
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cu(0)-RDRP of styrene: balancing initiator efficiency and dispersity†
Richard
Whitfield
 ,
Athina
Anastasaki
,
Glen R.
Jones
and
David M.
Haddleton
*
,
Athina
Anastasaki
,
Glen R.
Jones
and
David M.
Haddleton
*
University of Warwick, Chemistry Department, Library Road, CV4 7AL, Coventry, UK. E-mail: D.M.Haddleton@warwick.ac.uk
First published on 3rd August 2018
Abstract
Cu(0)-RDRP is a powerful technique to synthesise a wide range of polymeric materials and architectures with controlled molecular weight, low dispersities and high end group fidelity. The vast majority of reports using this technique focus on the polymerisation of acrylates or methacrylates, with very limited examples on styrene, which is surprising as this is one of the most important vinyl monomers. Herein we present the first Cu(0)-wire mediated polymerisation of styrene with enhanced initiator efficiency and dispersity. The structure of the ligand, the type of the initiator, the nature of the solvent and the catalyst concentration have been systematically varied to afford polystyrene at relatively high molecular weights (∼50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) with excellent agreement between theoretical and experimental number average molecular weight values and good control over the molecular weight distributions (Đ ∼ 1.15).
000 g mol−1) with excellent agreement between theoretical and experimental number average molecular weight values and good control over the molecular weight distributions (Đ ∼ 1.15).
Introduction
Over the past decades reversible deactivation radical polymerisation (RDRP) has greatly advanced the field of controlled polymerisation. The development of reversible addition fragmentation chain-transfer (RAFT)1–3 polymerisation, nitroxide mediated polymerisation (NMP)4–6 and atom transfer radical polymerisation (ATRP)7–11 have allowed the synthesis of complex polymeric materials with controlled architecture and molecular weight, narrow molecular weight distributions and high end group functionality.12–20Among these techniques, Cu(0)-wire RDRP21 (single electron transfer living radical polymerisation (SET LRP)22 or supplemental activator and reducing agent (SARA) ATRP)23 has attracted considerable attention as a versatile and robust methodology demonstrating broad monomer scope, yielding polymers with high end group fidelity even at near-quantitative conversions.24,25 Perhaps the most significant advantage of Cu(0)-RDRP is its simplicity26 as the reactions can often be carried out in a disposable vial (rather than Schlenk tubes) with simple deoxygenation via nitrogen bubbling for a few minutes being sufficient for a controlled polymerisation, rather than time-consuming freeze–pump–thaw cycles. In addition, the majority of the Cu(0)-wire catalyst can be removed post-polymerisation by simply removing the wire and stirrer it is wrapped around. This results in a polymerisation product mixture with only ppm concentrations of copper, which can subsequently be simply removed, circumventing the perceived issues of product metal contamination and any associated residual colour.27,28
To date, Cu(0)-RDRP has been extensively explored for the synthesis of poly(acrylates) demonstrating an impressive monomer scope, initiator, ligand and solvent choice.21 Importantly, polyacrylates can be easily prepared over a wide range of molecular weights and architectures which is exemplified by the synthesis of high-ordered complex materials.29–31 Whittaker, Haddleton and Junkers were the first to effectively use this technique in the preparation of high-order multiblock copolymers with unprecedented control and minimal loss of end-group fidelity.32–36 Significantly, this methodology involves no purification between successive blocks as each step is taken to full monomer conversion, paving the way for the design and synthesis of a new generation of materials. More recently, similar advancements have been accomplished with polyacrylamides by exploiting the rapid disproportionation (usually < 1 minute) of CuBr/Me6Tren into Cu(0) particles and CuBr2 in either aqueous or mixtures of aqueous and alcoholic media.37–40 In contrast to monomers with relatively high kp such as acrylates and acrylamides, monomer with lower kp such as methacrylates are more rarely explored, due to additional problems associated with low rates of propagation of this monomer class. Nevertheless, the controlled polymerisation of methacrylates via Cu(0)-RDRP has been reported in both aqueous and organic media with an acceptable level of control.41–43
Interestingly, the synthesis of polystyrene by Cu(0)-RDRP has received very little attention to date which is rather surprising given the importance of this material from both an engineering and technological standpoint.44 Due to the low kp of this monomer, reaction times are significantly longer and reaction temperatures typically higher in comparison to acrylate polymerisations. The only detailed report, to the best of our knowledge, is by Perrier, Harrison and co-workers who have successfully synthesised polystyrene via Cu(0)-RDRP with dispersity 1.2.45 However, the maximum molecular weight attained was 24600 g mol−1 and the catalyst employed was Cu(0)-particles which have been reported to be a less effective when compared to Cu(0)-wire.46,47 In addition, different types of solvents and initiators were not investigated. A few other reports demonstrate higher dispersities (Đ = 1.40–4) which really indicate a lack of control and these reports should be considered as unsuccessful attempts, thus highlighting that optimal conditions for the polymerisation of styrene by Cu(0)-RDRP have yet to be found.48,49
Herein we report the first comprehensive study of Cu(0)-RDRP of styrene utilising copper wire as a more efficient Cu(0) source. A wide range of initiators, ligands and solvents are employed to identify optimal conditions and obtain well-defined polystyrene in a facile manner (Fig. 1). The effect of these components on the control over the molecular weight distribution and the initiator efficiency will be investigated and critically discussed.
