
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly-ordered onion micelles made from amphiphilic highly-branched copolymers†
Sarah L.
Canning
 ab,
Joseph M. F.
Ferner
a,
Natalie M.
Mangham
a,
Trevor J.
Wear
c,
Stuart W.
Reynolds
c,
Jonathan
Morgan
c,
J. Patrick A.
Fairclough
d,
Stephen M.
King
e,
Tom
Swift
f,
Mark
Geoghegan
*b and
Stephen
Rimmer
*af
ab,
Joseph M. F.
Ferner
a,
Natalie M.
Mangham
a,
Trevor J.
Wear
c,
Stuart W.
Reynolds
c,
Jonathan
Morgan
c,
J. Patrick A.
Fairclough
d,
Stephen M.
King
e,
Tom
Swift
f,
Mark
Geoghegan
*b and
Stephen
Rimmer
*af
aDepartment of Chemistry, University of Sheffield, S3 7HF, UK. E-mail: s.rimmer@bradford.ac.uk
bDepartment of Physics and Astronomy, University of Sheffield, S3 7RH, UK. E-mail: mark.geoghegan@sheffield.ac.uk
cDomino UK Ltd, Bar Hill, Cambridge, CB23 8TU, UK
dDepartment of Mechanical Engineering, University of Sheffield, S3 7HQ, UK
eISIS Pulsed Neutron & Muon Source, STFC Rutherford Appleton Laboratory, Didcot, OX11 0QX, UK
fDepartment of Chemistry and Biosciences, University of Bradford, Bradford BD7 1DP, UK
First published on 12th November 2018
Abstract
Uniform onion micelles formed from up to ten nano-structured polymer layers were produced by the aqueous self-assembly of highly-branched copolymers. Highly-branched poly(alkyl methacrylate)s were chain extended with poly(acrylic acid) in a two-step reversible addition–fragmentation chain transfer-self-condensing vinyl polymerization (RAFT-SCVP) in solution. The resulting polymers were dispersed into water from oxolane (THF) using a self-organized precipitation-like method and the self-assembled particles were studied by phase-analysis light scattering, small-angle neutron scattering, and electron microscopy techniques. The relative hydrophobicity of the blocks was varied by changing the alkyl methacrylate (methyl, butyl, or lauryl) and this was found to affect the morphology of the particles. Only the poly(butyl methacrylate)-containing macromolecule formed an onion micelle structure. The formation of this morphology was observed to depend on: the evaporation of the good solvent (THF) during the self-assembly process causing kinetic trapping of structures; the pH of the aqueous phase; and also on the ratio of hydrophobic to hydrophilic segments within the copolymer. The lamellar structure could be removed by annealing the dispersion above the glass transition temperature of the poly(butyl methacrylate). To exemplify how these onion micelles can be used to encapsulate and release an active compound, a dye, rhodamine B (Rh B), was encapsulated and released. The release behaviour was dependent on the morphology of the particles. Particles formed containing the poly(methyl methacrylate) or poly(lauryl methacrylate) core did not form onions and although these materials absorbed Rh B, it was continuously released at room temperature. On the other hand, the lamellar structure formed from branch-poly(butyl methacrylate)-[poly(butyl methacrylate)-block-poly(acrylic acid)] allowed for encapsulation of approximately 45% of the dye, without release, until heating disrupted the lamellar structure.
Introduction
Onion-like micelles were first observed in bulk block copolymer blends.1 More recently, onion-like micelles have been formed during the self-assembly of linear block copolymers, for example, the stepwise aggregation of AB and BC diblock copolymers in solution.2,3 It was also found they could be formed from a single block copolymer through applying shear to Pluronic® systems,4 and later by self-organized precipitation (SORP)5 for hydrophobic,6–8 or solvent exchange for amphiphilic, block copolymers.9,10 Triblock copolymers obtained by nitroxide-mediated polymerization in miniemulsion have also been shown to be capable of forming onion-like structures.11 Onion-like micelles have shown promise in drug delivery,12,13 since they provide high loading capacity,13 and also find application in the fabrication of templates for inorganic compounds14 and the production of hybrid functional nanoparticles.15An important means of controlling the formation of particles is that of SORP,5 in which two miscible solvents (one of which is a good solvent and the other is poor) are used. The faster drying component is a good solvent, which, when evaporated, allows spherical particles of controlled structure to be formed as a dispersion in the poor solvent. Block copolymers with immiscible components are ideal candidate materials5 for SORP because order is inherent in their structure. Nevertheless, a two-solvent process has been shown to create particles of a hyperbranched polymer relatively uniform in size.16 In the case of block copolymers, most examples of the formation of particles in a poor solvent are of concentric layered (onion) micelles,5–8 but hollow spheres have been observed,17 and other structures can be formed, for example by blending.5
Branched polymers, encompassing dendritic, multi-branched and highly-branched architectures, exhibit unique properties in terms of solution behaviour and rheology in comparison to linear analogues.18,19 Additionally, the large number of chain ends per molecule offers the possibility of adding further chemical functionality to the polymer.20–23 These branched materials can be produced by chain growth polymerization via the use of a branching monomer which acts as either monomer and transfer agent24 or monomer and initiator,25 in a process known as self-condensing vinyl polymerization (SCVP). A similar approach uses monomers that both polymerize and undergo addition–fragmentation with the propagating chain end.26 An extension of SCVP uses reversible addition–fragmentation chain transfer (RAFT) polymerization,27 a type of controlled radical polymerization which has been widely used to produce polymers with well-defined architectures, such as block copolymers, graft copolymers, and star polymers.28–33
Highly-branched polymers do not entangle in solution,34 which means that such materials are liable to form films quickly on the evaporation of their solvent,35 and may require careful control if there are specific requirements for molten structures. Perhaps surprisingly given the heterogeneous nature of highly-branched polymers, it has been possible to create ordered macroscopic objects from them,36,37 and micellar37–39 or vesicular37,40,41 structures of various sizes have also been possible.
