
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
MALDI-LID-ToF/ToF analysis of statistical and diblock polyacrylate copolymers†
James S.
Town
 ,
Glen R.
Jones
and
David M.
Haddleton
*
,
Glen R.
Jones
and
David M.
Haddleton
*
Department of chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: d.m.haddleton@warwiick.ac.uk
First published on 7th September 2018
Abstract
We report the use of MALDI-LID-ToF/ToF utilising the laser induced dissociation (LID) fragmentation technique, which has been almost exclusively applied to protein/peptide analysis to date. MALDI-LID-ToF/ToF is used as a powerful tool to give information of monomer distributions in synthetic co-poly(acrylates). Analysis of a range of poly(acrylate) homopolymers by LID have found slight improvement over CID, with less rearrangements in the poly(butyl acrylate) fragmentation and more backbone cleavages. Analysis of both block and statistical copolymers, when compared to homopolymer analogues, yield markedly different spectra which can be correlated to give some monomer sequence information, thus achieving a wealth of structural information from two samples which would otherwise be identical by single stage MALDI-ToF analysis. The determination of fragmentation pathways of polymers with varying end group lability and monomer structure has allowed for high-resolution microstructural determination at the block boundary, showing even small amounts of mixing at the block boundary.
Introduction
Matrix assisted laser desorption ionization (MALDI) is an ionization technique that has shown wide use in the analysis of polymers,2–4 including acrylates,5 and polymers prepared by living radical polymerization.6–16 MALDI-ToF has been utilized to give values for the molecular weight,17,18 information on polymer end groups,19–24 as well as to probe the mechanisms of polymer synthesis.25–28 It is a soft ionization technique where fragmentation and double charged species are rare,29,30 allowing for good ionization of low to medium weight non-volatile compounds. For matrix assisted laser desorption time of flight mass spectrometry (MALDI-ToF-MS) obtaining a clear spectra of single oligomers, with isotopic resolution, in a polymer mixture can be achieved with relative ease. Copolymers, however, often present a much more significant challenge with several difficulties which require more complex analysis to overcome.27,31 Such difficulties include; overlapping peaks, different ionization regimes between co-monomers32 and determining the orientation and position of the monomers along the backbone and hence the type of copolymer.33 It is also noted that the experiment is highly mass sensitive due to issues with both ionization and detector mass discrimination effects in an ion detector and other causes for shifts in polymer mass distributions. This leads to a loss of sensitivity as mass increases which is exacerbated as the experiment detects numbers as opposed to weights of molecules leading to number average distributions.34 MALDI-ToF/ToF is a tandem mass spectrometry technique which uses two stages of acceleration voltage to produce fragmented daughter ions of isolated precursor ions within a selected m/z window.35 Tandem mass spectrometry techniques have been applied extensively to homopolymer compounds and the determination of fragmentation patterns of many polymers have been studied.36 When applied to homopolymers it can be used to further confirm certain structural information such as end groups,37 post polymerization backbone modifications38 and differentiate between linear and cyclic polymers.39 When applied to copolymers, however, it has mostly been applied to block copolymers,40–44 usually determining the length of the blocks.45,46 There have been previous studies, which have used various MS/MS techniques to determine the microstructure of polymers. Previous studies have used techniques including MALDI – post source decay,47 ESI – electron capture dissociation,48 MALDI – collision induced dissociation,49 and ESI – collision induced dissociation.50–52 However, nothing has been published on the use of laser-induced dissociation with polymer compounds.Previously MALDI-LID-ToF/ToF has been utilized for biomacromolecules, such as proteins1 and oligosaccharides.53 The technique utilizes the metastable nature of compounds containing a charged adduct, by accelerating the species slowly at first the molecules undergo fragmentation and rearrangement as they exist beyond their metastable lifetime. Subsequently the fragments are isolated and reaccelerated, they are then detected in the same way as any ToF technique.54 This technique has been compared to the collision induced dissociation (CID) technique, which is much more popular for polymers.55 In this comparison it was found that laser induced dissociation (LID) produced similar structural fragments to collision induced dissociation, however less non-structural fragments were observed in the laser induced dissociation when compared to the collision induced dissociation.
Herein, we describe an approach for the analysis of diblock and statistical poly(methyl acrylate-co-ethyl acrylate) copolymers using matrix assisted laser desorption/ionization laser induced dissociation tandem time of flight mass spectrometry (MALDI-LID-ToF/ToF). We demonstrate the power of MALDI-LID-ToF/ToF as a technique for the characterization of copolymers. We have investigated how labile end groups affect fragmentation patterns, how their fragmentation patterns differ based on their co-monomer arrangement and observed the complexities of copolymer analysis by tandem mass spectrometry. Copolymers have been synthesized via a copper mediated photo-controlled radical polymerization, this technique was chosen due to the; high conversion, high end group fidelity, good molecular weight control, and very low polydispersity, associated with photomediated controlled radical polymerizations.56,57 This is the first report of the use of MALDI-LID-ToF/ToF in synthetic copolymer analysis to the best of our knowledge.
Methods
Mass spectrometry
MALDI-ToF and MALDI-LID-ToF/ToF experiments were carried out using an UltrafleXtreme MALDI-ToF/ToF analyser (Bruker, Coventry, UK), equipped with a nitrogen 337 nm laser and a high resolution ion gate for isolation in the collision cell. The MALDI-ToF experiments were carried out in reflectron positive ion mode, with an acceleration voltage of 19 kV. All measurements were recorded with 500 shots per measurement, with each spectra being the average of 5000 shots. Calibration was achieved with PEG 2000 (Sigma-Aldrich), with an average mass accuracy of 0.02 Daltons.MALDI-LID-ToF/ToF experiments were carried out in reflectron positive mode, with an ion source voltage of 8 kV, and a post fragmentation cell acceleration voltage of 19 kV. Each spectra contains a single 200 shot precursor ion measurement, and 20, 500 shot fragment ion measurements, for a total of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 fragment ion measurements averaged into the spectra. The precursor ion selector (PCIS) window was set between −2 and 10 Daltons.
200 fragment ion measurements averaged into the spectra. The precursor ion selector (PCIS) window was set between −2 and 10 Daltons.
Sample preparation
Poly(methyl acrylate), poly(ethyl acrylate), poly(n-butyl acrylate), poly(iso-butyl acrylate), poly(methyl acrylate-b-ethyl acrylate) and poly(methyl acrylate-co-ethyl acrylate) were all prepared in a tetrahydrofuran (THF) matrix solution at 10 mg ml−1 concentrations. The THF matrix solution contained; 40 mg ml−1trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB), 0.1 mg ml−1 sodium iodide. 0.5 μl of each sample was then spotted onto a MTP 384 ground steel target plate (Bruker, Coventry, UK).Results and discussion
Synthesis of polymers
In order to ascertain whether tandem MALDI-ToF analysis could reveal structural information on copolymer composition a number of model polymers were synthesized via a photomediated polymerization in the presence of Cu(II)Br2 and Me6Tren (Scheme 1).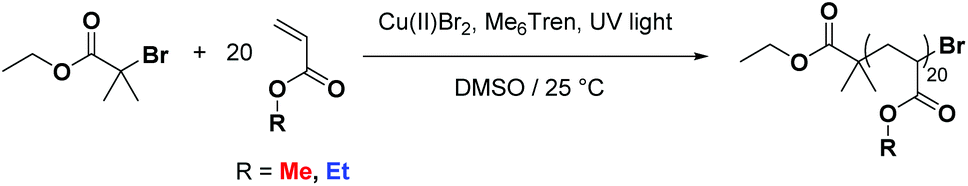 | ||
| Scheme 1 General scheme showing the photomediated polymerization of acrylate monomers as used in this study. | ||
The co-monomer system comprising of methyl acrylate (MA) and ethyl acrylate (EA) was chosen as they were expected to have reactivity ratios of ∼1, thus a statistical copolymerization should yield an essentially random monomer distribution. In addition to this controlled polymerization of these monomers is well-reported, thus well-defined block copolymers can be readily prepared. The targeted degree of polymerization for all polymers was selected as DP20, as this is in an ideal mass range for MALDI-ToF MS analysis, Fig. 1: homopolymers of both methyl acrylate and ethyl acrylate, as well as a statistical copolymer of the two and a block copolymer. The statistical copolymer was prepared by polymerizing a 1:1 molar ratio of MA and EA. The block copolymer was prepared by first polymerizing a homopolymer of MA followed by in situ addition of EA once conversion was determined to be over 98%. Full experimental details can be found in the ESI.†
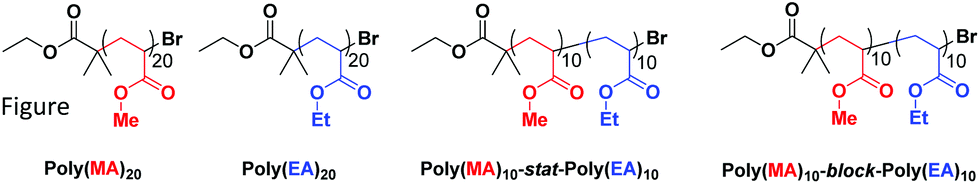 | ||
| Fig. 1 Polymers used in this study: poly(methyl acrylate)20 homopolymer, poly(ethyl acrylate)20 homopolymer, poly(methyl acrylate)10-stat-poly(ethyl acrylate)10 statistical copolymer, poly(methyl acrylate)10-block-poly(ethyl acrylate)10 block copolymer. | ||
MALDI-ToF and MALDI-LID-ToF/ToF data
MALDI-ToF spectra of the homopolymer displays the lability of the bromine ω-end group. The highest intensity series of peaks throughout the majority of the spectra corresponds to a bromine terminated poly(methyl acrylate) as a Na+ adduct as expected, Fig. 2. However, there are other peaks present in the spectra, which are assigned as H terminated poly(methyl acrylate). There is also some evidence of K+ adducts of both the bromine and hydrogen terminated polymers. It has been shown previously that conventional MALDI-ToF MS has no effect on the labile end groups and we have previously reported many spectra of polymers prepared by copper mediated polymerization with similar end groups which are stable under these conditions in the mass spectrometer.22,58–65 Thus, the H terminated polymers are present in the sample prior to the measurement due to side reactions in the chemistry. For the purposes of this work the presence of these impurities is of no concern. MALDI-LID-ToF/ToF spectra of the peak at m/z = 2107 (Fig. 2B), corresponding to a bromine terminated poly(methyl acrylate) homopolymer with DP = 22, was recorded (Fig. 2C & D).