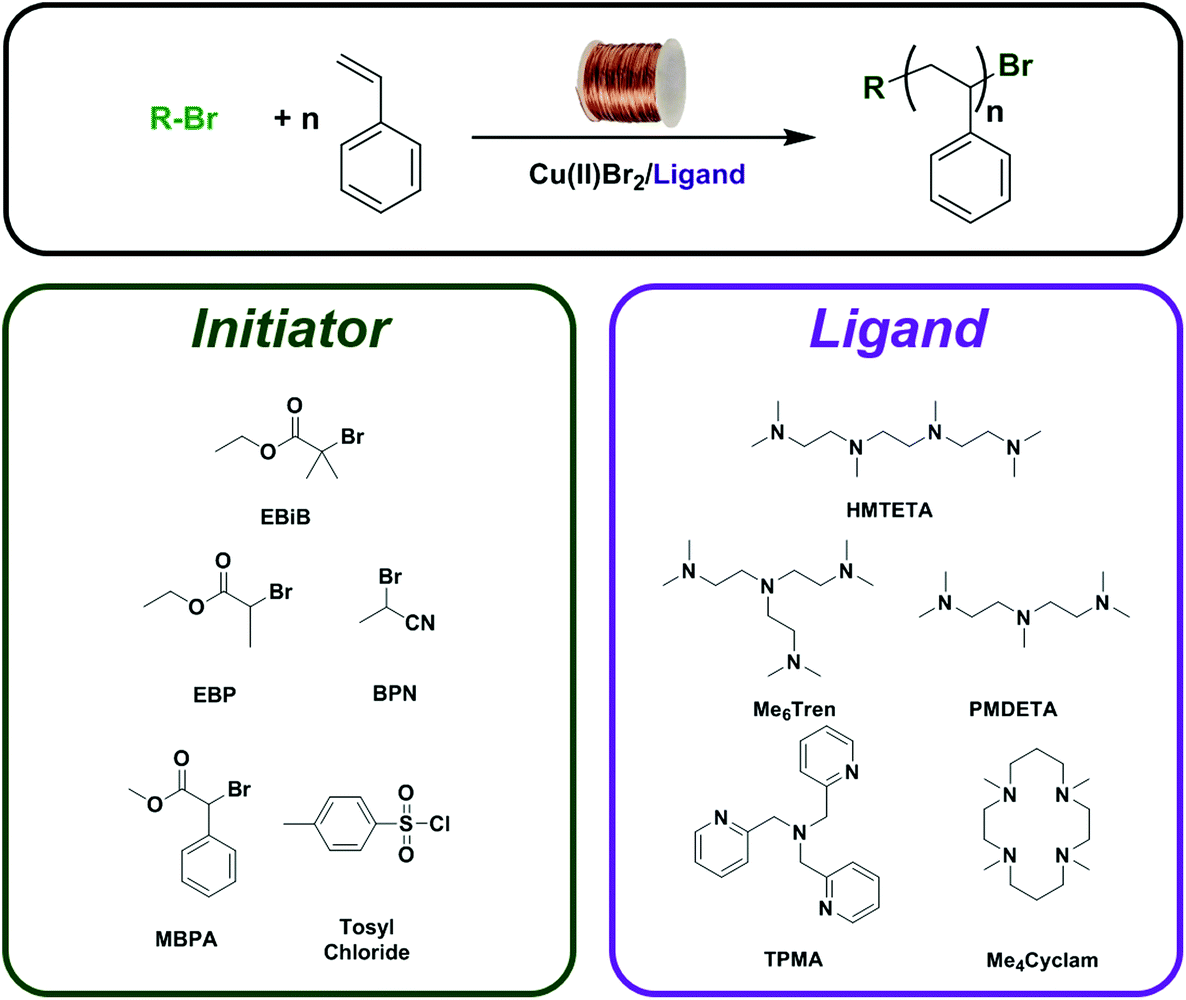 | ||
| Fig. 1 A schematic representation of the Cu(0)-wire RDRP of styrene, illustrating the structures of initiators and ligands utilised in the optimisation. | ||
Experimental
Materials and instrumentation
All chemicals were purchased from Sigma Aldrich (Merck) or VWR and used as received unless otherwise stated. All monomers were used as received, without subsequent purification. Solvents and initiators were used as purchased. Tris-(2-(dimethylamino)ethyl)amine (Me6Tren) was synthesised according to previously reported literature50 and distilled prior to use. Tris-(2-Aminoethyl)amine (Tren) and N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) were distilled prior to use. Cu(0) (gauge 0.25 mm) wire was purchased from Comax Engineered wires and purified by immersion in concentrated hydrochloric acid for 15 minutes and subsequently rinsed with water and dried prior to use. 1H NMR spectra were recorded on Bruker DPX-300 MHz or DPX-400 MHz spectrometers in CDCl3. Chemical shifts are given in ppm downfield from the internal standard tetramethylsilane. Monomer conversions were determined via1H NMR spectroscopy by comparing the integrals of monomeric vinyl protons to polymer signals. Fig. S1† illustrates a 1H NMR of polystyrene synthesised. Size exclusion chromatography (SEC) measurements were conducted using an Agilent 390-LC MDS instrument fitted with differential refractive index (DRI), viscometer (VS), dual angle light scatterer (LS) and dual wavelength UV detectors. The system was equipped with two PLgel 5 mm mixed-C columns (300 × 7.5 mm), one PLgel 5 μm guard column and autosampler. Narrow linear polystyrene standards (Agilent EasyVials) with PS molecular weights ranging from calibration between 550 g mol−1 and 1568000 g mol−1 were used as calibrants and fitted with a 3rd order polynomial. Samples were run at a flow rate of 1.0 mL min−1 at 30 °C. All samples were passed through a 0.22 μm GVHP membrane prior to analysis. The mobile phase was THF with 2% TEA and 0.01% BHT (butylated hydroxytoluene) as additives. Experimental molar mass (Mn, SEC) and dispersity (Đ) values were analysed using Agilent GPC/SEC software (version 1.2). Initiator efficiency was calculated by finding the ratio of theoretical to actual molecular weight via SEC analysis.
