
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePoly(meth)acrylate-PVDF core–shell particles from emulsion polymerization: preferential formation of the PVDF β crystal phase†
Florian
Brandl
a,
Andreas F.
Thünemann
 b and
Sabine
Beuermann
*a
b and
Sabine
Beuermann
*a
aClausthal University of Technology, Institute of Technical Chemistry, Arnold-Sommerfeld-Straße 4, 38678 Clausthal-Zellerfeld, Germany. E-mail: sabine.beuermann@tu-clausthal.de
bFederal Institute for Materials Research and Testing (BAM), Unter den Eichen 87, 12205 Berlin, Germany
First published on 17th October 2018
Abstract
A facile and convenient approach for the synthesis of core–shell particles via emulsion polymerization is presented. The shell consists of poly(vinylidene fluoride) (PVDF) and the core of poly(methyl methacrylate) (PMMA), poly(glycidyl methacrylate) (PGMA) or poly(methyl acrylate) (PMA). In a first step, a non-fluorinated (meth)acrylate monomer is polymerized in the emulsion to produce poly(meth)acrylate core particles. Secondly, vinylidene fluoride (VDF) is directly added to the reactor and polymerized for shell formation. Small-angle X-ray scattering (SAXS) was employed to characterize the structure of the core–shell particles. Interestingly, the particles’ core contains fluorinated and non-fluorinated polymers, whereas the shell of the particles consists only of PVDF. The resulting particles with a diameter of around 40 nm show a significantly higher PVDF β phase content than the PVDF homopolymer obtained by emulsion polymerization.
Introduction
Poly(vinylidene fluoride) (PVDF) is a thermoplastic fluoropolymer, which shows extraordinary properties like thermal stability, chemical inertness, resistance to acids, and low flammability.1 PVDF is used in the chemical and automotive industries as well as in electric, medical, and other high-tech applications.2 Due to its ferro-, piezo-, and pyroelectric properties, PVDF is attractive for sensors and switches.3–5 These electroactive properties are associated with the all-trans conformation of the PVDF β crystalline phase, which is one of five PVDF crystalline phases (α, β, γ, δ, and ε).6,7 Most frequently PVDF shows α phase crystallinity. Several processes, e.g., such as mechanical stretching of the α polymorph, melting under high pressure, ultrafast cooling, solvent casting, and addition of fillers, were reported to yield β phase PVDF.4,8–11 Moreover, blends of PVDF and poly(methyl methacrylate) (PMMA) as well as block copolymers of PVDF with PMMA or other fluoropolymers may yield β phase PVDF.12–15Recently, the complex interactions between PVDF and PMMA in polymer blends were studied in detail.16–19 Moreover, the interactions in systems consisting of a PVDF core and a PMMA shell in the form of Janus particles were addressed.20,21 Pan et al. investigated systems with a PVDF core.21,22 Apart from these publications, reports on core–shell architectures with PVDF are scarce.23,24 Moreover, the introduction of particulate materials into a PVDF matrix25–27 to impact the crystallinity and the electroactive properties was studied.28,29
Based on reports that in particular the presence of PMMA either in blends or block copolymers with PVDF is favorable for the formation of β phase PVDF, it appeared interesting to investigate the crystallinity, if a PMMA or more general a poly(meth)acrylate core is covered with a PVDF shell. It was speculated that such a core–shell structure with PVDF as the shell may influence the morphology and may affect the amount of β phase material. Due to expected favorable interactions between fluorine atoms in PVDF and the carbonyl groups in poly((meth)acrylates), a directive force may be expected to occur.30 Thus, the concept of affecting the crystallinity of PVDF in core–shell systems appeared very attractive to investigate.
Generally, for the preparation of core–shell particles (CSPs), techniques such as emulsion, dispersion, and precipitation polymerization were applied.31 The synthetic routes encompass several process steps. Examples are two stage emulsion polymerization, CSP formation in the emulsion by using reactive surfactants, stepwise hetero-coagulation of smaller cationic polymer particles onto larger anionic polymer particles, and self-aggregation of block copolymers. In this contribution, a rather facile synthetic route is introduced. Firstly, a PMMA core is obtained by emulsion polymerization. Secondly, after MMA is fully consumed VDF is added to the reactor to continue with the VDF polymerization. Thus, there is no work-up required after the first step. The synthesis of the CSPs is achieved in a single process, which is of course followed by the work-up of the emulsion. Following this approach, various materials with core–shell architecture with PMMA, poly(glycidyl methacrylate) (PGMA) or poly(methyl acrylate) (PMA) as the core and PVDF as the shell were prepared. The core–shell structure was characterized by TEM and SAXS and the crystallinity was investigated via FTIR and XRD.
Experimental section
Materials
Vinylidene fluoride (VDF) and ammonium 4,8-dioxa-3H-perfluorononanoate (ADONA) were kindly provided by 3 M/Dyneon GmbH and used as received. The monomers methyl methacrylate (MMA, 99% Sigma-Aldrich), glycidyl methacrylate (GMA, ≥97%, Sigma-Aldrich) and methyl acrylate (MA, 99%, Sigma-Aldrich) were distilled under reduced pressure to remove inhibitors. Ammonium persulfate (APS, ≥98% ACROS ORGANICS) as the initiator, dimethyl acetamide (DMAc, 99%, ACROS ORGANICS) and LiBr (≥99%, Riedel-de-Häen) serving as the eluent for size-exclusion chromatography were used as received. As the reaction medium ultrapure and deionized water (electric conductivity: 0.055 μS cm−1) was used.Polymer characterization
For CSP characterization, the following techniques and equipment are used:Size-exclusion chromatography (SEC) measurements of the core were carried out in DMAc, which contains 0.1% LiBr as the eluent at a column temperature of 45 °C. The SEC set-up consists of an Agilent 1200 isocratic pump, an Agilent 1200 refractive index detector, and four PSS GRAM columns (guard, 100 Å, 3000 Å, and 3000 Å) from Polymer Standard Service (PSS). Measurements were carried out at a flow rate of 1 mL min−1. Polystyrene standards (PSS) of narrow dispersity were used for calibration.
For FT-IR measurements, a Vertex 70 spectrometer (Bruker Optik GmbH, Bremen, Germany) equipped with a globar source and a photoacoustic cell (PA301) was used. Spectra were recorded with a resolution of 4 cm−1 using 20 scans.
Differential scanning calorimetry (DSC) for thermal analysis measurements was performed with a DSC 1/500658/200 W STARe system by Mettler Toledo, Columbus, OH, USA. This system is equipped with a FRS5 sensor and liquid nitrogen cooling. Each sample passes through a complete heating and cooling cycle before the second heating run is used for analysis. The heating and cooling rate is 10 °C min−1 for all measurements.
The particle size distribution (PSD) of the core- and later also the core–shell latex particles was determined by dynamic light scattering (DLS). The system consists of an ALV-CGS-3 compact goniometer system (ALV-GmbH) and an ALV-LSE-5003 correlator. The measurements were carried out at 25 °C at a scattering angle of 90°.
For the assessment of the particle morphology of the latex particles, a Helios Nanolab 600 (FEI, USA) field emission scanning microscope (FESEM) was used. The measured sample is obtained by film formation. For a better conductivity of the sample, a thin layer of carbon was sputter-coated under vacuum.
Transmission electron microscopy (TEM) for imaging the core–shell structure was performed using a JEM-2100 (Fa. JEOL) TEM, operating with a LaB6 electron source at 200 kV and standard carbon-coated copper grids.
Wide-angle X-ray diffraction (XRD) for phase determination of the PVDF shell was conducted with a Bruker AXS D8 Discover diffractometer using Cu–Kα (graphite monochromator) radiation (λ = 0.1540 nm) equipped with a General Area Diffraction System (GADDS, Bremen, Germany) as the detector.
