
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganocatalysis for depolymerisation
Coralie
Jehanno
 abc,
Maria M.
Pérez-Madrigal
c,
Jeremy
Demarteau
a,
Haritz
Sardon
*a and
Andrew P.
Dove
*c
abc,
Maria M.
Pérez-Madrigal
c,
Jeremy
Demarteau
a,
Haritz
Sardon
*a and
Andrew P.
Dove
*c
aPOLYMAT, University of the Basque Country UPV/EHU, Joxe Mari Korta Center, Avda. Tolosa 7, 20018 Donostia-San Sebastian, Spain
bDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK
cSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: a.dove@bham.ac.uk
First published on 23rd October 2018
Abstract
Polymeric materials have been accumulating in the environment for decades as a result of the linear way of consuming plastics. Unfortunately, the current approaches followed to treat such a large amount of plastic waste, mainly involving physical recycling or pyrolysis, are not efficient enough. Recently, chemical degradation has emerged as a long-term strategy towards reaching completely sustainable cycles where plastics are polymerised, depolymerised, and then re-polymerised with minimal changes in their quantity or final properties. Organocatalysts, which are promising “green” substitutes for traditional organometallic complexes, are able to catalyse depolymerisation reactions yielding highly pure small molecules that are adequate for subsequent polymerisations or other uses. Moreover, by varying several reaction parameters (e.g. solvent, temperature, concentration, co-catalyst, etc.), the depolymerisation products can be tuned in innumerable possibilities, which further evidences the versatility of depolymerisation. In this review, we highlight the recent advances made by applying organocatalysts, such as organic bases, organic acids, and ionic compounds, to chemically degrade the most commonly used commercial polymers. Indeed, organocatalysis is envisaged as a promising tool to reach a circular and environmentally friendly plastic economy.
 Coralie Jehanno | Coralie Jehanno earned her master's degree in 2015 from the graduate school of Chemistry, Biology and Physics of Bordeaux, France. She is currently a PhD student between the university of the Basque country in San Sebastian, Spain, under the supervision of Dr Haritz Sardon and Dr Fernando Ruipérez, and the university of Warwick, UK, under the supervision of Pr Andrew Dove. Her PhD has also involved a period in the IBM Almaden research centre, CA, USA, with Dr James Hedrick, in 2017. Coralie is working on the synthesizing and modelling of innovative organocatalysts, mainly for the recycling of commodity polymers. |
 Maria M. Pérez-Madrigal | Maria M. Pérez-Madrigal is a postdoctoral researcher at the Dove group (University of Birmingham). Under the supervision of Prof. Carlos Alemán and Dr Elaine Armelin (IMEM group), she obtained her PhD in 2015 from Universitat Politècnica de Catalunya (UPC). Her research focused on combining conducting polymers (CPs) with conventional polymers to obtain biointerfaces at the nanoscale for bioapplications. After a post-doctoral year in the IMEM group working on supercapacitor devices based on CPs and hydrogels, she joined the Dove group in 2016 as a Marie-Curie Fellow for developing polymer-based hydrogels as scaffolds for load-bearing soft tissue regeneration. |
 Jeremy Demarteau | Jeremy Demarteau received his undergraduate education in chemistry in Belgium. In 2013, he started his Ph.D. at the laboratory of CERM (Liège, Belgium). During his Ph.D. supervised by Dr Christophe Detrembleur, he investigated the controlled radical polymerization of less-activated monomers using organocobalt complexes, notably with the synthesis of block copolymers based on vinyl acetate and ethylene. In 2017, he joined the Innovative Polymers Group of POLYMAT (San Sebastian, Spain) as a post-doctoral researcher. With Dr Harritz Sardon, he is working on the organocatalytic depolymerization of commodity polymers, such as polyesters, into valuable monomers and their use in a circular economy. |
 Haritz Sardon | Haritz Sardon received his Ph.D. in 2011 at the University of Basque Country under the supervision of Prof. Lourdes Irusta and Prof. M. J. Fernandez-Berridi before joining the group of Dr James Hedrick at IBM-Almaden Research Center, USA, as a post-doc. In 2014, Haritz returned to Spain with a Ministry grant and joined POLYMAT institute as junior group leader before starting his independent research career as Associate Prof. for the University of Basque Country in 2017. His work is focused on organocatalysis for polymerization/depolymerization reactions, including step growth polymerization of polyurethanes, polycarbonates, polyesters and polyethers as well as depolymerization of commodity plastics. |
 Andrew P. Dove | Andrew. P. Dove is a Professor of Chemistry at the University of Birmingham, UK. He obtained his Ph.D. under Prof. Vernon Gibson from Imperial College London in 2003 before postdoctoral study at both Stanford University and IBM, under the supervision of Prof. Robert Waymouth and Dr James Hedrick. Andrew returned to the UK as a RCUK Fellow at the University of Warwick in 2005 before being promoted to Assistant (2006), Associate (2009) and Full Professor (2014). Andrew moved to Birmingham in 2018. Andrew was awarded the Macro Group UK Young Researcher Medal in 2009, RSC Gibson-Fawcett Award in 2014, Biomacromolecules/Macromolecules Young Researcher Award in 2016 and RSC Norman Heatley Award in 2018. |
Introduction
Polymers have become ubiquitous materials in our daily life on account of their low cost production and safety combined with their remarkable functional properties. Many of the materials that we use, however, have extremely short lifetimes and are commonly limited to a single use. Consequently, plastic waste has been accumulating in the environment for years, and it is only very recently that this linear way of consuming plastics has raised concerns. Hence, the treatment of plastic waste, which is a global societal and environmental problem, requires innovative solutions to sort, degrade, and re-process these materials.Despite economic and environmental incentives to promote plastic waste treatment, current alternatives are very limited. The Ellen MacArthur foundation recently suggested three different strategies for a sustainable plastic packaging economy based on (i) reusing 20% of the packaging items in the long-term; (ii) re-designing 30% of them; and (iii) recycling the remaining 50%.1 This last category mainly concerns the most commonly used plastics, such as polyethylene (PE), polyesters, polycarbonates, or polyurethanes. Currently, only 14% of these polymers are being recycled, mostly by mechanical methods that involve grinding and re-processing of the material into low-value plastic products. The inferior properties of the resulting material compared to the initial polymer has been cited as “downcycling”, referring to loss of strength, chemical or food contamination, discoloration, or decrease in molecular weight, for example.2 Another approach for treating post-consumed plastics relies on their direct conversion into high calorific value fuels through pyrolysis, a treatment that requires elevated pressure and temperature. However, this thermal degradation only postpones their unsustainable end-of-life since the resulting combustible will typically be burnt to produce energy, releasing undesirable gases into the environment. Therefore, chemical degradation, another recycling approach, represents an attractive long-term strategy to create a sustainable polymer supply chain. Recently, the chemical recycling of polymers has attracted a lot of attention among the scientific community,3–6 mainly driven by the current public awareness of this problem.
Chemical recycling involves transforming polymers from plastic waste into small molecules with high yield and purity. Specifically, chemical depolymerisation either produces the initial monomers that can be subsequently re-polymerised into high quality polymers (circular economy – Fig. 1), or innovative small molecules that can be used as high added-value building blocks for creating unique polymeric materials or other chemicals (added-value plastic economy – Fig. 1). However, as a consequence of the high stability of polymeric materials, forcing conditions, such as microwave assistance,7–9 supercritical conditions,10,11 or the use of catalysts12–14 are usually required to enhance the efficiency of the depolymerisation reactions. In particular, stable and highly active organometallic catalysts, such as zinc or lead acetates, sodium/potassium sulphate, or titanium phosphate, which are already well-known for organic chemistry reactions, have been largely applied to depolymerisation processes. Despite their advantages, these metal-based catalysts display several drawbacks: (i) they are challenging to separate from the crude product, thus leading to lower-quality final materials; (ii) they have poor selectivity during the depolymerisation process, which results in a mixture of oligomers that are difficult to re-process; and (iii) the use of metal-based catalysts entails a high environmental and economic cost – some widely used metals risk complete disappearance in the next 100 years (e.g. zinc or silver), while others will be seriously threated in the future if their consumption continues to increase (e.g. ruthenium, lithium, or copper).15
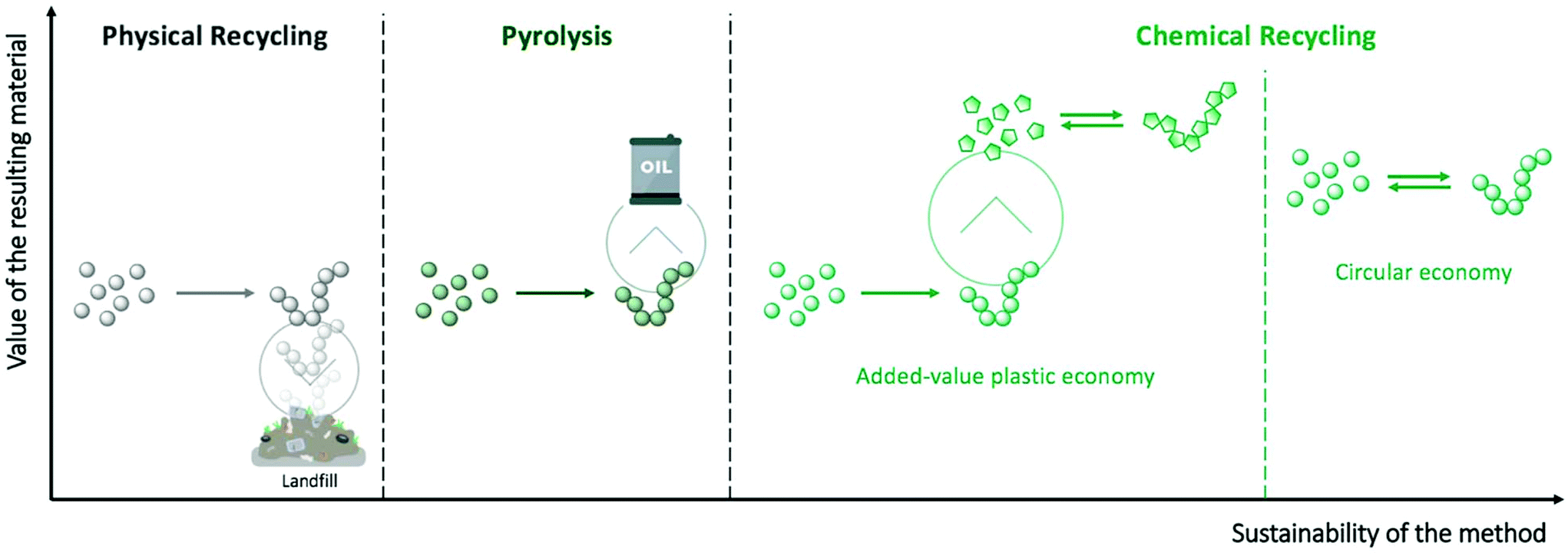 | ||
| Fig. 1 The different methodologies of polymer recycling. | ||
As an emerging alternative, organocatalysts have appeared as promising “green” substitutes for traditional organometallic complexes. While a wide range of organic catalysts are being applied in an increasing number of polymerisations,16–18 to date, the translation to depolymerisation processes is limited. When applied to polymer degradation, in particular to transesterification reactions, organocatalysts can promote mechanisms that lead to highly pure small molecules, which are in turn suitable for subsequent polymerisations. In many cases, hydrogen-bonding interactions involved in the depolymerisation mechanism promoted by organocatalysts play an important role in controlling the catalytic activity and selectivity of the depolymerisation reaction, as well as the architecture of the resulting polymer.19–21 Herein, the contribution of organocatalysts to the field of chemical recycling is reviewed, outlining the advances made by using organic base, organic acid, and ionic compound catalysts, as well as comparing their performance to that displayed by typically applied organometallic catalysts.
