
A versatile synthetic platform for amphiphilic nanogels with tunable hydrophobicity†

Abstract
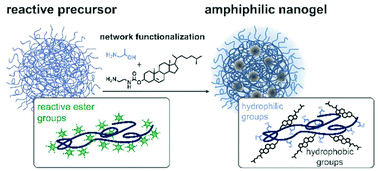
We present a new versatile synthetic strategy for amphiphilic nanogels with tunable amphiphilicity. Our approach is based on crosslinked reactive precursor particles, which serve as a platform for secondary functionalization with hydrophilic and hydrophobic moieties: the functionalization of reactive poly(pentafluorophenyl methacrylate) (PPFPMA) nanogels with different amines allows precise adjustment of the internal network composition. Since nanogels with varying amphiphilicity originate from the same uniform precursor particles, they exhibit similar particle sizes, size distributions and homogeneous morphologies. This comparability between nanogels allows accurate investigation of their structure–property relationships. For example, by tuning the amphiphilic network composition, the loading and release profiles for Nile red as a hydrophobic model compound can be controlled through adjusting the hydrophobic interactions with the network. Consequently, our strategy represents a powerful platform for the generation of a new type of amphiphilic nanocarrier that combines the exceptional biocompatibility of hydrophilic nanogels with the transport of hydrophobic cargoes.
 Please wait while we load your content...
Please wait while we load your content...

