
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHybrid block copolymers of polyesters/polycarbonates and polypeptides synthesized via one-pot sequential ring-opening polymerization†
Špela
Gradišar
ab,
Ema
Žagar
a and
David
Pahovnik
 *a
*a
aNational Institute of Chemistry, Department of Polymer Chemistry and Technology, Hajdrihova 19, 1000 Ljubljana, Slovenia. E-mail: david.pahovnik@ki.si
bUniversity of Ljubljana, Faculty of Chemistry and Chemical Technology, Večna pot 113, 1000 Ljubljana, Slovenia
First published on 28th August 2018
Abstract
An efficient approach toward one-pot sequential ring-opening polymerization (ROP) of cyclic esters/carbonates and N-carboxyanhydride (NCA) monomers, differing in reactivity and type of propagating group, is presented. In the first step, a polyester/polycarbonate is synthesized using methanesulfonic acid as a catalyst. After the completion of polymerization NCA is added to the reaction mixture. Methanesulfonic acid successfully catalyzes the initiation step of ROP of NCA and simultaneously prevents the chain propagation by protonation of the formed amine groups. After the completion of NCA initiation, the propagation is started by addition of N-ethyldiisopropylamine as a base to prepare the hybrid block copolymers of polyester/polycarbonate and polypeptide in a one-pot manner.
Introduction
A one-pot synthetic approach is a convenient procedure for preparation of block copolymers since time consuming and expensive steps of isolation, purification, and end-group transformation of the first block can be avoided. While one-pot synthesis of block copolymers via a sequential addition of monomers of the same type, polymerized by the same initiating/catalytic system, is well established, sequential polymerization of the monomers requiring different initiating/catalytic systems is still challenging. If the polymerization mechanisms of the two different monomers are orthogonal, e.g. ring-opening polymerizations (ROP) and controlled radical polymerization,1–3 one-pot synthesis by using a difunctional initiator is feasible as long as the initiating and the propagating species do not interfere with each other. On the other hand, sequential one-pot polymerization of the monomers, which all polymerize by the ROP mechanism, usually requires different approaches. One-pot sequential ROP of ethylene oxide (EO), ε-caprolactone (CL) and L-lactide (LLA) catalyzed by a relatively mild phosphazene base (t-BuP2) as a common catalyst was proved to be possible, though a sequence of monomer additions is crucial for the controlled synthesis of well-defined polyether–polyester block copolymers due to different reactivities of the monomers/polymers.4 Unfortunately, the synthesis of both blocks of the copolymer by the same catalyst is highly unlikely since the catalyst's activity has to be high enough for the least reactive monomer, but not too high for the most reactive monomer/polymer to prevent uncontrolled polymerization and side reactions. Recently, several strategies have been developed for one-pot synthesis of block copolymers by ROP. These involve either the switch of the catalyst and/or adjusting the experimental conditions to perform polymerization of each block under optimal conditions.5 In the case of organocatalysts various acid-to-base,6–8 base-to-acid,9–11 and base-to-base12,13 approaches, allowing the preparation of well-defined block copolymers based on epoxides, lactones and lactides, have been reported. In all these cases, the monomers significantly differ in reactivity; however, they all propagate through a hydroxyl group. In contrast, the N-carboxyanhydride (NCA) monomers, from which the synthetic polypeptides are prepared by ROP, propagate through a primary amine group when they are polymerized by a normal amine mechanism.14 Even though NCAs are known to react with the hydroxyl group, the rate of propagation by the released amine group is significantly faster as compared to the rate of initiation by the hydroxyl group, leading to uncontrolled NCA polymerization.15 On the other hand, the initiation of the cyclic ester monomers by the amine group is just as efficient as the initiation by the hydroxyl group,16 so polypeptides are able to act as the macroinitiators for ROP of the cyclic ester monomers. In this way, block copolymers based on poly(sarcosine) polypeptoid and PCL (P(Sar)-b-PCL) were prepared in a one-pot manner, where P(Sar) was first synthesized in ACN, which was then removed by evaporation, followed by addition of the CL monomer with Sn(Oct)2 as a catalyst to polymerize the second polyester block in the bulk.17,18 Unfortunately, this approach is not suitable for the preparation of hybrid block copolymers based on the most commonly applied polyglutamate and polyaspartate polypeptides since they bear the ester side group in the structure, which can participate in side reactions like transesterification, leading to a loss of the control over ROP of the second block. Therefore, the preparation of polypeptide-based hybrid block copolymers, which proved to be very interesting materials for various biomedical applications,19 most commonly proceeds via the primary amine-functionalized macroinitiators (the first block).14,20 Block copolymers composed of polypeptide and polyester or polycarbonate blocks are usually prepared by using difunctional initiators bearing the protected amine group. In this way, the PLLA, PCL, or PTMC blocks were first prepared by initiation of polymerization of the corresponding monomers by the hydroxyl group, followed by the removal of the Boc/Fmoc protective group21–23 or reduction of the nitro group24 to release the initiator amine functional group, which was after purification of the thus prepared first block used to initiate ROP of the benzyl L-glutamate (BLG) or benzyl L-aspartate (BLA) NCA. Such block copolymers in the deprotected form have been used to prepare polymer micelles21 and polymersomes22 as promising drug delivery systems.25Approaches to overcome slow initiation of ROP of NCA by the hydroxyl group involve the use of thiourea-26 and guanidine-based27 organocatalysts, however, they are not suitable for one-pot synthesis of polypeptide-based hybrid block copolymers since thioureas only work in combination with aminoalcohols as the initiators, while guanidine superbase catalysts can lead to an undesirable activated monomer polymerization mechanism. Recently, we reported on the synthetic method28 where hydroxyl-functionalized (macro)initiators have been successfully applied for ROP of NCA in the presence of an acid catalyst (methanesulfonic acid, MSA). During the initiation step, MSA catalyzes the opening of the NCA ring by the hydroxyl group, and simultaneously suppresses further chain propagation by a protonation of the formed amine group. Only after the completion of initiation, the chain propagation is started by the addition of a base that deprotonates the ammonium groups. This synthetic procedure has enabled us to prepare well-defined homopolypeptides and polypeptide-based hybrid block copolymers by using the hydroxyl functionalized (macro)initiators. Herein, we report on a sequential synthetic procedure that combines the sulfonic acid-based organocatalyzed ROP of cyclic esters or carbonates29–35 and our synthetic method for ROP of NCA initiated by the hydroxyl group in order to synthesize the polyester/polycarbonate-b-polypeptide hybrid block copolymers in a one-pot manner.