Depolymerisation of commercial polymers
Commodity polymers, which are extensively used across a wide range of sectors including packaging, building, automotive, or electronics, are seldom recycled. Hence, despite being a technological challenge, the design of suitable pathways for their depolymerisation is of the utmost necessity. An important aspect of such a challenge is found in the diversity of the plastic waste that requires treatment (Fig. 2). Because of the internal chemical differences among the various polymeric families, each type of polymer needs to be treated in a different way. Herein, the different organic catalysis methods that have been studied to date have been compiled to provide an overview of the progress for each class of polymers.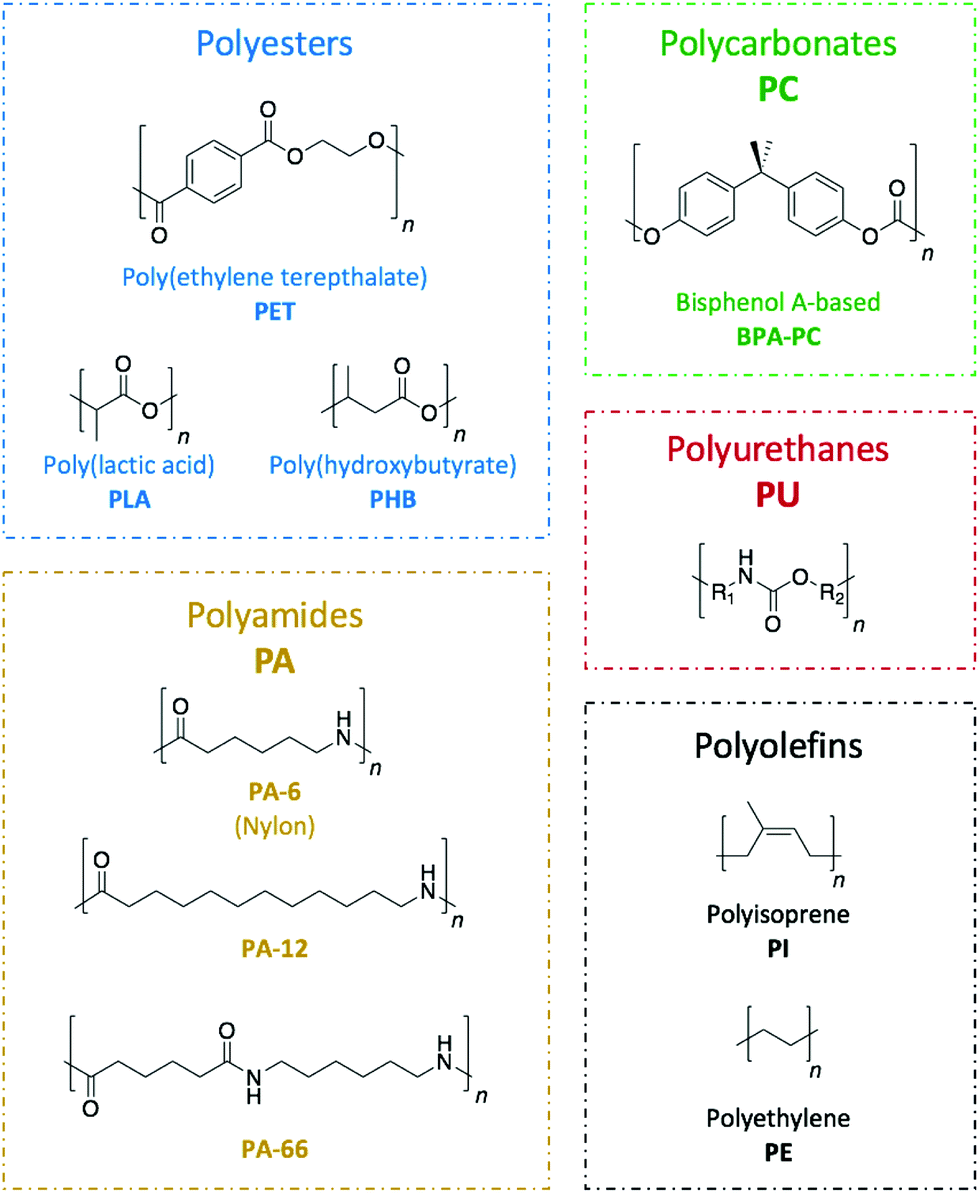 | ||
| Fig. 2 Commercial polymers examined in this review. | ||
Poly(ethylene terephthalate), PET
Poly(ethylene terephthalate) (PET) is the most commonly used thermoplastic from the polyester family (i.e. 13% of the world plastic production), and it is used in a large variety of applications, from clothing to food and liquid packaging. It is also the most recycled polymer in the world, with current industrial applications mainly in Europe and USA. According to the European Environment Agency (EEA), the rate of recycled PET bottles reached 57% in 2017 in Europe,22 an encouraging number only clouded by the high portion of mechanical recycling processes that lead to low-value materials, mainly fibres for carpet or clothes. As a result, PET is also one of the most studied polymers for depolymerisation, and organocatalysts have been often tested first on PET before applying them on other type of polymers.23–25 Hence, designing chemical recycling technologies for PET would impact the cyclic production of other polymers.In 2011, Hedrick and co-workers reported the glycolysis of PET using a strong guanidine base, 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD, Fig. 3).26 In a large excess of ethylene glycol (EG) (16 eq.) at 190 °C, pellets of waste PET beverage bottles degraded in 3.5 h. The major product (78% after crystallisation from water) of this reaction was bis(2-hydroxyethyl)terephthalate (BHET), a convenient monomer for the subsequent polymerisation into high quality PET (Scheme 1, I). Insoluble impurities were identified as short oligomers of PET and additives (i.e. isophtalic acid, diethylene glycol, and cyclohexane dimethanol). For coloured PET bottles, glycolysis took place at a slower rate with a lower BHET yield (64%). These results are comparable to those obtained when organometallic catalysts, such as acetate or alkoxide salts, are employed for this reaction.27,28 A complementary density functional theory (DFT) computational study demonstrated that while EG was modelled as co-activator of the depolymerisation, the system was energetically favoured, emphasising that both TBD and EG played an important role in the depolymerisation mechanism by activating the transesterification reaction via H-bonding.29
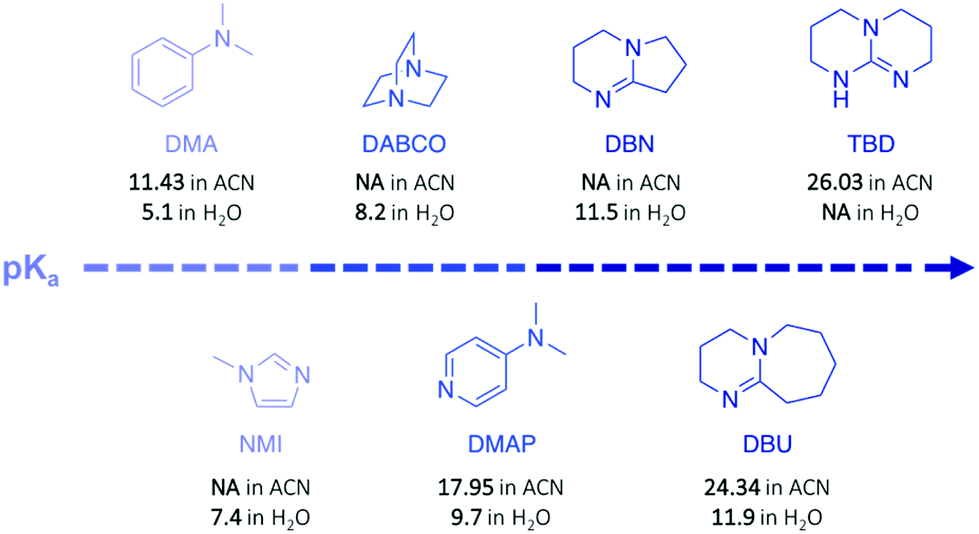 | ||
| Fig. 3 Organic bases described in this review. | ||
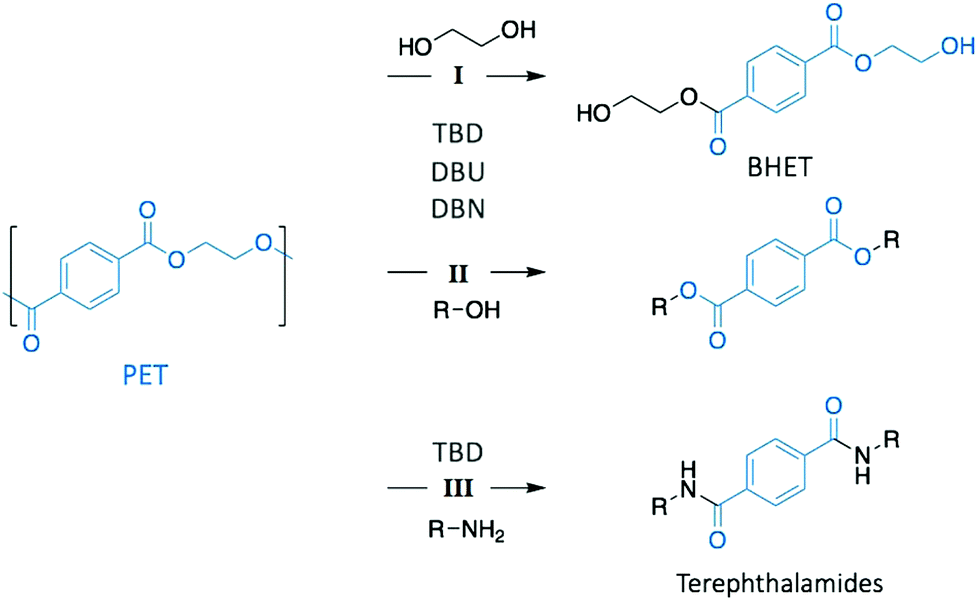 | ||
| Scheme 1 Depolymerisation of PET through glycolysis (I), alcoholysis (II), and aminolysis (III). | ||
In a subsequent work, the efficiency of a range of other nitrogen bases was investigated to establish the correlation between their basicity (pKa) and their catalytic activity (Fig. 3).30 Glycolysis appeared to be more efficient (i.e. more rapid with lower undesirable oligomers content) when strong bases, such as TBD, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), or 1,5-diazabicyclo[4.3.0]non-5-ene (DBN) were used compared to bases with a lower pKa, such as 1-methylimidazole (NMI) or dimethylaniline (DMA) (Fig. 3). Surprisingly, DBU showed higher efficiency than TBD despite its slightly lower pKa. Moreover, the authors observed differences in reactivity by conducting an experiment using different chain-length diols, ranging from EG to 1-octanol (Scheme 1, II). For alcohols with a 4-carbon or longer chain, TBD exhibited higher catalytic activity than DBU since the bifunctional acid/base character of TBD provides a simultaneous activation of the carbonyl group of the ester and the nucleophilic group of the reactant, thus leading to a faster reaction, as computational studies revealed. In contrast, for short-chain alcohols in large excess, the diol activates the carbonyl of the polymer, undermining the bifunctionalilty of TBD and increasing the reaction rate.
Extension of this approach to other nucleophiles has led to the study of the aminolysis of PET to create a range of crystalline terephtalamide compounds as high added-value materials (Scheme 1, III).31 Moreover, the thermal and mechanical properties of such building blocks depended on the amine that was used as reagent. In a typical depolymerisation, terephtalamides were synthesised in reasonable yields at temperatures from 110 to 190 °C for 1 to 18 h, using TBD as catalyst. Again, the bifunctionality of TBD was crucial to activate the carbonyl group via H-bonding, making it more efficient for aminolysis than other organic bases.
The use of salt-based organocatalysts for PET depolymerisation has also received some attention. Although slower than DBU alone for depolymerising PET, DBU-based salts (specifically the organic salts DBU![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :benzoic acid and DBU:phenol, in a 1:1 molar ratio) were more stable in air-rich atmospheres, thus facilitating the depolymerisation procedure.30 Similarly, we recently reported the glycolysis of PET using a thermally stable acid:base salt catalyst (Fig. 4a).32 An equimolar mixture of TBD and methane sulfonic acid (MSA) produced a very stable protic ionic salt that resists thermal degradation up to 400 °C, an uncommon property since most organic catalysts usually degrade at much lower temperatures, thus making them impractical for bulk depolymerisation processes (Fig. 4b). At 180 °C, TBD:MSA (1:1) efficiently depolymerised post-consumed PET bottle waste in less than 2 h in the presence of EG. Under optimised conditions, over 90% of monomeric BHET was recovered with high purity through simple crystallisation in water. Additionally, the TBD:MSA catalyst was able to mediate the polymerisation of BHET back to PET showing comparable properties to the industrially produced one. Finally, the catalyst was recycled up to 6 times without any activity loss. Hence, TBD:MSA is proposed as an interesting catalytic platform for the industrially viable depolymerisation/polymerisation cycle of PET.
:benzoic acid and DBU:phenol, in a 1:1 molar ratio) were more stable in air-rich atmospheres, thus facilitating the depolymerisation procedure.30 Similarly, we recently reported the glycolysis of PET using a thermally stable acid:base salt catalyst (Fig. 4a).32 An equimolar mixture of TBD and methane sulfonic acid (MSA) produced a very stable protic ionic salt that resists thermal degradation up to 400 °C, an uncommon property since most organic catalysts usually degrade at much lower temperatures, thus making them impractical for bulk depolymerisation processes (Fig. 4b). At 180 °C, TBD:MSA (1:1) efficiently depolymerised post-consumed PET bottle waste in less than 2 h in the presence of EG. Under optimised conditions, over 90% of monomeric BHET was recovered with high purity through simple crystallisation in water. Additionally, the TBD:MSA catalyst was able to mediate the polymerisation of BHET back to PET showing comparable properties to the industrially produced one. Finally, the catalyst was recycled up to 6 times without any activity loss. Hence, TBD:MSA is proposed as an interesting catalytic platform for the industrially viable depolymerisation/polymerisation cycle of PET.