Small-angle X-ray scattering (SAXS) measurements were performed with a Kratky-type instrument (SAXSess from Anton Paar, Austria) at (21 ± 1) °C. The SAXSess has a low sample-to-detector distance of 0.309 m, which is appropriate for investigation of dispersions with low scattering intensities.32 Each sample was measured as delivered for 360 × 10 s in a flow-through capillary. The measured intensity was converted to the absolute scale according to Orthaber et al.33 The scattering vector is defined in terms of the scattering angle θ and the wavelength λ of the radiation (λ = 0.154 nm): thus θ = 4π n/λ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ. Deconvolution (slit length desmearing) of the SAXS curves was performed with the SAXS-Quant software. Samples analyzed by SAXS were used as prepared.
sinθ. Deconvolution (slit length desmearing) of the SAXS curves was performed with the SAXS-Quant software. Samples analyzed by SAXS were used as prepared.
Scattering length densities of the polymers were determined with the scattering length density calculator implemented in small-angle scattering data evaluation tool SASfit34 using an X-ray energy of 8041 eV (corresponding to a wavelength of copper Kα radiation of 0.15418 nm). At 20 °C, the calculated scattering length densities are bPVDF = 1.518 × 1011 cm−2 for poly(vinylidene fluoride) (PVDF, C2H2F2, density ρ = 1.78 g cm−3), bPMA = 1.110 × 1011 cm−2 for poly(methacrylate) (PMA, C4H6O2, density ρ = 1.22 g cm−3), bPMMA = 1.084 × 1011 cm−2 for poly(methyl methacrylate) (PMMA, C5H2O2, density ρ = 1.18 g cm−3), bPGMA = 7.328 × 1010 cm−2 for poly(glycidyl methacrylate) (PGMA, C7H11O3, density ρ = 0.805 g cm−3) and for the medium bwater = 9.446 × 1010 cm−2 for water (H2O, density ρ = 0.9982 g cm−3).
Polymerization reactor
The preparation of CSPs with a PVDF shell in aqueous emulsion polymerizations requires a polymerization set-up, which allows for elevated pressure. Because of the gaseous monomer VDF and its poor solubility in water a continuous monomer feed (semi-batch mode) is advantageous for reasonable polymerization rates. Due to these requirements, a reactor set-up for VDF semi-batch emulsion polymerization as shown in Fig. 1 was used.35,36 The core part is a 2 L double-jacketed stainless steel reactor with a bottom outlet (Büchi AG, Switzerland, Typ 3, max. 60 bar, 220 °C). The reactor is closed by a cover plate (Büchi AG, Switzerland, polyclave, 60 bar), which holds installations for pressure and temperature measurement as well as fittings and valves for, e.g., addition of liquids or purging with nitrogen. All the fittings, connections and valves were purchased from Swagelok®. The temperature control is established by a circulation thermostat (ministat 240w, Huber Kältemaschinenbau AG, Germany). The pressure is measured using a Keller PA-21S/80400 pressure transducer.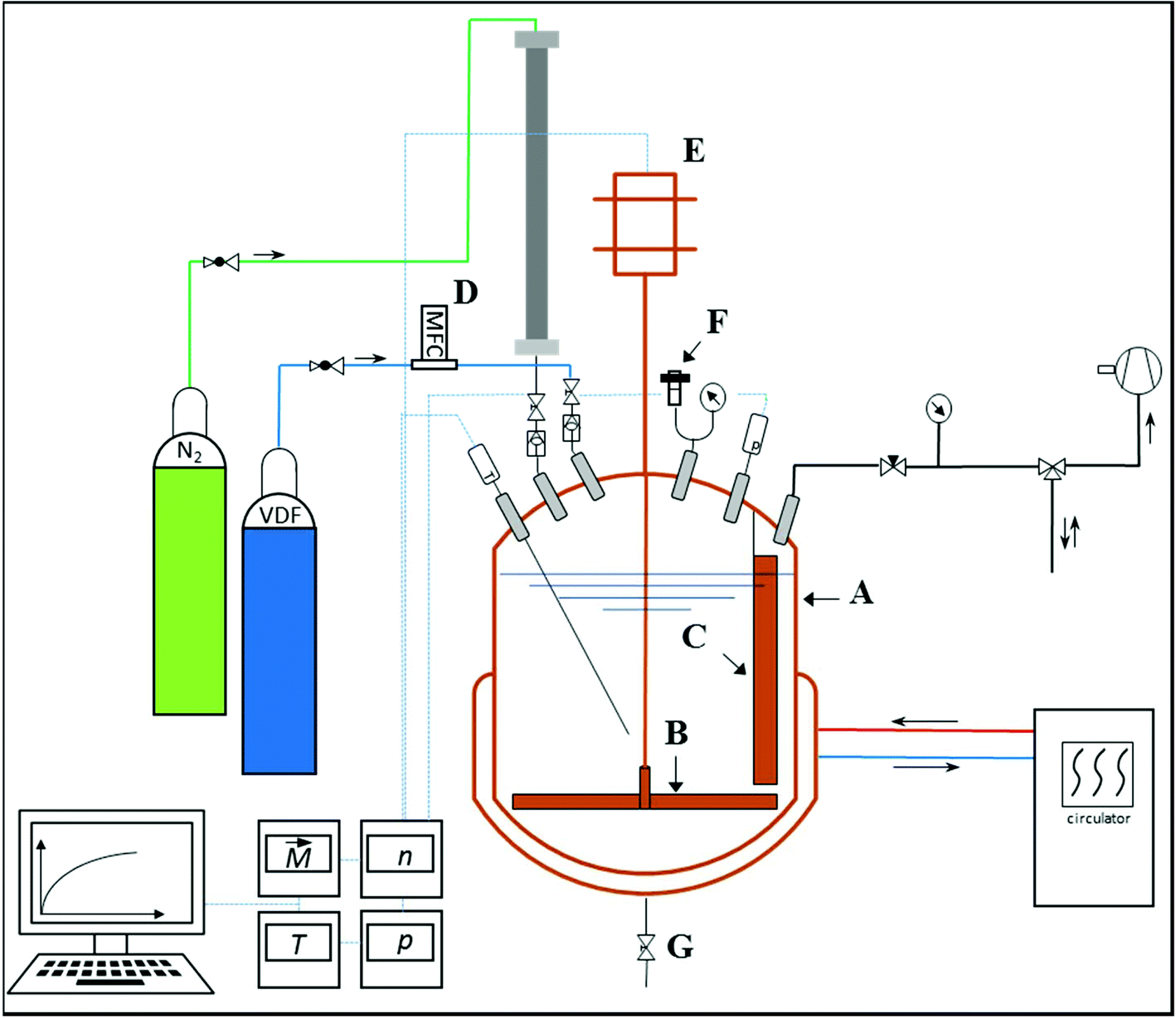 | ||
| Fig. 1 Schematic representation of the polymerization reactor. A: Reactor, B: stirrer, C: baffle, D: mass flow controller, E: stirrer motor with torque measurement, F: rupture disc, and G: bottom outlet. | ||
During the reaction, temperature and pressure were measured, as well as stirring speed and the required torque. VDF is supplied to the reactor via a flexible 1/8′′ copper pipe in combination with a pressure regulator (C200/1 TP B, Linde AG, Germany). This regulator is independent of the bottle pressure and ensures a constant reaction pressure during the semi-batch operation. The measurement of the continuously flowing VDF is carried out with a mass flow controller (MFC) (EL-FLOW®, Bronkhorst High-Tech BV, Netherlands). The MFC is calibrated in the range from 1.2 to 60 g h−1 with an accuracy of ±0.5%. Data of the VDF mass flow required to keep a constant pressure directly provide information on the solid content in the reactor and the polymerization rate. To achieve good mixing, a pitched blade agitator and a baffle are installed. For security reasons, the reactor is equipped with a rupture disc and check valves in all pipes. Furthermore, through a pressure sluice, liquids like initiator solution can be added during the reaction.