Since SORP works readily with linear block copolymers, it is pertinent to question the kind of structures that can be formed with highly-branched block copolymers. Here, the conditions for the SORP-induced formation of multi-lamellar onion micelles from copolymers of poly(acrylic acid) (PAA) and different highly-branched poly(alkyl methacrylates)s are presented. Three different polymers were prepared, with hydrophobic highly-branched poly(methyl methacrylate) (PMMA), poly(butyl methacrylate) (PBMA), or poly(lauryl methacrylate) (PLMA). A SORP-like procedure was used to disperse these molecules into water, a good solvent for the hydrophilic poly(acrylic acid) (PAA) blocks but a poor solvent for the hydrophobic poly(alkyl methacrylate) (PnMA). Gradual addition of a selective solvent to a solution of copolymer in a good solvent for both blocks, followed by slow evaporation of the good solvent, led to the formation of self-assembled structures. This self-assembly was studied by a combination of transmission electron microscopy (TEM), particle-sizing and zeta-potential measurements using phase-analysis light scattering (PALS), small-angle neutron scattering (SANS), and scanning electron microscopy (SEM). It is generally accepted that a deterioration of solvent quality leads to the formation of aggregates.42–44 Indeed onion micelles are formed under specific conditions and only for branch-poly(butyl methacrylate)-[poly(butyl methacrylate)-block-poly(acrylic acid)], which is abbreviated here as HB-PBMA-PAA. (The nomenclature used here is extended to the other PnMA highly-branched cores: HB-PMMA-PAA and HB-PLMA-PAA.) The requirement for the poor solvent evaporation step and the effect of varying the ratio of hydrophilic to hydrophobic blocks were investigated. A mechanism for the formation of these self-assembled structures is postulated. Finally, these onion micelles are investigated for application delivery by studying the encapsulation and release of a model compound, rhodamine B (Rh B).
Materials and methods
RAFT polymerization was used to prepare highly-branched alkyl methacrylate polymers with pyrrole dithioester groups at the chain ends, using a previously-demonstrated synthetic strategy.43 The polymerization was performed in dioxane at 60 °C using the RAFT chain-transfer agent (CTA) 4-vinylbenzyl 1H-pyrrole-1-carbodithioate, a dithioate ester that also possesses alkene functionality. The dual action of the CTA enabled branching to occur during the polymerization, due to the occurrence of both copolymerization with the styryl double bond and reversible addition–fragmentation chain transfer with the dithioate group. Polymerizations were stopped at intermediate conversion to preserve RAFT chain-end functionality. These hydrophobic macromolecular chain transfer agents (macro-CTAs) were then chain-extended (again using RAFT solution polymerization in dioxane) with a hydrophilic monomer, acrylic acid (AA), to yield amphiphilic block copolymers with a highly-branched architecture, as shown in Scheme 1. Each AA polymerization proceeded to high conversion (>90% as determined by 1H NMR spectroscopy, see Table 1).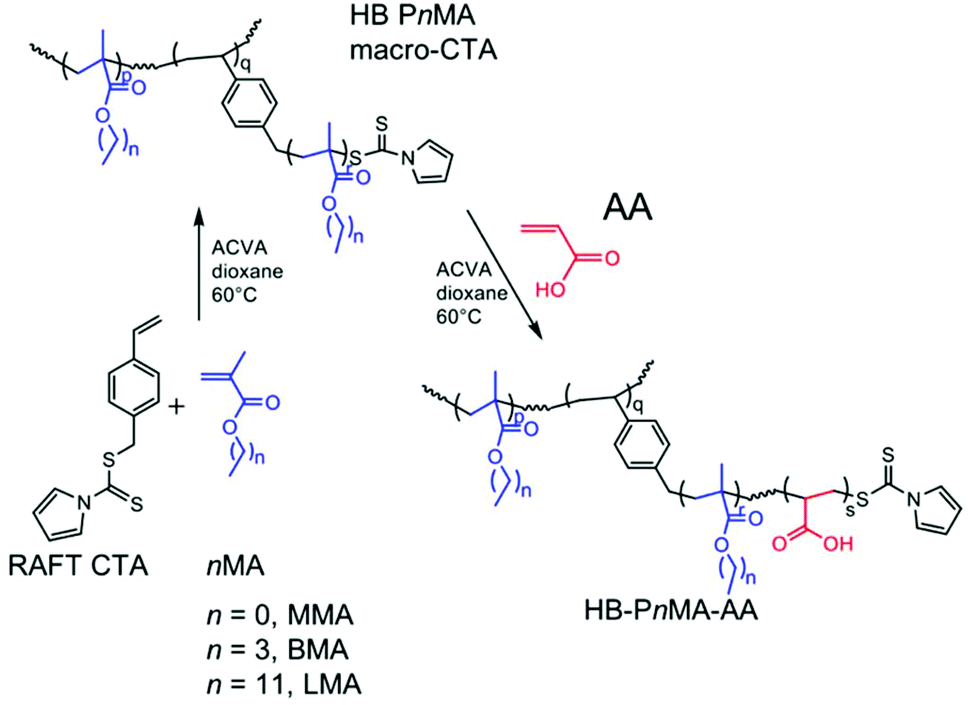 | ||
| Scheme 1 Synthesis of HB-PnMA-PAA block copolymers with pyrrole chain ends in a two-step RAFT-SCVP polymerization. | ||
| Polymer | Conversiona /% | Molar feed ratio nMA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AA :AA |
Polymer molar ratio PnMA:PAA |
DBa |
M
nb/kg mol−1 |
M
wb/kg mol−1 |
Đ |
|---|---|---|---|---|---|---|---|
| a Monomer conversion and degree of branching determined by 1H NMR. b Molar masses and dispersity (Đ) determined by GPC (THF, PMMA standards). | |||||||
| HB-PMMA | 55 | — | — | 0.059 | 13.5 | 27.3 | 2.0 |
| HB-PBMA | 88 | — | — | 0.030 | 18.0 | 35.8 | 2.0 |
| HB-PLMA | 77 | — | — | 0.062 | 30.8 | 64.7 | 2.1 |
| HB-PMMA-PAA | 93 | 0.7:1.0 |
1.1:1.0 |
0.029 | 29.5 | 118.3 | 4.0 |
| HB-PBMA1.4-PAA1.0 | 97 | 0.5:1.0 |
1.4:1.0 |
0.013 | 40.0 | 139.9 | 3.5 |
| HB-PLMA-PAA | 99 | 0.3:1.0 |
0.7:1.0 |
Unknown | 34.8 | 328.0 | 9.4 |
| HB-PBMA0.9-PAA1.0 | 92 | 0.5:1.0 |
0.9:1.0 |
0.014 | 2.7 | 7.6 | 2.8 |
| HB-PBMA1.3-PAA1.0 | 91 | 0.75:1.0 |
1.3:1.0 |
0.011 | 4.3 | 16.7 | 3.9 |
| HB-PBMA1.55-PAA1.0 | 94 | 1.0:1.0 |
1.55:1.0 |
0.018 | 2.6 | 8.0 | 3.1 |
| HB-PBMA1.67-PAA1.0 | 96 | 1.5:1.0 |
1.67:1.0 |
0.029 | 7.5 | 30.1 | 4.0 |
| HB-PBMA1.71-PAA1.0 | 97 | 2.0:1.0 |
1.71:1.0 |
0.033 | 14.4 | 126.8 | 8.8 |
Materials
Sodium hydride (60% in mineral oil dispersion, Aldrich), carbon disulfide (>99%, Aldrich), 4-vinylbenzyl chloride (90%, Aldrich), 4,4′-azobis(4-cyanovaleric acid) (ACVA, >98%, Aldrich), methanol (Fisher), diethyl ether (Fisher), hexane (Fisher), 1,4-dioxane (Aldrich, sure-seal, anhydrous 99.8%) and petroleum ether 40–60 (Fisher) were used as purchased. Trimethylsilyldiazomethane (2 M solution in diethyl ether, Aldrich) was used as received. Pyrrole (99%, Aldrich) was distilled over calcium hydride (95%, Aldrich) under reduced pressure to give a colourless liquid. Acrylic acid (99%, Aldrich) was distilled under reduced pressure to remove inhibitors. N,N-Dimethylformamide (DMF) was obtained from Fisher Scientific and dried using a Grubbs dry solvent method. MEHQ inhibitors were removed from methyl methacrylate (99%, Aldrich), butyl methacrylate (99%, Aldrich) and lauryl methacrylate (96%, Lancaster) by running through a column packed with inhibitor removing beads (Aldrich). Deionized water was used in all experiments.Synthesis of 4-vinylbenzyl 1H-pyrrole-1-carbodithioate