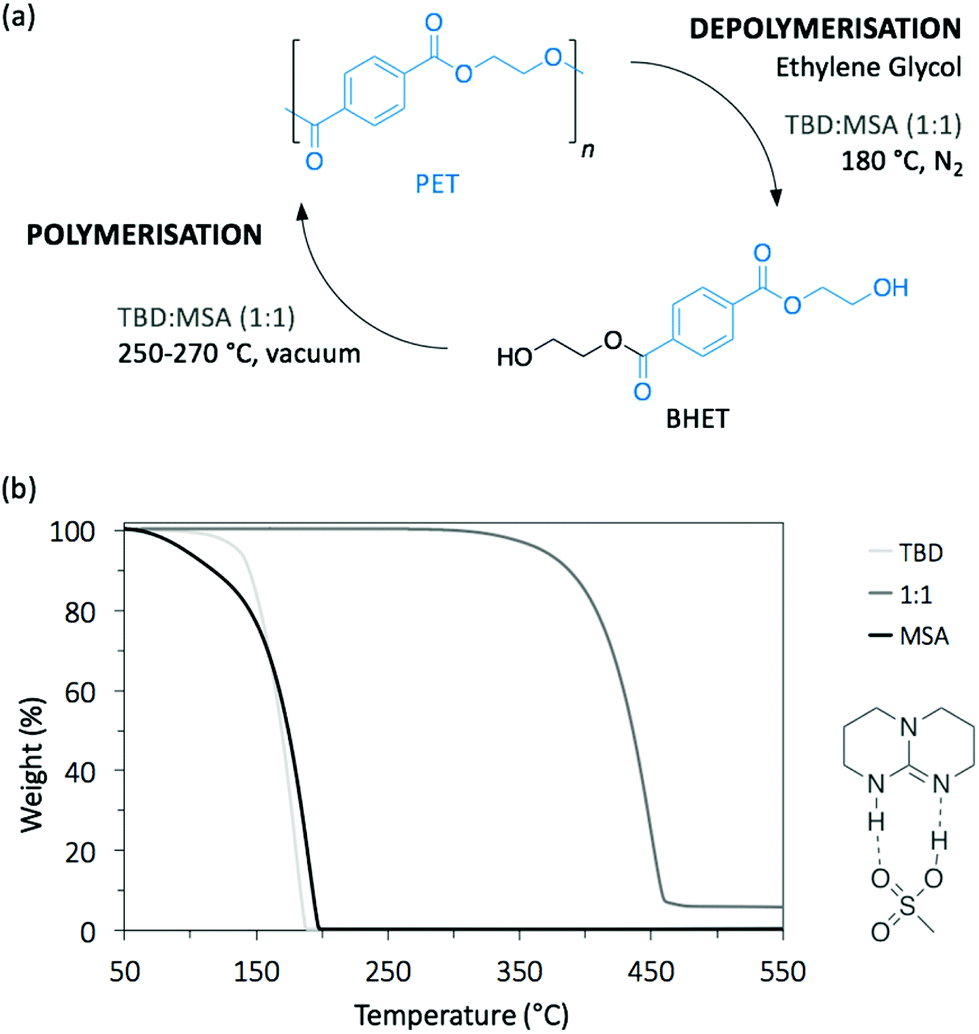 | ||
| Fig. 4 (a) The depolymerisation of PET and re-polymerisation of monomeric products (BHET) using TBD:MSA (1:1) catalyst and (b) the thermogravimetric analysis (TGA) comparison between TBD:MSA (1:1) and TBD and MSA alone. | ||
Ionic liquids (ILs) are classically defined as mixtures completely composed of ions with a melting point below 100 °C. As such, they display many similarities with the acid:base salts outlined above. The use of ILs for the depolymerisation of PET was firstly reported in 2009 by Wang et al. and involved imidazolium-based ILs. This family has been increasingly studied for the last couple of decades, especially as green solvents or catalysts for organic synthesis or separation techniques.33,34 In this work, 1-butyl-3-methylimidazolium ([Bmim]) was investigated with different anions (Cl, Br, BF4, PF6) as solvent for the glycolysis of PET.35 Even though some of the ILs ([Bmin][Cl] and [Bmin][Br]) solubilised the polymeric material, depolymerisation required additional catalysts, such as zinc acetate, tetrabutyltitanate, or solid superacid, to be completed. The authors indicated the depolymerisation product to be an oligomeric mixture without identifying its nature or yield. Nevertheless, the possibility of recycling the IL, as well as its easy separation from the product, made the study an encouraging starting point for the use of ILs for depolymerisation.
More recently, Al-Sabagh et al. showed that a basic imidazolium-based IL (i.e. 1-butyl-3-methylimidazolium acetate [Bmim][Ac]) fully degraded PET without requiring any additional catalyst.36 At 190 °C, PET depolymerised through glycolysis in 3 h giving a BHET monomeric yield of 58%, as opposed to neutral 1-butyl-3-methylimidazolium chloride ([Bmim][Cl]) or 1-butyl-3-methylimidazolium bromide ([Bmim][Br]), which did not display any catalytic activity (Scheme 2a). These results suggest that basic imidazolium ILs can promote the desired depolymerisation, while neutral ones can only be used as solvent and require the adding of a catalyst. In another approach to improve the depolymerisation of PET with neutral [Bmim][BF4], Nunes et al. used supercritical ethanol (255 °C).37 Under the best conditions, diethyl terephtalate, the monomer resulting from the ethanolysis of PET, was obtained at 98% yield in 45 min.
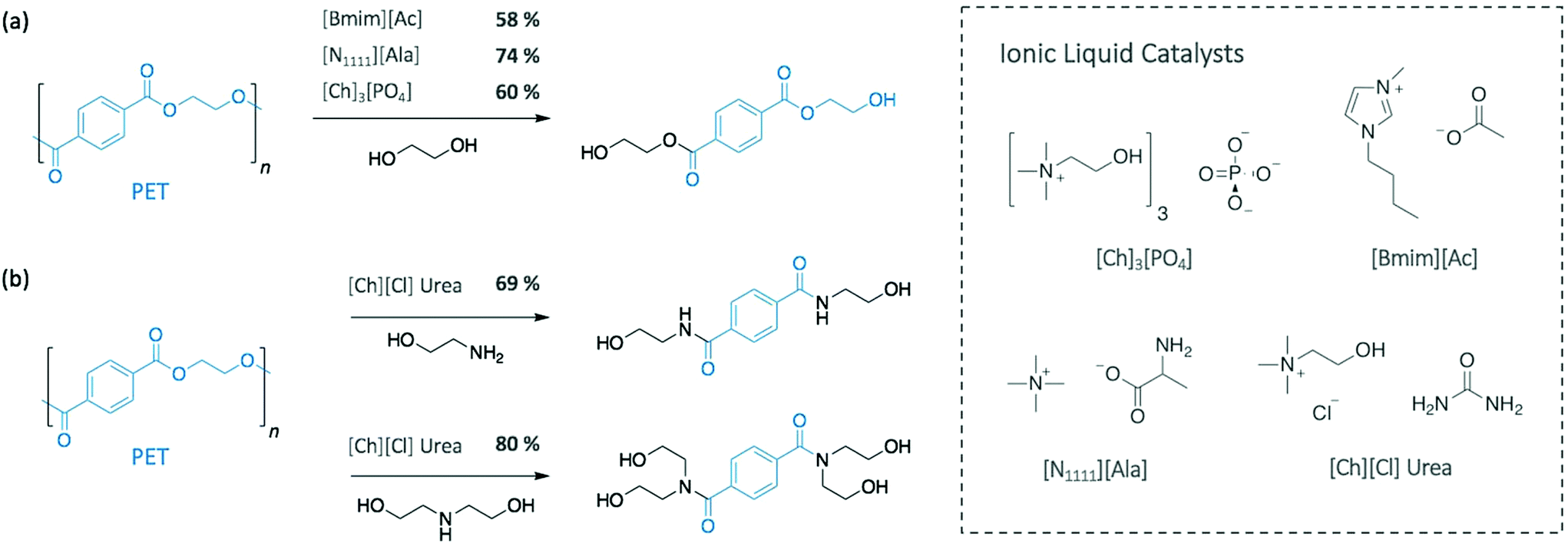 | ||
| Scheme 2 (a) Glycolysis and (b) aminolysis of PET using different ionic liquids as catalyst. | ||
Similarly, urea-based ILs have been applied as efficient catalysts for both glycolysis and aminolysis of PET. Using EG as nucleophile and solvent at 170 °C, 73.9% of BHET was collected after 190 min with urea alone catalysing the reaction.38 Residual EG and urea were recycled up to 10 times with no loss of catalytic activity, while in situ IR spectroscopy and DFT calculations emphasised the predominant role of the H-bonding between urea and EG in the efficiency of the system. With this observation, tetraalkylammonium-based amino acid-functionalised ILs were then used as catalyst to further enhance the efficiency of the depolymerisation, as they are known to form H-bond with alcohol groups. Compared to urea, the reaction catalysed by tetramethylammonium alaninate [N1111][Ala] gave a similar monomer yield (74.3%) but in a reduced time, 50 min at 170 °C (Scheme 2a).
Urea and urea-based ILs have also been applied as highly efficient catalysts for the depolymerisation of PET using amines.39 In particular, Musale and Shukla showed that choline chloride ([Ch][Cl]) urea salt completely degraded PET in less than 30 min, under reflux. In this study, the urea-based catalyst was more efficient than urea itself. When using [Ch][Cl] urea, ethanolamine (EA) and diethanolamine (DEA) provided the corresponding amides with 69% and 80% monomeric yields, respectively, whereas yields of 55% and 66% were obtained for urea alone (Scheme 2b).
In the perspective of using completely sustainable strategies, Sun et al. very recently employed low-cost, biocompatible, and recyclable ILs (cholinium phosphate ([Ch]3[PO4]) to solubilise and depolymerise PET into BHET (60% yield) at a relatively low temperature (120 °C) (Scheme 2a).40 In this case, NMR and IR spectroscopic analysis suggested a bifunctional activation of the system, where the cation activates the carbonyl group of PET, while the anion simultaneously activates one hydroxyl group of EG, which resembles the TBD:MSA (1:1) mechanism previously presented.
BPA – polycarbonate
Polycarbonates (PC) are thermoplastic polymers that contain more hydrolytically stable carbonate linkages (compared to esters). The main advantage of this family of polymers is that their adequate balance of features, such as thermal resistance, excellent mechanical properties, and optical transparency, makes them suitable for both commodity and engineering plastics. Bisphenol A-based polycarbonate (BPA-PC) is the most widely used polycarbonate with a world production exceeding 5 million tons in 2016.41 Moreover, BPA-PC is a potential reservoir of Bisphenol A (BPA), which has been recently suspected to be a xenoestrogen.42–44 Thus, BPA-PC depolymerisation entails an additional issue since the release of BPA through uncontrolled polymer hydrolysis involves serious disruptions for both humans and the environment.Similarly to PET, research on the depolymerisation of BPA-PC has been recently carried out using organic base catalysts. For instance, BPA-PC was degraded into BPA and the corresponding organic carbonate in good yield within 30 min at 100 °C in an excess of ethanol or methanol using DBU (10 mol%) (Scheme 3a).45 The ability of DBU to catalyse the depolymerisation reaction for several cycles by subsequently adding BPA-PC in situ was proven, albeit the reaction time increased with the successive loads of fresh polymer, from 30 min to 4 h between the 1st and the 5th cycle. Such response is ascribed to the formation of a DBU-BPA adduct in the crude reaction product which is less active than DBU itself, and thus induces a sequential decline of the catalytic activity. Further investigation showed that weaker bases, such as 1,4-diazabicyclo[2.2.2]octane (DABCO, Fig. 3) and 4-dimethylaminopyridine (DMAP, Fig. 3), are less active than DBU requiring from 4- to 6-fold longer reaction times to complete depolymerisation. Indeed, as previously mentioned, the catalytic performance of organic bases for transesterification reactions seems to improve with their basicity.