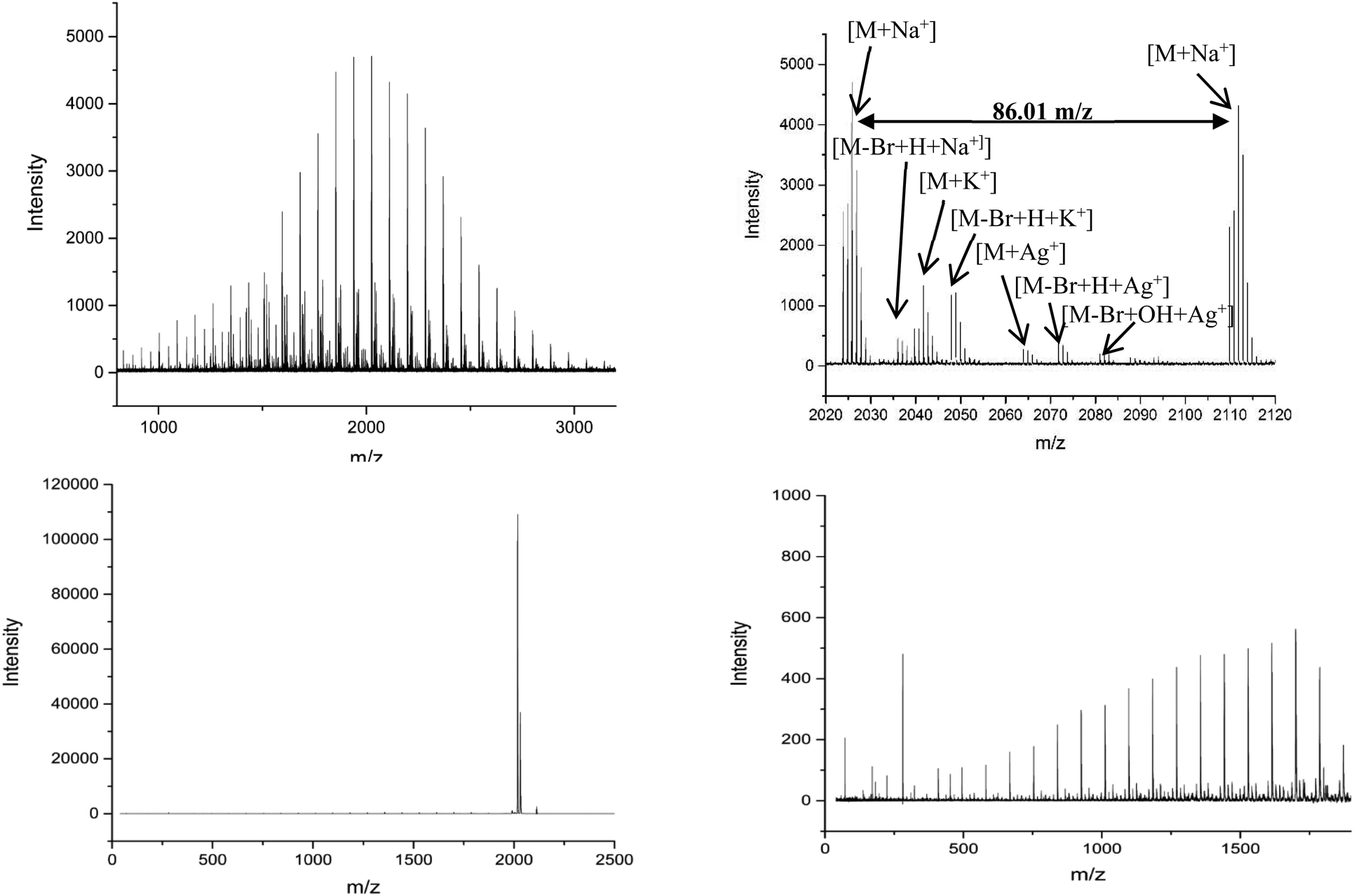 | ||
| Fig. 2 (A) (Top Left): Full MALDI-ToF spectra of poly(methyl acrylate) homopolymer. (B) (Top Right) Zoom of the MALDI-ToF results in the 2020–2120 m/z range showing issues with end group fidelity and K+ adducts. (C) (Bottom Left) Full spectra of the MALDI-LID-ToF/ToF, the large peak shows the loss of the end group but very little else. (D) (Bottom Right) Zoom in on the spectra, excluding the large peak for the loss of end group, showing the backbone cleavages described using Scheme 1. | ||
The lability of the bromine end group results in a spectrum which only contains △ chains (mechanism shown in Scheme 2, full analyzed spectra shown in Fig. 3), and some secondary fragments in the low molecular weight region (<500 Da). The main series of fragments, labelled △, is indicative of a vinyl CH2 terminated fragment. Due to these chains arising from secondary fragmentation, this implies that secondary fragment ions prefer the form shown in Scheme 2 as ○. This would lead to the radical on the –CH2 form on the △ chains via a proposed mechanism shown in Scheme 2; note this might only apply for polyacrylates with labile ω end groups such as secondary bromides. These chains then lose a hydrogen radical to produce a vinyl bond with the extra CH2. The peaks of significantly less intensity assigned to extra CH2 chains forming from a transfer of the radical to other acrylate molecules on the backbone, leading to a radical acrylate leaving the backbone and hence giving chains of CH2 double bonded to CH. This process seems to be more energetically unfavorable to the loss of a hydrogen radical. The peaks corresponding to the secondary fragment ○, at 281.156 and 453.047 m/z, are of particularly high intensity. While ○ fragments do exist at higher masses in the poly(methyl acrylate), they are much lower intensity peaks, which could imply that many of our smaller ○ chains are actually the product of three fragmentations or more.