Synthesis of the core–shell polymers
In order to prepare the CSP in an emulsion polymerization in a single process step without the need for work-up after synthesis of the core, already the core must be polymerized using a stabilizer also suitable for the polymerization of VDF.37 Here, ADONA, a commercially used fluorinated stabilizer for VDF emulsion polymerizations, is employed. Firstly, the (meth)acrylic polymer is prepared in a batch emulsion polymerization in the reactor described above. Secondly, the emulsion polymerization of VDF is performed in the semi-batch mode. Due to the low VDF solubility in the emulsion, it is fed continuously into the reactor. The polymerization strategy is illustrated in Fig. 2.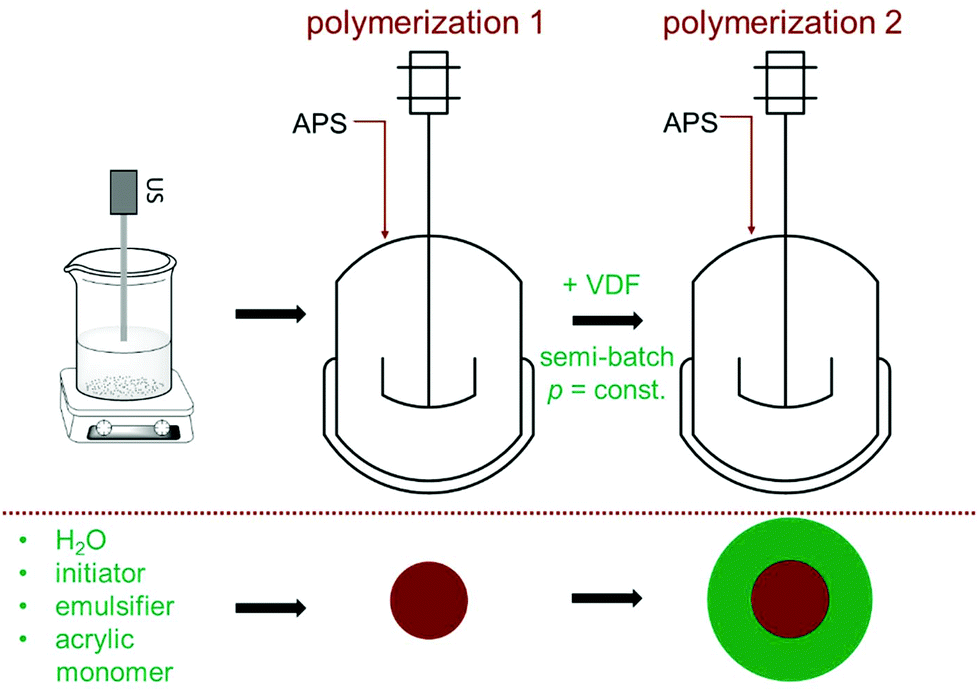 | ||
| Fig. 2 Concept of the core–shell formation by two subsequently carried out emulsion polymerizations in batch for the (meth)acrylate monomer and in the semi-batch mode for VDF. | ||
The core is typically synthesized as follows: 1100 ml deionized water is boiled for 45 minutes under a nitrogen flow to reduce the oxygen content. After cooling to room temperature under nitrogen, 10 g of the emulsifier ADONA and the acrylic monomer (MMA, MA, or GMA) are added while stirring. The amount of monomer added is given in Table 1. The mixture is treated with an ultrasonic sonotrode (UP200S, Fa. Hielscher Ultrasonics GmbH), which dips into the mixture. This emulsion is transferred into the polymerization reactor, already heated to the reaction temperature of 80 °C. After sealing, purging the reactor with N2 and testing for leakage, 0.0175 mol APS dissolved in 10 mL of water are added through the sluice. The reaction is carried out with a stirring speed of 600 rpm and yields at least 95% of monomer conversion (checked gravimetrically) in 3 hours. For further analyses via DLS and SEC, a sample of 2 mL is taken.
| Sample | Core polymer | m mon/g (for core) | T/°C | m VDF/g (for shell) | p VDF/bar | T/°C |
|---|---|---|---|---|---|---|
| a An amount of 2.0 g dimethyl ether was added as the chain transfer agent. | ||||||
| Core-1a | PMMA | 26 | 75 | — | — | — |
| CSP-1 | PMMA | 15 | 80 | 80 | 25 | 75 |
| CSP-2 | PMMA | 5 | 80 | 200 | 25 | 75 |
| CSP-3 | PGMA | 7.5 | 80 | 150 | 25 | 75 |
| CSP-4 | PMA | 7.5 | 80 | 150 | 25 | 75 |
| CSP-5 | PMA | 15 | 80 | 100 | 25 | 75 |
Subsequently, with the intention to encapsulate the (meth)acrylate core VDF is filled into the reactor while stirring (400 rpm) until a pressure of 25 bar is reached (marked with (1) in Fig. 3). Then, pressure and temperature are adjusted to the desired values and to start the VDF polymerization 0.0175 mol of initiator dissolved in 10 mL are added. Typically, the polymerization starts with a delay of about 20 to 30 minutes. The consumption of the VDF monomer is indicated by an increase in the VDF feed (Fig. 3). For PMMA as the core polymer, the stirring speed had to be reduced to 300 rpm (at (2) in Fig. 3) to reduce the amount of VDF introduced into the system and, thus, to limit the polymerization rate. To increase the amount of PVDF formed, an additional initiator may be fed to the reactor at a later stage (at (3) in Fig. 3).
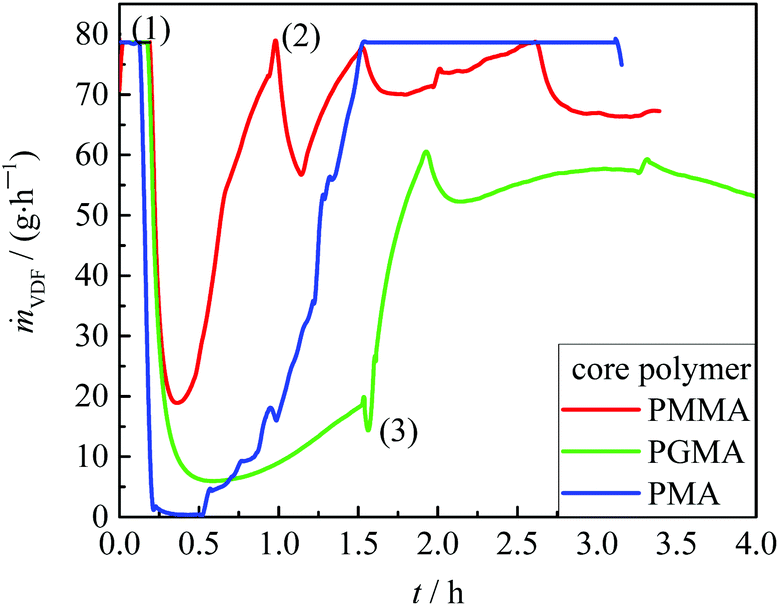 | ||
| Fig. 3 VDF mass flow mVDF during the VDF polymerization with the core polymers indicated. (1) Filling VDF into the reactor until reaction pressure is reached; (2) reduction of stirring speed to 300 rpm; (3) addition of the initiator. For further details, refer to the text. | ||
The reaction time mainly depends on the conversion rate and the desired shell thickness. Typically, the VDF polymerization is carried out for 3 to 4 h. The reaction time was chosen based on the DLS results of samples taken during the VDF polymerization.
Table 1 summarizes the reaction conditions for both stages of the CSP synthesis with MMA, GMA, and MA as monomers for the core. Sample core-1 refers to a MMA polymerization. The product was analyzed via FESEM. The associated image is shown in Fig. 4a. Dimethyl ether was used as the chain transfer agent to limit the molar masses and avoid gelling and to obtain a soluble material. To reduce the reaction time after the first polymerization, the reaction temperature was increased to 80 °C for all other (meth)acrylate polymerizations. The amounts of (meth)acrylate and VDF were varied to achieve different core and shell sizes.
 | ||
| Fig. 4 FESEM images of the resulting particles: (a) PMMA core (core-1), (b) and (c) PMMA-PVDF core–shell particles (CSP-1). | ||
Results and discussion
The CSPs obtained were insoluble in dimethyl formamide and dimethyl acetamide, the solvents generally used for SEC analyses of PVDF. This finding suggests that very high molar mass PVDF was obtained in the absence of chain transfer agents. Thus, information on the molar mass distributions is not accessible via SEC. The analytical methods were restricted to characterization of the particles in the dispersion and in the dried state.Analyses of the size and structure of the core–shell particles
FESEM analyses of dried samples were carried out to obtain an overview on the shape and surface morphology of particles consisting only of the core material and particles with a core–shell structure. The hydrodynamic radii of the particles at different stages of polymerization were determined by DLS. The structure and size of the final particles were analyzed via SAXS and TEM.