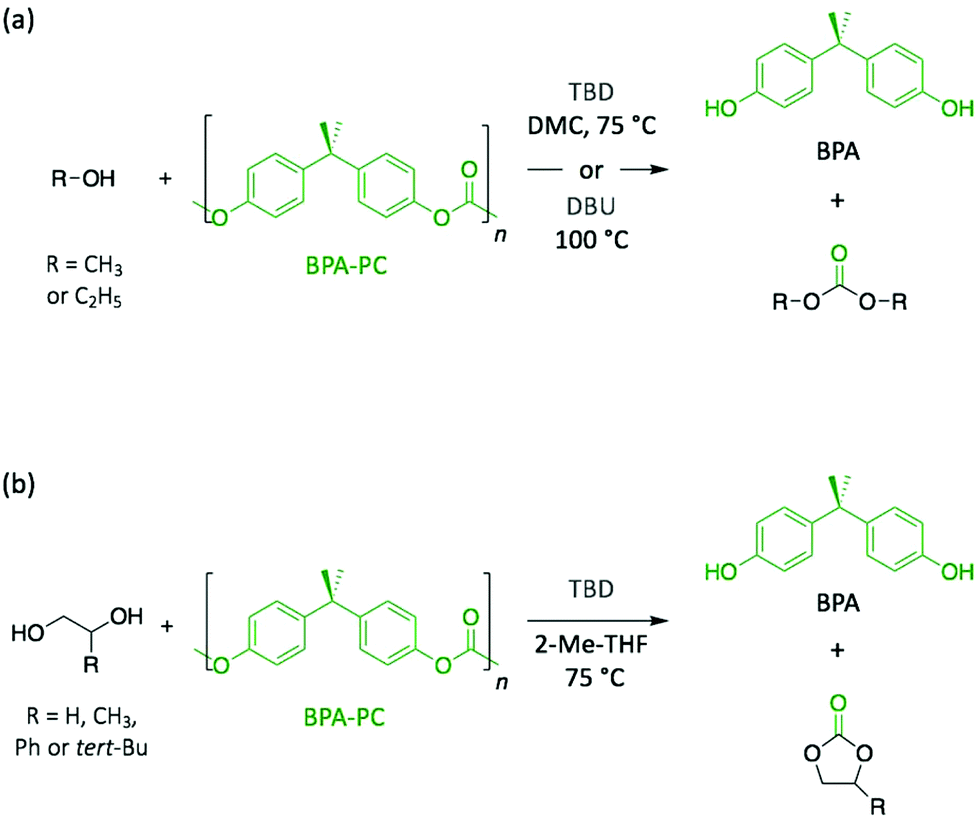 | ||
| Scheme 3 Depolymerisation of BPA-PC using (a) alcohols and (b) diols as reagents. | ||
In another very recent publication, the use of TBD as catalyst was reported for the methanolysis of BPA-PC.46 The best yields (>96%) for both depolymerisation products (i.e. BPA and dimethyl carbonate (DMC)) were obtained using DMC as solvent at 75 °C with 2 mol% of TBD. The use as solvent of DMC, which is one of the depolymerisation products, leads to an easier separation, thus simplifying the purification process. Additionally, the depolymerisation of BPA-PC to obtain cyclic carbonates was investigated in 2-methyltetrahydrofuran (2-Me-THF) using small diols (Scheme 3b). The resulting 5-membered cyclic carbonates, which were identified in the crude product using GC analysis, were obtained with good yields (89–97% for carbonates, and 93–99% for BPA).
In 2010, Liu et al. investigated imidazolium-based ILs as catalysts for the depolymerisation of BPA-PC through methanolysis.47 These ILs, which were already described for the depolymerisation of PET,35–37 were synthesised displaying N-alkyl imidazolium moieties with different chain nature and inorganic anions (i.e. Cl, Br, BF4, PF6, or acetate). Most of the ILs did not display any catalytic activity, but the reaction employing 1-butyl-3-methylimidazolium chloride ([Bmim][Cl]) completely depolymerised BPA-PC in 2.5 h at 105 °C (Table 1). Moreover, BPA and dimethyl carbonate (DMC) were isolated in high yields (up to 96%), and the IL was reused up to 8 times with no significant decrease in the catalytic activity being observed.
Following this promising result, [Bmim][Cl] and [Bmim][Ac] were further investigated for the methanolysis and hydrolysis of BPA-PC.48,49 For both catalysts, the polymer fully depolymerised in 3 h with very good yields (>95%) (Table 1). Notably, higher temperature and catalyst loading were required to complete the depolymerisation by hydrolysis (i.e. 140 °C and 165 °C) in comparison to methanolysis (i.e. 90 °C and 105 °C). For [Bmim][Ac], the depolymerisation reaction through hydrolysis followed first-order kinetics with an activation energy of 228 kJ mol−1, which is higher than for methanolysis (168 kJ mol−1), thus validating that more forcing conditions were required to complete the depolymerisation when water was employed. Furthermore, the authors suggested that the enhanced performance of [Bmim][Ac] for catalysing the depolymerisation of BPA-PC under milder conditions is partially attributed to the better solubility of the polymer in this IL. Indeed, at 105 °C, 5 g of [Bmim][Ac] solubilised 1 g of BPA-PC, whereas, in the same quantity of [Bmim][Cl], the polymer barely swelled.
Polyamides
Polyamides are polymers that can be either found in nature (e.g. proteins, silk, or wool) or prepared synthetically (e.g. nylons, aramids, or sodium polyaspartate). They are characterised by amide bonds linking their repeating units. Most typically with respect to plastic waste, polyamides are found in the form of nylons which have several applications from biomedicine to clothing. As a consequence of their excellent mechanical strength, artificially made polyamides are widely used in textile, automotive, and transportation industries.Typical aqueous acidic solutions (e.g. formic acid, hydrochloric acid, or sulfuric acid) were firstly applied as acid catalysts for the chemical degradation of discarded polyamide-6 (PA-6) waste fibres, which were completely dissolved in the concentrated solutions.50 After different times up to 20 h, the degradation products, which displayed different molecular weight, were recovered through fractional precipitation. Interestingly, for hydrochloric acid and sulfuric acid, aminocaproic acid was the major component of the crude product and it was isolated in high purity, which eventually allowed for subsequent polymerisation. More recently, Kamimura and co-workers reported the depolymerisation of polyamide-66 (PA-66) into high-added value chemicals combining supercritical methanol as solvent and organic acids as catalysts (Scheme 4).51 Specifically, depolymerisation with 8 eq. of glycolic acid in methanol yielded 75% of dimethyl adipate and 50% of 1,6-hexanediol after 6 h of reaction at 270 °C. Other organic acids, such as lactic or benzoic acid, also catalysed the depolymerisation of PA-66 with similar yields, while esters or weaker acids provided comparable yield values for dimethyl adipate, but lower for 1,6-hexanediol. In this case, the authors proposed that the acid catalyst favoured the scission of the amide bond to yield dimethyl adipate and hexamethylenediamine before supercritical methanol promoted the subsequent degradation of the diamine into various compounds, including 1,6-hexanediol. Hence, the resulting diol, which is a monomer of interest in the polymerisation of polyesters and polyurethanes, can be obtained from commercial PA-66, thus turning this depolymerisation process into an economically viable technology.
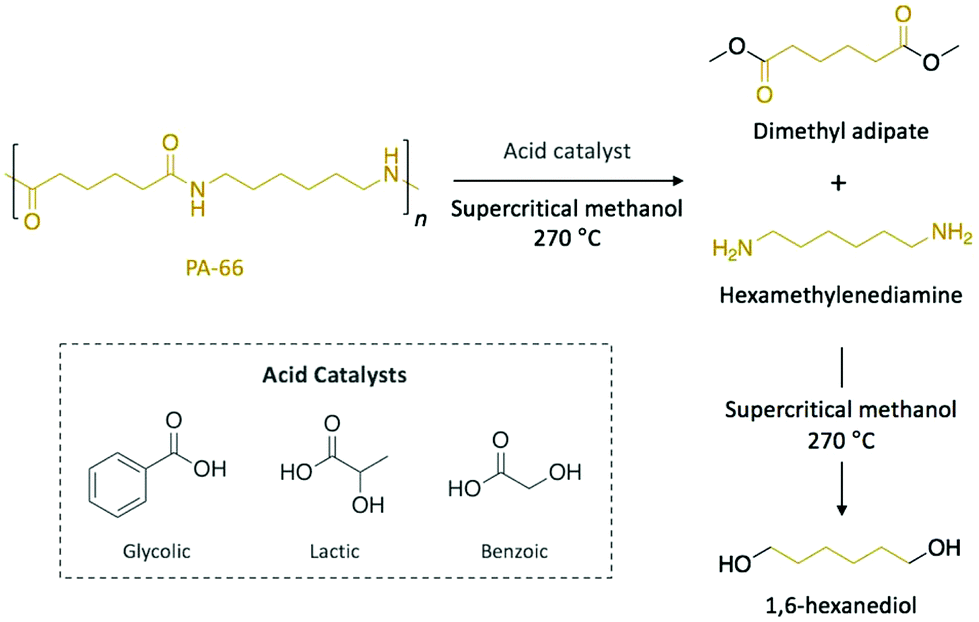 | ||
| Scheme 4 Depolymerisation of PA-66 catalysed by organic acids in supercritical methanol. | ||
For this family of polymers, ILs have also been applied as efficient catalytic media for the depolymerisation of PA-6 (Scheme 5). Quaternary ammonium salts, such as N-methyl-N-propylpiperidinium (PP13) and N,N,N-trimethyl-N-propylammonium (TMPA), together with bis(trifluoromethane sulphonyl)imide (TFSI) as counter anion, successfully depolymerised PA-6 into ε-caprolactam (43–55% in yield) in 5–6 h at 300 °C without requiring any additional catalyst.52 However, adding 5 wt% of DMAP improved the depolymerisation efficiency up to 86% monomeric yield. Further optimisation of the reaction conditions demonstrated the important role played by temperature, with 7%, 55%, and 6% of the monomer being isolated at 270 °C, 330 °C, and 360 °C, respectively. For temperatures lower than 300 °C, a large portion of oligomeric polyamides were found in the crude product, which explains the poor recovery value of ε-caprolactam, whereas ILs decomposed at temperatures higher than 300 °C, as evidenced by the formation of N-methyl- and N-propyl lactams by-products, which prevented depolymerisation from reaching good yield values. Later, this methodology was extended to polyamide-12 (PA-12), for which the corresponding laurolactam was isolated; however, the yield did not exceed 17%, most likely as a consequence of the closing of the 12-member ring being energetically disfavoured.53 Furthermore, the ILs were recycled up to 5 times without losing their depolymerisation efficiency. Finally, the same research group depolymerised PA-6 in these quaternary ammonium-based ILs using DMAP salts prepared with iodine and imidazolium as counter anions.54 This system catalysed the reaction several times, reaching up to 79% of monomer recovery; however, since their performances in terms of catalytic activity and selectivity were poorer than the ones displayed in previous experiments using DMAP alone, this catalyst was not further investigated.
 | ||
| Scheme 5 Depolymerisation of PA-6 into ε-caprolactam using ILs as catalyst. | ||
Polyurethanes
Polyurethanes (PUs) are generally prepared by the condensation reaction between isocyanates and polyols. Owing to their distinctive feature to be processed as flexible foam, rigid, or elastomeric materials, PUs have found applications as diverse as insulation panels, wheels and tires, adhesives, surface coatings, sealants, synthetic fibres, or hard-plastic electronic components for the consumer goods, automotive, and construction industries. This variety of applications requires clear distinctive properties, which originate from the distinctive polymer structures obtained from the multiple combinations of polyols and isocyanates. As PU is the 6th most produced polymer in the world, with an annual production above 16 million tons in 2016, the sorting and the treatment of this polymeric material is of very special interest.41In 1993, Xue et al. successfully depolymerised discarded PU rigid foams through glycolysis using diethylene glycol (DEG) as reagent and EA as catalyst. The resulting product, glycolysate, was used as additive for the further synthesis of epoxy resins.55 However, all recent studies have reported the use of alkaline salts or metal complexes as catalysts to complete the depolymerisation,56,57 thus suggesting that low molecular-weight glycols and alkanolamines may be unable to depolymerise PU rigid foams by themselves. The depolymerisation of PU flexible foams using alkanolamines was also reported, a year later, by Kanaya and Takahashi.58 Contrary to the previous example, a diphenyl isocyanate-based PU was depolymerised into a pure polyether polyol using EA, without adding any further catalyst, and the authors suggested alcoholysis rather than aminolysis as the reaction mechanism involved (Scheme 6a). Similarly, in 2000, Borda et al. studied the glycolysis of PU flexible foams and elastomers using DEA, and EG, 1,2-propylene glycol, triethylene glycol, or low molecular weight poly(ethylene glycol) as reagents.59 Specifically, using an equimolar quantity of EG and DEA, PU-based waste depolymerised in less than 2 h at 180 °C, and the resulting polyol phase was directly employed to synthesise an industrial adhesive mixture (Scheme 6b). In this study, although alcoholysis was assumed to be the favoured pathway, the presence of CO2 in the gases released during the reaction suggested decarboxylation as a competing process.