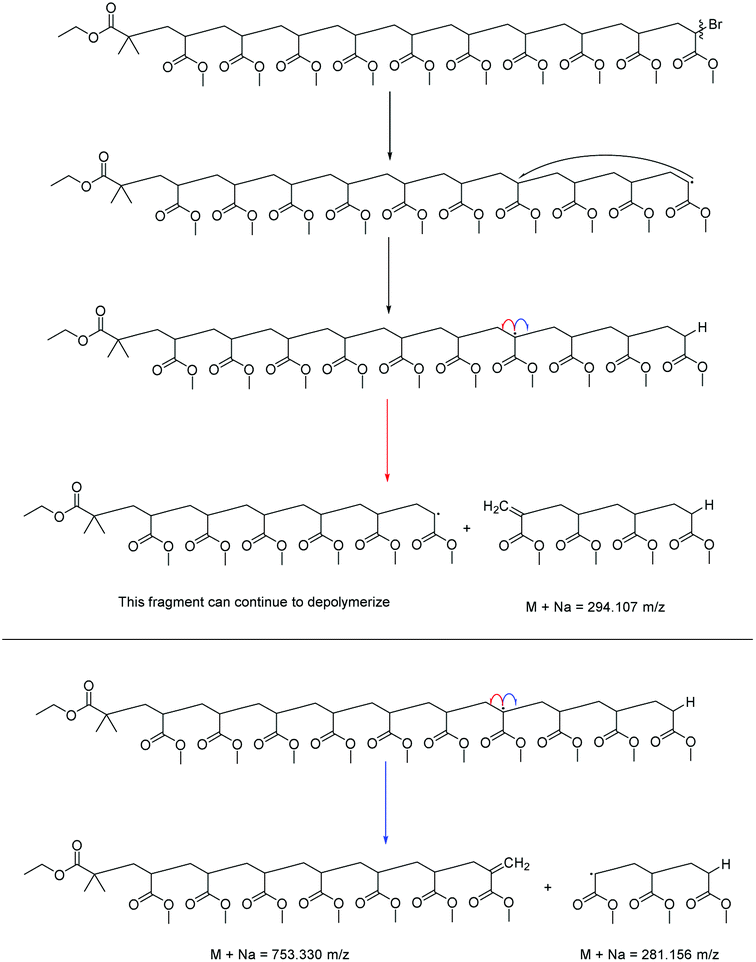 | ||
| Scheme 2 Fragmentation pathways observed for the MALDI-LID-ToF/ToF of acrylate homopolymers and copolymers. The △ and ○ species are the only two observed in the spectra, as they are the only “none radical”, and hence stable, species that can come from the radical backbiting regime, which drives the fragmentation. | ||
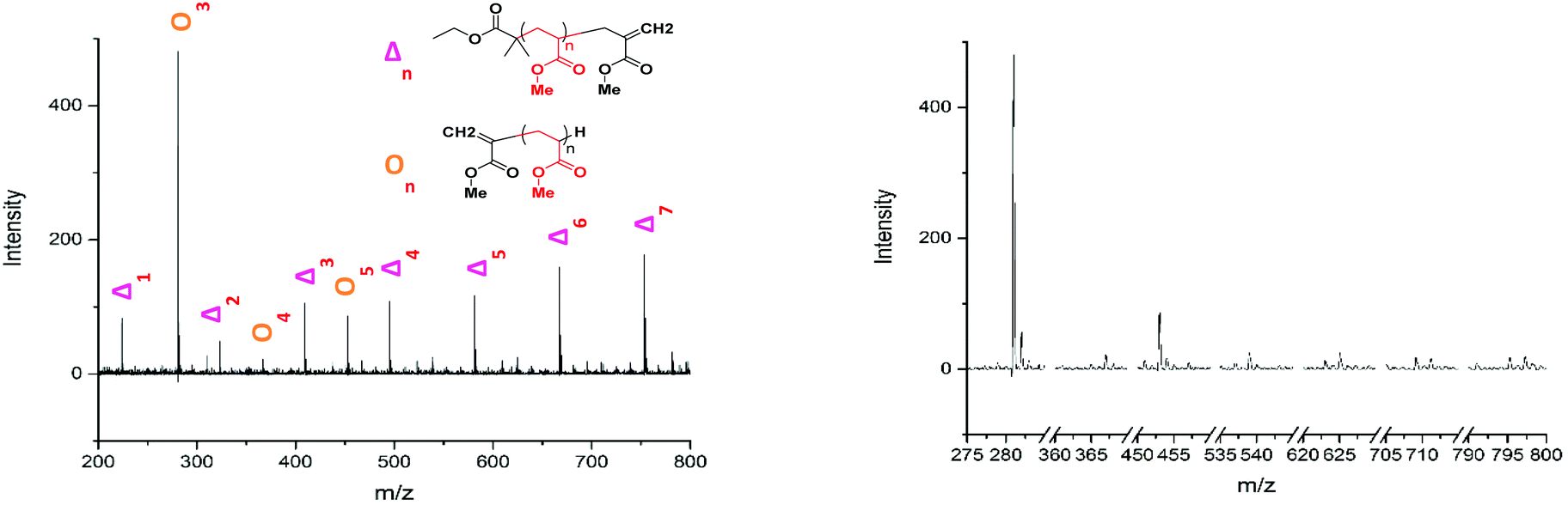 | ||
Fig. 3 (A) Assignment of the 200 to 800 m/z range of the poly(methyl acrylate) homopolymer. Beyond this point the peaks continue as a major series of 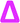 fragments of increasing repeat unit. (B) Comparison of all the fragments of increasing repeat unit. (B) Comparison of all the  fragment chains from one and other. fragment chains from one and other. | ||
A poly(ethyl acrylate) homopolymer was then examined to confirm that the fragmentation pathway is the same as the poly(methyl acrylate). The conventional MALDI-ToF spectra, Fig. 4A, showed only bromine terminated poly(ethyl acrylate). The peak at m/z = 2618 was isolated and a MALDI-LID-ToF/ToF spectrum taken. The pathway is the same as the poly(methyl acrylate), Scheme 2, however, the ○ fragments of the poly(ethyl acrylate) homopolymer tend to be larger, as there are high intensity peaks at 323, 523, 723 and 923 m/z, with slightly lower intensity peaks at 423, 623 and 823 m/z (Fig. 5B & D). These peaks express that the radical backbiting process tends to pass the radical further down the chain when compared to the poly(methyl acrylate) homopolymer. This is believed to be due to the increased steric hindrance the longer alkyl (R) chain (Fig. 1) provides, and hence it is therefore more energetically unfavorable to undergo the backbiting process close to the original radical formation.
 | ||
| Fig. 4 (A) Full MALDI-ToF spectra of the poly(ethyl acrylate). (B) Expansion of the 2400–2550 m/z, showing the repeat unit and that this is the only series in the sample. This means that there have been no changes to the end group configuration, such as those seen in the poly(methyl acrylate). | ||
 | ||
Fig. 5 (A) (Top Left) Full MALDI-LID-ToF/ToF spectra of the poly(ethyl acrylate) homopolymer, the large few peaks are loss of bromine end group. (B) (Top Right) Expansion to exclude the large end group peak and showing the backbone cleavages. (C) (Bottom Left) expansion of the 200–1000 m/z region, showing the assignment as similar as poly(methyl acrylate) but with higher mass  fragments. (D) (Bottom Right) isolating those fragments only. fragments. (D) (Bottom Right) isolating those fragments only. | ||
In an attempt to characterize this MALDI-ToF and MALDI-LID-ToF/ToF spectra of two different poly(butyl acrylate) homopolymers was recorded. Both poly(n-butyl acrylate) and poly(iso-butyl acrylate) were used so as to investigate any steric effects from the R group while keeping the other chemical properties as similar as possible.
These polymers were synthesized with a chlorinated end group from a chlorine containing initiator, this is due to the 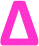 end group of the EBIB (ethyl α-bromoisobutyrate) synthesized polymers has the same empirical formula as a repeat unit of butyl acrylate. This causes the secondary fragment
end group of the EBIB (ethyl α-bromoisobutyrate) synthesized polymers has the same empirical formula as a repeat unit of butyl acrylate. This causes the secondary fragment  to overlap with the first
to overlap with the first 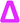 fragment, and hence methyl-2-chloropropionate was used as an initiator to synthesize these two poly(butyl acrylate) polymers.
fragment, and hence methyl-2-chloropropionate was used as an initiator to synthesize these two poly(butyl acrylate) polymers.
Both MALDI-ToF spectra showed that the polymers were similar in terms of mass distribution. Chlorine terminated polymer chains form the highest intensity peaks in the spectra, with a smaller secondary distribution of hydrogen-terminated chains. The 2578 m/z peak, was isolated from both the poly(n-butyl acrylate) and poly(iso-butyl acrylate) polymers, for MALDI-LID-ToF/ToF measurement.
The fragmentation pathways were the same as found for poly(methyl acrylate), Scheme 2, indicating that the ω end group has little effect on the overall fragmentation pathway. However, examining the ○ fragments of the two spectra, there does not appear to be the high intensity m/z peaks which were found in the poly(ethyl acrylate) homopolymer. Also, if we compare the intensity of the ○ fragments in the poly(n-butyl acrylate) and the poly(iso-butyl acrylate) spectra, we find that there is no notable difference between the ○ fragments of the two different structural isomers of poly(butyl acrylate), Fig. 6B & D. Therefore we are unable to confirm that the steric hindrance is what causes the increase in secondary fragment size found in the poly(ethyl acrylate). This does not affect copolymer analysis, but is something to take into consideration during assignment.