Pyrrole (5.0 g, 74.5 mmol) and DMF (10 ml) were added dropwise over 30 min to a rapidly stirred suspension of sodium hydride (2.98 g, 124.2 mmol) in DMF (80 ml) to produce a yellow foam. The solution was stirred at room temperature for 30 min then cooled to 0 °C using an ice bath. Carbon disulfide (5.68 g, 4.50 ml, 74.6 mmol) and DMF (10 ml) were added dropwise over 10 min to create a dark red solution. This was stirred at room temperature for 30 min and then cooled to 0 °C. 4-Vinylbenzyl chloride (11.37 g, 10.50 ml, 74.5 mmol) and DMF (10 ml) were then added dropwise over 20 min. The brown solution was stirred overnight at room temperature. The solution product was placed in a separating funnel with diethyl ether (80 ml) and distilled water (80 ml). The organic layer was recovered and the aqueous layer was extracted with diethyl ether (3 × 160 ml). The organic extracts were combined and dried over magnesium sulfate, before gravity filtration. The solvent was then removed by rotary evaporation to give a brown oil. The oil was purified by silica chromatography using 100% hexane as the eluent, then the solvent was removed by rotary evaporation to give a bright yellow oil (5.93 g).RAFT polymerization of methacrylates and subsequent chain extension with acrylic acid
A typical procedure for the synthesis of HB-PMMA was as follows: MMA (1 g, 9.99 mmol), 4-vinylbenzyl 1H-pyrrole-1-carbodithioate (0.0852 g, 0.329 mmol), ACVA (0.0184 g, 66 μmol, CTA/ACVA molar ratio = 5.0), and dioxane (10 g, 10% w/w) were mixed together until the solid initiator had dissolved. The resulting solution was transferred into a glass ampoule and freeze–pump–thawed on a high vacuum line (10−4 mbar, three cycles) then flame-sealed and heated in a water bath set to 60 °C by a thermostat for up to 36 h to undergo polymerization. Products were precipitated into rapidly stirred methanol. The methanol was removed by decanting and the polymer was dried under vacuum at room temperature for 24 h. The precipitation procedure was repeated once more to remove any traces of residual monomer, giving polymer products as yellow solids.The HB-PMMA macro-CTA (0.4 g) was dissolved in 7.2 g dioxane, then AA (0.4 g, 5.55 mmol) and ACVA (0.0022 g, 80 μmol, CTA/ACVA molar ratio = 5.0) were added and allowed to mix until the solid initiator had dissolved. The resulting solution was transferred into a glass ampoule, freeze–pump–thawed on a high vacuum line, flame-sealed, and heated in a water bath as described above to undergo polymerization. Products were then precipitated into rapidly stirring ice-cold petroleum ether 40–60 °C. The petroleum ether was removed by decanting and the polymer was dried under vacuum at room temperature for 24 h. The procedure was repeated once more to remove any traces of residual monomer, giving polymer products as a pale-yellow powder. HB-PBMA-PAA and HB-PLMA-PAA were synthesized following the same method.
Preparation of dispersions
Dispersions were prepared using a solvent-switch method. The copolymer was dissolved in oxolane (THF) at a concentration of 5 mg ml−1 (0.5% w/v) and stirred overnight. A Razel R-99 syringe pump was used to add an equal volume of ultrapure H2O at a constant rate of 0.1 ml min−1. The dispersion was then stirred uncovered for 3 h to allow the THF to evaporate. 1H NMR analysis showed that no residual THF was present after this time. These dispersions were sealed in sample tubes to prevent evaporation and placed into an oil bath set to 45 °C whilst stirring for 12 h.Annealing of dispersions
Dispersions were prepared using the solvent switch method, as described above. These were then sealed in sample tubes to prevent evaporation and placed into an oil bath set to 45 °C whilst stirring for 12 h.Encapsulation and release of rhodamine B
Copolymer dispersions were prepared as above but with a solution of Rh B in deionized water at a concentration of 0.2 mg cm−3 added dropwise to allow Rh B encapsulation. Dispersions were injected into pre-hydrated 3500 MWCO dialysis cassettes and were dialysed against deionized water for approximately 48 h with regular water changes until a negligible concentration of Rh B was observed in UV-vis absorption spectra. The cassette was then transferred into a fresh volume of deionized water heated to 45 °C and rhodamine B release was monitored by UV-vis spectroscopy to a maximum wavelength of λmax = 553 nm (extinction coefficient = 94198 l mol−1 cm−1).
1H NMR spectroscopy
All NMR spectra were recorded at ambient temperature on a Bruker AV-400 at 400 MHz (64 scans averaged per spectrum). Samples of mass 20–40 mg were dissolved in CDCl3 (4-vinylbenzyl 1H-pyrrole-1-carbodithioate, HB-PnMA macro-CTA) or 1:1 CDCl3:d-DMSO (HB-PnMA-PAA), filtered and placed in 7 mm NMR tubes.