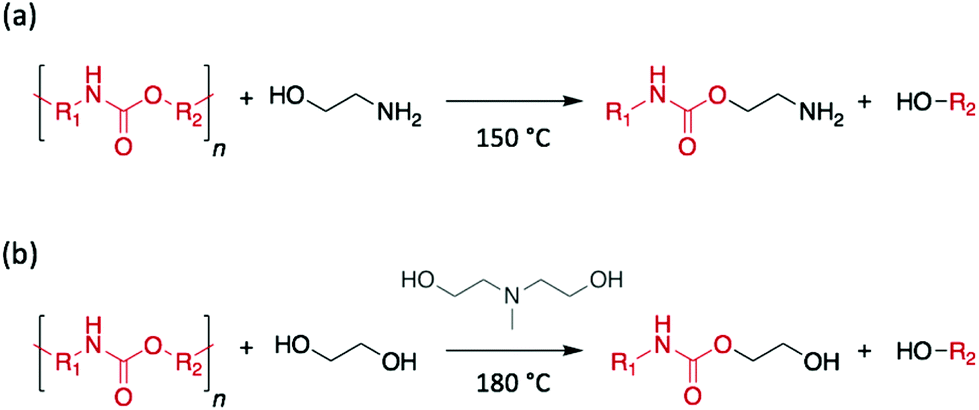 | ||
| Scheme 6 Depolymerisation of PU (a) using EA as reagent and catalyst or (b) using EG as reagent and DEA as catalyst. | ||
More recently, the efficiency of DEA and EA to degrade PU flexible foams was compared with that displayed by metal catalysts, such as acetate metallic salts, tin oxides, or tin laurates.60 At 200 °C, even though alkanolamines significantly enhanced the depolymerisation rate compared to the control reaction performed without adding catalyst, the highest efficiency was still obtained for a metallic catalyst, namely zinc acetate. Notably, DEA showed the same performance as barium acetate, while EA performed similarly to potassium salt. In contrast, other metallic catalysts (i.e. dibutyl tin dilaurate, butyl tin oxide, and hydroxy butyl tin oxide) performed extremely poorly.
In another example, the glycolysis of a thermoplastic PU elastomer (TPU) based on 4,4′-diphenylmethane diisocyanate and polyether polyol was also conducted using low-weight glycols (i.e. DEG or EG), and EA and lithium acetate as catalyst and co-catalyst, respectively.61 When treated with a DEG:EG:EA (9:9:2) mixture at a temperature range from 160 °C to 190 °C, the elastomeric TPU depolymerised into two distinct layers in 3 h. In the resulting crude product, the upper liquid phase contained a polyether polyol that corresponded to the starting industrial polyol used to polymerise TPU.
Polyolefins: polyisoprene and polyethylene
Polyolefins, which are produced from polymerising alkenes, can be prepared as liquid or rigid solids depending on their molecular weight and degree of crystallinity. For instance, polyethylene (PE), which is one of the most commonly used plastic with an annual production of around 80 million tons, is mainly used as a packaging item manufactured as bags, films, or bottles. Degrading polyolefins represents one of the major challenges in polymer recycling for two main reasons. Chemically, their C–C backbone is extremely stable and as such harsh conditions are usually required to afford and control the depolymerisation reaction. Furthermore, economically the production of polyolefins is a cheap and well-controlled process. Hence, chemical depolymerisations are far from competitive with their preparation using low-cost petroleum-based monomers. However, some attempts in depolymerising polyolefins have been undertaken, giving different kind of products, from a mixture of light alkanes, to novel materials, and among them, organocatalysts have been an even more rare methodology.62–66Firstly, polyisoprene-based materials, which are more accessible owing to the reactivity of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond that remains in the polymer structure, have been depolymerised with m-chlorobenzoic acid,67 phenylhrazine,68 or potassium persulfate.69 In all cases, the molecular weight of the polymer decreased drastically, producing telechelic oligomers as degradation products. However, among the acid catalysts explored for the depolymerisation of both natural and synthetic rubber (i.e. poly(cis-isoprene)), periodic acid (i.e. H5IO6 or HIO4) has been the most widely studied. For instance, in 2005, Phinyocheep et al. reported the oxidative depolymerisation of epoxidised natural rubber (ENR) catalysed by periodic acid.70 Indeed, periodic acid, which in an excess of water is a weak acid, releases periodate anions, which are able to cleave 1,2-difunctional compounds. Specifically, the molecular weight of ENR was reduced from 15 kg mol−1 to 0.4 kg mol−1 after 8 h of reaction at low temperature (30 °C). Moreover, the same process applied to natural rubber (NR) also resulted in the depolymerisation of the polymer, the molecular weight decreasing from 20 kg mol−1 to 1 kg mol−1 in 30 h. In the case of NR, the authors formulated that periodic acid catalyses the epoxidation of NR into ENR in the first step of the reaction, which explains the longer reaction time (30 h instead of 8 h) (Scheme 7). Subsequently, Sadaka et al. adapted this method to directly depolymerise ground waste tyres through an oxidative mechanism,71 which first involved the epoxidation of the double bond of polyisoprene before cleaving the in situ formed oxirane (Scheme 7). As a result, a range of low molecular-weight telechelic polymers with aldehyde end-groups were obtained by controlling the quantity of periodic acid employed. Interestingly, these oligomers were then used as precursors for the synthesis of other relevant materials for the rubber industry.
C double bond that remains in the polymer structure, have been depolymerised with m-chlorobenzoic acid,67 phenylhrazine,68 or potassium persulfate.69 In all cases, the molecular weight of the polymer decreased drastically, producing telechelic oligomers as degradation products. However, among the acid catalysts explored for the depolymerisation of both natural and synthetic rubber (i.e. poly(cis-isoprene)), periodic acid (i.e. H5IO6 or HIO4) has been the most widely studied. For instance, in 2005, Phinyocheep et al. reported the oxidative depolymerisation of epoxidised natural rubber (ENR) catalysed by periodic acid.70 Indeed, periodic acid, which in an excess of water is a weak acid, releases periodate anions, which are able to cleave 1,2-difunctional compounds. Specifically, the molecular weight of ENR was reduced from 15 kg mol−1 to 0.4 kg mol−1 after 8 h of reaction at low temperature (30 °C). Moreover, the same process applied to natural rubber (NR) also resulted in the depolymerisation of the polymer, the molecular weight decreasing from 20 kg mol−1 to 1 kg mol−1 in 30 h. In the case of NR, the authors formulated that periodic acid catalyses the epoxidation of NR into ENR in the first step of the reaction, which explains the longer reaction time (30 h instead of 8 h) (Scheme 7). Subsequently, Sadaka et al. adapted this method to directly depolymerise ground waste tyres through an oxidative mechanism,71 which first involved the epoxidation of the double bond of polyisoprene before cleaving the in situ formed oxirane (Scheme 7). As a result, a range of low molecular-weight telechelic polymers with aldehyde end-groups were obtained by controlling the quantity of periodic acid employed. Interestingly, these oligomers were then used as precursors for the synthesis of other relevant materials for the rubber industry.
 | ||
| Scheme 7 Depolymerisation of polyisoprene using periodic acid. | ||
While PE is one of the simplest and cheapest polymers to synthesise, its depolymerisation requires breaking the very energetically stable C–C single bond, which makes the depolymerisation of PE technologically demanding. Despite this fact, Bäckström et al. undertook an original initiative to recycle low density PE (LDPE) into value-added functional chemicals via a microwave-assisted fast and effective oxidative process.72 Through this approach, LDPE powder was totally degraded after just 1 h of microwave irradiation at 180 °C in relatively dilute nitric acid solution, which led to well-defined water-soluble products, such as succinic, glutaric, and adipic acids, as well as longer dicarboxylic acids, acetic acid, and propionic acid. Noteably, the length of the dicarboxylic acids could be adjusted by varying the reaction conditions (i.e. time, temperature, and the amount of oxidizing agent). Finally, the authors validated their strategy by recycling LDPE freezer bags, which were depolymerised into dicarboxylic acids with a good yield (71%).
Finally, ILs have also been used as solvent and catalyst to depolymerise polyolefins. In 2000, Adams et al. described the use of chloroaluminate(III) ILs for cracking both high and low density PE (HDPE and LDPE, respectively).73 At temperatures below 200 °C, the reaction led to low-volatility alkanes. However, although this catalyst is metal-based, the novelty of this example opened the way to other initiatives for polymer recycling. Indeed, more recently, waste tyres were depolymerised through metathesis in hydrophobic ILs (i.e. trihexyl-(tetradecyl)phosphonium chloride (Cyphos101) and N,N-dioctylimidazolium bromide (C8 C8 ImBr)), which produced low molecular weight telechelic polymers of controlled lengths.74 These resulting oligomers, as pointed out above, are notably interesting intermediates for the synthesis of innovative polymers by methathesis.
Biodegradable polyesters
The polyester family gathers polymers containing an ester functional group in their main chain, PET being the most commonly used. However, other polymers, such as polylactide (PLA) and poly(hydroxy butyrate) (PHB) are relatively new commodity polymers and, in the case of PHB, still to make a significant impact. Both polymers attract high interest as a consequence of their production from renewable sources and their ability to decompose in relatively mild conditions. Thus, they have been extensively investigated as decomposable materials, mainly for packaging applications. While PLA has begun to be successfully translated into these areas (with an expected PLA production growth by 300% between 2015 and 2020 in Europe, for example ref. 22), they have been also applied as polymeric scaffolds in the biomedical and biotechnological fields on account of their ready compatibility with biological systems.For both polymers, organic bases have gained a lot of attention as catalysts for their depolymerisation. Indeed, Leibfarth et al. reported the efficiency of TBD to degrade PLA into valuable building blocks at room temperature (Scheme 8).87 Similarly to what was observed for the depolymerisation of PET, degrading PLA with a range of alcohols led to a mixture of products of lactate esters and their dimers. Specifically, more than 95% of ethyl lactate was recovered with ethanol as reagent and using 2.5 mol% TBD.
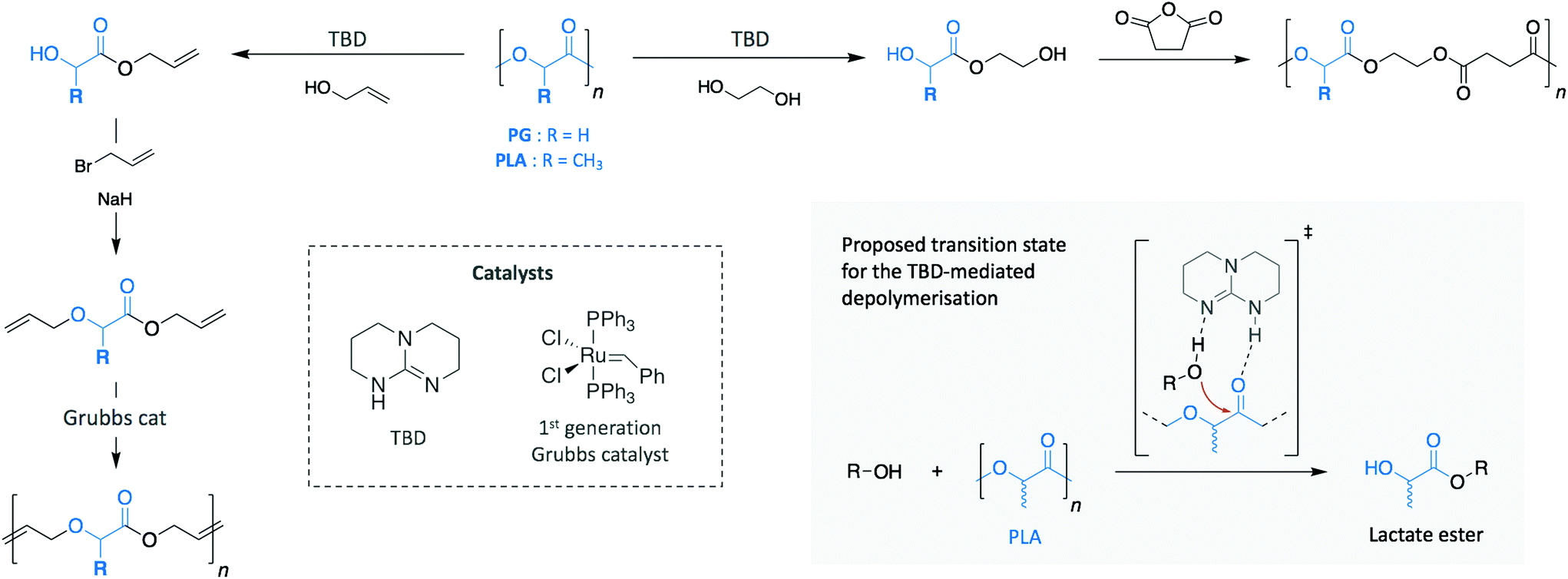 | ||
| Scheme 8 Depolymerisation and subsequent polymerisation of PG and PLA for the synthesis of new materials. | ||
Additionally, when poly(L-lactide) was depolymerised with benzyl alcohol, the product retained the stereochemistry of the polymer (i.e. >95% of the product was S-benzyl lactate). Moreover, the possibility to introduce various polymerisable groups to the ester products allows new polymers to be produced by step growth methods. For instance, the monomers resulting from the depolymerisation of PLA and polyglycolide (PG) with EG and allyl alcohol were polymerised into new materials. Specifically, the diols produced using EG were condensed with succinic anhydride, while the dialkenes obtained via allyl alcohol were alkylated with allyl bromide before polymerising them through acyclic diene metathesis. These two different examples highlight the wide spectrum of possibilities that recycled monomers present to create new materials.