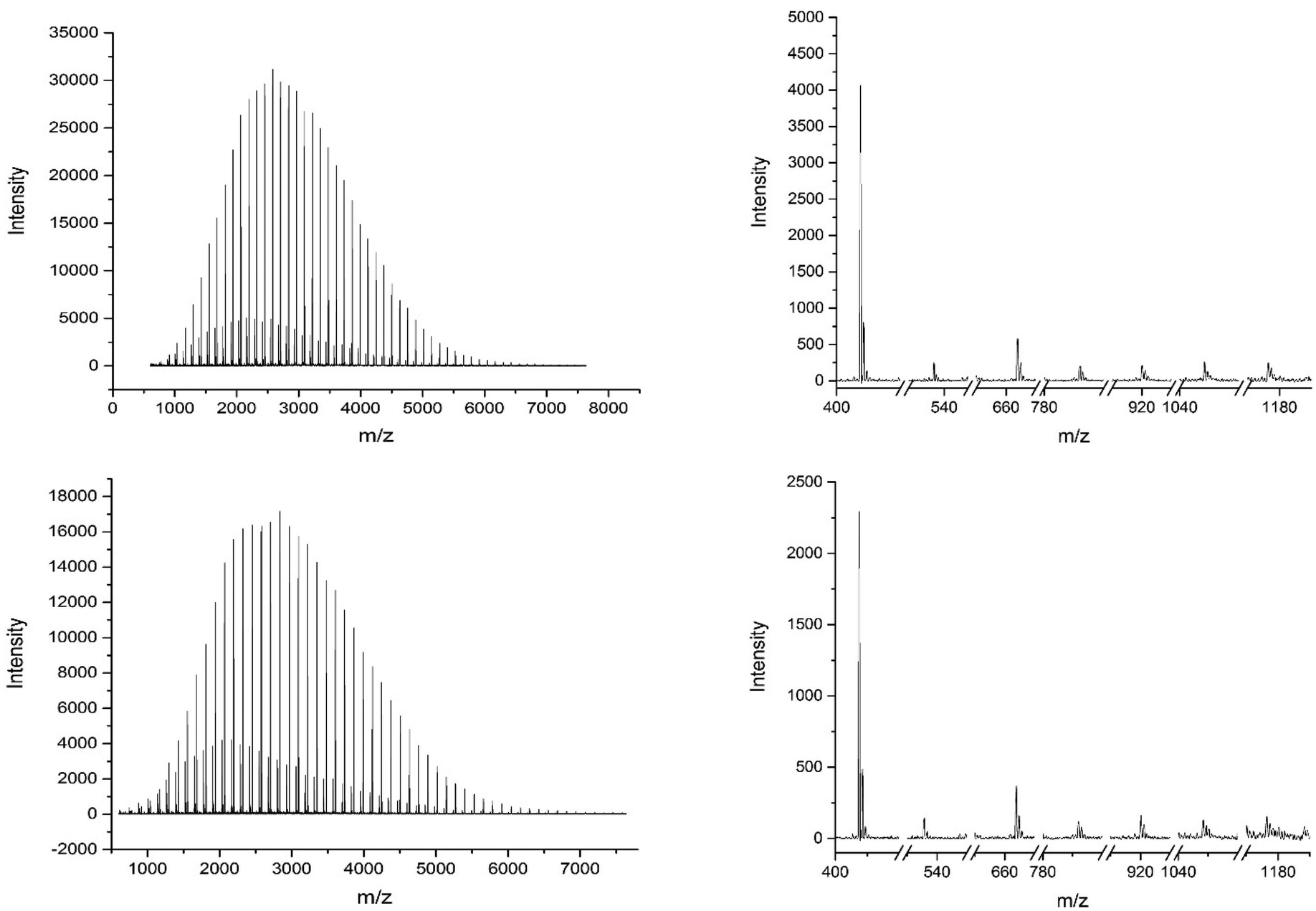 | ||
Fig. 6 (A) (Top Left) MALDI-ToF of the poly(n-butyl acrylate) homopolymer. (B) (Top Right) The  fragments from the MALDI-LID-ToF/ToF of the poly(n-butyl acrylate) homopolymer. (C) (Bottom Left) MALDI-ToF of the poly(iso-butyl acrylate) homopolymer. (D) (Bottom Right) The fragments from the MALDI-LID-ToF/ToF of the poly(n-butyl acrylate) homopolymer. (C) (Bottom Left) MALDI-ToF of the poly(iso-butyl acrylate) homopolymer. (D) (Bottom Right) The  fragments from the MALDI-LID-ToF/ToF of the poly(iso-butyl acrylate) homopolymer. fragments from the MALDI-LID-ToF/ToF of the poly(iso-butyl acrylate) homopolymer. | ||
The homopolymers can be compared to those found in literature for polyacrylates. The break down pattern for poly(methyl acrylate) is similar to that found by collision induced dissociation (CID), with more of an emphasis on the secondary fragments due to the labile end group and the focus on radical backbiting. The poly(butyl acrylates), however, undergo much fewer rearrangements and instead provide more backbone cleavages.66
 | ||
| Fig. 7 (A) Full MALDI-ToF spectra of the poly (methyl acrylate-b-ethyl acrylate) copolymer. (B) Expansion of the MALDI-ToF spectra in the 2220–2340 m/z region, showing the comonomer distribution for DP 22. | ||
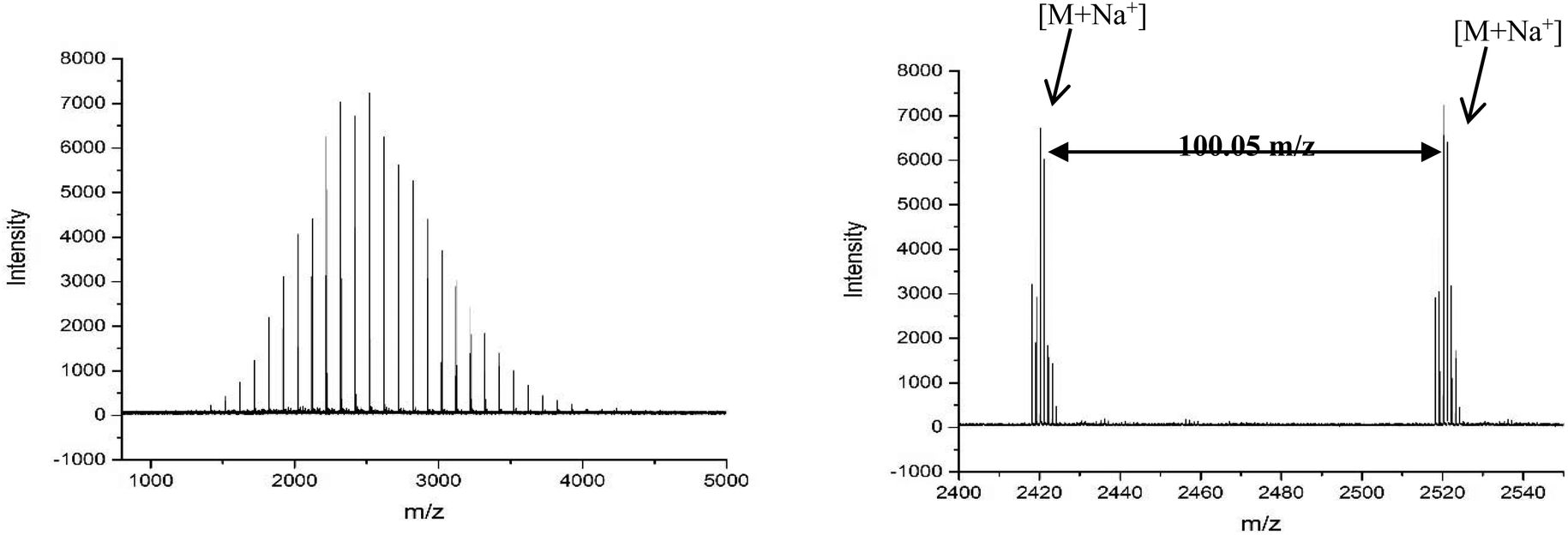
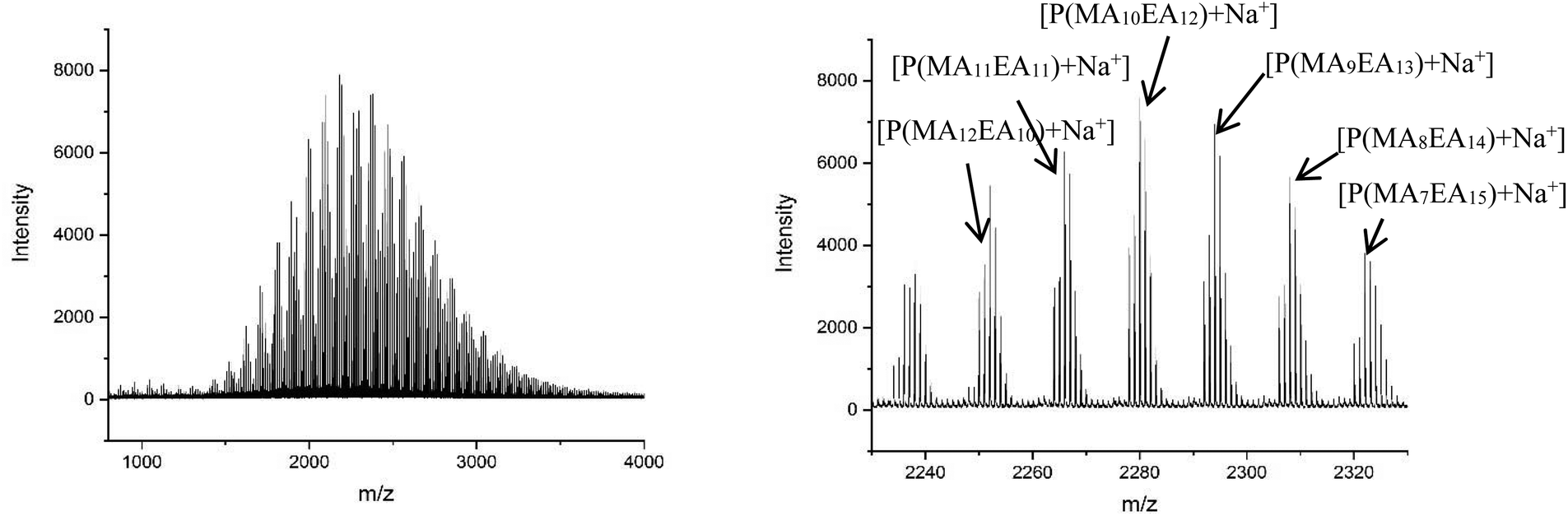
The MALDI-LID-ToF/ToF was taken of the 2278 m/z peak (Fig. 8), which assigns as poly(methyl acrylate10-b-ethyl acrylate12). The fragmentation pathways appear to be the same as for the homopolymers. However, there are two distinct distributions found in the spectra, where the difference between the △ chains as the m/z increases changes from 86 Daltons to 100 Daltons. These distributions represent two distinct monomer blocks, which are present in the polymer chains, one from methyl acrylate, and one of ethyl acrylate. This indicates that the co-monomers in the sample are arranged with a diblock structure. It is also possible to determine the length of each monomer block, as the difference between the △ chains changes at the m/z representing 10 repeat units and 11 repeat units, indicating that the original assignment is correct and this arises from a poly(methyl acrylate10-b-ethyl acrylate12) chain.