General procedure: PS (DP800) optimised bulk
Styrene (8 mL or 7.28 g, 800 equiv.), CuBr2 (0.98 mg, 0.05 equiv.) and Me6Tren (4.2 μL, 0.18 equiv.) were sonicated for 20 minutes in a glass vial so as to achieve saturated solutions of Cu(II)Br2. A stirrer bar wrapped with 5 cm of pre-activated copper wire was subsequently added to the reaction mixture and the vial sealed with a septum and subsequently deoxygenated by bubbling with nitrogen for 20 minutes. EBiB (12.8 μL or 17.0 mg, 1 equiv.) was then introduced in the vial via a gas-tight syringe and the polymerisation was allowed to commence at 60 °C for 36 h. Samples were taken periodically under a nitrogen blanket and passed through a short column of neutral alumina to remove dissolved copper salts prior to analysis by 1H NMR and SEC.Results and discussion
The effect of temperature
We recently reported one set of conditions to synthesise well-defined polymethacrylates, polyacrylates and polystyrene via Cu(0)-RDRP.51 Although this system is ideal to provide universal conditions for three different monomer classes and allows for simplicity in terms of procedure for non-experts, several compromises were sought for each individual monomer class (e.g. styrene). For acrylates and methacrylates to some extent, very well-optimised conditions via Cu(0)-RDRP are well reported and established.21 On the other hand, the polymerisation of styrene via Cu(0)-RDRP remains poorly explored.In the reported universal procedure and upon targeting a degree of polymerisation of 50, methyl-α-bromophenylacetate (MBPA) was used as the initiator, N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) as the ligand and isopropanol (IPA) as the solvent while the ideal temperature was illustrated to be 60 °C.54 Interestingly, at lower temperatures (25–50 °C) much slower polymerisation rates were observed with the final conversion never exceeding 70% after Indeed, ideal conditions for a specific monomer class (e.g. acrylates) would not be ideal for the polymerisation of a different monomer class (e.g. styrene). For acrylates and methacrylates to some extent, very well-optimised conditions via Cu(0)-RDRP are well reported and established.21 On the other hand, the polymerisation of styrene via Cu(0)-RDRP remains poorly explored.
In the reported universal procedure and upon targeting a degree of polymerisation of 50, methyl-α-bromophenylacetate (MBPA) was used as the initiator, N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) as the ligand and isopropanol (IPA) as the solvent while the ideal temperature was illustrated to be 60 °C.52 Interestingly, at lower temperatures (25–50 °C) much slower polymerisation rates 36 hours of reaction time (Table 1, entries 1.1–1.6 and Fig. 3 & S2†). However, upon increasing the temperature to 60 °C, very high conversions could be obtained (∼ 98%) without compromising the control over the molecular weight distributions (Đ ∼1.15) (Table 1, entries 1.7–1.8 and Fig. 3 & S3†). When further increasing the temperature to 70 °C a gradual broadening of the molecular weight distribution was evident (Đ ∼1.25) with the final dispersity greater than 1.4 when 80 °C was employed (Table 1, entries 1.9–1.12 and Fig. 3 & S4†). This is rather surprising as traditional ATRP of styrene typically operates well at higher temperatures and as such the higher dispersities could be attributed to the low boiling point of IPA.53,54
| Entry number | Temp. (°C) | Reaction time (h) | Conversion (%) | M n (Theo.) (g mol−1) | M n (SEC) | Đ |
|---|---|---|---|---|---|---|
|
a 5 cm of Cu(0) wire and 0.05% equivalents of CuBr2 and 0.36 equivalents of PMDETA with respect to MBPA initiator were utilised for all polymerisations, and the volume ratio of monomer to IPA was maintained at 1:1 throughout. The target DP was 50 and conversion was calculated via1H NMR.
|
||||||
| 1.1 | 25 | 18 | <10 | — | — | — |
| 1.2 | 25 | 36 | 31 | 1800 | 1900 | 1.22 |
| 1.3 | 40 | 18 | 21 | 1300 | 1400 | 1.19 |
| 1.4 | 40 | 36 | 67 | 3700 | 4300 | 1.19 |
| 1.5 | 50 | 18 | 35 | 2000 | 2400 | 1.13 |
| 1.6 | 50 | 36 | 73 | 4000 | 4900 | 1.14 |
| 1.7 | 60 | 18 | 47 | 2600 | 3800 | 1.14 |
| 1.8 | 60 | 36 | 98 | 5300 | 8100 | 1.15 |
| 1.9 | 70 | 18 | 55 | 3100 | 5200 | 1.17 |
| 1.10 | 70 | 36 | >99 | 5600 | 8200 | 1.25 |
| 1.11 | 80 | 18 | 61 | 3500 | 6800 | 1.30 |
| 1.12 | 80 | 36 | >99 | 5600 | 7800 | 1.42 |
Nevertheless, even at 60 °C, very poor initiator efficiency was observed (Ieff = 64%) which demonstrates that these universal conditions, although sufficient when low dispersities are required, were not ideal for the polymerisation of styrene. This deviation in initiator efficiency is even more pronounced when higher targeted degrees of polymerisations (DP) were attempted. For example, when targeting DP800 even lower initiator efficiency was evident (Ieff ∼54%) resulting in polystyrene with a molecular weight of 34300 (Mn theoretical 18500 g mol−1) (Table 2, entry 2.1 and Fig. S5†). Still, however, under these conditions well-defined polystyrene of relatively high molecular weight can be obtained with a dispersity as low as 1.2.
| Entry number | Ligand and equivalents wrt initiator | Conversion (%) | M n (Theo.) (g mol−1) | M n (SEC) | I eff (%) | Đ |
|---|---|---|---|---|---|---|
|
a 5 cm of Cu(0) wire and 0.05 equivalents of CuBr2 with respect to MBPA initiator were utilised for all polymerisations, and the volume ratio of monomer to IPA solvent was maintained at 1:1 throughout. The target DP was 800, reaction time was 36 hours and conversion was calculated via1H NMR.