Experimental
Materials
Chemicals: β-benzyl-L-aspartate (BLA, 99%, Iris Biotech GmbH), triphosgene (98%, Aldrich), calcium hydride (95%, Sigma-Aldrich), methanesulfonic acid (MSA, 99.5%, Sigma-Aldrich), trimethylene carbonate (TMC, 99%, Polysciences, Inc.), and solvents: tetrahydrofuran (THF, 99.9%, anhydrous, Sigma-Aldrich), n-hexane (99%, Merck), chloroform (99%, anhydrous, Sigma-Aldrich), diethyl ether (99.7%, Merck), methanol (99.8%, Merck) and trifluoroacetic acid (TFA, 99%, Aldrich) were used as received. N-Ethyldiisopropylamine (EDIPA, 98%, Sigma-Aldrich), 3-phenyl-1-propanol (PPA, 98%, Aldrich) and ε-caprolactone (CL, 97%, Sigma-Aldrich) were dried over calcium hydride and distilled under vacuum. 1,3-Propanediol was dried over potassium carbonate and distilled under vacuum.Instrumentation
1 H NMR spectra of samples were recorded on a Varian Unity Inova 300 MHz instrument (Varian, Inc., USA) in the pulse Fourier transform mode. All measurements were carried out at room temperature in DMSO-d6 with a few drops of TFA and both the relaxation delay and the acquisition time of 5 s. Tetramethylsilane (TMS, δ = 0) was used as an internal chemical-shift standard.Size-exclusion chromatography coupled to a multi-angle light-scattering photometer (SEC-MALS) measurements were performed using a Hewlett-Packard pump series 1100 coupled to a Dawn Heleos multi-angle light-scattering photometer with a GaAs linearly polarized laser (λ0 = 661 nm) and to an Optilab rEX interferometric refractometer (RI), operating at the same wavelength as the photometer (both instruments are from Wyatt Technology Corp., USA). The separations were carried out at 50 °C using successively coupled MIXED-E and Oligopore columns (Agilent, USA) with a precolumn in 0.1 M solution of LiBr in N,N-dimethylacetamide (DMAc) at a flow rate of 0.5 mL min−1. The masses of the samples injected onto the column were typically 1.0 × 10−3 g, whereas the solution concentration was 1.0 × 10−2 g mL−1. The samples’ dn/dc values needed for calculation of molecular weight characteristics were determined assuming 100% mass recovery of the samples from the columns. For the data acquisition and evaluation Astra 5.3.4 software (Wyatt Technology Corp., USA) was utilized.
Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) measurements were carried out on a Bruker UltrafleXtreme MALDI-TOF mass spectrometer (Bruker Daltonics, Bremen, Germany). Homopolymers were dissolved in THF (10 mg mL−1) or aliquots were diluted with THF and mixed with a solution of matrix, that is 2,5-dihydroxybenzoic acid in THF (30 mg mL−1) and sodium trifluoroacetate in THF (10 mg mL−1), in a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:3. 0.4 μL of thus prepared solution was spotted on the target plate (dried-droplet method). The reflective positive ion mode was used to acquire the mass spectra of the samples. Calibration was performed externally using a mixture of poly(methyl methacrylate) standards dissolved in THF (MALDI validation set PMMA, Fluka Analytical), covering the measured molecular weight range. The sample preparation for the standard mixture was the same as that for the samples. The standard mixture was spotted on the nearest neighbor positions.
:10:3. 0.4 μL of thus prepared solution was spotted on the target plate (dried-droplet method). The reflective positive ion mode was used to acquire the mass spectra of the samples. Calibration was performed externally using a mixture of poly(methyl methacrylate) standards dissolved in THF (MALDI validation set PMMA, Fluka Analytical), covering the measured molecular weight range. The sample preparation for the standard mixture was the same as that for the samples. The standard mixture was spotted on the nearest neighbor positions.
Synthesis of BLA NCA
In a flame-dried argon-purged flask BLA (5.00 g, 22.4 mmol) was suspended in dry THF (50 mL). A solution of triphosgene (3.52 g, 11.9 mmol) in dry THF (15 mL) was then slowly added. The reaction mixture was stirred and heated up in an oil bath to 55 °C for 75 min to obtain a clear solution. The reaction mixture was concentrated to approximately 25% of its original volume using a rotary evaporator and then n-hexane (approx. 80 mL) was slowly added to crystallize the product. The white product was filtered, washed with n-hexane, and recrystallized three times from the THF/n-hexane (20/80 mL). The resulting NCA monomer was dried under vacuum.Y (BLA NCA) = 90%. 1H NMR (DMSO-d6): δ = 2.91 (dd, J1 = 17.8 Hz, J2 = 4.3 Hz, 1H, CH2a), 3.08 (dd, J1 = 17.8 Hz, J2 = 4.3 Hz, 1H, CH2b), 4.70 (m, 1H, CH), 5.14 (s, 2H, benzyl CH2), 7.37 (m, 5H, aryl CH), 9.00 (s, 1H, NH). Tm = 126–128 °C.
One-pot synthesis of poly(ε-caprolactone)-b-poly(β-benzyl-L-aspartate) (PCL-b-PBLA)
All polymerizations were carried out with a freshly prepared batch of the NCA monomer in a flame-dried Schlenk flask under an argon atmosphere. CL (250 μL, 2.26 mmol) was dissolved in dry chloroform (4 mL, [CL]0 = 0.56 M). The PPA initiator (12.28 μL, 0.09 mmol) and MSA (17.57 μL, 0.27 mmol) were successively added. The reaction mixture was stirred at 40 °C for 1 h. Complete consumption of the CL was monitored by 1H NMR spectroscopy and the polymer formed was characterized by MALDI-TOF MS. When the polymerization had been finished a half of the reaction mixture was taken from the flask to isolate the first block. The aliquot was quenched with EDIPA (25 μL, 0.15 mmol) to neutralize the catalyst and then it was poured into a cold mixture of methanol and diethyl ether (1/1, v/v) to precipitate the product. The precipitate was isolated by centrifugation (8000 rpm, 1 min) and dried under vacuum for 24 h.The remaining half of the reaction mixture was diluted with 2 mL of dry chloroform and the BLA NCA (0.281 g, 1.13 mmol) was added and stirred at 40 °C for 24 h to ensure complete initiation, which was followed by MALDI-TOF MS. After complete initiation, the reaction mixture was cooled down in an ice bath to 0 °C and then the propagation was started by the addition of EDIPA (19.31 μL, 0.11 mmol). After 24 h, the reaction mixture became clear and full conversion of BLA NCA monomer was achieved as determined by 1H NMR. When the polymerization had been finished a half of the reaction mixture was taken from the flask to isolate the block copolymer. The reaction mixture was poured into a cold mixture of methanol and diethyl ether (1/9, v/v) to precipitate the product. The precipitate was isolated by centrifugation (8000 rpm, 1 min) and dried under vacuum for 24 h. The remaining reaction solution was used for the chain-extension experiment, where first the BLA NCA (0.084 g, 0.34 mmol, corresponding to additional 15 amino acids per chain) in 1 mL of dry chloroform was added to the remaining polymerization solution, and the mixture was stirred at 0 °C for 24 h to ensure complete conversion of the BLA NCA. Afterwards, 1.5 mL of the block copolymer solution was isolated as described before, whereas another aliquot of the BLA NCA (0.042 g, 0.17 mmol, corresponding to additional 15 amino acids per chain) in 1.5 mL of dry chloroform was added to the remaining solution and stirred at 0 °C for 24 h to obtain the final block copolymer, which was isolated as described above.