 | ||
| Fig. 8 (A) Full MALDI-LID-ToF/ToF spectra of the poly(methyl acrylate-b-ethyl acrylate) copolymer. (B) Expansion of the backbone cleavages to show the presence of a block of methyl acrylate monomers followed by a block of ethyl acrylate monomers. | ||
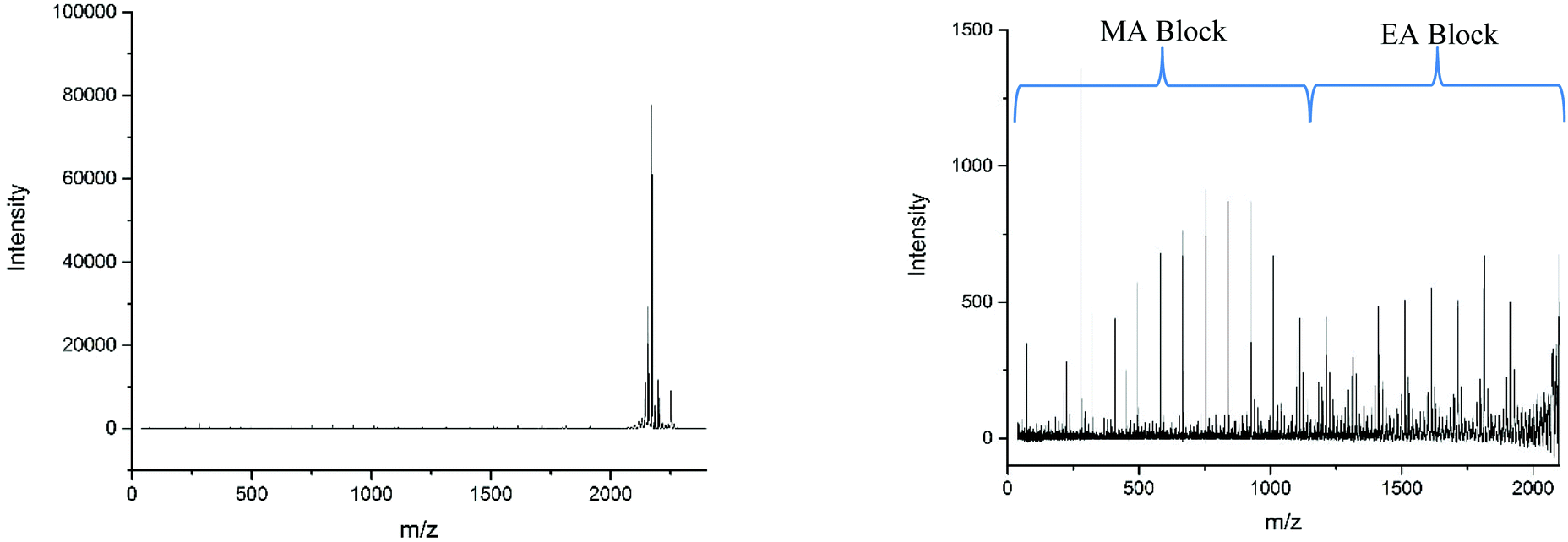
However, there is also evidence of an amount of undesired mixing, from the synthesis, in the region where the two polymer blocks were intended to switch, where we see fragments consisting of poly(methyl acrylate9-b-ethyl acrylate1) as a 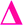 chain. This poly(methyl acrylate9-b-ethyl acrylaten) chain is found at higher m/z values, with a lower intensity, showing that these chains do contain the expected number of both monomers when not fragmented.
chain. This poly(methyl acrylate9-b-ethyl acrylaten) chain is found at higher m/z values, with a lower intensity, showing that these chains do contain the expected number of both monomers when not fragmented.
There is also a peak at 14 Da lower than the 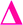 fragment, this peak is only found after where methyl acrylate polymerization was intended to be completed. We initially ascribed this to be a result of monomer mixing within the chains, however, the structure which would need to be present to form a
fragment, this peak is only found after where methyl acrylate polymerization was intended to be completed. We initially ascribed this to be a result of monomer mixing within the chains, however, the structure which would need to be present to form a 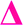 fragment with this m/z would have to be from poly(methyl acrylate11-b-ethyl acrylate1) which, according to the isolation procedure used should not be present in the spectrum. The isolation of the 2278 m/z has no overlapping peaks of similar m/z that would result in this intensity, when taking into account the lability of the bromine end group. In addition, a further explanation might be that the peak has taken the form of a
fragment with this m/z would have to be from poly(methyl acrylate11-b-ethyl acrylate1) which, according to the isolation procedure used should not be present in the spectrum. The isolation of the 2278 m/z has no overlapping peaks of similar m/z that would result in this intensity, when taking into account the lability of the bromine end group. In addition, a further explanation might be that the peak has taken the form of a 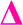 fragment, however
fragment, however 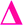 fragments at every other m/z in every other spectra are not present, as the radical undergoes a radical backbiting process to form the secondary fragments.
fragments at every other m/z in every other spectra are not present, as the radical undergoes a radical backbiting process to form the secondary fragments.
The most likely structural conformation which this equates to is a secondary fragment of poly(ethyl acrylate10) with a methyl acrylate unit as the terminal group. Interestingly this implies there is some special dependence on radical backbiting, as there are very little secondary fragments between those found at <500 m/z. Also the fragments found below 500 m/z have very little evidence of secondary fragments which are made up only of ethyl acrylate. As this was found in the homopolymer, this implies that there is some effect impeding the radical backbiting process from occurring closer to the original radical point with ethyl acrylate monomers. Whether a steric problem, or some other currently unknown effect, the differences found between the breakdown patterns of these two acrylate compounds, especially when in a diblock with each other, change how the radical backbiting occurs in a copolymer structure compared to either homopolymer.