|
||||||
| 2.1 | PMDETA (0.36) | 22 | 18500 |
34300 |
54 | 1.22 |
| 2.2 | PMDETA (0.72) | 32 | 26700 |
29200 |
91 | 1.20 |
| 2.3 | HMTETA (0.36) | 28 | 23600 |
29400 |
80 | 1.29 |
The effect of type and concentration of ligand
In our previous investigation, the concentration of PMDETA was kept constant at 0.36 equivalents with respect to the initiator (MBPA). Upon systematically varying the concentration of PMDETA from 0.18 to 0.72 equivalents, no change in the molecular weight distribution was observed with low dispersities being maintained for all polymerisations (Đ ∼1.2) (Table S2† and Fig. 4a). However, the initiator efficiency was significantly enhanced at higher ligand loading (0.72 equivalents). Kinetics of this polymerisation were performed illustrating an induction period of less than 2 hours and significant progression of the polymerisation by the time 4 hours had been reached. Importantly linear kinetics were observed as a proof of livingness (Table S2 and Fig. S6†). To better visualise this we targeted a higher degree of polymerisation (DP800) where the initiator efficiency was as high as 91% (Table 2, entry 2.2 and Fig. 2a & S7†). This is in stark contrast to when only 0.36 equivalents were utilised, where only 54% initiator efficiency was observed. This dramatic increase in efficiency with increased PMDETA concentration can be attributed to better solubility and complexation of CuBr2 when more ligand is present in solution. In an attempt to improve initiator efficiency in an alternative way, a number of ligands were subsequently screened. Me6Tren one of the highest activity ligands reported, exhibited a relatively controlled polymerisation at 0.36 equivalents (Đ = 1.35) although complete loss of control was observed at higher concentrations (Đ ∼3). In contrast, at lower concentrations (0.18 equivalents) a low dispersity of 1.12 could be obtained although a significantly lower polymerisation rate was evident as opposed to PMDETA. These results suggest that when Me6Tren is employed, lower concentrations are preferred and the controlled polymerisation of styrene to yield higher conversions is not possible with this ligand under the conditions studied (Table S3† and Fig. 4b). | ||
| Fig. 2 SEC chromatograms of well-defined polystyrene homopolymers synthesised via the optimised Cu(0)-RDRP conditions, namely (a) increasing ligand concentration, (b) optimising initiator and solvent and (c) the development of a bulk polymerisation system. In all cases polymerisations were carried out at 60 °C, with 5 cm of copper wire and 0.05 equivalents of CuBr2 deactivator with respect to the initiator utilised. | ||
 | ||
| Fig. 3 SEC chromatograms illustrating the effect of temperature on the polymerisation of styrene (Target DP50) with traces of the polymers synthesised at 25 °C, 60 °C and 80 °C respectively. | ||
 | ||
| Fig. 4 SEC chromatograms illustrating the effect of ligand concentration on the polymerisation of styrene (Target DP50) with (a) PMDETA and (b) Me6Tren via Cu(0)-RDRP. | ||
A similar behaviour was observed when tris(2-aminoethyl)amine (Tren)45 was instead employed with an even more pronounced loss of control (Table S4 and Fig. S8†). A range of other ligands were also explored including bipyridine (bpy), tris(2-pyridylmethyl)amine (TPMA), 1,4,8,11-tetraazacyclotetradecane (Cyclam) and Me4-Cyclam. However, in all cases this resulted in an absence of polymerisation or a significant loss of control, highlighting the incompatibility of these ligands to mediate the controlled polymerisation of styrene under the selected conditions (Table S5 and Fig. S9†). In contrast, 1,1,4,7,10,10-hexamethyltriethylenetetramine (HMTETA) generated much higher conversions while maintaining low dispersity values (Đ < 1.20) (Table S6 and Fig. S10†). To further investigate whether HMTETA is a better alternative, we pushed the system further by targeting DP800. This led to well-defined polystyrene with improved initiator efficiency (Ieff = 80%) although broader molecular weight distributions (∼1.29) were also observed (Table 2, entry 2.3 and Fig. S11†). The enhanced initiator efficiency could be due to the better solubility and complexation of CuBr2 with HMTETA which gave more efficient deactivation. Overall, we have shown that in IPA the initiator efficiency can be significantly improved from ∼50 to 80–90% by simply increasing the ligand concentration (PMDETA) or by employing HMTETA. However, PMDETA might be a better choice since it strikes a better balance between the highest molecular weight, dispersity and initiator efficiency.
The effect of the initiator
In the previous section, we concluded that PMDETA allows for the preparation of well-defined polystyrene. To explore different ways to improve the initiator efficiency, we also performed an initiator study by maintaining the ligand concentration at 0.36 equivalents with respect to the initiator. The temperature was maintained at 60 °C, as previously concluded. Interestingly, when ethyl-α-bromoisobutyrate (EBiB) was employed no polymerisation was detected, even when the reaction was left to proceed for one week (Table 3, entry 3.1). This is surprising since conventional ATRP conditions with CuBr often successfully employing this initiator.36,44| Entry number | Initiator | Conversion (%) | M n (Theo.) (g mol−1) | M n (SEC) | I eff (%) | Đ |
|---|---|---|---|---|---|---|
|
a 5 cm of Cu(0) wire and 0.05 equivalents of CuBr2 and 0.36 equivalents of PMDETA with respect to initiator were utilised for all polymerisations, and the volume ratio of monomer to IPA was maintained at 1:1 throughout. The target DP was 50, reaction time was 36 hours and conversion was calculated via1H NMR.