Mass spectroscopy
Electron ionization (EI) mass spectroscopy was carried out using a VG Autospec mass spectrometer.Gel permeation chromatography
Average molar masses and molar mass distributions were measured relative to polystyrene standards by gel permeation chromatography (GPC) with PL gel MIXED-B (10 μm particle size, 100–106 Å pore size, effective molar mass range 103–106 g mol−1, 3 × 30 cm plus a guard column; Polymer Laboratories, UK) on a refractive index detector. The mobile phase was THF (GPC grade) set at a flow rate of 1 ml min−1. The sample (concentration 2 mg ml−1) was filtered before injection (Gilson 234 auto injector). Samples containing PAA were methylated before analysis using trimethylsilyldiazomethane to prevent column interaction, following standard procedure.45Particle-size and zeta-potential measurements
Particle-size analysis was carried out on a Brookhaven Instruments Corporation ZetaPALS zeta-potential analyser with the 90Plus/BI-MAS multi-angle particle-sizing option. 15 μl of copolymer dispersion was added to 3 ml of 10 mmol KCl solution, sonicated for 20 seconds, and filtered through a 1 μm filter. Measurements were made at 25 °C. Ten analysis runs were made in triplicate for each sample. For zeta-potential measurements, 15 μl of copolymer dispersion was added to 1.5 ml of 1 mmol KCl solution. Measurements were made at 25 °C three times for each sample in 5 cycles of 2 min runs.Small-angle neutron scattering
SANS measurements were performed at the ISIS pulsed neutron and muon source (STFC Rutherford Appleton Laboratory Didcot, UK) using the fixed-geometry, time-of-flight LOQ diffractometer.46 LOQ has two fixed position-sensitive detectors at 0.5 and 4.1 m from the sample. Using incident neutron wavelengths from 2.2 to 10.0 Å, it is able to cover a scattering wave vector (Q) range of 0.009 to 1.3 Å−1. Polymer samples were prepared as 0.5% w/v solutions: 5 mg of polymer in 1 ml D2O. All samples were transferred to 2 mm path-length quartz cells (Hellma GmbH) and placed on a computer-controlled sample changer. The sample changer temperature was controlled by using circulating fluid baths and was set to 25 °C. Background (solvent) and calibration samples were also measured. Scattering data were reduced according to standard procedures using the Mantid framework to obtain the differential scattering cross section, dΣ/dΩ(Q), in absolute units (cm−1), which is referred to here as I(Q).47,48 Scattering data were fitted to models using the SasView software package (sasview.org).Transmission electron microscopy
TEM imaging was carried out using a Philips CM 100 instrument operating at 100 kV. Polymer samples dispersed in ultrapure H2O as described above were prepared for TEM by placing a 5 μl drop of sample onto a glow-discharged carbon-coated grid for 1 min. The grid was blotted, washed in a drop of distilled water and blotted again. The grid was then washed in a drop of uranyl formate, blotted and then stained by holding the grid in a drop of uranyl formate for 20 s before blotting. Particle diameters were obtained using ImageJ analysis software49 to measure enough particles to represent a statistically significant sample; where possible at least 100 particles were measured.Scanning electron microscopy
For the SEM imaging, a TEM grid (as above) with sample adsorbed was applied to an aluminium stub of 1.27 cm diameter using a carbonized sticky tab as an adhesive. Stubs were sputter-coated with gold using an Edwards S150b coater and viewed using a Philips XL-20 SEM operating at 20 kV.UV-visible absorption spectroscopy
UV-vis measurements were performed using a PerkinElmer Lambda 35 UV-visible spectrometer with a double beam set-up employing deuterium and tungsten–halogen lamps. Glass sample cells were used with a path length of 1 cm. Deionized water was used as the reference. Spectra were recorded over a wavelength range of 500–600 nm. The absorbance was measured at λmax = 553 nm. A calibration curve was constructed using solutions of known concentration to determine the molar extinction coefficient.Results and discussion
Characterization of HB-PnMA-PAA copolymers
The expected structures for these polymers are shown in Scheme 1, with the pyrrole groups situated at the chain ends whereas the styryl groups create a branching point within the structure. Analysis of these polymers by 1H NMR showed the presence of the pyrrole groups at the ends of the polymer branches at chemical shifts δ = 6.34 and 7.72 ppm. Broad peaks due to the styryl units were also observed between δ = 7.30 and 7.45 ppm. Average values of the degrees of branching (DB) were calculated by first comparing the methacrylate protons at around 0.90 ppm to the aromatic protons from the styryl and pyrrole groups in the region δ = 6.34 to 7.72 ppm to give the average number of monomers per branch (MB). DB is then the reciprocal of MB. Here, the degree of branching is the number of branch points, i.e. the aryl units, divided by the total number of repeat units including those of acrylic acid. The associated error in the measurement of the integration of NMR signals has been estimated to be ±5%.50Table 1 displays the results of the syntheses of both the homopolymer macro-CTAs, and also the copolymers. 1H NMR spectra peaks of the copolymers at δ = 1.63 and 2.22 ppm were due to PAA. The ratio of methacrylate to acrylic acid within the copolymers could also be calculated from the 1H NMR spectra. Equal mass fractions of PnMA and PAA were targeted for each copolymer, in addition to equal molar masses for all three copolymers, with the intention that any observed differences in self-assembly could be attributed to the change in hydrophobicity. The amount of PAA in all three copolymers was less than the feed ratio. However, all three copolymerizations of AA proceeded to high conversion, indicating the formation of PAA homopolymer, as expected for RAFT and RAFT-SCVP polymerizations.21 Homopolymer of the monomer used to form the second block is generally formed during the RAFT polymerization reaction. This lower molar mass peak in the chromatogram is due to the presence of a small amount of PAA homopolymer. DB was calculated for all of the macro-CTAs and the block copolymers, except for HB-PLMA-PAA where the pyrrole and styryl groups could not be clearly seen in the 1H NMR spectrum. DB values for the PnMA cores were close to the 0.06 targeted, then values decreased following PAA addition as the branch lengths increased.