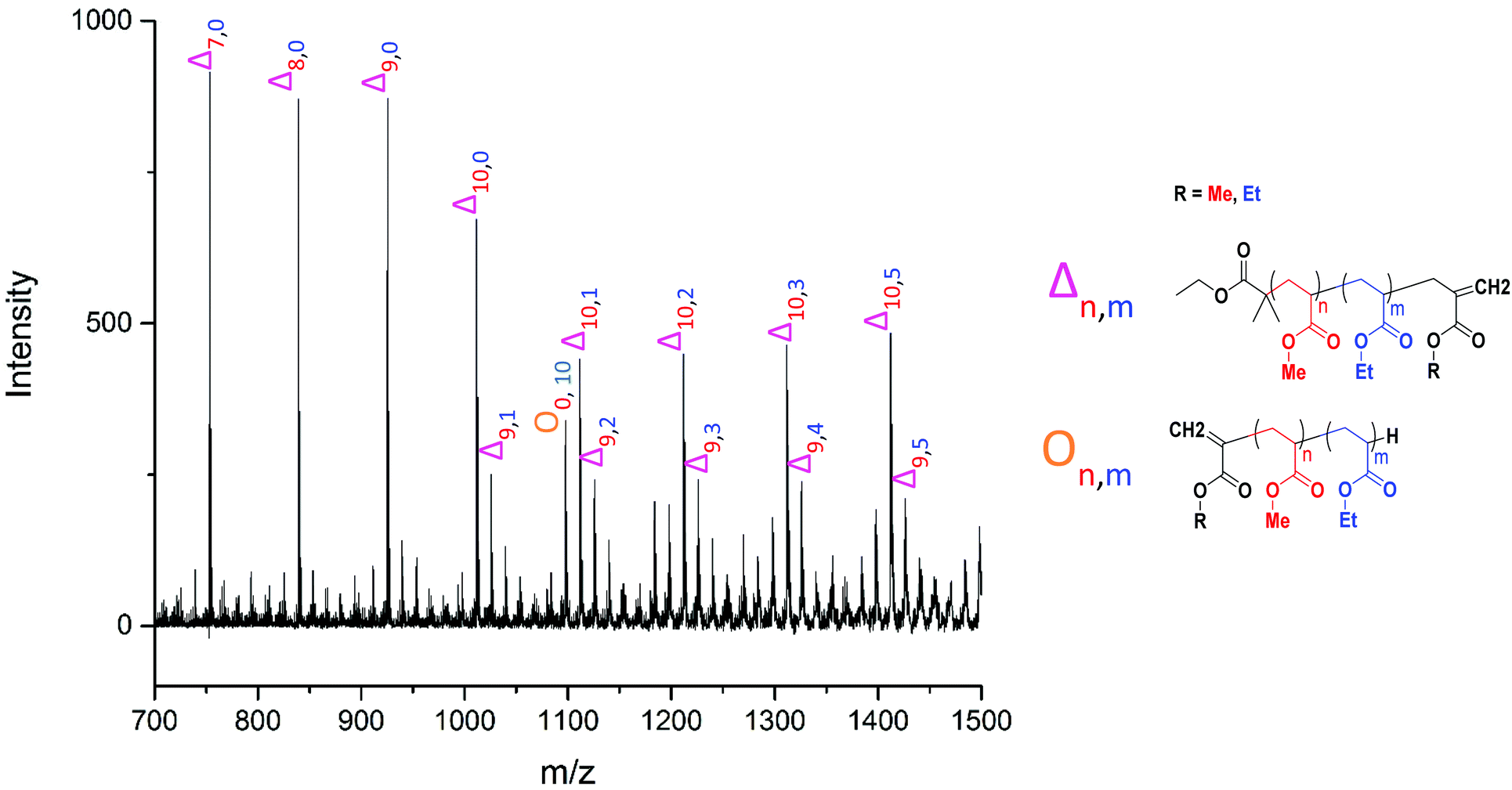 | ||
Fig. 9 Poly(methyl acrylate-b-ethyl acrylate) MALDI-LID-ToF/ToF at the block boundary region, showing the mixing peak 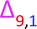 as well as the large secondary fragment as well as the large secondary fragment  . . | ||
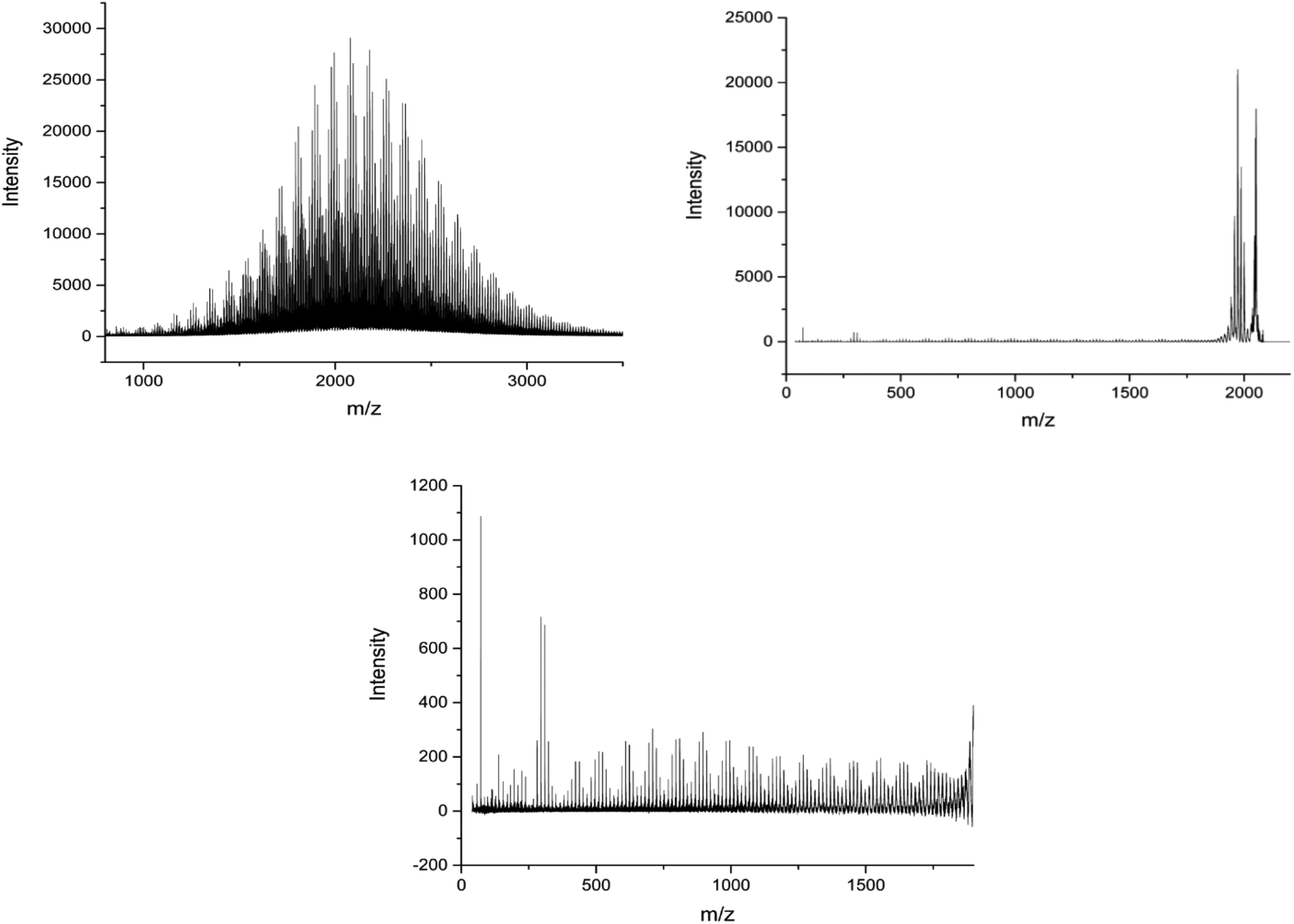 | ||
| Fig. 10 (A) Full MALDI-ToF spectra of the poly(methyl acrylate-co-ethyl acrylate) copolymer, (B) full MALDI-LID-ToF/ToF spectra of the poly(methyl acrylate-co-ethyl acrylate) copolymer. (C) Expansion to show the backbone cleavages of the poly(methyl acrylate-co-ethyl acrylate) copolymer, note its co-monomer distribution throughout the spectra indicating its statistical nature. | ||
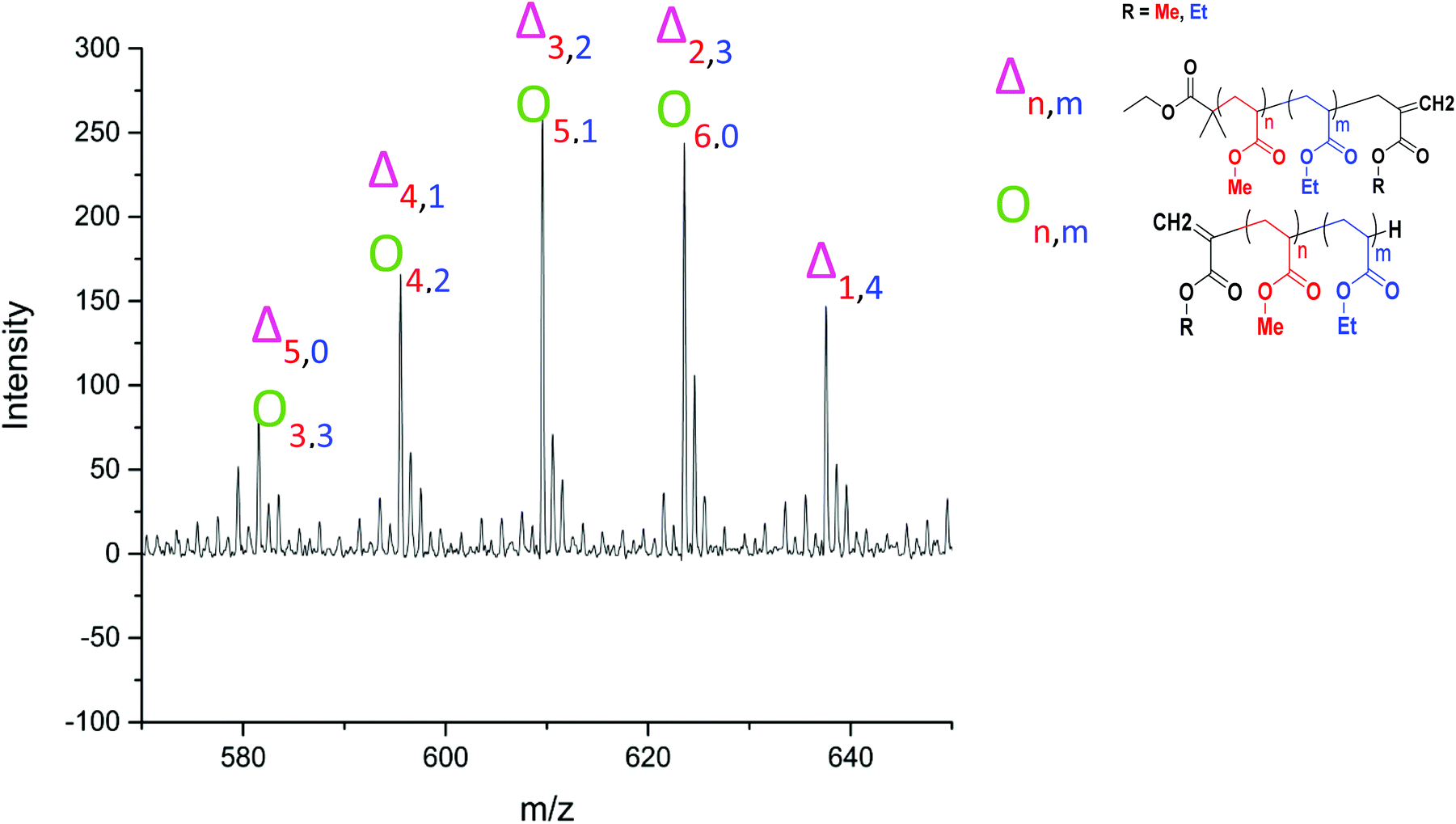 | ||
| Fig. 11 Assignment of a series of peaks in the MALDI-LID-ToF/ToF spectra of the poly(methyl acrylate-co-ethyl acrylate) copolymer, between 570–650 m/z, highlighting the overlap between the species. | ||
Applying the assignment of the homopolymer does not cover all of what we observe in the MALDI-LID-ToF/ToF spectra. As we observe in the 300–500 m/z region, several of the peaks in the pattern we have previously seen at higher molecular weights these cannot be accounted for as △ chains. We assigned these as ○ chains as previously observed in the secondary fragmentation patterns found in the homopolymer spectra (Fig. 2D), these ○ chains also can be assigned to several of the △ chain peaks.
As observed for poly(ethyl acrylate) homopolymer, a peak which we are only able to assign to the ○ has a high intensity at 581.533 m/z, higher mass than any of the other higher intensity secondary fragment peaks in the poly(methyl acrylate) homopolymer. This peak indicates that the effect causing the larger secondary fragments containing ethyl acrylate repeat units is also occurring, at least to some extent in this case. It is difficult to discern to what extent the peaks overlap, with 581 m/z being the highest m/z chain we can find which is only assigned as an ○ chain. The downside of this is that many of the higher molecular weight peaks found in the statistical copolymer spectra, which mathematically could be assigned as ○ as well as △ chains, cannot be discounted based on the MALDI-LID-ToF/ToF spectra for the homopolymer.
Conclusions
MALDI-LID-ToF/ToF has provided in depth analysis into the structure of poly(methyl acrylate), poly(ethyl acrylate), poly(n-butyl acrylate) and poly(iso-butyl acrylate) homopolymers, and poly(methyl acrylate-co-ethyl acrylate) diblock and statistical copolymers. Clear differences are seen between the diblock copolymer and the statistical copolymer spectra. Despite the simplicity of the acquisition, using the same sample preparation as the MALDI-ToF, determining the co-monomer arrangement of a copolymer is fully within the reach of MALDI-LID-ToF/ToF. The lability of the bromine terminating end group simplified the homopolymer spectra to allow only a single major series of fragments to form, alongside low molecular weight, high