One-pot synthesis of poly(trimethylene carbonate)-b-poly(β-benzyl-L-aspartate) (PTMC-b-PBLA)
TMC (0.200 g, 1.96 mmol) was dissolved in dry chloroform (2 mL, [TMC]0 = 0.98 M). The PPA initiator (8.88 μL, 0.07 mmol) and MSA (12.71 μL, 0.20 mmol) were successively added. The reaction mixture was stirred at 40 °C for 1 h. Complete consumption of the TMC was monitored by 1H NMR and the polymer formed was characterized by MALDI-TOF MS. When the polymerization had been finished a half of the reaction mixture was taken from the flask to isolate the first block. This aliquot was quenched with EDIPA (20 μL, 0.12 mmol) to neutralize MSA catalyst and poured into cold methanol to precipitate the product. The precipitate was isolated by centrifugation (8000 rpm, 1 min) and dried under vacuum for 24 h.The remaining half of the reaction mixture was diluted with 1 mL of dry chloroform. Then, the BLA NCA (0.203 g, 0.82 mmol) was added and stirred at 40 °C for 24 h to ensure complete initiation, which was followed by MALDI-TOF MS. After completion of the initiation, the reaction mixture was cooled down in an ice bath to 0 °C and then the chain propagation was started by the addition of EDIPA (13.97 μL, 0.08 mmol). After 24 h, the reaction mixture became clear and a full conversion of the BLA NCA monomer was achieved as determined by 1H NMR. When the polymerization had been finished a half of the reaction mixture was taken from the flask to isolate the block copolymer. The reaction mixture was poured into a cold mixture of methanol and diethyl ether (1/4, v/v) to precipitate the product. The precipitate was isolated by centrifugation (8000 rpm, 1 min) and dried under vacuum for 24 h. The remaining reaction solution was used for the chain-extension experiments, where the first portion of the BLA NCA (0.061 g, 0.24 mmol, corresponding to additional 15 amino acids per chain) together with 1 mL of dry chloroform was added to the remaining polymerization solution which was stirred at 0 °C for 24 h to ensure complete conversion of the BLA NCA. Afterwards, 1 mL of the block copolymer solution was isolated as described above, while another portion of the BLA NCA (0.031 g, 0.12 mmol, corresponding to additional 15 amino acids per chain) together with 1 mL of dry chloroform was added again to the remaining polymerization solution, and stirred at 0 °C for another 24 h to obtain the final block copolymer, which was isolated as described above.
One-pot synthesis of poly(β-benzyl-L-aspartate)-b-poly(trimethylene carbonate)-b-poly(β-benzyl-L-aspartate) (PBLA-b-PTMC-b-PBLA)
TMC (0.200 g, 1.96 mmol) was dissolved in dry chloroform (2 mL, [TMC]0 = 0.98 M). 1,3-Propanediol (4.72 μL, 0.07 mmol) and MSA (16.95 μL, 0.26 mmol) were successively added. The reaction mixture was stirred at 40 °C for 1 h. Complete consumption of the TMC was monitored by 1H NMR and the polymer formed was characterized by MALDI-TOF MS. When the polymerization had been finished a half of the reaction mixture was taken from the flask to isolate the first block. This aliquot was quenched with the EDIPA (25 μL, 0.15 mmol) to neutralize the catalyst, and then the mixture was poured into cold methanol to precipitate the product. The precipitate was isolated by centrifugation (8000 rpm, 1 min) and dried under vacuum for 24 h.The remaining half of the reaction mixture was diluted with 1 mL of dry chloroform. Then, the BLA NCA (0.325 g, 1.31 mmol) was added and the mixture stirred at 40 °C for 44 h to ensure complete initiation which was followed by MALDI-TOF MS. After completion of the initiation, the reaction mixture was cooled down in an ice bath to 0 °C, and then 2 mL of dry chloroform was added. The propagation was started by the addition of EDIPA (16.77 μL, 0.10 mmol). After 24 h, the reaction mixture became clear and a full conversion of the BLA NCA monomer was achieved as determined by 1H NMR. The reaction mixture was poured into a cold mixture of methanol and diethyl ether (1/4, v/v) to precipitate the product. The precipitate was isolated by centrifugation (8000 rpm, 1 min) and dried under vacuum for 24 h.