In another example, Coulembier et al. noticed the chemical degradation of poly(L-lactide) catalysed by organic bases. Specifically, the presence in the polymer of residual DBU from the polymerisation favoured its thermal degradation. Indeed, the salt formed by the organocatalyst, DBU, and benzoic acid, which was used to quench the activity of the organic base, acted as a catalyst for the depolymerisation of poly(L-lactide) under high temperatures (i.e. melt process).75
As described in previous sections, basic ILs have been widely investigated for the alcoholysis of oxygen-containing polymers, such as PET or BPA-PC. Similarly, the efficiency of different imidazolium ILs (i.e. [Bmim][Cl], [Bmim][PF6], [Bmim][Ac] and [Bmim][HSO4]) to catalyse the methanolysis of PLA into methyl lactate was compared (Scheme 9a).76 Likewise PET, neutral ILs ([Bmim][Cl] and [Bmim][PF6]) were inactive, whereas basic [Bmim][Ac] and acidic [Bmim][HSO4] completely degraded PLA in 3 h at 115 °C, which is not a surprising result since the efficacy of acid and base catalysts for PLA depolymerisation by transesterification is well known. [Bmim][Ac] appeared to be slightly more active than its acidic homologue, reaching 96% of PLA conversion and producing methyl lactate in 91% yield.
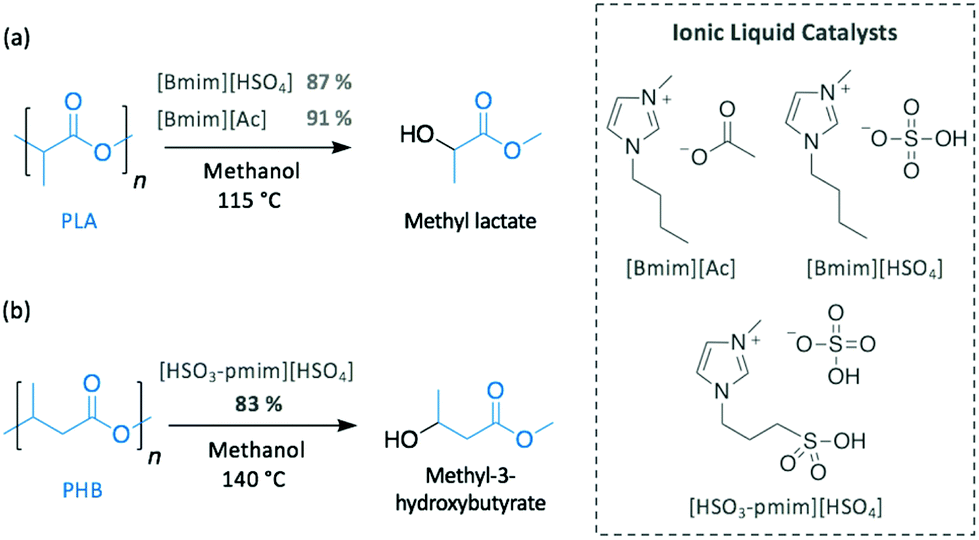 | ||
| Scheme 9 PLA and PHB depolymerisation using ILs. | ||
In contrast, although acidic ILs have received much less attention, one notable work explored the efficient methanolysis of PHB into methyl 3-hydroxybutyrate (83% yield) in 3 h at 140 °C using 1-methyl-3-(3-sulfopropyl)imidazolium hydrogen sulfate [HSO3-pmim][HSO4] as catalyst (Scheme 9b).77 Interestingly, the ionic liquid containing the same cation but different anion (i.e. dihydrogen phosphate (H2PO4−)) was inactive, most likely as a consequence of the less acidic nature of H2PO4− (pKa = 7.21)78 compared to HSO4− (pKa = 1.99).78
With a clear vision towards sustainable production cycles, as soon as in 1996, Melchiors et al. already depolymerised PHB into the corresponding cyclic trimer ((R,R,R)-4,8,12-trimethyl-1,5,9-trioxacyclododeca-2,6,10-trione, TBL), at 80% yield, in the melt and in solution using p-toluenesulfonic acid (PTSA) as catalyst (Scheme 10).79 Although bomb calorimetry attested the non-feasibility of the reversible reaction, authors were practically able to polymerise TBL to obtain high quality PHB using dibutyltin dimethoxide. This difference between the thermodynamic and the experimental results was attributed to polymer crystallinity and polymer–monomer interactions.
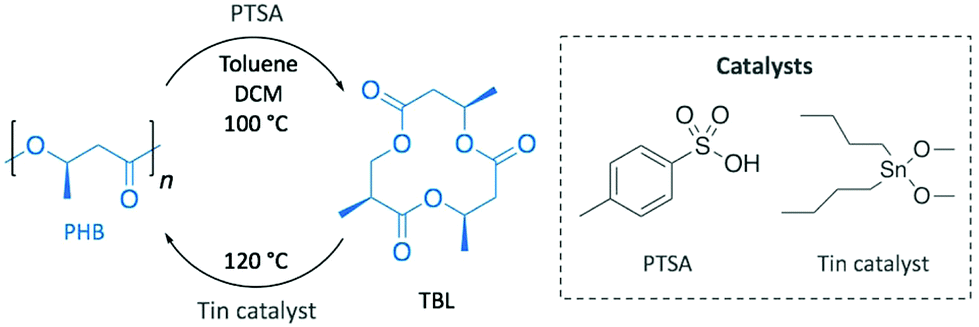 | ||
| Scheme 10 Depolymerisation of PHB into TBL. | ||
Emerging opportunities
Although the depolymerisation of commodity polymers is an urgent matter for managing the current accumulation of plastic waste, the next challenge involves the design of innovative polymers together with their recycling pathways. In this perspective, recent publications have demonstrated the possibility of synthesising polymerisation/depolymerisation circular routes for novel polymers.In 2016, Hong and Chen polymerised γ-butyrolactone (γ-BL) into poly(γ-butyrolactone) (PγBL) with a fairly high molecular weight (Mn of 30 kg mol−1) via ROP at −40 °C and using a powerful lanthanum-based catalyst.80 Despite being considered until then as a “non-polymerisable” monomer as a consequence of its very low ring strain, the polymerisation reached high monomer conversion, up to 90%, and led to both linear and cyclic structures. Then, PγBL was depolymerised back to γ-BL within minutes using either the same catalyst or TBD at room temperature (Scheme 11a). In a similar manner, the phospazene superbase tert-Bu-P4 was also explored as catalyst for this cyclic polymerisation/depolymerisation process.81 However, although tert-Bu-P4 initiated the polymerisation reaction, the resulting polymer was achieved in 30% yield with a lower molecular weight (Mn of 26.4 kg mol−1). These results improved when benzyl alcohol was added as initiator, with both the conversion and molecular weight of PγBL increasing to 90% and 27.1 kg mol−1, respectively, depending on the reaction conditions (Scheme 11b). Importantly, the starting monomer and initiator were completely recovered by heating the polymer at 260 °C. Therefore, the authors designed a completely sustainable cycle where PγBL is polymerised via ROP using an organocatalyst, and its depolymerisation is carried out by thermolysis. Chen and co-workers have recently extended the infinite recycling concept to a more thermally-robust PγBL derivative that shows a promising potential as a thermoplastic material.82
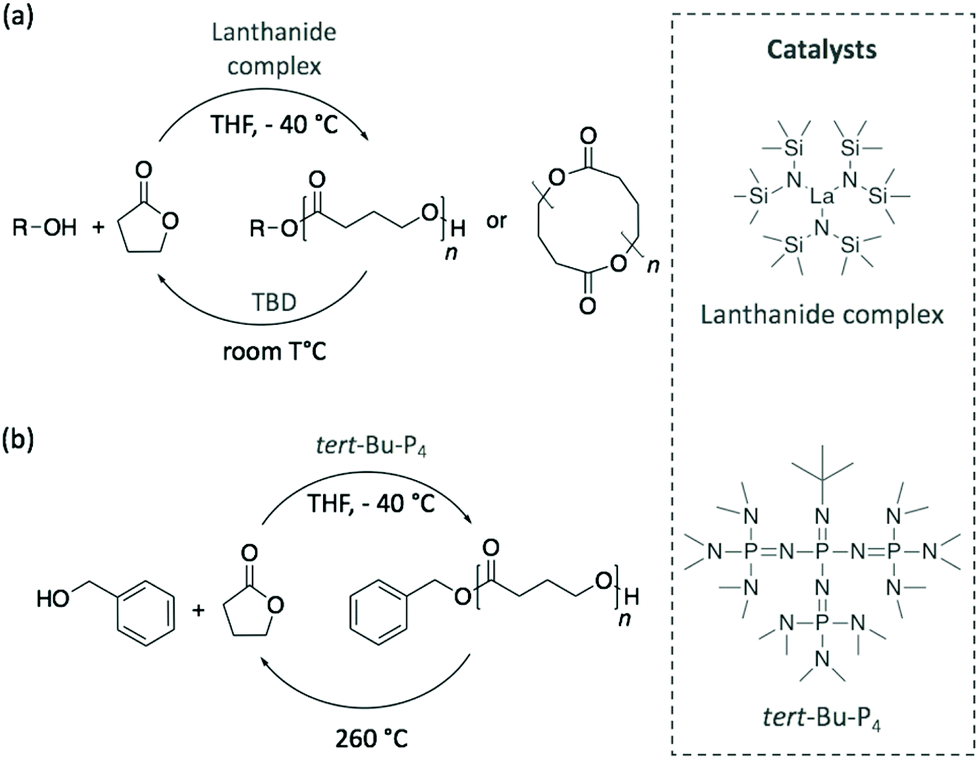 | ||
| Scheme 11 Polymerisation of γ-BL into PγBL and its depolymerisation pathway using (a) a metal catalyst and TBD or (b) tert-Bu-P4 superbase and thermolysis. | ||
Other investigations were focused on contriving sustainable cycles involving completely new polymers. Thus, in 2017, a degradable polycarbonate was synthesised from the copolymerisation of CO2 with 1-benzyloxycarbonyl-3,4-epoxy pyrrolidine (BEP) using a dinuclear chromium complex (salen) or a superbase, bis(triphenylphosphine)iminium salts, (PPN-Y), (Scheme 12a).83 Both polymerisation and depolymerisation reactions, which were achieved in less than 24 h, required the two catalysts to be added in an equimolar quantity. In the presence of CO2 at 60 °C, BEP polymerised into a polycarbonate with a molecular weight close to 10 kg mol−1 and a narrow dispersity (ĐM < 1.35). By increasing the temperature up to 100 °C, the polycarbonate depolymerised releasing CO2, and the starting epoxide was obtained as unique monomer. DFT calculations corroborated these experimental results and demonstrated that the alkoxide chain backbiting, which led to the formation of the epoxide monomer, corresponded to the energetically favoured mechanism.