intensity, secondary fragmentation peaks. Comparing the LID results to the CID results from literature, there is very little difference for the poly(methyl acrylate). However, poly(butyl acrylate) shows more backbone cleavages and less rearrangement.66 The diblock polymers gives a spectrum enabling the microstructure to be examined. When the block boundary region was examined there was found to be a certain level of polymer mixing occurring between the two monomer blocks. Clear differences are observed between the diblock copolymer and the statistical copolymer. Determining the co-monomer arrangement of a copolymer is fully within the reach of this simple MALDI-LID-ToF/ToF technique.Conflicts of interest
There are no conflicts to declare.Acknowledgements
James S. Town thanks EPSRC for a PhD studentship through the EPSRC Centre for Doctoral Training in Molecular Analytical Science, grant number EP/L015307/1. Additional funding granted by Syngenta and AstraZeneca.Notes and references
- C.-K. Lai, D. C. M. Ng, H. F. Pang, J. C. Y. Le Blanc, J. W. Hager, D.-C. Fang, A. S.-C. Cheung and I. K. Chu, Rapid Commun. Mass Spectrom., 2013, 27, 1119–1127 CrossRef PubMed.
- M. W. F. Nielen, Mass Spectrom. Rev., 1999, 18, 309–344 CrossRef.
- K. H. Hunt, M. C. Crossman, D. M. Haddleton, P. M. Lloyd and P. J. Derrick, Macromol. Rapid Commun., 1995, 16, 725–732 CrossRef.
- M. J. Deery, K. R. Jennings, C. B. Jasieczek, D. M. Haddleton, A. T. Jackson, H. T. Yates and J. H. Scrivens, Rapid Commun. Mass Spectrom., 1997, 11, 57–62 CrossRef.
- M. Bednarek, T. Biedroń and P. Kubisa, Macromol. Chem. Phys., 2000, 201, 58–66 CrossRef.
- A. Moreno, S. Grama, T. Liu, M. Galia, G. Lligadas and V. Percec, Polym. Chem., 2017, 8, 7559–7574 RSC.
- M. Enayati, R. B. Smail, S. Grama, R. L. Jezorek, M. J. Monteiro and V. Percec, Polym. Chem., 2016, 7, 7230–7241 RSC.
- M. Enayati, R. L. Jezorek, R. B. Smail, M. J. Monteiro and V. Percec, Polym. Chem., 2016, 7, 5930–5942 RSC.
- M. Enayati, R. L. Jezorek and V. Percec, Polym. Chem., 2016, 7, 4549–4558 RSC.
- M. Enayati, R. L. Jezorek, M. J. Monteiro and V. Percec, Polym. Chem., 2016, 7, 3608–3621 RSC.
- M. J. Monteiro, P. Virgil Percec, P. V. Bernhardt, T. J. Zerk and M. Gavrilov, Polym. Chem., 2015, 7, 933–939 Search PubMed.
- A. Favier, C. Ladavière, M.-T. Charreyre and C. Pichot, Macromolecules, 2004, 37, 2026–2034 CrossRef.
- Q. Fu, K. Xie, T. G. McKenzie and G. G. Qiao, Polym. Chem., 2017, 8, 1519–1526 RSC.
- M. Chen, Q. Quan and h. gong, Polym. Chem., 2018, 9, 4161–4171 RSC.
- L. Martin, R. Peltier, A. Kuroki, J. S. Town and S. Perrier, Biomacromolecules, 2018 DOI:10.1021/acs.biomac.8b00146.
- A. Simula, M. Aguirre, N. Ballard, A. Veloso, J. R. Leiza, S. van Es and J. M. Asua, Polym. Chem., 2017, 8, 1728–1736 RSC.
- J. Wu, C. Tan, X. Zhou, Y. Tan, P. Yang and Y. Jiang, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2537–2543 CrossRef.
- P. M. Lloyd, K. G. Suddaby, J. E. Varney, E. Scrivener, P. J. Derrick and D. M. Haddleton, Eur. Mass Spectrom., 1995, 1, 293–300 CrossRef.