Results and discussion
For the preparation of polypeptide-based hybrid block copolymers, the cyclic ester or carbonate monomer (CL or TMC) was polymerized first using PPA as a monofunctional initiator together with 3 equiv. of MSA with respect to initiator (Scheme 1). MSA is an effective organocatalyst for ROP of both CL and TMC as it has been demonstrated previously.32–35 The ROP of CL and TMC was performed in chloroform at 40 °C. After a reaction time of 1 hour, the conversion of both monomers reached 97% as determined by 1H NMR. The PCL and PTMC homopolymers withdrawn at the end of the polymerization were analyzed by 1H NMR, MALDI-TOF MS and SEC-MALS.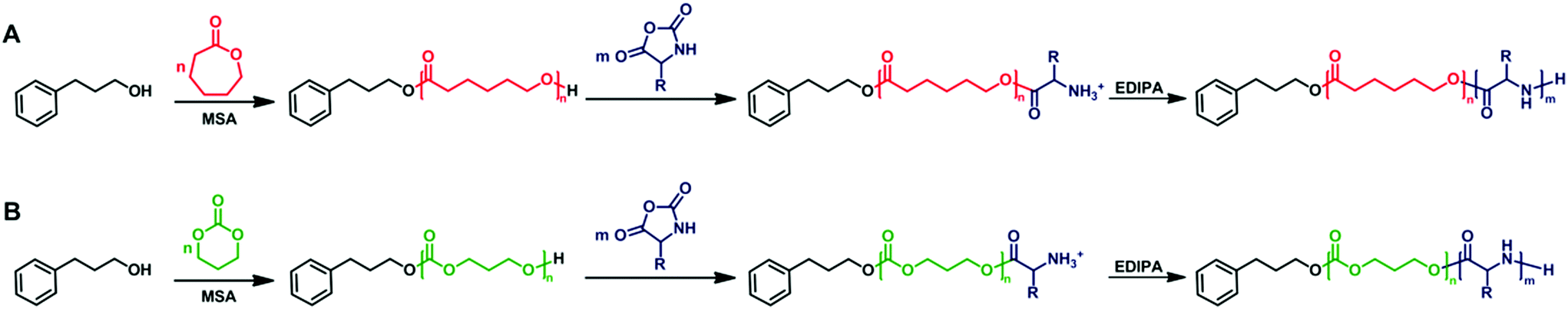 | ||
| Scheme 1 General reaction scheme for the synthesis of polyester-b-polypeptide (A) and polycarbonate-b-polypeptide (B) copolymers via the one-pot sequential ROP. | ||
1H NMR (Fig. S1†) and MALDI-TOF MS (Fig. 1A) confirm the expected structure of the PCL homopolymer; that is, all PCL chains bear PPA and hydroxyl end-groups. The number average molar mass (Mn) of PCL was determined from 1H NMR from the integral ratio of the signals for the main chain methylene groups (–CH2OCO–) and the terminal methylene groups (–CH2OH) at the chemical shifts of 3.99 and 3.38 ppm, respectively. Such a determined Mn of 2.8 kg mol−1 is in good agreement with the PCL theoretical molar mass calculated from the monomer/initiator feed ratio (Table 1; sample A) as well as with the molecular weight at the peak apex (Mp = 2.9 kDa) in the MALDI-TOF mass spectrum of PCL. The molar mass distribution of PCL is narrow as indicated by a low molar mass dispersity determined by SEC-MALS (Table 1).
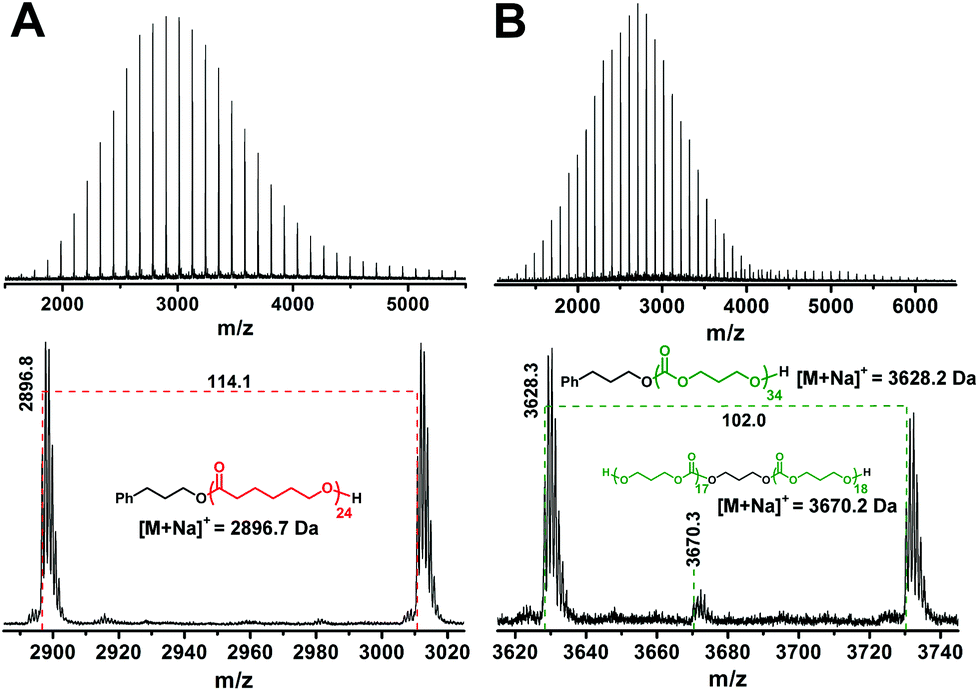 | ||
| Fig. 1 MALDI-TOF mass spectra of the PCL (A) and PTMC (B) homopolymers prepared by ROP of CL and TMC, respectively. Both homopolymers were initiated by PPA in the presence of MSA. The measured monoisotopic signals are denoted in the enlarged regions of the mass spectra together with the calculated exact masses ionized with the sodium ion for the proposed structures. | ||
| Sample | [I]0:[M1]0:[M2]0a |
M
n,theorb (kg mol−1) |
M
n,NMRc (kg mol−1) |
M w,SEC-MALS (kg mol−1) |
M
w/Mnd |
Y (%) |
|---|---|---|---|---|---|---|
| a [I]0 is PPA (samples A–H) or 1,3-propandiol (samples I–J). [M1]0 is CL (samples A–D) or TMC (samples E–J). [M2]0 is BLA NCA. b Calculated from the monomer(s) to initiator ratio. c Calculated from the signals of repeating units and end-groups for the homopolymers and the ratio of repeating units of each block for the block copolymers. d Determined by SEC-MALS. e Isolated yields after precipitation, and the monomer conversion was >99% in all cases as determined by 1H NMR. | ||||||
| A | 1:25:0 |
3.0 | 2.8 | 3.1 | 1.03 | 75 |
| B | 1:25:25 |
8.1 | 8.1 | 8.1 | 1.02 | 78 |
| C | 1:25:40 |
11.2 | 10.6 | 10.7 | 1.01 | 85 |
| D | 1:25:55 |
14.3 | 13.4 | 13.8 | 1.02 | 81 |
| E | 1:30:0 |
3.2 | 2.9 | 3.1 | 1.05 | 85 |
| F | 1:30:25 |
8.3 | 7.8 | 11.9 | 1.16 | 75 |
| G | 1:30:40 |
11.4 | 10.9 | 14.6 | 1.13 | 72 |
| H | 1:30:55 |
14.5 | 14.7 | 17.9 | 1.11 | 62 |
| I | 1:30:0 |
3.1 | 2.8 | 3.2 | 1.05 | 70 |
| J | 1:30:40 |
11.3 | 10.1 | 11.5 | 1.10 | 85 |
The MALDI-TOF mass spectrum of the PTMC homopolymer (Fig. 1B) shows beside the expected distribution of peaks typical of the PTMC chains initiated by the PPA, an additional distribution of peaks of much lower intensity with approximately double molecular weights, which match with the dihydroxyl-functionalized PTMC originating either from the initiation of ROP of TMC with the traces of water or from the polymerization of TMC according to the active chain end polymerization mechanism.33
After the completion of CL/TMC polymerization, the BLA NCA monomer was added to the reaction mixture to perform the initiation step of ROP of NCA by the hydroxyl terminated PCL or PTMC. Full conversion of the hydroxyl groups of the PCL or PTMC into the ester bonds was achieved in chloroform at 40 °C in 20 hours as revealed by MALDI-TOF MS (Fig. 2). Full MALDI-TOF mass spectra recorded after completion of the BLA NCA initiation do not show significant broadening of the peak distributions as compared to those of the homopolymers, indicating the absence of transesterification or transcarbonation reactions as the side reactions during the initiation step. A mass difference between the peaks of the main distributions in the mass spectra of the PTMC before and after the BLA NCA initiation is 1 Da, leading to strongly overlapping isotopic patterns (Fig. 2B). Nevertheless, the mass difference of +1 Da still makes it possible to follow the initiation step of BLA NCA by monitoring a decrease in the intensity of the monoisotopic signal of the uninitiated PTMC chains (Fig. 2B).