|
||||||
| 3.1 | EBiB | 0 | — | — | — | — |
| 3.2 | MBPA | 98 | 5300 | 8100 | 65 | 1.15 |
| 3.3 | EBP | 77 | 4200 | 5300 | 79 | 1.11 |
| 3.4 | 2-BPN | 77 | 4200 | 5400 | 78 | 1.10 |
| 3.5 | Tosyl chloride (CuBr2) | 66 | 3600 | 7600 | 47 | 1.26 |
| 3.6 | Tosyl chloride (CuCl2) | 67 | 3700 | 8200 | 45 | 1.29 |
This also highlights that high ppm copper systems can often exhibit significantly different behaviour to low ppm systems. When tosyl chloride was used as the initiator, in combination with CuBr2 or CuCl2 deactivator very poor initiator efficiency was observed (Ieff < 50%) (Table 3, entries 3.5–3.6 and Fig. S12†). Impressively, however, upon employing either ethyl-2-bromopropionate (EBP) or 2-bromopropiontrile (BPN) the initiator efficiency was significantly enhanced (Ieff = 80%) while narrow molecular weight distributions could also be achieved (∼1.10) (Table 3, entries 3.3–3.4 and Fig. 5). These results together demonstrate that secondary radical forming initiators (except phenylacetate which has extra stabilisation) are much more advantageous for the controlled polymerisation of styrene via Cu(0)-RDRP when compared to tertiary forming radical initiators.
 | ||
| Fig. 5 SEC chromatograms of polystyrene homopolymers (Target DP50) with narrow molecular weight distributions synthesised with our optimal initiators, (a) MBPA, (b) EBP and (c) BPN. | ||
To further probe the potential of these initiators to improve the initiator efficiency we targeted polystyrene of DP800. In agreement with our previous observations, BPN showed Ieff = 76% and EBP showed Ieff = 72%. Therefore, both initiators exhibited higher initiator efficiency as opposed to the highly active MBPA (Ieff = 54%) (Table S7 and Fig. S13†). MBPA's low initiator efficiency is related to the slow addition of some radicals to styrene resulting in termination and a lower number of polymer chains.
The effect of the solvent
To expand the scope of this system we also explored the potential of other solvents to mediate the controlled polymerisation of styrene. Polar solvents such as DMSO, DMF and ethanol (Table 4, entries 4.1–4.3 and Fig. S14†) yielded uncontrolled polymerisation and polystyrene with broad molecular weight distributions while acetone, methanol and trifluoroethanol resulted in no polymerisation (Table 4, entries 4.4–4.6). However, upon using the more hydrophobic solvent tert-butanol a controlled polymerisation took place although the initiator efficiency was comparable to IPA (Table 4, entries 4.7–4.8 and Fig. S15†). This could be attributed to the fact that both solvents form biphasic mixtures during polymerisation which may affect the initiator efficiency.54,55–59 Finally, toluene, acetonitrile and dioxane, were also explored (Table 4, entries 4.9–4.11 and Fig. S16†). Interestingly, all three solvents were compatible with the controlled polymerisation of styrene and demonstrated improved initiator efficiencies (Ieff > 85% in all cases).| Entry number | Solvent | Conv. (%) | M n (Theo.) (g mol−1) | M n (SEC) | Đ |
|---|---|---|---|---|---|
|
a 5 cm of Cu(0) wire, 0.05 equivalents of CuBr2 and 0.36 equivalents of PMDETA with respect to MBPA initiator were utilised for all polymerisations, and the volume ratio of monomer to solvent was maintained at 1:1 throughout. The target DP was 50, the reaction time was 36 hours and conversion was calculated via1H NMR.
|
|||||
| 4.1 | DMSO | 74 | 4100 | 5500 | 1.57 |
| 4.2 | DMF | 79 | 4300 | 8000 | 1.48 |
| 4.3 | Ethanol | 75 | 4100 | 6200 | 1.58 |
| 4.4 | Acetone | — | — | — | — |
| 4.5 | Methanol | 0 | — | — | — |
| 4.6 | TFE | — | — | — | — |
| 4.7 | IPA | 98 | 5300 | 8100 | 1.15 |
| 4.8 | tBuOH | 96 | 5100 | 6500 | 1.23 |
| 4.9 | Toluene | 90 | 4800 | 5600 | 1.12 |
| 4.10 | Acetonitrile | 65 | 3600 | 4200 | 1.24 |
| 4.11 | Dioxane | 77 | 4300 | 4400 | 1.10 |
| 4.12 | IPA:Tol 1:1 |
90 | 4800 | 7600 | 1.18 |
| 4.13 | IPA:Tol 1:4 |
89 | 4700 | 7500 | 1.15 |
As with other optimised conditions, these three solvents were subsequently tested upon targeting polystyrene with DP 800 (Table S8, Fig. S17†). Surprisingly, the polymerisation in acetonitrile resulted in loss of control (dispersity ∼2) while the polymerisation in dioxane and toluene both demonstrated improved initiator efficiencies over IPA (Ieff = 80% and Ieff = 68% respectively in comparison to Ieff = 54%), thus highlighting the superiority of solvents that solubilise copper species less well. The loss of control observed in the case of acetonitrile might be due to the better stabilisation of CuBr species in this solvent which may lead to faster polymerisation rates and subsequent loss of control.60 Interestingly, even the addition of small amounts of IPA to a toluene polymerisation resulted in a dramatic decrease of the initiator efficiency, similar to that of IPA, thus suggesting this solvent should be avoided (Table 4, entries 4.12–4.13 and Fig. S18†).