THF GPC analysis of the copolymers following methylation with trimethylsilyldiazomethane demonstrated an increase in molar mass in each case compared to that of the macro-CTA. A large increase in dispersity was also observed for all three copolymers compared to their macro-CTA, which indicates a considerable degree of branching within the copolymer structure (Fig. 1). The large dispersities obtained here are not unusual for copolymerizations of this nature.25,50
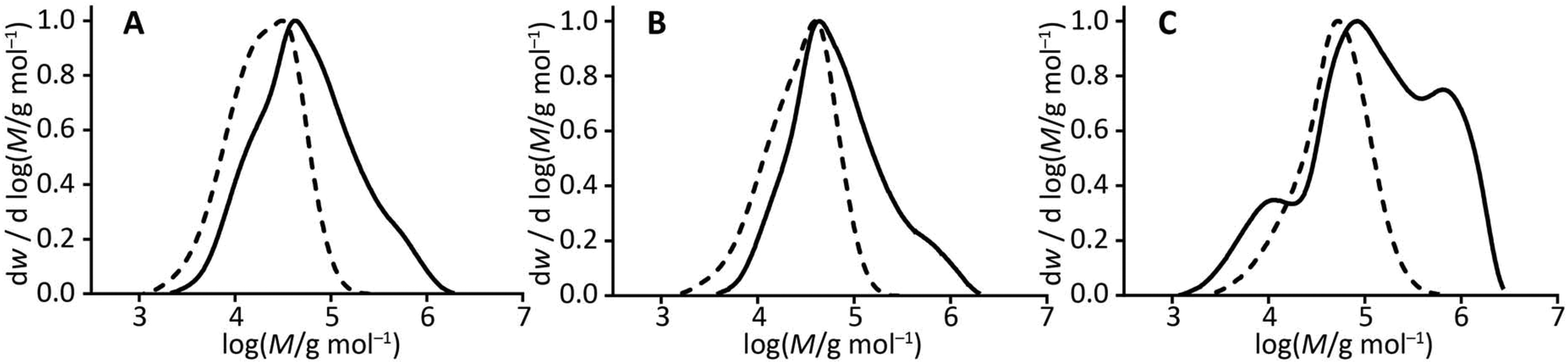 | ||
| Fig. 1 A comparison of molar mass distributions obtained from GPC: (A) HB-PMMA (dashed line) and HB-PMMA-PAA (solid line), (B) HB-PBMA (dashed line) and HB-PBMA1.4-PAA1.0 (solid line), and (C) HB-PLMA (dashed line) and HB-PLMA-PAA (solid line). | ||
Self-assembly of HB-PnMA-PAA in water
These branched copolymers were dispersed in water using a solvent-switch method. This involved the dropwise addition of water, a block-selective solvent for PAA, at a controlled rate using a syringe pump into a stirring solution of the amphiphilic copolymer in THF, a good solvent for both blocks. This was followed by evaporation of the THF to form an aqueous dispersion of self-assembled copolymer particles. The structures formed are expected to be governed by several factors including solution pH, ionic strength, and polymer concentration.51The three HB-PnMA-PAA copolymers self-assembled into very different structures when dispersed into water, as shown by the TEM images in Fig. 2. Spherical micelles with a rough appearance to the particle surface were formed from HB-PMMA-PAA. These are consistent with the granular appearance of large particles formed by the aggregation of unimicellar aggregates.38 The BMA analogue, however, formed lamellar onion micelles. The HB-PLMA-PAA copolymer formed much smaller particles which were more elliptical in shape with a “dimple” in the centre. These results clearly demonstrate the strong effect that changing the alkyl group on the methacrylate monomer has on the self-assembly of these polymers. The architecture, degree of branching, and molar masses were similar and thus should not account for the differences in the self-assembled structures observed.
 | ||
| Fig. 2 TEM micrographs of dispersions of HB-PnMA-PAA copolymers in water, stained with uranyl formate: (A) HB-PMMA-PAA, (B) HB-PBMA1.4-PAA1.0 (with inset magnified image of an onion of diameter 188 nm) and (C) HB-PLMA-PAA. The scale bar in each case is 200 nm. | ||
A combination of TEM, particle-sizing, and zeta-potential measurements (made by PALS) and SANS (by fitting a simple sphere model to scattering data) was used to characterize the different dispersions. The results are summarized in Table 2. Diameters measured from TEM images are expected to be smaller than those determined by PALS and SANS due to the effect of drying, whereas PALS and SANS measure particles in their hydrated state. This is true for the HB-PLMA-PAA sample. However, in the case of HB-PMMA-PAA and HB-PBMA1.4-PAA1.0 there is no clear trend in size measured by the different techniques. It must be noted that the diameter obtained using TEM represents the particles measured in a limited number of images analysed, whereas the SANS and PALS techniques give an ensemble average and therefore are more representative of the whole sample. Particle-size measurements of the HB-PBMA1.4-PAA1.0 dispersion indicated the presence of a bimodal population of larger and smaller onion structures which correlated with the TEM images (see Fig. 2). The HB-PLMA-PAA dispersion was notably uniform. All three dispersions were shown to be stable by the zeta-potential results, which indicate a negative charge on the particle surfaces due to ionization of the carboxylic acid groups of the PAA segments. At the native pH of the deionized water (pH ∼ 5) the PAA is partially ionized.
| Sample | Mean diameter (PALS)/nm | PDI (PALS)a | Mean zeta potential/mV | Mean diameter (TEM)/nm | Mean diameter (SANS)b/nm | DI (SANS)c |
|---|---|---|---|---|---|---|
| a Dispersity of particles measured by PALS. b Using a simple sphere model. c Dispersity of particles determined by SANS. | ||||||
| HB-PMMA-PAA | 159 ± 1 | 0.13 ± 0.01 | −55 ± 1 | 169 ± 5 | 137 ± 6 | 0.07 ± 0.02 |
| HB-PBMA1.4-PAA1.0 | 82 ± 1 | 0.07 ± 0.02 | −50 ± 1 | 164 ± 6 | 171 ± 2 | 0.06 ± 0.01 |
| 191 ± 1 | ||||||
| HB-PLMA-PAA | 60 ± 1 | 0.02 ± 0.02 | −41 ± 10 | 44 ± 1 | 63 ± 1 | 0.18 ± 0.01 |
Scanning electron microscopy (SEM) was used to study the surface of the sample and TEM to visualize the internal structure. SEM was used to confirm that the onion micelles were spherical. A TEM grid to which a stained HB-PBMA1.4-PAA1.0 dispersion was adsorbed was sputter-coated with gold and used for SEM imaging to allow direct comparison between the TEM and SEM images. The SEM images in Fig. 3 show that the onion micelles were indeed spherical, and the lamellar structure (Fig. 2B) from TEM was a series of internal concentric shells.