Fig. 4 depicts FESEM images of pure PMMA cores (a) and the PMMA core with a PVDF shell (b and c). The images were taken from dried particles. The data for the pure PMMA indicate that emulsion polymerization of MMA with the fluorinated surfactant ADONA yields well-defined spherical particles with a narrow particle size distribution. The image indicates that the PMMA particles have a diameter of around 50 nm. The number average molar mass of this polymer is 29300 g mol−1 with a dispersity of 1.7 as determined by SEC. Similar to the PMMA sample, the images of the composite particles (CSP1 from Table 1) shown in Fig. 4b and c are very uniform with respect to the spherical shape and size of the particles. The magnification in Fig. 4c indicates that the particle diameters are around 70 nm and that the VDF polymerization does not change the shape of the original core particles.
The development of the intensity-weighed distributions of the hydrodynamic radii in the course of core–shell particle formation is shown exemplarily for the synthesis of CSP-2 in Fig. 5. The distribution of the PMMA cores displays a peak at rH = 10 nm (red curve, polymerization of 5 g of MMA). During the VDF polymerization, the position of the peak is shifted to higher values of rH. After polymerization of 100 g VDF, the peak occurs at rH = 50 nm and after polymerization of additional 100 g of VDF (total 200 g VDF) the peak is shifted to rH = 68 nm (blue and green curves, respectively). The shift of the unimodal distribution strongly suggests that all core particles grew due to the addition of VDF in the second stage of the polymerization and that core–shell particles were obtained. To verify this assumption, SAXS and TEM measurements were carried out.
 | ||
| Fig. 5 Hydrodynamic radii distributions obtained from DLS for a pure PMMA core (red curve) and the resulting PSDs for the core–shell particles (CSP-2) after addition of 100 g VDF (blue curve) and adding additional 100 g VDF (green curve). The given radii are the mean peak positions of rH. | ||
While SAXS and TEM will be very important for identification of the anticipated core–shell structure, it has to be noted that the results from the three methods, e.g., the particle diameter, may differ significantly. The reason may be seen in the fact that very different types of measurements are carried out and that physical effects may alter the results, which may hamper direct comparability of data derived from DLS, TEM, and SAXS.38,39 As will be shown below the intensity-weighed hydrodynamic radii derived from DLS are typically larger than the radii derived from SAXS and especially from TEM. This finding is suggested to be due to the formation of an electric double layer40 on the surface of the particles, which affects Brownian motion of the particles. Since the hydrodynamic radii are derived from diffusion coefficients, the radii appear to be larger than that obtained by TEM, which monitors number-weighed radii.
SAXS measurements of the samples dispersed in water result in scattering curves which are typical of spherical particles as shown in Fig. 6. The scattering intensity of five of the six samples can be described by the scattering of core–shell particles and will be described in detail in the following. By contrast, attempts to fit the curves with the model of spherical particles with a homogeneous density distribution were not successful (not shown). An analytical expression of particles with a spherical core of radius rc surrounded by a shell of thickness Δ was derived by Bartlett and Ottewill41 which is briefly summarized here. The assumption in this model is that the shell thickness is uniform and the core radii have a Schulz distribution defined as
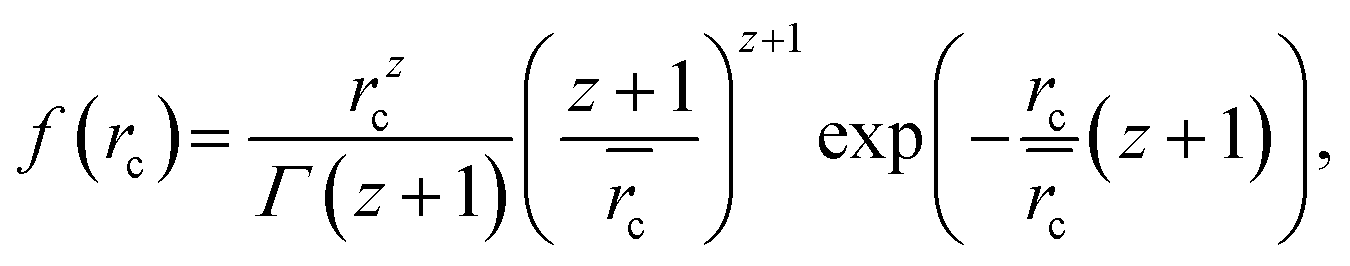 | (1) |
 . For scattering length densities of core, ρc, shell ρs, and dispersing medium ρm, respectively, the scattering intensity of the particles is given as
. For scattering length densities of core, ρc, shell ρs, and dispersing medium ρm, respectively, the scattering intensity of the particles is given as | (2) |
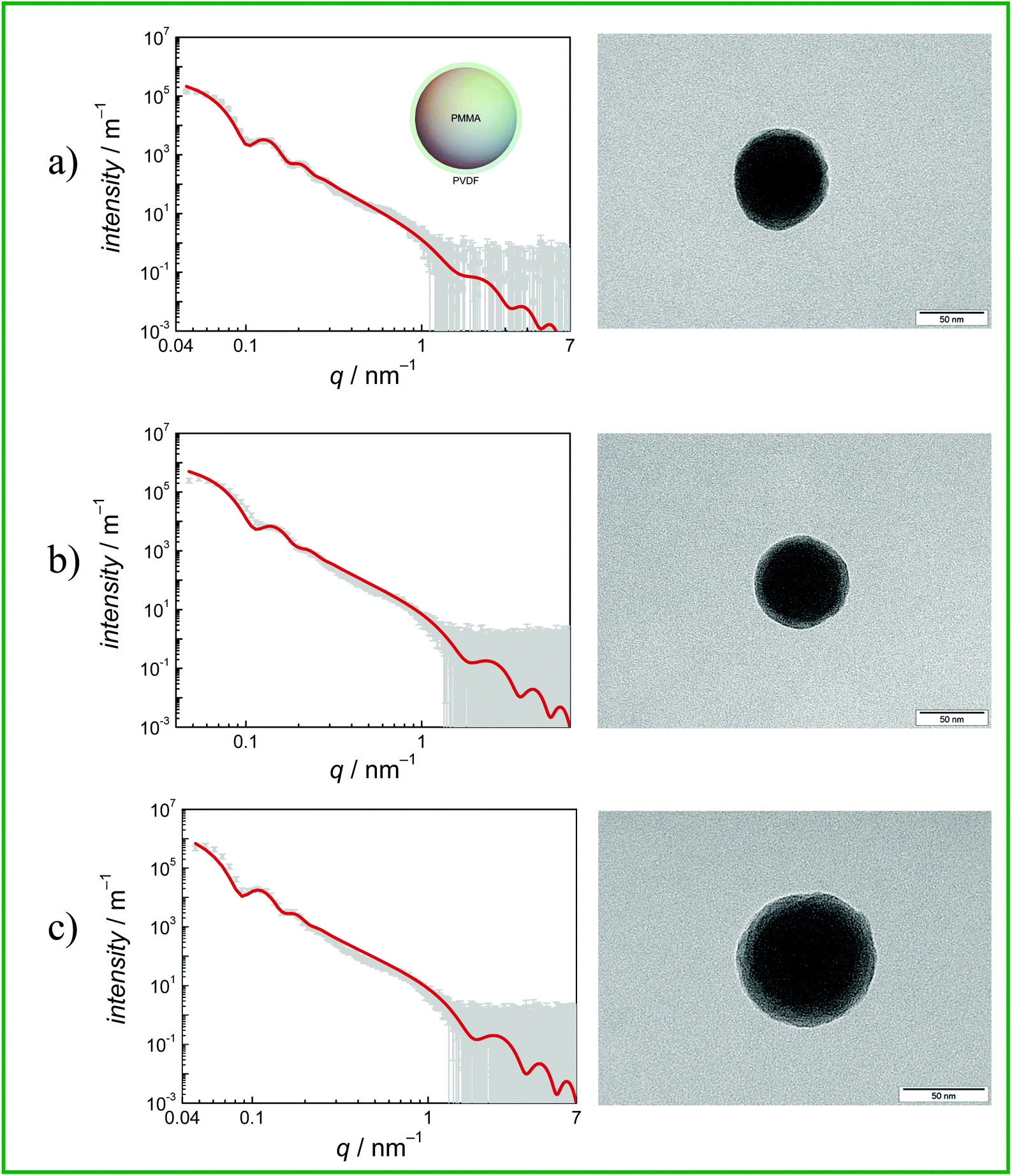 | ||
| Fig. 6 SAXS data and curve fits (left) and TEM images (right, the scale bars in the right corners represent 50 nm) of CSPs with (a) PMMA (CSP-1) (b) PGMA (CSP-3) and (c) PMA (CSP-4) core. The shell polymer is always PVDF. | ||
The k is a scaling constant. The q-dependent functions B(q) and D(q) are defined as

The coefficients c1 to c9 are
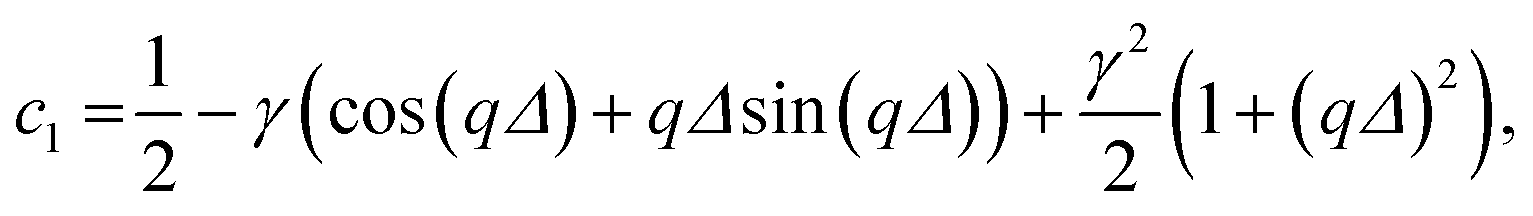
| c2 = γqΔ(γ − cos (qΔ)), |

| c4 = γ2[qΔ cos(qΔ) − sin(qΔ)]2 − c1, |
| c5 = 2γ sin(qΔ)[1 − γ(qΔ sin(qΔ) + cos(qΔ))] + c2, |
| c6 = c3 − γ2 sin2 (qΔ), |


| c9 = γ sin(qΔ[1 − γ cos(qΔ)]). |
The γ is the ratio of the contrast between medium and shell, ρm − ρs, and between the core and the shell, ρc − ρs, i.e. γ = (ρm − ρs)/(ρc − ρs). If the shell has the same electron density as the dispersing medium ρm = ρs, the γ = 0 and only the core contributes to the scattering. If the core has the same density as the dispersing medium ρc = ρm, the γ = 1 and only the shell contributes to the scattering.