Combining optimal conditions
Our findings that toluene and dioxane are much better solvents in mediating the controlled polymerisation of styrene while maintaining high initiator efficiency were further confirmed by replacing MBPA with the most effective initiators; EBP and BPN. Both initiators exhibited improved initiator efficiency in dioxane and toluene (when compared to MBPA) with EBP achieving dispersities as low as 1.13 and Ieff = 82% (Table 5, entries 5.1–5.2 and Fig. S19†). On the contrary, BPN yielded polystyrene with initiator efficiencies >92% although the dispersities were ∼ 1.2 (Table 5, entries 5.3–5.4 and Fig. 2b & S20†). Thus, depending on the application of the polymers required, any of the initiators/solvents presented in this subsection (EBP and BPN in combination with dioxane or toluene) could be successfully employed.| Entry number |
Initiator | Solvent | Conversion (%) | M n (Theo.) (g mol−1) | M n (SEC) | I eff (%) | Đ |
|---|---|---|---|---|---|---|---|
|
a In all polymerisations, 5 cm of Cu(0) wire and 0.05 equivalents of CuBr2 with respect to initiator were utilised, and the volume ratio of monomer to solvent was maintained at 1:1 throughout. The target DP was 800, the reaction time was 36 hours and conversion was calculated via1H NMR.
|
|||||||
| 5.1 | EBP | Dioxane | 28 | 23600 |
29200 |
81 | 1.14 |
| 5.2 | EBP | Toluene | 28 | 23600 |
28800 |
82 | 1.13 |
| 5.3 | BPN | Dioxane | 26 | 21900 |
23800 |
92 | 1.20 |
| 5.4 | BPN | Toluene | 31 | 26000 |
25800 |
100 | 1.25 |
Exploring polymerisation in bulk
Conventional ATRP in the absence of solvent (bulk) has been well explored, however Cu(0)-RDRP in bulk has rarely been investigated.47 Since it was demonstrated that the nature of the solvent can have such a dramatic effect on the initiator efficiency, we decided to further simplify our system and eliminate any solvent effects. A targeted degree of polymerisation of 800 was again chosen for this study. To our surprise, 0.36 equivalents of PMDETA with respect to initiator yielded well controlled polystyrene (Mn = 31900) with perfect initiator efficiency (∼100%) (Table 6, entry 6.1 and Fig. S21a†). This is in contrast to when IPA or toluene were used, where 54% and 68% initiator efficiencies were observed respectively. EBP also gave rise to excellent initiator efficiencies (∼100%) and narrow molecular weight distributions (Đ = 1.14) (Table 6, entry 6.2 and Fig. S21b†). Interestingly, although EBiB was unable to successfully polymerise styrene in solution, under bulk conditions it allowed for the controlled polymerisation of styrene (Đ = 1.13) also with very good initiator efficiency (∼97%) and a final Mn of 45100 (Table 6, entry 6.3 and Fig. S21c†). This is attributed to the higher concentration of monomer, which results in more efficient initiation and polymerisation. Bulk systems have also been previously illustrated to reduce termination events.61 Similarly to the solvated system, an induction period of less than 2 hours was observed (Table S9 and Fig. S22†).
| Entry number | Ligand | Initiator | Conversion (%) | M n (Theo.) (g mol−1) | M n (SEC) | I eff (%) | Đ |
|---|---|---|---|---|---|---|---|
| a In all polymerisations, 5 cm of Cu(0) wire and 0.05 equivalents ofCuBr2 with respect to initiator were utilised. The target DP was 800, the reaction time was 36 hours and conversion was calculated via1H NMR. | |||||||
| 6.1 | PMDETA (0.36 wrt [I]) | MBPA | 38 | 31900 |
31900 |
100 | 1.13 |
| 6.2 | EBP | 39 | 32700 |
32700 |
100 | 1.14 | |
| 6.3 | EBiB | 59 | 46900 |
45100 |
96 | 1.13 | |
| 6.4 | Me6Tren (0.18 wrt [I]) | MBPA | 30 | 25200 |
28800 |
88 | 1.10 |
| 6.5 | EBP | 31 | 26000 |
26100 |
100 | 1.10 | |
| 6.6 | EBiB | 60 | 49900 |
48200 |
97 | 1.16 | |
Given the great success achieved with these bulk experiments, we hypothesised that the absence of solvent might also increase the tolerance of the system to other components. To validate our hypothesis, Me6Tren was employed as an alternative ligand. The bulk reactions of MBPA, EBP and EBiB all resulted in controlled polymerisations with low dispersity values (<1.16) and exceptional initiator efficiencies (∼88–99%) (Table 6, entries 6.4–6.6 and Fig. 2c & 6). The greater versatility of this system compared to high copper systems and also polar solvated conditions can be attributed to the relative solubility of Cu(I) and Cu(II) complexes, with low copper concentration systems having much better relative solubilities. These results demonstrate the superiority of bulk conditions for the controlled polymerisation of styrene while maintaining a balance between low dispersities and excellent initiator efficiencies for a range of initiators and ligands.