- B. Zhang, H. Zhang, B. K. Myers, R. Elupula, J. Jayawickramarajah and S. M. Grayson, Anal. Chim. Acta, 2014, 816, 28–40 CrossRef PubMed.
- A. R. Bottrill, A. E. Giannakopulos, C. Waterson, D. M. Haddleton, K. S. Lee and P. J. Derrick, Anal. Chem., 1999, 71, 3637–3641 CrossRef PubMed.
- D. M. Haddleton, C. Topping, D. Kukulj and D. Irvine, Polymer, 1998, 39, 3119–3128 CrossRef.
- T. S. Fischer, J. Steinkoenig, H. Woehlk, J. P. Blinco, K. Fairfull-Smith and C. Barner-Kowollik, Polym. Chem., 2017, 8, 5269–5274 RSC.
- G. Moriceau, G. Gody, M. Hartlieb, J. Winn, H. Kim, A. Mastrangelo, T. Smith and S. Perrier, Polym. Chem., 2017, 8, 4152–4161 RSC.
- J. C. Brendel, G. Gody and S. Perrier, Polym. Chem., 2016, 7, 5536–5543 RSC.
- C. Barner-Kowollik, T. P. Davis and M. H. Stenzel, Polymer, 2004, 45, 7791–7805 CrossRef.
- M. D. Zammit, T. P. Davis and D. M. Haddleton, Macromolecules, 1996, 29, 492–494 CrossRef.
- K. G. Suddaby, K. H. Hunt and D. M. Haddleton, Macromolecules, 1996, 29, 8642–8649 CrossRef.
- Y. Zhou, Q. Liu, Z. Zhang, J. Zhu and X. Zhu, Polym. Chem., 2017, 8, 6909–6916 RSC.
- L. Zhu, G. R. Parr, M. C. Fitzgerald, C. M. Nelson and L. M. Smith, J. Am. Chem. Soc., 1995, 117, 6048–6056 CrossRef.
- C. Chendo and L. Charles, Int. J. Mass spectrom., 2017, 416, 46–52 CrossRef.
- P. Terrier, W. Buchmann, G. Cheguillaume, B. Desmazières and J. Tortajada, Anal. Chem., 2005, 77, 3292–3300 CrossRef PubMed.
- R. Giordanengo, S. Viel, M. Hidalgo, B. Allard-Breton, A. Thévand and L. Charles, J. Am. Soc. Mass. Spectrom., 2010, 21, 1075–1085 CrossRef PubMed.
- M. S. Montaudo, Mass Spectrom. Rev., 2002, 21, 108–144 CrossRef PubMed.
- J. Axelsson, E. Scrivener, D. M. Haddleton and P. J. Derrick, Macromolecules, 1996, 29, 8875–8882 CrossRef.
- Z. Liu and K. L. Schey, J. Am. Soc. Mass. Spectrom., 2005, 16, 482–490 CrossRef PubMed.
- C. Wesdemiotis, N. Solak, M. J. Polce, D. E. Dabney, K. Chaicharoen and B. C. Katzenmeyer, Mass Spectrom. Rev., 2011, 30, 523–559 CrossRef PubMed.
- T. Yalcin, W. Gabryelski and L. Li, Anal. Chem., 2000, 72, 3847–3852 CrossRef PubMed.
- S. Sallam, Y. Luo, M. L. Becker and C. Wesdemiotis, Eur. J. Mass Spectrom., 2017, 23, 402–410 CrossRef PubMed.
- A. M. Yol, D. E. Dabney, S.-F. Wang, B. A. Laurent, M. D. Foster, R. P. Quirk, S. M. Grayson and C. Wesdemiotis, J. Am. Soc. Mass. Spectrom., 2013, 24, 74–82 CrossRef PubMed.
- A. Maciejczek, V. Mass, K. Rode and H. Pasch, Polymer, 2010, 51, 6140–6150 CrossRef.
- K. Knop, B. O. Jahn, M. D. Hager, A. Crecelius, M. Gottschaldt and U. S. Schubert, Macromol. Chem. Phys., 2010, 211, 677–684 CrossRef.
- L. Charles, G. Cavallo, V. Monnier, L. Oswald, R. Szweda and J.-F. Lutz, J. Am. Soc. Mass. Spectrom., 2017, 28, 1149–1159 CrossRef PubMed.
- A. Alalwiat, S. E. Grieshaber, B. A. Paik, K. L. Kiick, X. Jia and C. Wesdemiotis, Analyst, 2015, 140, 7550–7564 RSC.
- A. Baumgaertel, E. Altuntaş, K. Kempe, A. Crecelius and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5533–5540 CrossRef.
- A. C. Crecelius, C. R. Becer, K. Knop and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4375–4384 CrossRef.
- L. Przybilla, V. Francke, H. J. Räder and K. Müllen, Macromolecules, 2001, 34, 4401–4405 CrossRef.
- P. Rizzarelli, C. Puglisi and G. Montaudo, Rapid Commun. Mass Spectrom., 2005, 19, 2407–2418 CrossRef PubMed.
- A. T. Jackson, J. H. Scrivens, J. P. Williams, E. S. Baker, J. Gidden and M. T. Bowers, Int. J. Mass spectrom., 2004, 238, 287–297 CrossRef.
- C. Wesdemiotis, F. Pingitore, M. J. Polce, V. M. Russell, Y. Kim, C. M. Kausch, T. H. Connors, R. E. Medsker and R. R. Thomas, Macromolecules, 2006, 39, 8369–8378 CrossRef.
- R. Giordanengo, S. Viel, M. Hidalgo, B. Allard-Breton, A. Thévand and L. Charles, Anal. Chim. Acta, 2009, 654, 49–58 CrossRef PubMed.
- M. Girod, T. N. T. Phan and L. Charles, J. Am. Soc. Mass. Spectrom., 2008, 19, 1163–1175 CrossRef PubMed.
- T. Fouquet, C. Chendo, V. Toniazzo, D. Ruch and L. Charles, Rapid Commun. Mass Spectrom., 2013, 27, 88–96 CrossRef PubMed.
- U. Lewandrowski, A. Resemann and A. Sickmann, Anal. Chem., 2005, 77, 3274–3283 CrossRef PubMed.
- D. Suckau, A. Resemann, M. Schuerenberg, P. Hufnagel, J. Franzen and A. Holle, Anal. Bioanal. Chem., 2003, 376, 952–965 CrossRef PubMed.
- N. Shenar, N. Sommerer, J. Martinez and C. Enjalbal, J. Mass Spectrom., 2009, 44, 621–632 CrossRef PubMed.
- S. Shanmugam, J. Xu and C. Boyer, Macromol. Rapid Commun., 2017, 38, 1700143 CrossRef PubMed.
- X. Pan, M. A. Tasdelen, J. Laun, T. Junkers, Y. Yagci and K. Matyjaszewski, Prog. Polym. Sci., 2016, 62, 73–125 CrossRef.
- L. Charles, Mass Spectrom. Rev., 2014, 33, 523–543 CrossRef PubMed.
- V. Nikolaou, A. Anastasaki, F. Brandford-Adams, R. Whitfield, G. R. Jones, G. Nurumbetov and D. M. Haddleton, Polym. Chem., 2016, 7, 191–197 RSC.
- A. Anastasaki, V. Nikolaou and D. M. Haddleton, Polym. Chem., 2016, 7, 1002–1026 RSC.
- V. Nikolaou, A. Simula, M. Droesbeke, N. Risangud, A. Anastasaki, K. Kempe, P. Wilson and D. M. Haddleton, Polym. Chem., 2016, 7, 2452–2456 RSC.
- A. Anastasaki, J. Willenbacher, C. Fleischmann, W. R. Gutekunst and C. J. Hawker, Polym. Chem., 2017, 8, 689–697 RSC.
- G. Nurumbetov, N. Engelis, J. Godfrey, R. Hand, A. Anastasaki, A. Simula, V. Nikolaou and D. M. Haddleton, Polym. Chem., 2017, 8, 1084–1094 RSC.
- I.-H. Lee, E. H. Discekici, A. Anastasaki, J. R. de Alaniz and C. J. Hawker, Polym. Chem., 2017, 8, 3351–3356 RSC.
- M. W. Jones, M. I. Gibson, G. Mantovani and D. M. Haddleton, Polym. Chem., 2011, 2, 572–574 RSC.
- K. Chaicharoen, M. J. Polce, A. Singh, C. Pugh and C. Wesdemiotis, Anal. Bioanal. Chem., 2008, 392, 595–607 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8py00928g. The research data supporting this publication can be accessed at http://wrap.warwick.ac.uk/106723 |
| This journal is © The Royal Society of Chemistry 2018 |