Eqn (2) was employed for interpretation of the scattering data of five of the samples and results in fit curves shown in Fig. 6 (grey and red lines, respectively). Note that the shell thickness was used as the fixed parameter during curve fitting to avoid ambiguous results. Also the density of the shell material was held constant at curve fitting using the density of PVDF in the bulk material of 1.78 g cm−3 to avoid ambiguous fits. Fit results are given in the figure captions and in Table 2.
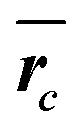 , relative width of distribution of core sizes σ, the thickness of the shell Δ and the volume fraction, ϕc, according to eqn (3). The density of the dispersant matrix (water) ρm = 0.998 g cm−3 and the shell (PVDF) ρs = 1.78 g cm−3 was held constant at curve fitting
, relative width of distribution of core sizes σ, the thickness of the shell Δ and the volume fraction, ϕc, according to eqn (3). The density of the dispersant matrix (water) ρm = 0.998 g cm−3 and the shell (PVDF) ρs = 1.78 g cm−3 was held constant at curve fitting
| Sample | ρ c, bulk [g cm−3] | ρ c [g cm−3] |
|
σ [%] | Δ [nm] | ϕ c |
|---|---|---|---|---|---|---|
| CSP-1 | 1.18 | 1.60 ± 0.15 | 38.2 ± 0.7 | 12 | 4.0 | 0.30 |
| CSP-2 | 1.18 | 1.60 ± 0.05 | 45.1 ± 0.3 | 10 | 3.5 | 0.30 |
| CSP-3 | 0.805 | 1.50 ± 0.21 | 33.9 ± 1.0 | 14 | 3.5 | 0.29 |
| CSP-4 | 1.22 | 1.46 ± 0.04 | 45.2 ± 0.3 | 12 | 3.5 | 0.56 |
| CSP-5 | 1.22 | 1.60 ± 0.06 | 33.2 ± 0.4 | 12 | 3.8 | 0.30 |
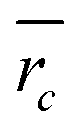
The density of the cores was in all cases found to be higher than that of the core material in the bulk form. This leads to the assumption that PVDF may be partially incorporated into the core. Then the volume fractions ϕc of PMMA, PGMA, and PMA, respectively, as the core material are less than 1. Their volume fractions were estimated according to eqn (3),
 | (3) |
The data in Table 2 show that the fraction of the acrylate or methacrylate monomer in the core is rather similar for all samples with the exception of sample CSP-4. The finding is rather surprising since the amounts of the (meth)acrylate monomer and VDF used during polymerization are quite different. The data may be interpreted, if it is assumed that VDF dissolves in the core particles at the beginning of the second stage of the polymerization. At this point, VDF is fed into the emulsion until a constant pressure is reached. It is known that in particular VDF and MMA units may show favorable interactions, which is the reason for rather good miscibility of PVDF and PMMA blends.18,19,42 Thus, it appears reasonable to assume VDF and PVDF solubility in the methacrylate core. The SAXS and TEM data strongly suggest that the core constitutes of a mixture of both polymers since no signs of segregation are seen. It may be anticipated that gaseous VDF enters into the particles. Upon entry of a radical formed in the aqueous phase, the VDF monomer in the core is polymerized leading to a mixture of PMMA and PVDF. In addition, small amounts of a statistical copolymer consisting of VDF and residual (meth)acrylate monomer are formed. The shell of the particle is clearly visible and suggests that the shell consists only of PVDF. This observation may be explained by reaching a solubility limit for VDF inside the core material.
Initially, within the experiments the amount of monomer for the core was varied to allow for the formation of different core sizes. The resulting core sizes range from 33 to 45 nm, with PVDF volume fractions inside the core of around 30%. Only in the case of CSP-4, a deviating PVDF volume fraction of 56% was reached. Thus, generally the core sizes do not scale with the amount of the (meth)acrylate monomer used.
Fig. 6 gives the SAXS results (scattering curves and curve fits applying a core shell model (grey symbols and red solid line, respectively)) and TEM images for CSP-1, CSP-3 and CSP-4. The systems may be described as follows: (a) the core radius formed by PMMA is rc = (38.2 ± 0.7) nm with a dispersity of 0.12. The shell formed by PDVF has a thickness of 4.0 nm and was held constant at curve fitting to avoid ambiguous results. The relative volume of the core is (74 ± 6) %. The TEM picture confirms a broad distribution of shell thicknesses. (b) The core radius formed by PGMA is rc = (33.9 ± 1.0) nm with a dispersity of 0.14. The shell formed by PDVF has a thickness of 3.5 nm and was held constant at curve fitting to avoid ambiguous results. The relative volume of the core to the total volume is (74 ± 11) %. The TEM picture displays a core–shell structure. (c) The core radius in the second system with PMA is rc = (45.2 ± 0.30) nm with a dispersity of 0.12 and the shell formed by PDVF has a thickness of 3.5 nm, which was held constant at curve fitting to avoid ambiguous results. The relative volume of the core to the total volume is (80 ± 2) %. The TEM picture displays a core–shell structure.