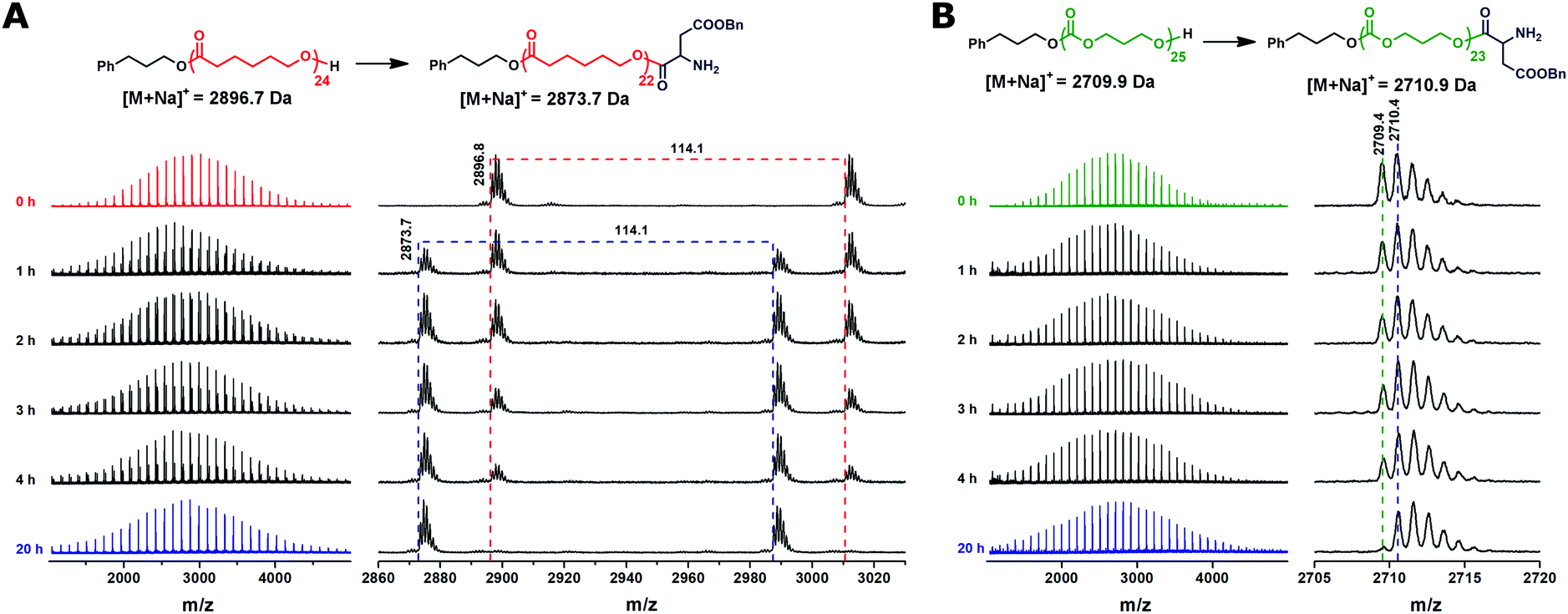 | ||
| Fig. 2 MALDI-TOF mass spectra of the reaction aliquots withdrawn at different times of initiation of BLA NCA with the hydroxyl-terminated PCL (A) or PTMC (B). Measured monoisotopic signals are denoted in the enlarged regions of the mass spectra together with the calculated exact masses ionized with the sodium ion for the proposed structures. | ||
After the completion of initiation, the chain propagation was started by addition of EDIPA as a base (2.5 equiv. with regard to PPA) to the reaction mixture which was cooled to 0 °C. EDIPA deprotonates the ammonium groups and starts the propagation step of ROP of NCA. After the reaction time of 24 hours, a full conversion of BLA NCA monomers was achieved as determined by 1H NMR. The 1H NMR spectra of the PCL-b-PBLA25 (Fig. S4†) and PTMC-b-PBLA25 (Fig. S5†) copolymers show the signals characteristic of both blocks after their isolation. In order to confirm the controlled/living nature of the polymerization, we carried out chain-extension experiments which were performed by a sequential BLA NCA monomer addition to the reaction mixture, each time after a full conversion of the previous batch of the NCA monomer added. Consumption of the NCA monomers was followed by 1H NMR, whereas the increase in the copolymer molar mass was confirmed by SEC-MALS of the aliquots taken from the reaction mixture before each addition of the fresh NCA batch. The results of SEC-MALS analysis reveal the monomodal SEC traces of the PCL-b-PBLA block copolymers which clearly shifted toward a lower elution volume with each NCA batch addition, indicating the increase in polypeptide block length (Fig. 3, Table 1: samples B–D). The number average molar masses of the block copolymers determined by SEC-MALS fit very well with their theoretical values, whereas the narrow molar mass distributions of the block copolymers are reflected in their low molar mass dispersities.