 | ||
| Fig. 3 SEM images of the particles formed from HB-PBMA-PAA on a TEM grid. The adsorbed HB-PBMA1.4-PAA1.0 particles were stained with uranyl formate, sputter-coated with Au, and then imaged by SEM. These images demonstrate the spherical structure, a conclusion inaccessible to TEM. The image in A is a magnification of part of that in B. | ||
SANS was used to study dispersions of the copolymers in D2O. Diverse scattering profiles were obtained for the three samples, as shown in Fig. 4. A clear Bragg peak was observed for the HB-PBMA1.4-PAA1.0 sample which is characteristic of a multilayered structure. The layer spacing can be obtained from Q = 2π/d. Using the Q value associated with the Bragg peak, an average spacing of 12 nm is predicted for HB-PBMA1.4-PAA1.0. Despite the apparent differences in the profiles, the scattering of all three polymers was successfully fitted to a lamellar paracrystal model,52 and indicated a structure composed of layers with the hydrophobic PnMA on the inside to avoid the D2O and PAA segments on the outer surfaces. This model (which is more sophisticared than that used to calculate the particle sizes) is used to calculate the scattering from a stack of ordered lamellae, which is considered as a paracrystal to account for the repeat spacing. The model has been used previously to model the scattering from large multilamellar vesicles.52,53
 | ||
| Fig. 4 SANS scattering profiles of I(Q) against Q for the reduced data obtained from HB-PnMA-AA copolymers dispersed in D2O at 0.5% w/v (red: HB-PMMA-PAA, green: HB-PBMA1.4-PAA1.0, blue: HB-PLMA-PAA). Solid lines show fits to the lamellar paracrystal model. | ||
The values of the fitting parameters obtained from the model are summarized in Table 3. The model fit gives a layer spacing of 11.5 nm for HB-PBMA1.4-PAA1.0, which is consistent with the spacing obtained from the Bragg peak, and shows some associated dispersity which can be observed in the TEM images. The model suggests an average of ten layers for this sample in agreement with the TEM results. The scattering length density (SLD) of the layers is approximately 2.2 × 10−6 Å−2, which indicates that there is water within the layered structure, as the SLD for the copolymer alone is 1.4 × 10−6 Å−2. A water content of 16.1% by volume can therefore be calculated using the calculated SLDs of the copolymer and the D2O, and the SLD of the layers determined from the model fit. Indeed, contrast in the experiment is due to trapped D2O within the onion micelle. This is likely to be in contact with the PAA.
| Model parameter | HB-PMMA-PAA | HB-PBMA1.4-PAA1.0 | PLMA-PAA |
|---|---|---|---|
| Number of layers | 2.4 ± 0.4 | 10.3 ± 0.2 | 2.6 ± 0.1 |
| Thickness/nm | 19 ± 2 | 4.2 ± 0.2 | 14.5 ± 0.2 |
| Spacing/nm | 18 ± 2 | 11.5 ± 0.5 | 16.9 ± 0.1 |
| Dispersity of spacing | 0.26 ± 0.09 | 0.22 ± 0.04 | 0.22 ± 0.02 |
| SLD layers/Å−2 (×10−6) | 2.1 ± 0.2 | 2.20 ± 0.02 | 1.9 ± 0.2 |
| % D2O within layers | 14.4 | 16.1 | 10.2 |
Lamellar structures were also indicated for HB-PMMA-PAA and HB-PLMA-PAA, but with fewer layers. The results suggest a mixture of bi- and tri-layered micelles, with much thicker layers than obtained for HB-PBMA1.4-PAA1.0. Increased SLD again indicates the presence of D2O within the micelle structure. Because of the greater amount of noise in the scattering of the HB-PMMA-PAA particles, the model fit is much less reliable than those of the other copolymers and therefore the parameters in Table 3 are subject to greater uncertainty.
The stability of the onion micelles under ambient conditions was also investigated. A sample of HB-PBMA1.4-PAA1.0 dispersed in water was prepared and analysed by TEM and PALS to confirm that onion micelles were formed. This sample was then kept at room temperature for a period of 5 weeks and subsequently re-analysed. Fig. 5 shows TEM images of the sample before and after storage. It is clear that the lamellar structure remains intact during this period. A bimodal distribution was again observed, with an increase in diameter of both the smaller and larger particles, accompanied by an increase in dispersity. This suggests some growth or swelling of the onion micelles had occurred during the storage period; but significantly the lamellar structure was maintained.
 | ||
| Fig. 5 TEM images of onion micelles formed from HB-PBMA1.4-PAA1.0 dispersed in water (A and B) immediately following preparation and (C) and (D) after 5 weeks storage at ambient conditions. The scale bar in each case is 200 nm. | ||
Annealing was carried out on the dispersions to investigate temperature-dependent behaviour. Sample tubes containing copolymer dispersions in water were sealed and immersed in an oil bath heated to 45 °C for 12 h. Samples were removed and imaged by TEM before and after the annealing period (Fig. 6). Particle-sizing measurements were also performed. The HB-PMMA-PAA and HB-PLMA-PAA structures underwent a small increase in size and associated uncertainty, and their shapes appeared to become slightly more irregular. The HB-PBMA1.4-PAA1.0 micelles also underwent a small increase in particle size and the uncertainty increased. More significantly, the lamellar structure was no longer present, with a more vesicle-like structure observed. These results can be rationalized by considering the glass transition temperature (Tg) of the three alkyl methacrylate polymers. PMMA has a high Tg of 105 °C, similar to that of PAA (106 °C), and PLMA has a low Tg of −65 °C.54 PBMA, on the other hand, has Tg = 25 °C,55i.e. close to ambient temperature at which the onion micelles are assembled. When the temperature of the dispersion is increased to 45 °C, this has little effect on the self-assembled HB-PMMA-PAA and HB-PLMA-PAA structures since they remain well above or well below their respective Tg. However, the PBMA segment of HB-PBMA1.4-PAA1.0 is heated to above its Tg during the annealing process and hence the lamellae are able to coalesce to form an amorphous ‘vesicular’ structure.
 | ||
| Fig. 6 Representative TEM images of copolymers A: HB-PMMA-PAA (164 ± 2 nm), B: HB-PBMA1.4-PAA1.0 (90 ± 1 nm), and C: HB-PLMA-PAA (57 ± 1 nm) self-assembled in water and D: HB-PMMA-PAA (170 ± 3 nm), E: HB-PBMA1.4-PAA1.0 (96 ± 2 nm), and F: HB-PLMA-PAA (61 ± 2 nm) following annealing at 45 °C for 12 h. The parenthetical length scales represent the results of particle-sizing measurements. Samples were stained with uranyl formate prior to imaging. The scale bar in each case is 200 nm. | ||
The rate of self-assembly of the onion micelles was investigated by varying the rate of water addition from 0.05 to 1.00 ml min−1. No differences in micelle structure were observed (see ESI Fig. S4 and S5†). One possibility is that, if equilibrium structures were being formed, then the evaporation of the good solvent would not be important. On the other hand, if solvent evaporation proved to be important then this would confirm the formation of dynamic, non-ergodic structures. In order to test this, the micelle preparation procedure was carried out as usual but instead of allowing the THF to evaporate once the water was added, the solution was injected into a dialysis cassette and the THF removed by dialysis against water. After 24 h, the dispersion was removed from the cassette and imaged by TEM; Fig. 7 shows some of the images obtained. Instead of onion micelles, many small spherical micelles were formed. The mean diameter of these micelles was 18 ± 4 nm. This was comparable to the thickness of the individual layers of the onion micelles formed when the THF was evaporated. Some coalescence of small spheres to form worm-like structures was observed, and in the final image an onion micelle apparently in the process of formation was observed. However, this represents a very small population of the overall sample, the majority of which are present as small spheres. These results suggest that the evaporation step is a significant part of the process of onion micelle formation from this HB-PBMA1.4-PAA1.0 copolymer, and removal of this step impedes the formation of the kinetically-trapped onion structures.