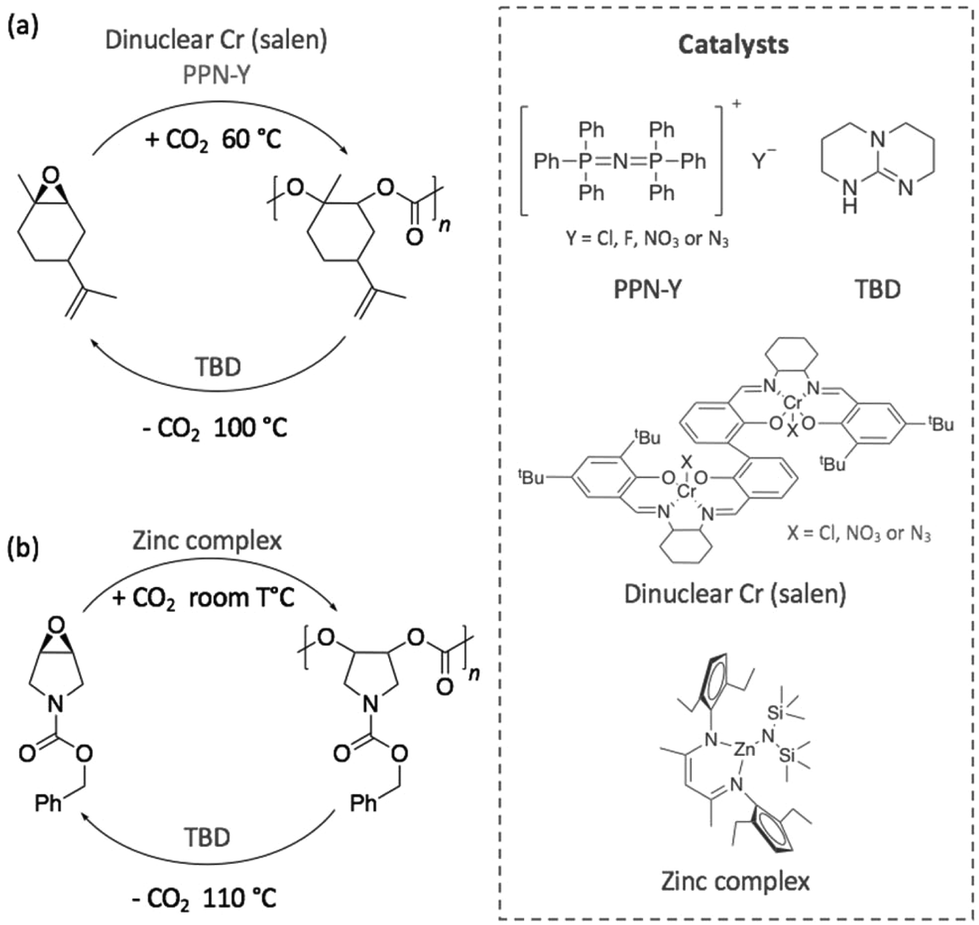 | ||
| Scheme 12 Sustainable cycle for the polymerisation and depolymerisation of (a) a degradable polycarbonate synthesised from BEP and (b) a limonene-derived based polycarbonate. | ||
With the same objective of building a sustainable polymer cycle, another recent example involved the depolymerisation of an innovative limonene-based polycarbonate into a limonene-derivative epoxide, using TBD as catalyst.84 In this study, Li et al. demonstrated that the scission of the polymer chain was initiated by the deprotonation of the hydroxyl chain ends of the polymer before a chain transfer process released one molecule of monomer (Scheme 12b). This mechanism selectively transformed the polymer into its corresponding limonene-based epoxide. Then, the re-polymerisation of this monomer was possible through copolymerisation with CO2 under different reaction conditions and using a zinc complex as catalyst at room temperature.
Similarly, Olsén et al. reported an innovative polycarbonate developed following a sustainable cycle (Scheme 13). Specifically, a 6-member carbonate bearing an allyl group (AOMEC) was polymerised by ROP and subsequently ring-closing depolymerised (RCDP) using DBU as catalyst. The authors investigated the kinetic of these two reactions using different solvents (i.e. toluene, acetonitrile and dichloromethane), as well as varying the concentration of the monomer (from 0.125 to 6 mol L−1) and temperature (from 30 to 90 °C). The optimized conditions for the ROP reaction were found to be 30 °C in dichloromethane with a high monomer concentration (4 mol L−1), whereas complete depolymerisation was achieved in 10 h in acetonitrile at 82 °C with 0.5 mol L−1 monomer concentration.85
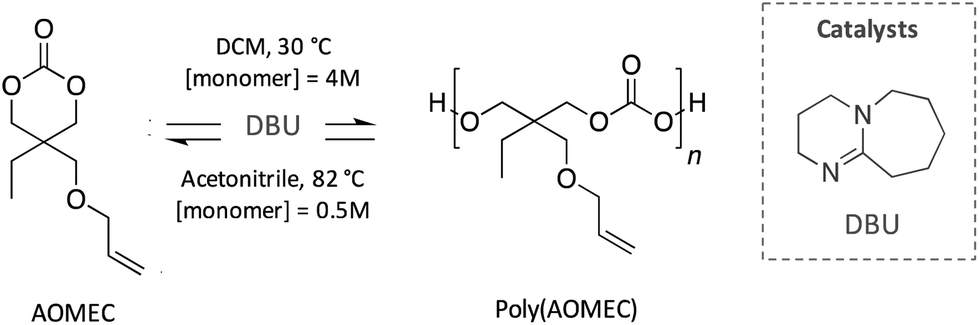 | ||
| Scheme 13 Sustainable cycle for the polymerisation and depolymerisation of a degradable polycarbonate synthesised from an innovative 6-member-ring carbonate (AOMEC). | ||
As a final example, we highlight the work by Fahnhorst et al., who recently described the “divergent chemical recycling” of a malic acid-based polyvalerlactone.86 In a two-step synthesis, 4-carbomethoxyvalerolactone (1) was firstly polymerised into an innovative polyester (P1), which was then depolymerised using two different catalysts (Scheme 14). Specifically, P1 returned to the initial monomer 1 via a transesterification reaction promoted by tin octanoate, while a methacrylate-like monomer (2) was obtained through an elimination mechanism using DBU. The subsequent polymerisation of this distinct monomer provided the corresponding polymethacrylate analogous (P2). Therefore, this “divergent” depolymerisation illustrates the inherently different mechanisms available depending on the choice of catalyst, which impacts the nature of the final product, in particular for depolymerisation reactions where the polymer backbone can be cleaved at multiple sites.
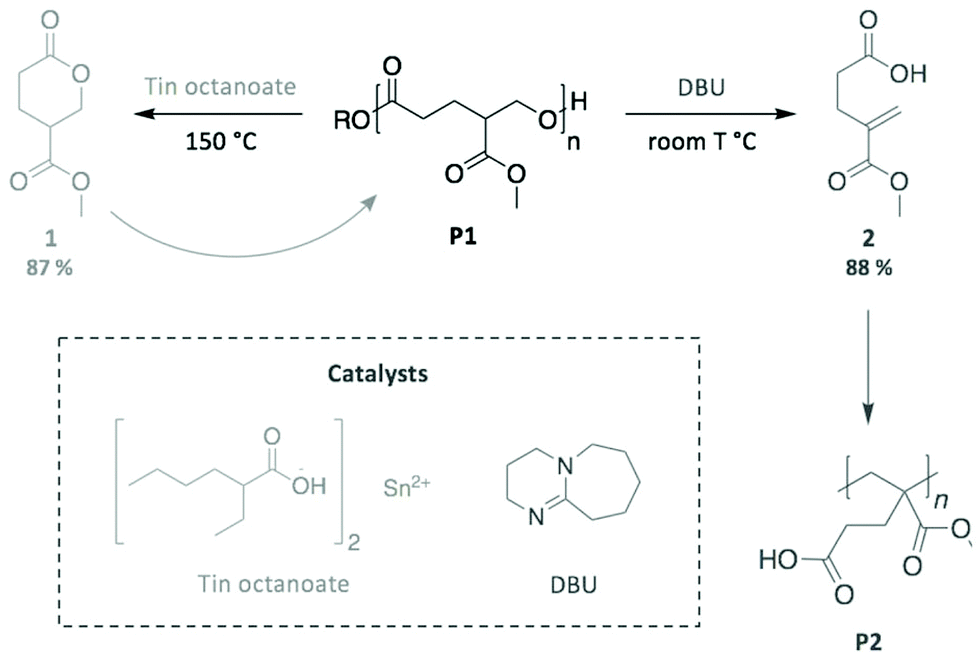 | ||
| Scheme 14 Two different pathways for the chemical recycling of a carbomethoxylated polyvalerolactone. | ||
Conclusions
Polymers display a wide range of chemical structures, thus making the depolymerisation of plastic waste a technically difficult task that is complicated further by the waste streams being mixed. In addition to that, plastic products usually contain several additives, such as plasticisers, dyes, or fibres, to improve their performances or reduce their cost, which further complicates their recyclability. Indeed, there is no “one size fits all” solution that can be applied to degrade all plastics and, consequently, each polymer family requires specific strategies. Currently, the equilibrium between the production of manufactured polymer-based products and their recycling options is very unbalanced, with few studies exploring their chemical degradation, and even fewer initiatives being translated to the industrial level.For instance, examples involving the depolymerisation of PET using organocatalysts are still rare despite being the most recycled and studied polymer. In these works, a range of guanidine bases, as well as some common ILs and ionic salts, are generally chosen as catalysts. On the one hand, the glycolysis of PET is largely described in most of these publications; however, with the equilibrium between BHET and its dimer being very fast, the selectivity of the reaction never reaches 100%, and studies rarely present the re-polymerisation of BHET. On the other hand, although the aminolysis of PET typically leads to a faster depolymerisation and higher yield values, the resulting monomers lack application within a sustainable recycling process. Finally, other polyesters, such as PLA, PG, or PHB, are easily degradable, albeit the control over the depolymerisation process greatly influences the nature of the added-value building blocks obtained for subsequent polymerisations.
In contrast, the chemical recycling of other types of polymers using organocatalysts have attracted considerably less attention in this field, regardless of their production being as abundant as that displayed by PET. For example, PC materials are used in a large variety of applications, from packaging to electronics, from building to medical devices. In particular, although commercial BPA-PC has been depolymerised into its starting monomer, BPA, through ILs and DBU, methods based on organometallic complexes or energy intensive supercritical approaches remain the more frequent in the literature. Similarly, few examples exploring the organocatalytic depolymerisation of PA exist, and those found exploit the used of acids or ILs in combination with DMAP as catalysts. More surprisingly, research on the depolymerisation of PU is extremely disproportionate in comparison to its synthesis, especially considering the global production of PU-based products. Indeed, only a few, old examples involving the chemical degradation of PU with simple alcohols and amines as catalysts, often in combination with metals, have been reported. As stated above, the large variety of manufactured PU products prevents the design of a universal depolymerisation technology, turning the depolymerisation of PU a topic of study on its own.
Finally, the high stability and easy processability of polyolefins, like PE or polypropylene, limits their recyclability, which illustrates the actual paradox of polymer recycling. These materials are designed to resist harsh conditions, such as high temperature and pressure, mechanical forces (i.e. stretching), and water or chemical treatment, and to conserve these features their entire service life; however, depolymerisation methods involve those exact same conditions. Hence, organocatalysis may take a key role in contriving sustainable polymerisation/depolymerisation cycles for such polymers.
Until now, organocatalysis has been approached as a tool for depolymerising oxygen-containing polymers, such as polyesters, polycarbonates, and polyamides, mainly through the activation of the carbonyl group, which facilitates the breakage of the C–O or C–N bond, and thus degrades the material. Nevertheless, emerging technologies, in combination with innovative organocatalysts, based on ILs, protic ionic salts, or eutectic solvents, could become powerful agents to catalyse a larger range of depolymerisation techniques.
Using organic catalysts not only is an alternative to metal complexes for depolymerising plastic waste accomplishing environmental and economic purposes, but also allows the production of innovative materials. Notably, many publications highlight the role played by H-bonding on the depolymerisation mechanism. Thus, by varying several parameters (e.g. the nature of the solvent, including a co-catalyst, the quantity of reagent, the temperature, etc.), the features of the degradation products can be tuned in innumerable possibilities.
From a practical point of view, although the number of studies in academia for recycling commodity polymers have recently increased, industrial examples of chemical degradation processes, specifically, depolymerisations involving organocatalysts are rare. The high cost of the methodologies (i.e. dry conditions, supercritical solvents, inert atmosphere, etc.), as well as the reagents used (i.e. phosphazene superbases, ionic liquids or organic bases) are key limiting factors for this technology at present. Beyond this, technical challenges for scaling up these processes have not been addressed and there remains a lack of appropriate infrastructure for collecting, sorting and storing the different plastic waste streams and the resultant degradation products. However, even though most of the procedures summarised in this review are still far from the large-scale industrial production, the huge potential for the field to create practical solutions is clear.
Overall, from a more general perspective, as a battery can be charged and discharged theoretically infinitely, polymer chemists should also conceive polymers together with their recycling route in the same way. Hence, towards reaching completely sustainable cycles, plastics require to be polymerised, depolymerised, and then re-polymerised with minimal changes in their quantity or final properties. To that end, organocatalysis represents a promising strategy to achieve this goal, which will highly benefit the transition from the current linear way of consuming plastics to a more environmentally friendly circular one.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by EU through project SUSPOL-EJD 642671 and the Gobierno Vasco/Eusko Jaurlaritza (IT 999-16). Haritz Sardon gratefully acknowledges financial support from MINECO through projects SUSPOL and FDI 16507. The authors thank for technical and human support provided by IZO-SGI SGIker of UPV/EHU and European funding (ERDF and ESF).Notes and references
- Catalysing action, https://newplasticseconomy.org/publications, (accessed May 9, 2018).
- W. McDonough and M. Braungart, Cradle to Cradle - Remaking the Way We Make Things, North Point Press., 2002 Search PubMed.
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2017, 50, 3733–3749 CrossRef CAS.
- M. Hong and E. Y.-X. Chen, Green Chem., 2017, 19, 3692–3706 RSC.
- A. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 41570 Search PubMed.
- P. Sharma, B. Lochab, D. Kumar and P. K. Roy, ACS Sustainable Chem. Eng., 2016, 4, 1085–1093 CrossRef CAS.