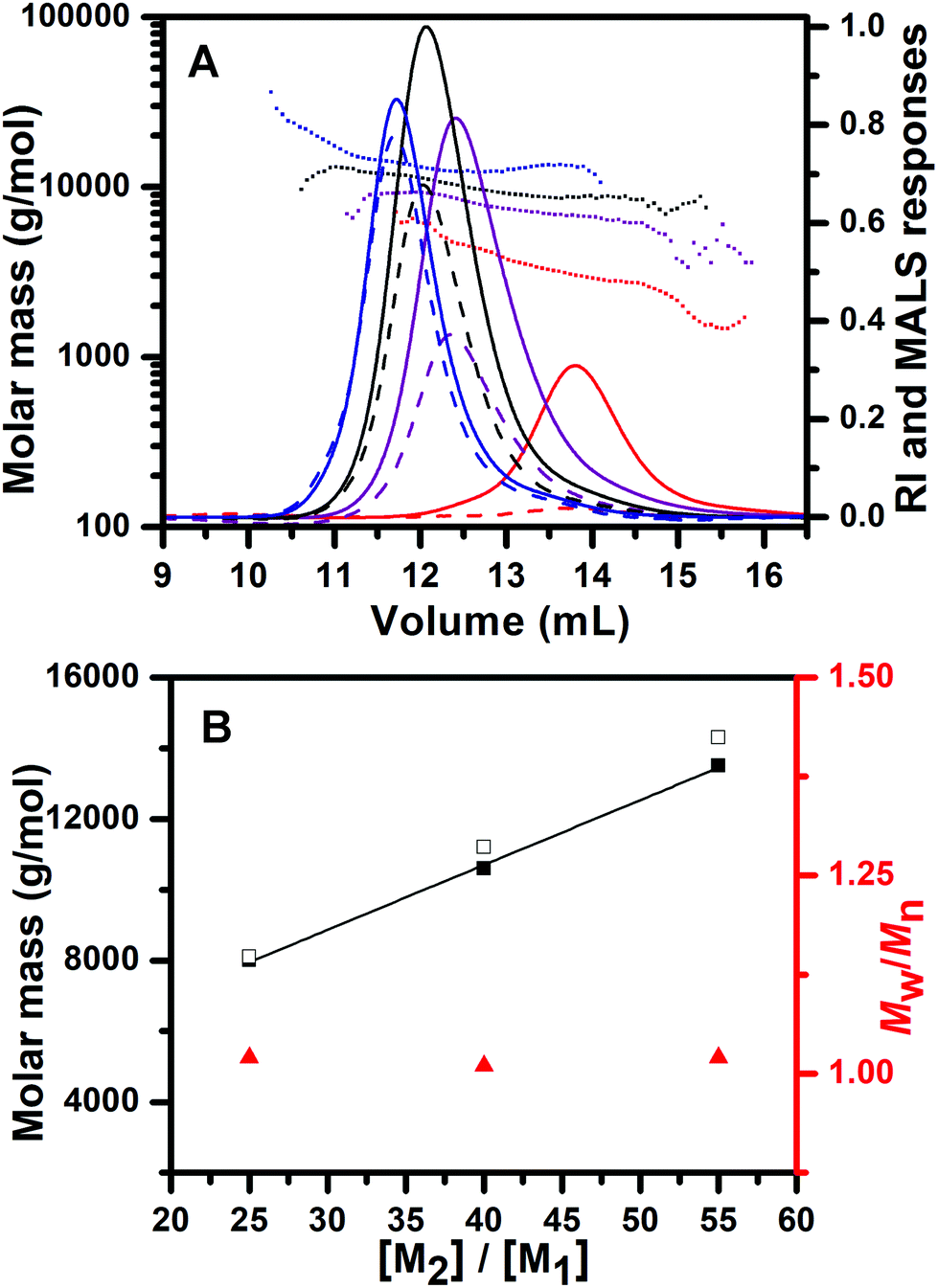 | ||
Fig. 3 (A) SEC-MALS chromatograms of PCL (red) and PCL-b-PBLA25 (purple), PCL-b-PBLA40 (black), PCL-b-PBLA55 (blue) block copolymers. Solid curves: refractive index detector responses; dashed curves: light-scattering detector responses at 90° angle; dotted curves: molar mass as a function of elution volume. (B) Chain length control and molar mass dispersity of the PCL-b-PBLA copolymers as a function of BLA to CL feed ratio ([M2]/[M1]) as determined by SEC-MALS: experimental Mn (■), theoretical M (□), and molar mass dispersity (Mw/Mn,  ). ). | ||
Contrary to the PCL homopolymer, the PTMC homopolymer consisted of not only the monohydroxyl terminated chains, but also a low amount of the dihydroxyl terminated PTMC chains which show double molar masses as discussed above. Therefore, SEC traces of the PTMC homopolymer as well as the resulting PTMC-b-PBLA copolymers obtained in chain-extension experiment show shoulders in the high molar mass ends of the distribution curves, especially at the highest copolymer molar mass targeted. For the same reason, the curve representing the molar mass vs. elution volume for the PTMC-b-PBLA55 copolymer also shows the presence of two populations of different molar masses corresponding to di- and tri-block copolymers (Fig. 4). As a consequence, the molar mass distributions of the PTMC-b-PBLA copolymers are somewhat broader as compared to those of the PCL-b-PBLA copolymers as indicated by their higher molar mass dispersities (Table 1: samples F–H). Nevertheless, the chain-extension experiments for both block copolymer types (PCL-b-PBLA and PTMC-b-PBLA) reveal the increase in the molar mass of the polypeptide block at each addition of the fresh NCA monomer batch.
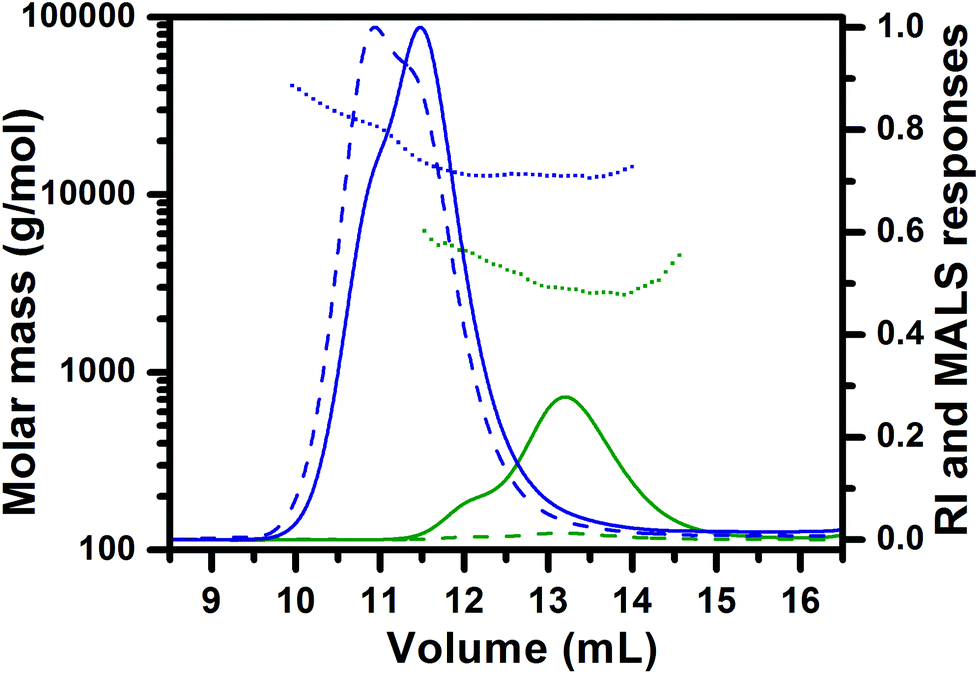 | ||
| Fig. 4 SEC-MALS chromatograms of PTMC (green) and PTMC-b-PBLA55 (blue) block copolymer. Solid curves: refractive index detector responses; dashed curves: light-scattering detector responses at 90° angle; dotted curves: molar mass as a function of elution volume. | ||
To confirm the effectiveness of the disclosed method, ROP of TMC was initiated with 1,3-propanediol as a difunctional initiator in order to prepare the triblock copolymer. After the completion of initiation, sequential ROP of BLA NCA was performed under similar experimental conditions to those in the case of diblock copolymers (samples E and G), except that MSA was added in 4 equiv. (instead of 3 equiv. for the monohydroxy initiator) with regard to 1,3-propanediol since 2 MSA equiv. are consumed for the protonation of the formed amine group on each side of the difunctional initiator, while another 2 MSA equiv. are necessary for the activation of the NCA rings. Consequently, the amount of EDIPA base used for the chain propagation was 3 equiv. with respect to initiator. Almost complete NCA