 | ||
| Fig. 7 Representative TEM images of HB-PBMA1.4-PAA1.0 copolymer self-assembled in water. Following water addition, samples were dialysed against water for 24 h to remove residual THF, instead of the evaporation step. Samples were stained with uranyl formate prior to imaging. Predominantly small spherical micelles are observed, with some evidence of coalescence into worm-like structures. The scale bar is 200 nm in each case. | ||
The ratio of hydrophilic to hydrophobic segments in a block copolymer is known to play an important role in self-assembly. Consequently, the ratio of poly(alkyl methacrylate) to poly(acrylic acid) in these copolymers was expected to affect their self-assembly behaviour. To test this, HB-PBMA-PAA copolymers with varying ratios of BMA to AA monomer were synthesized. Five different polymers were prepared in all, with BMA:AA ratios (from NMR) of 0.9:1.0, 1.3:1.0, 1.55:1.0, 1.67:1.0 and 1.71:1.0. Dispersions in water were prepared as previously and imaged by TEM; Fig. 8 shows the images obtained.
 | ||
| Fig. 8 Representative TEM images of HB-PBMA-PAA copolymers with varying ratios of BMA to AA self-assembled in water: (A) 0.9:1.0, (B) 1.3:1.0, (C) 1.55:1.0, (D) 1.67:1.0, and (E) 1.71:1.0. Samples were stained with uranyl formate prior to imaging. The scale bars are 200 nm in each case. | ||
The results shown in Fig. 8 confirm that the ratio of hydrophobic to hydrophilic segments within these copolymers has a significant effect on their self-assembly behaviour. Large spheres are seen when less AA is present relative to the amount of BMA, whereas very small spheres are formed where copolymers contain more AA relative to BMA. A longer hydrophilic ‘stabilizer’ block disfavours micelle fusion, whilst a shorter stabilizer block is more likely to lead to micelle fusion. In the case of linear block copolymers, this leads to the formation of fibres or ‘worms’ when the stabilizer block is shorter. In the case of these highly branched polymers, when there is less hydrophilic polymer the small unimolecular micelles fuse to form more energetically favourable large multimicellar aggregates, whereas when more hydrophilic polymer is present it can adequately stabilize the smaller micelles. Onion micelles are observed at only one of these ratios, 1.32:1.0 PBMA:PAA, indicating the presence of an optimum composition with an intermediate hydrophilic component where onion micelle formation is favourable.
The pH dependence of onion micelle formation was also studied. Dispersions were prepared in buffer solutions at pH 4, 7 and 10, with NaCl added as required to maintain constant ionic strength. Representative TEM images are shown in Fig. 9. These experiments showed that pH was not a strong factor in the formation of the self-assembled structures except for pH 10. The HB-PBMA1.4-PAA1.0 onion micelles were formed at all pH but were somewhat smaller at pH 10. At pH 4 the PAA is not expected to be significantly charged (PAA has a pKa of 4.5,56–59 which will shift to higher pH for dense systems such as these described here to avoid Coulombic frustration) but at pH 7 more than half the acidic groups would be expected to be charged, and most would be charged by pH 10; yet onion micelles are still formed. That pH does not play a significant role in the micelle formation indicates that the PAA may not become fully solvated when the water is added and that the THF promotes hydrogen bonding between PAA branches. Such a consideration would dictate that it is the hydrophobic PnMA that is driving the self-assembly.
 | ||
| Fig. 9 Representative TEM images of HB-PBMA1.4-PAA1.0 copolymers self-assembled in water at different pH: (A) pH 4, (B) pH 7, and (C) pH 10. Samples were stained with uranyl formate prior to imaging and the scale bar is 200 nm in each case. | ||
The critical role of solvent evaporation indicates that the formation of the onions is due to SORP. Amphiphilic block copolymers have not been observed to form onions,17 but nevertheless there is no reason why the mechanism cannot apply to specific systems. The resultant morphology is driven by a balance between the minimization of interfacial energies and the repulsive interactions generated by swelling of the PAA segments. As indicated in earlier work,60 the attainment of the observed morphologies almost certainly involves distortion of the segments within the hydrophobic phase; and clearly the distortion of highly branched polymers has a greater relative energy cost compared to that of linear polymers. Block copolymer worm-like micelles and vesicles can be considered as the products of the fusion of spherical micelles or single polymer chains.61,62 However, fusion of the unimolecular micellar spheres reported here is limited because of the branched architecture.63,64 The limitations on fusion will impose a restriction on the system and can contribute to the formation of unusual morphologies.
THF is a good solvent for both PAA and PBMA, so the copolymers are initially present as random coils in solution. Water, however, is a good solvent for PAA and a poor solvent for PBMA (meaning it is a traditional block-selective solvent for amphiphilic block copolymer self-assembly). As water is added and THF begins to evaporate, PAA becomes more soluble and PBMA less so. The PBMA segments are driven inside the micelle to minimize the contact with water, with PAA on the outside acting to stabilize the micelles. It is proposed that the presence of some remaining THF in the mixture promotes the hydrogen bonding between PAA segments rather than the occurrence of hydrogen bonding between PAA and water. Particle coalescence leads to the formation of lamellar phases. Curvature is induced to reduce PBMA contact with water and increase PAA–PAA contacts. For the onion micelles, as polymers are added to the micelle the entropic cost of coating each layer increases because the curvature decreases with each added layer. This curvature effect could explain the limit on micelle size of ∼10 layers.
Encapsulation and release of model compound
The layered nature of these onion micelles indicated that they may have potential application for the encapsulation of small molecule targets which could subsequently be released on heating. Heating would trigger release following the rearrangement and loss of the lamellar structure within the micelles. Rh B, a fluorescent dye molecule, was selected as a model compound for the encapsulation and release studies for two reasons: its positive charge should promote binding with the carboxylic groups on the PAA segments, and additionally, its release can be easily monitored by UV-vis absorption spectroscopy.A solution of Rh B in water was added to the HB-PBMA1.4-PAA1.0 in THF solution, following the previous methodology. Once the THF had evaporated, the solution was injected into a dialysis cassette with a 3500 MWCO membrane. Firstly, the solution was dialysed against water at room temperature to remove any unbound dye. The water was changed regularly, at which point a sample of this water was analysed for Rh B. After 48 h, a negligible amount of Rh B was being released so the cassette was placed into a sealed vessel containing fresh deionized water heated to 45 °C. Rh B release was monitored by UV-visible absorption spectroscopy.