The analytical results given in Table 3 indicate that at least 75% of the CSP is given by the core. As discussed above the core is a mixture of both polymers. The pure PVDF shells show just very small differences. It has to be noted that the volume increase of the shell is not directly proportional to its thickness, but to the third power of the thickness (Δ3). These results indicate that DLS measurements can only provide a very general picture of the variation in hydrodynamic radii of the particles in total. For example, fine details such as growth of the core and the shell during the second stage of the polymerization were not accessible. All analytical methods discussed indicate that narrow particle size distributions were obtained, and indicated a uniform growth of all composite particles.
| Sample | F (β) |
|---|---|
| PVDF homopolymer | 0.31 |
| CSP-1 (PMMA core) | 0.58 |
| CSP-3 (PGMA core) | 0.45 |
| CSP-4 (PMA core) | 0.60 |
To obtain further information on the particles, DSC measurements were carried out. The data presented in Fig. 7 refer to the second heating cycle. The DSC curves show that the thermal behavior of the CSPs is rather close to that of the PVDF homopolymer, with the closest similarity observed for the PVDF homopolymer and the CSP system with PMMA. In all cases, a main melting peak is found, which is located between 167 and 169 °C for the CSP samples and at 171 °C for the PVDF homopolymer. In addition, the homopolymer and the system with PMMA show a smaller peak at 162 and 160 °C, respectively. The sample containing PGMA shows a small shoulder in the low temperature side of the peak, while the system with PMA shows a monomodal melting peak. It appeared important to consider the thermal behavior of the original material. As indicated by Fig. S1 of the ESI† samples CSP-2 and CSP-4 show only minor differences in the first and second heating curves. In contrast, Fig. 8 exhibits significant differences in both DSC curves for sample CSP-3 with PGMA. The first heating curve shows a well-resolved peak at 65 °C and a shoulder at 91 °C. The main peak occurs at 168 °C. Upon cooling, a strong crystallization peak is observed at 136 °C with a small shoulder at 129 °C and at 58 °C a minor peak may be identified. The second heating curve shows only a single peak at 168 °C. The material obtained under the same conditions as CSP-3, but with 15 g GMA, resulted in a first heating curve with an even stronger peak at around 68 °C, given by the dashed red line in the upper part of Fig. 8. The cooling and second heating curves are not significantly different from sample CSP-3.
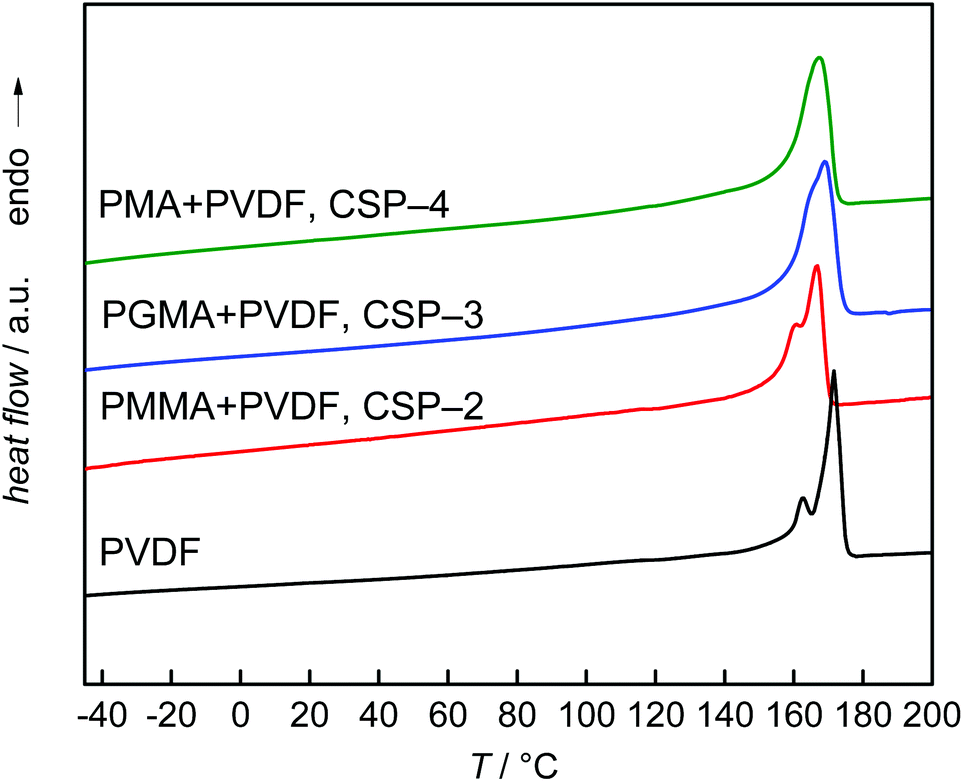 | ||
| Fig. 7 Second DSC heating curves of the PVDF homopolymer and samples CSP-2 to CSP-4. The DSC trace of pure PVDF is given for comparison. | ||
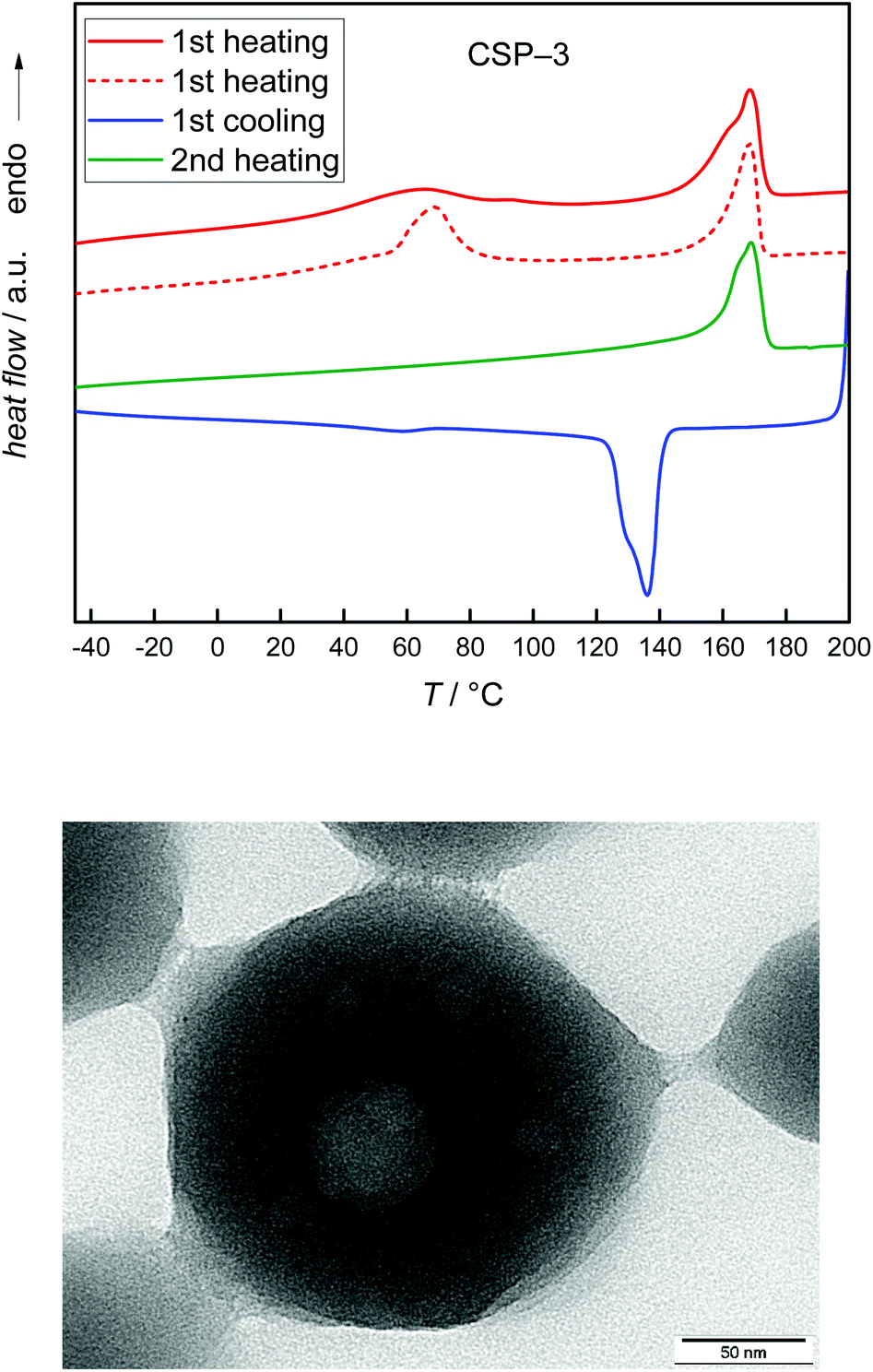 | ||
| Fig. 8 DSC curves of sample CSP-3 (top). The dashed line and the TEM image (bottom) refer to a CSP system synthesized with a higher amount of GMA. Further details are given in the text. | ||
Previously, Li et al. observed a somewhat similar behavior in a system consisting of a PVDF core with a PMMA shell prepared via blending of PVDF and PMMA lattices.16 Blending of lattices resulted in heterogeneous mixtures of PVDF-rich and PMMA-rich domains being associated with two melting peaks at 88 °C and 160 °C in the first heating run. The lower melting temperature, Tm, was explained with melting of imperfect small PVDF crystals, the higher Tm with melting of pure PVDF crystals. The second run showed only a single peak at 156 °C. Following the explanation given in ref. 16, a single Tm at high temperatures suggests that only large crystals of pure PVDF are present. If these findings are transferred to sample CSP-3, it may be suggested that firstly, a rather heterogeneous system with pure well-crystallized PVDF domains and some small imperfect PVDF crystals is present. Upon heating, intermixing occurs and the small imperfect PVDF crystallites disappear. In this context, the DSC results for CSP-2 and CSP-4 with PMMA and PMA suggest that only large PVDF crystals are present. Imperfect mixing induced small crystallites do not occur.