- R. V. Shah, V. S. Borude and S. R. Shukla, J. Appl. Polym. Sci., 2013, 127, 323–328 CrossRef CAS.
- Y. Hongmei, M. Yongshuai, Z. Weilu and Z. Dong, Adv. Mater. Res., 2012, 550, 280–283 Search PubMed.
- Y. Yang, Y. Lu, H. Xiang, Y. Xu and Y. Li, Polym. Degrad. Stab., 2002, 75, 185–191 CrossRef CAS.
- A. Kamimura, Y. Oishi, K. Kaiso, T. Sugimoto and K. Kashiwagi, ChemSusChem, 2008, 1, 82–84 CrossRef CAS.
- Y. Chujo, H. Kobayashi and Y. Yamashita, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 2007–2014 CrossRef CAS.
- C.-H. Chen, C.-Y. Chen, Y.-W. Lo, C.-F. Mao and W.-T. Liao, J. Appl. Polym. Sci., 2001, 80, 943–948 CrossRef CAS.
- M. Ghaemy and K. Mossaddegh, Polym. Degrad. Stab., 2005, 90, 570–576 CrossRef CAS.
- A. J. Hunt and J. H. Clark, Element Recovery and Sustainability, Royal Society of Chemistry, 2013 Search PubMed.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS.
- W. N. Ottou, H. Sardon, D. Mecerreyes, J. Vignolle and D. Taton, Prog. Polym. Sci., 2016, 56, 64–115 CrossRef CAS.
- C. Thomas and B. Bibal, Green Chem., 2014, 16, 1687–1699 RSC.
- A. P. Dove, ACS Macro Lett., 2012, 1, 1409–1412 CrossRef CAS.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS.
- M. D. Schoenmakere, Y. Hoogeveen, J. Gillabel and S. Manshoven, The circular economy and the bioeconomy - Partners in sustainability, European Environmental Agency, 2018 Search PubMed.
- V. Sinha, M. R. Patel and J. V. Patel, J. Polym. Environ., 2010, 18, 8–25 CrossRef CAS.
- B. Geyer, G. Lorenz and A. Kandelbauer, eXPRESS Polym. Lett., 2016, 10, 559–586 CrossRef CAS.
- A. M. Al-Sabagh, F. Z. Yehia, G. Eshaq, A. M. Rabie and A. E. ElMetwally, Egypt. J. Pet., 2016, 25, 53–64 CrossRef.
- K. Fukushima, O. Coulembier, J. M. Lecuyer, H. A. Almegren, A. M. Alabdulrahman, F. D. Alsewailem, M. A. Mcneil, P. Dubois, R. M. Waymouth, H. W. Horn, J. E. Rice and J. L. Hedrick, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1273–1281 CrossRef CAS.
- R. López-Fonseca, I. Duque-Ingunza, B. de Rivas, S. Arnaiz and J. I. Gutiérrez-Ortiz, Polym. Degrad. Stab., 2010, 95, 1022–1028 CrossRef.
- D. E. Nikles and M. S. Farahat, Macromol. Mater. Eng., 2005, 290, 13–30 CrossRef CAS.
- H. W. Horn, G. O. Jones, D. S. Wei, K. Fukushima, J. M. Lecuyer, D. J. Coady, J. L. Hedrick and J. E. Rice, J. Phys. Chem., 2012, 116, 12389–12398 CrossRef CAS.
- K. Fukushima, D. J. Coady, G. O. Jones, H. A. Almegren, A. M. Alabdulrahman, F. D. Alsewailem, H. W. Horn, J. E. Rice and J. L. Hedrick, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1606–1611 CrossRef CAS.
- K. Fukushima, J. M. Lecuyer, D. S. Wei, H. W. Horn, G. O. Jones, H. A. Al-Megren, A. M. Alabdulrahman, F. D. Alsewailem, M. A. McNeil, J. E. Rice and J. L. Hedrick, Polym. Chem., 2013, 4, 1610–1616 RSC.
- C. Jehanno, I. Flores, A. P. Dove, A. J. Müller, F. Ruipérez and H. Sardon, Green Chem., 2018, 20, 1205–1212 RSC.
- D. Zheng, L. Dong, W. Huang, X. Wu and N. Nie, Renewable Sustainable Energy Rev., 2014, 37, 47–68 CrossRef CAS.
- R. L. Vekariya, J. Mol. Liq., 2017, 227, 44–60 CrossRef CAS.
- H. Wang, Z. Li, Y. Liu, X. Zhang and S. Zhang, Green Chem., 2009, 11, 1568–1575 RSC.
- A. M. Al-Sabagh, F. Z. Yehia, A-M. M. F. Eissa, M. E. Moustafa, G. Eshaq, A.-R. M. Rabie and A. E. ElMetwally, Ind. Eng. Chem. Res., 2014, 53, 18443–18451 CrossRef CAS.
- C. S. Nunes, M. J. V. da Silva, D. C. da Silva, A. dos, R. Freitas, F. A. Rosa, A. F. Rubira and E. C. Muniz, RSC Adv., 2014, 4, 20308–20316 RSC.
- Q. Wang, X. Yao, S. Tang, X. Lu, X. Zhang and S. Zhang, Green Chem., 2012, 14, 2559–2566 RSC.
- R. M. Musale and S. R. Shukla, Int. J. Plast. Technol., 2016, 1–15 Search PubMed.
- J. Sun, D. Liu, P. Young Robert, G. Cruz Alejandro, G. Isern Nancy, T. Schuerg, R. Cort John, A. Simmons Blake and S. Singh, ChemSusChem, 2018, 11, 781–792 CrossRef CAS.
- Plastic Insights, Plastics Insight - Market Intelligence Portal for Plastics Industry, https://www.plasticsinsight.com/, (accessed August 22, 2018).
- P. Palanza, L. Gioiosa, F. S. vom Saal and S. Parmigiani, Environ. Res., 2008, 108, 150–157 CrossRef CAS.
- S. Biedermann, P. Tschudin and K. Grob, Anal. Bioanal. Chem., 2010, 398, 571–576 CrossRef CAS PubMed.
- D. K. Tucker, S. Hayes Bouknight, S. S. Brar, G. E. Kissling and S. E. Fenton, Environ. Health Perspect., 2018, 126, 087003 CrossRef.
- E. Quaranta, D. Sgherza and G. Tartaro, Green Chem., 2017, 19, 5422–5434 RSC.
- T. Do, E. R. Baral and J. G. Kim, Polymer, 2018, 143, 106–114 CrossRef CAS.
- F. Liu, Z. Li, S. Yu, X. Cui and X. Ge, J. Hazard. Mater., 2010, 174, 872–875 CrossRef CAS.
- L. Li, F. Liu, Z. Li, X. Song, S. Yu and S. Liu, Fibers Polym., 2013, 14, 365–368 CrossRef CAS.
- F. Liu, L. Li, S. Yu, Z. Lv and X. Ge, J. Hazard. Mater., 2011, 189, 249–254 CrossRef CAS.
- S. R. Shukla, A. M. Harad and D. Mahato, J. Appl. Polym. Sci., 2006, 100, 186–190 CrossRef CAS.
- H. Matsumoto, Y. Akinari, K. Kaiso and A. Kamimura, J. Mater. Cycles Waste Manage., 2017, 19, 326–331 CrossRef CAS.
- A. Kamimura and S. Yamamoto, Org. Lett., 2007, 9, 2533–2535 CrossRef CAS.
- A. Kamimura and S. Yamamoto, Polym. Adv. Technol., 2008, 19, 1391–1395 CrossRef CAS.
- S. Yamamoto and A. Kamimura, Chem. Lett., 2009, 38, 1016–1017 CrossRef CAS.
- S. Xue, F. He, M. Omoto, T. Hidai and Y. Imai, Kobunshi Ronbunshu, 1993, 50, 847–853 CrossRef CAS.
- M. Murai, M. Sanou, T. Fujimoto and F. Baba, J. Cell. Plast., 2003, 39, 15–27 CrossRef CAS.
- M. M. A. Nikje and M. Nikrah, J. Macromol. Sci., Part A: Pure Appl. Chem., 2007, 44, 613–617 CrossRef.
- K. Kanaya and S. Takahashi, J. Appl. Polym. Sci., 1994, 51, 675–682 CrossRef CAS.
- J. Borda, G. Pásztor and M. Zsuga, Polym. Degrad. Stab., 2000, 68, 419–422 CrossRef CAS.
- L. M. Dos Santos, C. L. P. Carone, J. Dullius, R. Ligabue and S. Einloft, Polim.: Cienc. Tecnol., 2013, 23, 608–613 CAS.
- X. Wang, H. Chen, C. Chen and H. Li, Fibers Polym., 2011, 12, 857 CrossRef CAS.
- C. Gutiérrez, M. T. García, I. Gracia, A. de Lucas and J. F. Rodríguez, J. Mater. Cycles Waste Manage., 2012, 14, 308–316 Search PubMed.
- W. S. Fonseca, X. Meng and D. Deng, ACS Sustainable Chem. Eng., 2015, 3, 2153–2159 CrossRef CAS.
- A. A. Marek, J. Zawadiak, T. Piotrowski and B. Hefczyc, Waste Manage., 2015, 46, 62–67 CrossRef CAS PubMed.
- V. Andreoni, H. G. M. Saveyn and P. Eder, J. Environ. Manage., 2015, 158, 103–110 CrossRef CAS.
- B. Kunwar, B. R. Moser, S. R. Chandrasekaran, N. Rajagopalan and B. K. Sharma, Energy, 2016, 111, 884–892 CrossRef CAS.
- S. Gillier-Ritoit, D. Reyx, I. Campistron, A. Laguerre and R. Pal Singh, J. Appl. Polym. Sci., 2003, 87, 42–46 CrossRef CAS.
- A. El Hamdaoui, D. Reyx, I. Campistron and S. F. Tétouani, Eur. Polym. J., 1999, 35, 2165–2183 CrossRef CAS.
- C. W. Phetphaisit and P. Phinyocheep, J. Appl. Polym. Sci., 2003, 90, 3546–3555 CrossRef CAS.
- P. Phinyocheep, C. W. Phetphaisit, D. Derouet, I. Campistron and J. C. Brosse, J. Appl. Polym. Sci., 2005, 95, 6–15 CrossRef CAS.
- F. Sadaka, I. Campistron, A. Laguerre and J.-F. Pilard, Polym. Degrad. Stab., 2012, 97, 816–828 CrossRef CAS.
- E. Bäckström, K. Odelius and M. Hakkarainen, Ind. Eng. Chem. Res., 2017, 56, 14814–14821 CrossRef.
- C. J. Adams, M. J. Earle and K. R. Seddon, Green Chem., 2000, 2, 21–24 RSC.
- A. Mouawia, A. Nourry, A.-C. Gaumont, J.-F. Pilard and I. Dez, ACS Sustainable Chem. Eng., 2017, 5, 696–700 CrossRef CAS.
- O. Coulembier, S. Moins, J.-M. Raquez, F. Meyer, L. Mespouille, E. Duquesne and P. Dubois, Polym. Degrad. Stab., 2011, 96, 739–744 CrossRef CAS.
- X. Song, X. Zhang, H. Wang, F. Liu, S. Yu and S. Liu, Polym. Degrad. Stab., 2013, 98, 2760–2764 CrossRef CAS.
- X. Song, H. Wang, F. Liu and S. Yu, Polym. Degrad. Stab., 2016, 130, 22–29 CrossRef CAS.
- J. Bjerrum, Stability Constants of Metal-ion Complexes, with Solubility Products of Inorganic Substances: Inorganic ligands, Chemical Society, 1958 Search PubMed.
- M. Melchiors, H. Keul and H. Höcker, Macromolecules, 1996, 29, 6442–6451 CrossRef CAS.
- M. Hong and E. Y.-X. Chen, Nat. Chem., 2016, 8, 42–49 CrossRef CAS.
- M. Hong and E. Y.-X. Chen, Angew. Chem. Int. Ed., 2016, 128, 4260–4265 CrossRef.
- J.-B. Zhu, E. M. Watson, J. Tang and E. Y.-X. Chen, Science, 2018, 360, 398–403 CrossRef CAS.
- Y. Liu, H. Zhou, J.-Z. Guo, W.-M. Ren and X.-B. Lu, Angew. Chem., Int. Ed., 2017, 56, 4862–4866 CrossRef CAS.
- C. Li, R. J. Sablong, R. A. T. M. van Benthem and C. E. Koning, ACS Macro Lett., 2017, 6, 684–688 CrossRef CAS.
- P. Olsén, J. Undin, K. Odelius, H. Keul and A.-C. Albertsson, Biomacromolecules, 2016, 17, 3995–4002 CrossRef.
- G. W. Fahnhorst and T. R. Hoye, ACS Macro Lett., 2018, 7, 143–147 CrossRef CAS.
- F. A. Leibfarth, N. Moreno, A. P. Hawker and J. D. Shand, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4814–4822 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |