions (∼70%) in the benzoin condensation reactions could be maintained up to 3 catalytic cycles. Interestingly, the conversions could be enhanced up to 85% when 1 mol% catalyst was used, as a result of the confinement in the catalytic environment in THF. The latter was further evidenced when comparing SCNPs with both molecular and polymeric models, showing higher catalytic efficiency, probably due to the smaller catalyst inhibition effect in the SCNP catalytic pocket. Finally, post-modification of SCNPs by CS2 insertion confirms the NHC active species generation, as well as providing a simple SCNP unfolding route. Although synthetically challenging, opening the SCNP chemistry to an entirely new terrain based on non-covalent interactions using PIL structures can provide answers to major organocatalysis challenges.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by EU through project SUSPOL-EJD 642671. We thank Steven Huband from X-ray RTP in the University of Warwick (UK) for SAXS measurements and Anne-Laure Wirotius in the University of Bordeaux for DOSY NMR spectroscopy.Notes and references
- M. Ouchi, N. Badi, J.-F. Lutz and M. Sawamoto, Nat. Chem., 2011, 3, 917–924 CrossRef CAS PubMed.
- M. Gonzalez-Burgos, A. Latorre-Sanchez and J. A. Pomposo, Chem. Soc. Rev., 2015, 44, 6122–6142 RSC.
- S. Mavila, O. Eivgi, I. Berkovich and N. G. Lemcoff, Chem. Rev., 2016, 116, 878–961 CrossRef CAS PubMed.
- A. M. Hanlon, C. K. Lyon and E. B. Berda, Macromolecules, 2016, 49, 2–14 CrossRef CAS.
- O. Altintas and C. Barner-Kowollik, Macromol. Rapid Commun., 2016, 37, 29–46 CrossRef CAS PubMed.
- O. Altintas and C. Barner-Kowollik, Macromol. Rapid Commun., 2012, 33, 958–971 CrossRef CAS PubMed.
- A. Latorre-Sánchez and J. A. Pomposo, Polym. Int., 2016, 65, 855–860 CrossRef.
- J. Rubio-Cervilla, E. González and J. Pomposo, Nanomaterials, 2017, 7, 341 CrossRef PubMed.
- H. Rothfuss, N. D. Knöfel, P. W. Roesky and C. Barner-Kowollik, J. Am. Chem. Soc., 2018, 140, 5875–5881 CrossRef CAS PubMed.
- A. R. A. Palmans, in Self-assembling Biomaterials, Elsevier, 2018, pp. 563–583 Search PubMed.
- Y. Liu, S. Pujals, P. J. M. Stals, T. Paulöhrl, S. I. Presolski, E. W. Meijer, L. Albertazzi and A. R. A. Palmans, J. Am. Chem. Soc., 2018, 140, 3423–3433 CrossRef CAS PubMed.
- M. Artar, E. Huerta, E. W. Meijer and A. R. A. Palmans, in Sequence-Controlled Polymers: Synthesis, Self-Assembly, and Properties, ed. J.-F. Lutz, T. Y. Meyer, M. Ouchi and M. Sawamoto, American Chemical Society, Washington, DC, 2014, 1170, pp. 313–325 Search PubMed.
- J. A. Pomposo, J. Rubio-Cervilla, A. J. Moreno, F. Lo Verso, P. Bacova, A. Arbe and J. Colmenero, Macromolecules, 2017, 50, 1732–1739 CrossRef CAS.
- M. Seo, B. J. Beck, J. M. J. Paulusse, C. J. Hawker and S. Y. Kim, Macromolecules, 2008, 41, 6413–6418 CrossRef CAS.
- E. J. Foster, E. B. Berda and E. W. Meijer, J. Am. Chem. Soc., 2009, 131, 6964–6966 CrossRef CAS PubMed.
- T. Terashima, T. Mes, T. F. A. De Greef, M. A. J. Gillissen, P. Besenius, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2011, 133, 4742–4745 CrossRef CAS PubMed.
- T. Mes, R. van der Weegen, A. R. A. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2011, 50, 5085–5089 CrossRef CAS PubMed.
- F. Wang, H. Pu, M. Jin, H. Pan, Z. Chang, D. Wan and J. Du, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1832–1840 CrossRef CAS.
- N. Hosono, A. M. Kushner, J. Chung, A. R. A. Palmans, Z. Guan and E. W. Meijer, J. Am. Chem. Soc., 2015, 137, 6880–6888 CrossRef CAS PubMed.
- E. A. Appel, J. Dyson, J. del Barrio, Z. Walsh and O. A. Scherman, Angew. Chem., Int. Ed., 2012, 51, 4185–4189 CrossRef CAS PubMed.
- J. Willenbacher, B. V. K. J. Schmidt, D. Schulze-Suenninghausen, O. Altintas, B. Luy, G. Delaittre and C. Barner-Kowollik, Chem. Commun., 2014, 50, 7056–7059 RSC.
- B. V. K. J. Schmidt and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2017, 56, 2–22 CrossRef.
- B. T. Tuten, D. Chao, C. K. Lyon and E. B. Berda, Polym. Chem., 2012, 3, 3068–3071 RSC.
- A. Sanchez-Sanchez, D. A. Fulton and J. A. Pomposo, Chem. Commun., 2014, 50, 1871–1874 RSC.
- J. He, L. Tremblay, S. Lacelle and Y. Zhao, Soft Matter, 2011, 7, 2380–2386 RSC.
- H. Frisch, J. P. Menzel, F. R. Bloesser, D. E. Marschner, K. Mundsinger and C. Barner-Kowollik, J. Am. Chem. Soc., 2018, 140, 9551–9557 CrossRef CAS PubMed.
- H. Rothfuss, N. D. Knöfel, P. W. Roesky and C. Barner-Kowollik, J. Am. Chem. Soc., 2018, 140, 5875–5881 CrossRef CAS PubMed.
- R. Lambert, A.-L. Wirotius, S. Garmendia, P. Berto, J. Vignolle and D. Taton, Polym. Chem., 2018, 9, 3199–3204 RSC.
- K. Freytag, S. Säfken, K. Wolter, J. C. Namyslo and E. G. Hübner, Polym. Chem., 2017, 8, 7546–7558 RSC.
- A. Sanchez-Sanchez, A. Arbe, J. Kohlbrecher, J. Colmenero and J. A. Pomposo, Macromol. Rapid Commun., 2015, 36, 1592–1597 CrossRef CAS PubMed.
- N. D. Knöfel, H. Rothfuss, J. Willenbacher, C. Barner-Kowollik and P. W. Roesky, Angew. Chem., Int. Ed., 2017, 56, 4950–4954 CrossRef PubMed.
- Y. Bai, X. Feng, H. Xing, Y. Xu, B. K. Kim, N. Baig, T. Zhou, A. A. Gewirth, Y. Lu, E. Oldfield and S. C. Zimmerman, J. Am. Chem. Soc., 2016, 138, 11077–11080 CrossRef CAS PubMed.
- J. Yuan, D. Mecerreyes and M. Antonietti, Prog. Polym. Sci., 2013, 38, 1009–1036 CrossRef CAS.
- J. Yuan and M. Antonietti, Polymer, 2011, 52, 1469–1482 CrossRef CAS.
- A. Eftekhari and T. Saito, Eur. Polym. J., 2017, 90, 245–272 CrossRef CAS.
- M. Fevre, J. Pinaud, Y. Gnanou, J. Vignolle and D. Taton, Chem. Soc. Rev., 2013, 42, 2142 RSC.
- S. Naumann and M. R. Buchmeiser, Catal. Sci. Technol., 2014, 4, 2466 RSC.
- M. Fèvre, J. Pinaud, A. Leteneur, Y. Gnanou, J. Vignolle, D. Taton, K. Miqueu and J.-M. Sotiropoulos, J. Am. Chem. Soc., 2012, 134, 6776–6784 CrossRef PubMed.
- P. Coupillaud, J. Pinaud, N. Guidolin, J. Vignolle, M. Fevre, E. Veaudecrenne, D. Mecerreyes and D. Taton, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4530–4540 CAS.
- R. Lambert, P. Coupillaud, A.-L. Wirotius, J. Vignolle and D. Taton, Macromol. Rapid Commun., 2016, 37, 1143–1149 CrossRef CAS PubMed.
- R. Lambert, A.-L. Wirotius and D. Taton, ACS Macro Lett., 2017, 6, 489–494 CrossRef CAS.
- S. Garmendia, R. Lambert, A.-L. Wirotius, J. Vignolle, A. P. Dove, R. K. O'Reilly and D. Taton, Eur. Polym. J., 2018, 107, 82–88 CrossRef CAS.
- H. He, M. Zhong, B. Adzima, D. Luebke, H. Nulwala and K. Matyjaszewski, J. Am. Chem. Soc., 2013, 135, 4227–4230 CrossRef CAS PubMed.
- E. Blasco, B. T. Tuten, H. Frisch, A. Lederer and C. Barner-Kowollik, Polym. Chem., 2017, 8, 5845–5851 RSC.
- P. Coupillaud, J. Vignolle, D. Mecerreyes and D. Taton, Polymer, 2014, 55, 3404–3414 CrossRef CAS.
- D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS PubMed.
- W. N. Ottou, H. Sardon, D. Mecerreyes, J. Vignolle and D. Taton, Prog. Polym. Sci., 2016, 56, 64–115 CrossRef CAS.
- R. S. Menon, A. T. Biju and V. Nair, Beilstein J. Org. Chem., 2016, 12, 444–461 CrossRef CAS PubMed.
- A. Lu, D. Moatsou, D. A. Longbottom and R. K. O'Reilly, Chem. Sci., 2013, 4, 965–969 RSC.
- D. Kuzmicz, P. Coupillaud, Y. Men, J. Vignolle, G. Vendraminetto, M. Ambrogi, D. Taton and J. Yuan, Polymer, 2014, 55, 3423–3430 CrossRef CAS.
- A. D. Ievins, X. Wang, A. O. Moughton, J. Skey and R. K. O'Reilly, Macromolecules, 2008, 41, 2998–3006 CrossRef CAS.
- P. Cotanda, N. Petzetakis and R. K. O'Reilly, MRS Commun., 2012, 2, 119–126 CrossRef CAS.
- A. Lu, P. Cotanda, J. P. Patterson, D. A. Longbottom and R. K. O'Reilly, Chem. Commun., 2012, 48, 9699–9701 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, monomer/polymer synthesis and related characterization analysis. See DOI: 10.1039/c8py01293h |
| This journal is © The Royal Society of Chemistry 2018 |