In addition, TGA measurements were carried out. The results shown in Fig. S3 of the ESI† indicate only minor differences between the composite particles and pure PVDF. In the presence of the non-fluorinated polymers PGMA (CSP-3) and PMA (CSP-4) slight decomposition of around 4% is already seen at 300 °C.
Together with the findings from SAXS the DSC data suggest that in the case of CSP-2 and CSP-4 well mixed cores consisting of both polymers with a shell of pure PVDF occur. CSP-3 is suggested to consist of a more heterogeneous core consisting of PGMA and PVDF surrounded by a shell of pure PVDF. This explanation is in line with the finding that some samples consisting of PVDF and larger amounts of PGMA lead to TEM images that show a core with some round inclusions (see the lower part of Fig. 8). Again a shell of PVDF was found. Furthermore, as mentioned above SAXS analyses of these samples applying a core shell structure were not successful. These significant differences between systems with PGMA and systems with PMMA or PMA are suggested to be due to differences in interactions between PVDF and the (meth)acrylate component.
Crystallinity of the shell PVDF
To obtain information on the crystallinity of the PVDF shell, FT-IR and XRD measurements were carried out. The FT-IR results for various CSPs are shown in Fig. 9. The characteristic peaks for the different phases are known from the literature.4,43 In the wavenumber range shown, the absorptions at 855 and 976 cm−1 are characteristic of the α-phase. The differentiation between β and γ phases solely based on IR spectra is not unambiguous. The polymer chain conformations are rather similar: the β phase is associated with an all trans (TTT) and γ phase with an T3GT3G′ conformation.4,44 While both crystal phases show an absorption at 840 cm−1, only the IR spectrum of the γ-phase shows a peak at 833 cm−1. The enlargement on the right hand side of Fig. 9 depicts that only at 840 cm−1 a signal is observed for the PVDF shell suggesting the presence of the β-phase material. The intensity of the peak is affected by the (meth)acrylate monomer used. Furthermore, the characteristic β peak at 1279 cm−1 is present. However, all spectra also show additional peaks that may be assigned to the α phase. In contrast, the IR spectrum of the PVDF homopolymer obtained from emulsion polymerization shows no signal at 840 cm−1. | ||
| Fig. 9 FT-IR results of PMMA + PVDF (CSP-1, red), PGMA + PVDF (CSP-3, green), PMA + PVDF (CSP-4, blue) and PVDF homopolymer (black dotted). (a) The vertical lines indicate peaks that are representative of α, β and γ crystalline phases of the PVDF shell, and (b) gives the characteristic γ and β region in detail. | ||
In order to estimate the fraction of α and β phase materials, the IR spectra were deconvoluted. For the deconvolution, it was assumed that the spectra only contain contributions from α and β phase PVDF. The resulting fraction of β phase material F(β) was calculated according to Gregorio46 and is listed in Table 3. The data indicate that the PVDF homopolymer contains the lowest fraction of the β phase material. The largest fraction of around 0.6 is found for PMMA and PMA in the core. The PGMA core leads to an intermediate β phase content. Thus, the presence of a poly(meth)acrylate core leads to a significant enhancement of the fraction of the β phase material.
For unambiguous identification of the β phase domains, additional XRD measurements were performed. The data in Fig. 10 show clear differences for the PVDF homopolymer and the core shell materials. According to Martins et al.4 the following 2θ values are characteristic of α, β and γ phase PVDF.4,45,46
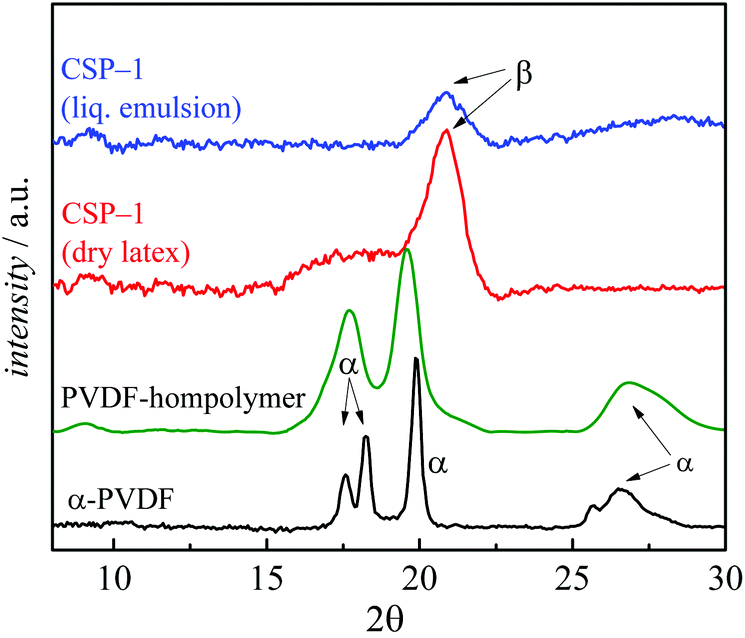 | ||
| Fig. 10 XRD results of CSP-1 measured in liquid emulsion (blue), as dry latex (red), PVDF homopolymer obtained by emulsion polymerization35,36 (green) and α phase PVDF-I13 as the reference obtained by polymerizations at high pressure in supercritical CO2 (black). | ||
2θ (α phase): 17.66°; 18.30°; 19.90°; 26.56°.
2θ (β phase): 20.26°.
2θ (γ phase): 18.5°; 19.2°; 20.04°.
While the PVDF spectrum shows only peaks characteristic of the α phase, the data for CSP-1 show a distinct peak at 20.26°, and the peak at around 26.56° typical of the α phase is absent. Especially clear is the formation of the β phase in the PVDF shell for the shown PMMA-PVDF CSP-1. The particles consist of ∼40% PMMA and 60% PVDF which leads to a thin shell. While the PVDF homopolymer still exhibits significant peaks from the α phase, the β phase dominates in the measurement of the liquid emulsion of CSP-1. This finding shows that the β phase is already present in the emulsion particles and is not due to film formation or drying of the emulsion. The XRD curve of the dried particles indicates that the peak for the β phase becomes clearer, while an additional broad peak occurs between 16.6° and 19.2° for 2θ. This additional is presumably a mixture of α and γ phases. The XRD results confirm that the particles contain mixtures of the different crystalline phases, with the β phase being dominant.