initiation by the dihydroxyl terminated PTMC was achieved at 40 °C in 44 hours as indicated by MALDI-TOF MS (Fig. 5A). In the case of the difunctional PTMC, a mass difference between the peaks of the main distributions before and after the full BLA NCA initiation is +2 Da, since the PTMC chains on both chain ends reacted with one BLA unit. Chain propagation was completed after 24 hours at 0 °C as determined by 1H NMR. In the case of the difunctional initiator no shoulder was observed neither in the SEC-MALS chromatogram of the dihydroxyl functionalized PTMC homopolymer nor in the SEC-MALS chromatogram of the triblock copolymer (Fig. 5B). SEC traces of the PTMC homopolymer and the PBLA-b-PTMC-b-PBLA triblock copolymer prepared by the difunctional initiator reveal narrow molar mass distributions with controlled molar masses, whereas molar mass dispersities are expectedly slightly higher as compared to those of the PCL-based (co)polymers prepared by the monofunctional initiator (Table 1; samples I and J).
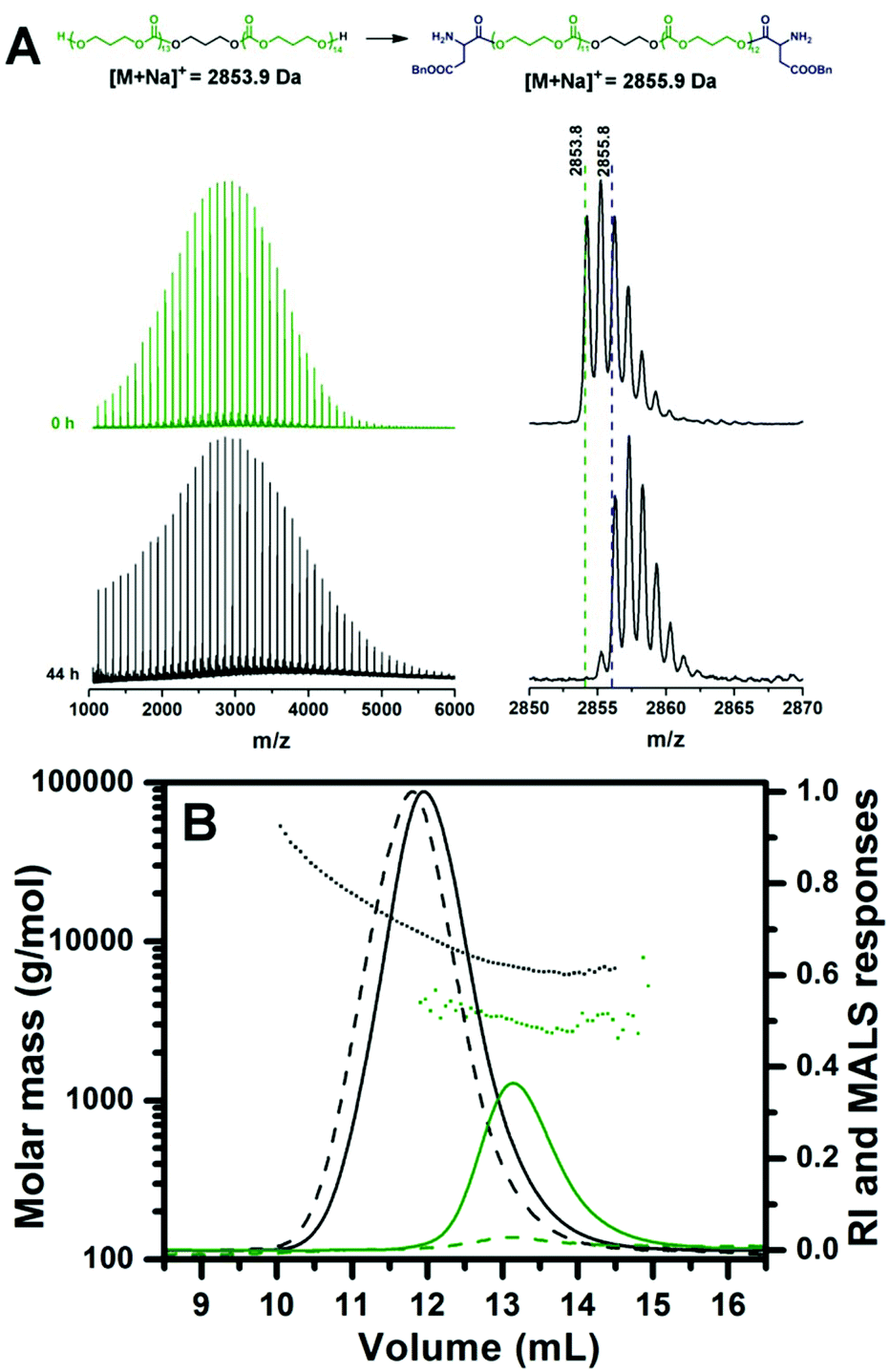 | ||
| Fig. 5 (A) Full MALDI-TOF mass spectra of the reaction aliquots withdrawn at the beginning and at the end of the initiation of BLA NCA with the 1,3-propanediol-initiated PTMC homopolymer. The measured monoisotopic signals are denoted in the enlarged regions of the mass spectra together with the calculated exact masses ionized with the sodium ion for the proposed structures. (B) SEC-MALS chromatograms of the PBLA20-b-PTMC-b-PBLA20 triblock copolymer (black) together with the corresponding PTMC homopolymer (green) prepared by ROP of TMC initiated by the 1,3-propanediol. Solid curves: refractive index detector responses; dashed curves: light-scattering detector responses at 90° angle; dotted curves: molar mass as a function of elution volume. | ||
Conclusions
One-pot sequential ROP of cyclic esters/carbonates and the NCA monomers has been conducted in a controlled manner. ROP of CL or TMC and, subsequently, the initiation step of ROP of the NCA were performed in the presence of MSA acid catalyst. After the completion of NCA initiation, the propagation step of ROP of NCA was started by addition of a base to synthesize the hybrid diblock and triblock copolymers of polyester/polycarbonate and polypeptide in a one-pot manner using the monohydroxy- and dihydroxy-initiator, respectively. The disclosed one-pot synthetic method significantly facilitates the preparation of hybrid block copolymers based on polypeptides and polyesters/polycarbonates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the Slovenian Research Agency (Research Core Funding no. P2-0145 and Project no. J2-9214) and the Centre of Excellence: Polymer Materials and Technologies for access to a MALDI-TOF mass spectrometer.References
- C. Fu, J. Xu, M. Kokotovic and C. Boyer, ACS Macro Lett., 2016, 5, 444–449 CrossRef.