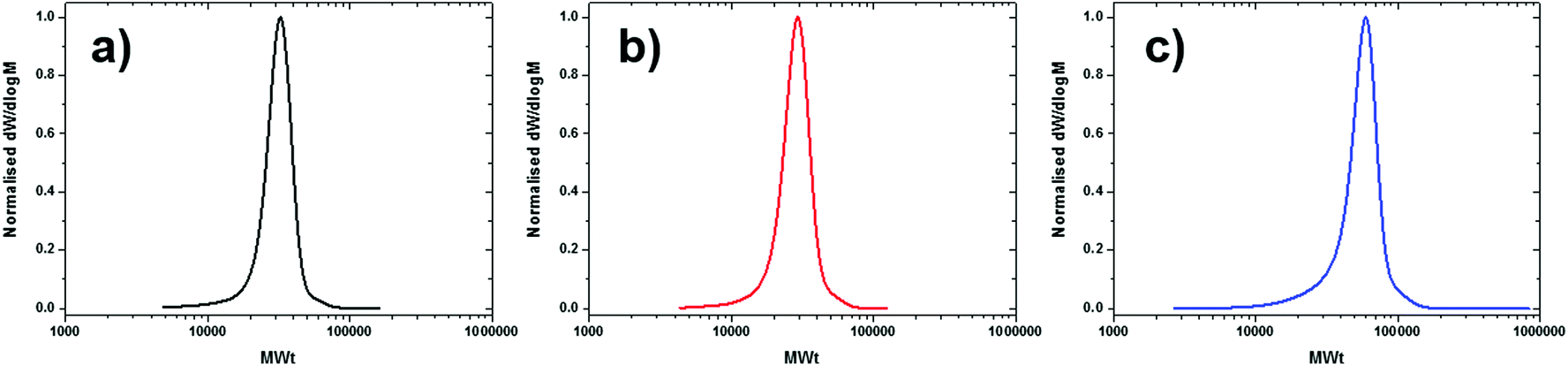 | ||
| Fig. 6 SEC chromatograms of well-defined polystyrene (Target DP800) synthesised in bulk utilising Me6Tren as the ligand and (a) MBPA, (b) EBP and (c) EBiB as the initiator. | ||
Conclusions
In conclusion, we have demonstrated a number of different conditions that allow access to the controlled polymerisation of styrene via Cu(0)-RDRP. Compared to the literature, we complete extensive optimisation, carefully adjusting the type and concentration of ligand, the initiator choice and the solvent, obtaining well-defined polystyrene of higher molecular weight low dispersity and very high initiator efficiencies. Using increased ligand concentrations (0.72 equiv.), specific solvents (toluene, dioxane) and secondary initiators (EBP, BPN) polystyrene can be made in a facile manner. Interestingly, our best results were obtained when performing the experiments in bulk where a number of initiators and ligand were shown to facilitate the controlled polymerisation of styrene without compromising the molecular weight distributions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would hereby like to acknowledge financial support from Syngenta (R. W.), Lubrizol (G. R. J.) and the University of Warwick (R. W. & G. R. J.). A. A. acknowledges the Global Marie Curie Fellowship for financial support. We gratefully appreciate the support provided by the University of Warwick Polymer Characterisation facility (RTP) and Dr Daniel Lester for providing access to SEC equipment.References
- J. Chiefari, Y. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. Le, R. T. Mayadunne, G. F. Meijs, C. L. Moad and G. Moad, Macromolecules, 1998, 31, 5559–5562 CrossRef.
- M. R. Hill, R. N. Carmean and B. S. Sumerlin, Macromolecules, 2015, 48, 5459–5469 CrossRef.
- C. Barner-Kowollik, T. P. Davis, J. Heuts, M. H. Stenzel, P. Vana and M. Whittaker, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 365–375 CrossRef.
- D. H. Solomon, E. Rizzardo and P. Cacioli, US Patent, 4581429, 1986 Search PubMed.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef PubMed.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef.
- C. Boyer, N. A. Corrigan, K. Jung, D. Nguyen, T. Nguyen, N. N. M. Adnan, S. Oliver, S. Shanmugam and J. Yeow, Chem. Rev., 2016, 116, 1803–1949 CrossRef PubMed.
- W. He, H. Jiang, L. Zhang, Z. Cheng and X. Zhu, Polym. Chem., 2013, 4, 2919–2938 RSC.
- R. Whitfield, A. Anastasaki, N. P. Truong, P. Wilson, K. Kempe, J. A. Burns, T. P. Davis and D. M. Haddleton, Macromolecules, 2016, 49, 8914–8924 CrossRef.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4, 2505 CrossRef PubMed.
- J. Tanaka, A. S. Gleinich, Q. Zhang, R. Whitfield, K. Kempe, D. M. Haddleton, T. P. Davis, S. Perrier, D. A. Mitchell and P. Wilson, Biomacromolecules, 2017, 18, 1624–1633 CrossRef PubMed.
- A. Anastasaki, V. Nikolaou, N. W. McCaul, A. Simula, J. Godfrey, C. Waldron, P. Wilson, K. Kempe and D. M. Haddleton, Macromolecules, 2015, 48, 1404–1411 CrossRef.
- N. G. Engelis, A. Anastasaki, G. Nurumbetov, N. P. Truong, V. Nikolaou, A. Shegiwal, M. R. Whittaker, T. P. Davis and D. M. Haddleton, Nat. Chem., 2017, 9, 171–178 CrossRef PubMed.
- N. G. Engelis, A. Anastasaki, R. Whitfield, G. R. Jones, E. Liarou, V. Nikolaou, G. Nurumbetov and D. M. Haddleton, Macromolecules, 2018, 51, 336–342 CrossRef.
- G. Nurumbetov, N. Engelis, J. Godfrey, R. Hand, A. Anastasaki, A. Simula, V. Nikolaou and D. M. Haddleton, Polym. Chem., 2017, 8, 1084–1094 RSC.
- R. Aksakal, M. Resmini and C. R. Becer, Polym. Chem., 2016, 7, 171–175 RSC.
- L. Barner, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2007, 28, 539–559 CrossRef.
- A. Anastasaki, V. Nikolaou, G. Nurumbetov, P. Wilson, K. Kempe, J. F. Quinn, T. P. Davis, M. R. Whittaker and D. M. Haddleton, Chem. Rev., 2015, 116, 835–877 CrossRef PubMed.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156–14165 CrossRef PubMed.
- D. Konkolewicz, Y. Wang, P. Krys, M. Zhong, A. A. Isse, A. Gennaro and K. Matyjaszewski, Polym. Chem., 2014, 5, 4396–4417 RSC.
- A. Anastasaki, V. Nikolaou and D. M. Haddleton, Polym. Chem., 2016, 7, 1002–1026 RSC.
- R. Aksakal, M. Resmini and C. R. Becer, Polym. Chem., 2016, 7, 6564–6569 RSC.
- E. Liarou, R. Whitfield, A. Anastasaki, N. G. Engelis, G. R. Jones, K. Velonia and D. Haddleton, Angew. Chem., Int. Ed., 2018, 57, 8998–9002 CrossRef PubMed.
- T. Pintauer and K. Matyjaszewski, Chem. Soc. Rev., 2008, 37, 1087–1097 RSC.
- R. Aksakal, M. Resmini and C. Becer, Polym. Chem., 2016, 7, 6564–6569 RSC.
- C. Boyer, P. B. Zetterlund and M. R. Whittaker, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2083–2098 CrossRef.