Conclusions
Core–shell polymer particles consisting of PVDF and PMMA, PGMA or PMA were prepared. Firstly, the non-fluorinated monomer was polymerized in the emulsion employing the fluorinated stabilizer ammonium 4,8-dioxa-3H-perfluorononanoate (ADONA). Dispersions with uniform spherical particles were accessible. Without any work-up, the VDF monomer was added to the reactor and the semi-batch polymerization of VDF was carried out leading to particles of a similar shape and size. TEM images clearly show a core shell structure of the material. SAXS analyses indicate that the core constitutes not only of the non-fluorinated polymer. The data suggest that a homogeneous mixture of both polymers is obtained in the core. The cores have a diameter of around 40 nm independent of the polymerization recipe. TEM images and SAXS results indicate that the core is surrounded by a shell consisting of PVDF, which is between 3.5 and 4.0 nm thick.The PVDF crystallinity was investigated via FTIR and XRD. The analyses show that the core–shell particles contain a large fraction of β phase PVDF. This holds true for the original latex, but also for the dried film.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge: Petra Fengler and Kerstin Brademann-Jock (Federal Institute for Materials Research and Testing (BAM)), for help in SAXS measurements, Dipl.-Min. Peggy Knospe (Institut für Mechanische Verfahrenstechnik, TU Clausthal) for TEM measurements, PD. Dr Jörg Adams (Institut für Physikalische Chemie, TU Clausthal) for XRD measurements and Dr Harald Kaspar (3 M/Dyneon GmbH) for provision of VDF and ADONA.References
- G. K. Kostov, L. Sauguet, B. Ameduri, H. Kaspar, T. Zipplies and K. Hintzer, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3964 CrossRef CAS.
- R. Dams and K. Hintzer, in Fluorinated Polymers, Volume 2: Applications, ed. B. Ameduri and H. Sawada, The Royal Society of Chemistry, Cambridge, 2017, vol. 24, pp. 1–31 Search PubMed.
- B. Ameduri, Chem. Rev., 2009, 109, 6632 CrossRef CAS PubMed.
- P. Martins, A. C. Lopes and S. Lanceros-Mendez, Prog. Polym. Sci., 2014, 39, 683 CrossRef CAS.
- T. Soulestin, V. Ladmiral, F. D. Dos Santos and B. Améduri, Prog. Polym. Sci., 2017, 72, 16 CrossRef CAS.
- R. Hasegawa, M. Kobayashi and H. Tadokoro, Polym. J., 1972, 3, 591 CrossRef CAS.
- A. Lovinger and D. Reed, Macromolecules, 1980, 13, 989 CrossRef CAS.
- P. Martins, A. Lasheras, J. Gutierrez, J. M. Barandiaran, I. Orue and S. Lanceros-Mendez, J. Phys. D: Appl. Phys., 2011, 44, 495303 CrossRef.
- P. Martins, X. Moya, L. C. Phillips, S. Kar-Narayan, N. D. Mathur and S. Lanceros-Mendez, J. Phys. D: Appl. Phys., 2011, 44, 482001 CrossRef.
- C. Muralidhar and P. Pillai, Mater. Res. Bull., 1988, 23, 323 CrossRef CAS.
- M. Sharma, J. K. Quamara and A. Gaur, J. Mater. Sci.: Mater. Electron., 2018, 29, 10875 CrossRef CAS.
- F. Bai, G. Chen, M. Nie and Q. Wang, RSC Adv., 2015, 5, 54171 RSC.
- N. Golzari, J. Adams and S. Beuermann, Polymers, 2017, 9, 306 CrossRef.
- K. Koga and H. Ohigashi, J. Appl. Phys., 1986, 59, 2142 CrossRef CAS.
- H. S. Nalwa and M. Dekker, Ferroelectric Polymers, Crc Press Inc, New York, 1995 Search PubMed.
- Y. Li, G. Zhang, S. Song, H. Xu, M. Pan and G.-J. Zhong, Polymers, 2017, 9, 448 CrossRef.
- D. Yang, H. Xu, W. Yu, J. Wang and X. Gong, J. Mater. Sci.: Mater. Electron., 2017, 28, 13006 CrossRef CAS.
- T. Nishi and T. T. Wang, Macromolecules, 1975, 8, 909 CrossRef CAS.
- C. Leonard, J. L. Halary and L. Monnerie, Macromolecules, 1988, 21, 2988 CrossRef CAS.
- W. Yan, M. Pan, J. Yuan, G. Liu, L. Cui, G. Zhang and L. Zhu, Polymer, 2017, 122, 139 CrossRef CAS.
- M. Pan, L. Yang, B. Guan, M. Lu, G. Zhong and L. Zhu, Soft Matter, 2011, 7, 11187 RSC.
- M. Pan, L. Yang, J. Wang, S. Tang, G. Zhong, R. Su, M. K. Sen, M. K. Endoh, T. Koga and L. Zhu, Macromolecules, 2014, 47, 2632 CrossRef CAS.
- N. Durand, P. Gaveau, G. Silly, B. Améduri and B. Boutevin, Macromolecules, 2011, 44, 6249 CrossRef CAS.
- N. Durand, B. Boutevin, G. Silly and B. Améduri, Macromolecules, 2011, 44, 8487 CrossRef CAS.
- N. Badi, R. Mekala, S. Khasim, A. S. Roy and A. Ignatiev, J. Mater. Sci.: Mater. Electron., 2018, 29, 10593 CrossRef CAS.
- T. Chen, Q. Tang, B. Wang, Y. Li and L. Liu, Mater. Lett., 2015, 159, 413 CrossRef CAS.
- D. Mondal, A. L. Gayen, B. K. Paul, P. Bandyopadhyay, D. Bera, D. S. Bhar, K. Das, P. Nandy and S. Das, J. Mater. Sci.: Mater. Electron., 2018, 321, 651 Search PubMed.
- L. Xie, X. Huang, K. Yang, S. Li and P. Jiang, J. Mater. Chem. A, 2014, 2, 5244 RSC.
- T. Chen and B. Liu, Mater. Lett., 2018, 210, 165 CrossRef CAS.
- M. S. Sebastian, A. Larrea, R. Gonçalves, T. Alejo, J. L. Vilas, V. Sebastian, P. Martins and S. Lanceros-Mendez, RSC Adv., 2016, 6, 113007 RSC.
- W.-H. Li and H. D. H. Stöver, Macromolecules, 2000, 33, 4354 CrossRef CAS.
- A. Bergmann, D. Orthaber, G. Scherf and O. Glatter, J. Appl. Crystallogr., 2000, 33, 869 CrossRef CAS.
- D. Orthaber, A. Bergmann and O. Glatter, J. Appl. Crystallogr., 2000, 33, 218 CrossRef CAS.
- I. Bressler, J. Kohlbrecher and A. F. Thünemann, J. Appl. Crystallogr., 2015, 48, 1587 CrossRef CAS PubMed.
- F. Brandl, M. Drache and S. Beuermann, Polymers, 2018, 10, 1008 CrossRef.
- F. Brandl and S. Beuermann, Chem. Ing. Tech., 2018, 90, 372 CrossRef CAS.
- S. Banerjee, J. Schmidt, Y. Talmon, H. Hori, T. Asai and B. Ameduri, Chem. Commun., 2018, 54, 11399 RSC.
- S. Pabisch, B. Feichtenschlager, G. Kickelbick and H. Peterlik, Chem. Phys. Lett., 2012, 521, 91 CrossRef CAS PubMed.
- R. Brydson, A. Brown, C. Hodges, P. Abellan and N. Hondow, J. Microsc., 2015, 260, 238 CrossRef CAS PubMed.
- S. Bhattacharjee, J. Controlled Release, 2016, 235, 337 CrossRef CAS PubMed.
- P. Bartlett and R. H. Ottewill, Colloidal Crystal, 1992, 96, 3306 CAS.
- W. Fan and S. Zheng, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 2580 CrossRef CAS.
- T. Boccaccio, A. Bottino, G. Capannelli and P. Piaggio, J. Membr. Sci., 2002, 210, 315 CrossRef CAS.
- A. J. Lovinger, Macromolecules, 1982, 15, 40 CrossRef CAS.
- R. Gregorio, J. Appl. Polym. Sci., 2006, 100, 3272 CrossRef CAS.
- L. Priya and J. P. Jog, J. Appl. Polym. Sci., 2003, 89, 2036 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8py01236a |
| This journal is © The Royal Society of Chemistry 2018 |