- L. Lei, F. Li, H. Zhao and Y. Wang, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 699–704 CrossRef.
- C. Aydogan, C. Kutahya, A. Allushi, G. Yilmaz and Y. Yagci, Polym. Chem., 2017, 8, 2899–2903 RSC.
- J. Zhao, D. Pahovnik, Y. Gnanou and N. Hadjichristidis, Polym. Chem., 2014, 5, 3750–3753 RSC.
- S. Hu, J. Zhao, G. Zhang and H. Schlaad, Prog. Polym. Sci., 2017, 74, 34–77 CrossRef.
- K. Makiguchi, Y. Ogasawara, S. Kikuchi, T. Satoh and T. Kakuchi, Macromolecules, 2013, 46, 1772–1782 CrossRef.
- K. Makiguchi, S. Kikuchi, K. Yanai, Y. Ogasawara, S. Sato, T. Satoh and T. Kakuchi, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1047–1054 CrossRef.
- X. Wang, J. Liu, S. Xu, J. Xu, X. Pan, J. Liu, S. Cui, Z. Li and K. Guo, Polym. Chem., 2016, 7, 6297–6308 RSC.
- J. Zhao, D. Pahovnik, Y. Gnanou and N. Hadjichristidis, Macromolecules, 2014, 47, 3814–3822 CrossRef.
- J. Zhao, D. Pahovnik, Y. Gnanou and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 304–312 CrossRef.
- J. Zhao and N. Hadjichristidis, Polym. Chem., 2015, 6, 2659–2668 RSC.
- Y. Xia, Y. Chen, Q. Song, S. Hu, J. Zhao and G. Zhang, Macromolecules, 2016, 49, 6817–6825 CrossRef.
- H. Alamri and N. Hajichristidis, Polym. Chem., 2016, 7, 3225–3228 RSC.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and G. Sakellariou, Chem. Rev., 2009, 109, 5528–5578 CrossRef PubMed.
- M. Szwarc, Adv. Polym. Sci., 1965, 4, 1–65 Search PubMed.
- A. Kowalski, J. Libiszowski, T. Biela, M. Cypryk, A. Duda and S. Penczek, Macromolecules, 2005, 38, 8170–8176 CrossRef.
- S. Cui, X. Wang, Z. Li, Q. Zhang, W. Wu, J. Liu, H. Wu, C. Chen and K. Guo, Macromol. Rapid Commun., 2014, 35, 1954–1959 CrossRef PubMed.
- S. Cui, X. Pan, H. Gebru, X. Wang, J. Liu, J. Liu, Z. Li and K. Guo, J. Mater. Chem. B, 2017, 5, 679–690 RSC.
- C. Deng, J. Wu, R. Cheng, F. Meng, H. A. Klok and Z. Zhong, Prog. Polym. Sci., 2014, 39, 330–364 CrossRef.
- H. R. Kricheldorf, Angew. Chem., Int. Ed., 2006, 45, 5752–5784 CrossRef PubMed.
- H. Arimura, Y. Ohya and T. Ouchi, Macromol. Rapid Commun., 2004, 25, 743–747 CrossRef.
- C. Sanson, C. Schatz, J.-F. Le Meins, A. Brulet, A. Soum and S. Lecommandoux, Langmuir, 2010, 26, 2751–2760 CrossRef PubMed.
- M. Le Hellaye, N. Fortin, J. Guilloteau, A. Soum, S. Lecommandoux and S. M. Guillaume, Biomacromolecules, 2008, 9, 1924–1933 CrossRef PubMed.
- G. Rong, M. Deng, C. Deng, Z. Tang, L. Piao, X. Chen and X. Jing, Biomacromolecules, 2003, 4, 1800–1804 CrossRef PubMed.
- L. Zhao, N. Li, K. Wang, C. Shi, L. Zhang and Y. Luan, Biomaterials, 2014, 35, 1284–1301 CrossRef PubMed.
- W. Zhao, Y. Gnanou and N. Hadjichristidis, Polym. Chem., 2015, 6, 6193–6201 RSC.
- B. A. Chan, S. Wuan, M. Horton and D. Zhang, Macromolecules, 2016, 49, 2002–2012 CrossRef.
- Š. Gradišar, E. Žagar and D. Pahovnik, ACS Macro Lett., 2017, 6, 637–640 CrossRef.
- M. Oshimura, T. Tang and A. Takasu, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1210–1218 CrossRef.
- M. Baśko and P. Kubisa, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 7071–7081 CrossRef.
- D. Bourissou, B. Martin-Vaca, A. Dumitrescu, M. Graullier and F. Lacombe, Macromolecules, 2005, 38, 9993–9998 CrossRef.
- S. Gazeau-Bureau, D. Delcroix, B. Martín-Vaca, D. Bourissou, C. Navarro and S. Magnet, Macromolecules, 2008, 41, 3782–3784 CrossRef.
- D. Delcroix, B. Martín-Vaca, D. Bourissou and C. Navarro, Macromolecules, 2010, 43, 8828–8835 CrossRef.
- A. Couffin, D. Delcroix, B. Martín-Vaca, D. Bourissou and C. Navarro, Macromolecules, 2013, 46, 4354–4360 CrossRef.
- J. M. Campos, M. R. Ribeiro, M. F. Ribeiro, A. Deffieux and F. Peruch, Macromol. Chem. Phys., 2013, 214, 85–93 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and MALDI-TOF MS spectra and SEC-MALS chromatograms. See DOI: 10.1039/c8py00835c |
| This journal is © The Royal Society of Chemistry 2018 |