- A. Anastasaki, V. Nikolaou, G. Nurumbetov, N. P. Truong, G. S. Pappas, N. G. Engelis, J. F. Quinn, M. R. Whittaker, T. P. Davis and D. M. Haddleton, Macromolecules, 2015, 48, 5140–5147 CrossRef.
- A. H. Soeriyadi, C. Boyer, F. Nyström, P. B. Zetterlund and M. R. Whittaker, J. Am. Chem. Soc., 2011, 133, 11128–11131 CrossRef PubMed.
- C. Boyer, A. Derveaux, P. B. Zetterlund and M. R. Whittaker, Polym. Chem., 2012, 3, 117–123 RSC.
- Q. Zhang, J. Collins, A. Anastasaki, R. Wallis, D. A. Mitchell, C. R. Becer and D. M. Haddleton, Angew. Chem., Int. Ed., 2013, 125, 4531–4535 CrossRef.
- Y.-M. Chuang, A. Ethirajan and T. Junkers, ACS Macro Lett., 2014, 3, 732–737 CrossRef.
- B. Wenn, A. Martens, Y.-M. Chuang, J. Gruber and T. Junkers, Polym. Chem., 2016, 7, 2720–2727 RSC.
- A. Anastasaki, C. Waldron, P. Wilson, C. Boyer, P. B. Zetterlund, M. R. Whittaker and D. Haddleton, ACS Macro Lett., 2013, 2, 896–900 CrossRef.
- Q. Zhang, P. Wilson, Z. Li, R. McHale, J. Godfrey, A. Anastasaki, C. Waldron and D. M. Haddleton, J. Am. Chem. Soc., 2013, 135, 7355–7363 CrossRef PubMed.
- C. Waldron, Q. Zhang, Z. Li, V. Nikolaou, G. Nurumbetov, J. Godfrey, R. McHale, G. Yilmaz, R. K. Randev and M. Girault, Polym. Chem., 2014, 5, 57–61 RSC.
- G. R. Jones, Z. Li, A. Anastasaki, D. J. Lloyd, P. Wilson, Q. Zhang and D. M. Haddleton, Macromolecules, 2016, 49, 483–489 CrossRef.
- G. R. Jones, A. Anastasaki, R. Whitfield, N. Engelis, E. Liarou and D. Haddleton, Angew. Chem., 2018, 130, 2–18 CrossRef.
- G. R. Jones, R. Whitfield, A. Anastasaki, N. Risangud, A. Simula, D. J. Keddie and D. M. Haddleton, Polym. Chem., 2018, 9, 2382–2388 RSC.
- S. R. Samanta, A. Anastasaki, C. Waldron, D. M. Haddleton and V. Percec, Polym. Chem., 2013, 4, 5563–5569 RSC.
- A. Simula, V. Nikolaou, F. Alsubaie, A. Anastasaki and D. M. Haddleton, Polym. Chem., 2015, 6, 5940–5950 RSC.
- I. Oral, H. Guzel and G. Ahmetli, Polym. Bull., 2011, 67, 1893–1906 CrossRef.
- J. Tom, B. Hornby, A. West, S. Harrisson and S. Perrier, Polym. Chem., 2010, 1, 420–422 RSC.
- N. H. Nguyen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5109–5119 CrossRef.
- X. Jiang, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2716–2721 CrossRef.
- S. Harihara Subramanian, R. Prakash Babu and R. Dhamodharan, Macromolecules, 2008, 41, 262–265 CrossRef.
- J. Gao, Z. Zhang, N. Zhou, Z. Cheng, J. Zhu and X. Zhu, Macromolecules, 2011, 44, 3227–3232 CrossRef.
- M. Ciampolini and N. Nardi, Inorg. Chem., 1966, 5, 41–44 CrossRef.
- R. Whitfield, A. Anastasaki, V. Nikolaou, G. R. Jones, N. G. Engelis, E. H. Discekici, C. Fleischmann, J. Willenbacher, C. J. Hawker and D. M. Haddleton, J. Am. Chem. Soc., 2017, 139, 1003–1010 CrossRef PubMed.
- J. F. Lutz and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 897–910 CrossRef.
- S. Perrier, D. Berthier, I. Willoughby, D. Batt-Coutrot and D. M. Haddleton, Macromolecules, 2002, 35, 2941–2948 CrossRef.
- A. Moreno, S. Grama, T. Liu, M. Galià, G. Lligadas and V. Percec, Polym. Chem., 2017, 8, 7559–7574 RSC.
- C. Boyer, A. Atme, C. Waldron, A. Anastasaki, P. Wilson, P. B. Zetterlund, D. Haddleton and M. R. Whittaker, Polym. Chem., 2013, 4, 106–112 RSC.
- S. Grama, J. Lejnieks, M. Enayati, R. B. Smail, L. Ding, G. Lligadas, M. J. Monteiro and V. Percec, Polym. Chem., 2017, 8, 5865–5874 RSC.
- R. L. Jezorek, M. Enayati, R. B. Smail, J. Lejnieks, S. Grama, M. J. Monteiro and V. Percec, Polym. Chem., 2017, 8, 3405–3424 RSC.
- R. B. Smail, R. L. Jezorek, J. Lejnieks, M. Enayati, S. Grama, M. J. Monteiro and V. Percec, Polym. Chem., 2017, 8, 3102–3123 RSC.
- W. Tang, A. K. Nanda and K. Matyjaszewski, Macromol. Chem. Phys., 2005, 206, 1171–1177 CrossRef.
- K. Matyjaszewski, S. Coca, S. G. Gaynor, M. Wei and B. E. Woodworth, Macromolecules, 1997, 30, 7348–7350 CrossRef.
- M. Zhong and K. Matyjaszewski, Macromolecules, 2011, 44, 2668 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional tables, figures and general procedures. See DOI: 10.1039/c8py00814k |
| This journal is © The Royal Society of Chemistry 2018 |