Measurement of the concentration of dye released during dialysis at room temperature allowed calculation of a ‘loading efficiency’ by comparing this to the initial amount of dye added. An average loading efficiency of 45% was obtained over the two release studies. Of this 45% encapsulated dye, 10% was released during 24 h of heating at 45 °C (Fig. 10). The same encapsulation procedure was carried out using the methyl and lauryl analogues, HB-PMMA-PAA and HB-PLMA-PAA. As expected, the encapsulation was significantly less efficient without the layered structure. The dye was steadily released from these micelles during dialysis at room temperature; after 15 days with regular water changes, 78% and 71% of the initial amounts of Rh B dye had been released for HB-PMMA-PAA and HB-PLMA-PAA respectively with release still ongoing so that it was not possible to obtain a loading efficiency.
 | ||
| Fig. 10 Release profile of encapsulated Rh B from HB-PBMA1.4-PAA1.0 onion micelles monitored by UV-visible absorption spectroscopy (λmax = 553 nm) during heating at 45 °C. | ||
The binding of Rh B with the PAA makes it likely that the rhodamine is located in the PAA layers of the micelle, even if the release does not occur in stages. This has also been described for other onion micelles.13
These data show the utility of the HB-PBMA-PAA onion micelles as vectors for encapsulation and release, initiated by thermal stimulus, of active compounds. Thermal release is applicable to a number of technologies involving the release of drugs or fragrances. Nanocapsules involving alkyl methacrylates and methacrylic acid have also been shown to release fragrances with a pH trigger.65
Conclusions
The synthesis of HB amphiphilic block copolymers via a two-step solution RAFT-SCVP has been reported. A hydrophobic HB poly(alkyl methacrylate) macro-CTA was first produced followed by chain extension with a hydrophilic monomer, acrylic acid. Copolymers with three different alkyl methacrylates (methyl, butyl, and lauryl) were chosen. These three copolymers were dispersed into water using a solvent switch in a SORP-like procedure. This caused the formation of self-assembled structures, which illustrated the effect of hydrophobicity on particle morphology. These particles were studied by PALS, TEM, and SANS.The HB-PBMA1.4-PAA1.0 copolymer self-assembled into unusual onion micelles with lamellar structure. SEM was used to show that the HB-PBMA1.4-PAA1.0 onion particles had a spherical 3D structure, which confirmed that the lamellar structure seen in the TEM images was internal. The lamellar structure was confirmed by SANS studies and the scattering data were fitted to a lamellar paracrystal model, with the parameters obtained from the fit in agreement with observations made by TEM. The onion particles were found to be stable when stored at room temperature over a period of five weeks. Annealing the sample at 45 °C for 24 h was found to remove the lamellar structure as this heating traversed the Tg of the PBMA segment, whilst no change was observed when the same procedure was carried out on the PMMA and PLMA analogues.
Several experiments were carried out to aid with elucidating the mechanism by which the onion micelles form. The evaporation of THF was found to play an important role in onion micelle formation as when this step was omitted, small spheres were formed instead. This is consistent with previous reports of SORP. It was also found that the ratio of PBMA to PAA within the polymer does affect the self-assembly behaviour, as expected. Onion micelles were only formed at one ratio of hydrophilic to hydrophobic component, while either small or large spheres were formed at the other ratios depending on the degree of steric stabilization.
A proof-of-concept study demonstrated that Rh B dye could be encapsulated during the onion micelle assembly process, and subsequently released by heating to 45 °C. This opens the system to potential applications in delivery of active agents.
Conflicts of interest
The work described in this manuscript is subject to a patent application by Domino UK Ltd: S. L. Canning, M. Geoghegan, J. Morgan, S. Reynolds, S. Rimmer, and T. Wear, Branched Block Copolymer, UK Patent Application GB1617168.8, filed October 10 2016 and World Parent Application WO2018069678 (A1) filed October 6 2017.Acknowledgements
The authors thank the EPSRC and Domino UK Ltd for a Nanotechnology KTN CASE studentship to support SLC. Experiments at the ISIS Pulsed Neutron and Muon Source were supported by a beam time allocation from the Science and Technology Facilities Council (experiment numbers RB1220108 and RB1320167).Notes and references
- D. Kinning, K. I. Winey and E. L. Thomas, Macromolecules, 1988, 21, 3502–3506 CrossRef CAS.
- K. Procházka, T. J. Martin, S. E. Webber and P. Munk, Macromolecules, 1996, 29, 6526–6530 CrossRef.
- M. R. Talingting, P. Munk, S. E. Webber and Z. Tuzar, Macromolecules, 1999, 32, 1593–1601 CrossRef CAS.
- J. Zipfel, P. Lindner, M. Tsianou, P. Alexandridis and W. Richtering, Langmuir, 1999, 15, 2599–2602 CrossRef CAS.
- H. Yabu, Bull. Chem. Soc. Jpn., 2012, 85, 265–274 CrossRef CAS.
- L. Li, K. Matsunaga, J. Zhu, T. Higuchi, H. Yabu, M. Shimomura, H. Jinnai, R. C. Hayward and T. P. Russell, Macromolecules, 2010, 43, 7807–7812 CrossRef CAS.
- H. Yabu, T. Higuchi, K. Ijiro and M. Shimomura, Chaos, 2005, 15, 047505 CrossRef PubMed.
- H. Yabu, T. Higuchi and M. Shimomura, Adv. Mater., 2005, 17, 2062–2065 CrossRef CAS.
- H. Fan and Z. Jin, Soft Matter, 2014, 10, 2848–2855 RSC.
- H. Shen and A. Eisenberg, Angew. Chem., Int. Ed., 2000, 39, 3310–3312 CrossRef CAS PubMed.
- J. Nicolas, A.-V. Ruzette, C. Farcet, P. Gérard, S. Magnet and B. Charleux, Polymer, 2007, 48, 7029–7040 CrossRef CAS.
- M.-C. Jones, Curr. Top. Med. Chem., 2015, 15, 2254–2266 CrossRef CAS PubMed.
- M.-K. Park, S. Jun, I. Kim, S.-M. Jin, J.-G. Kim, T. J. Shin and E. Lee, Adv. Funct. Mater., 2015, 25, 4570–4579 CrossRef CAS.
- S. Ren, R. M. Briber and M. Wuttig, Appl. Phys. Lett., 2009, 94, 113507 CrossRef.
- M. Pinna, S. Hiltl, X. Guo, A. Böker and A. V. Zvelindovsky, ACS Nano, 2010, 4, 2845–2855 CrossRef CAS PubMed.
- G. Jiang, L. Wang, T. Chen, H. Yu, X. Dong and C. Chen, Polymer, 2005, 46, 9501–9507 CrossRef CAS.
- H. Yabu, M. Shimomura and T. Higuchi, Int. J. Nanosci., 2006, 5, 195–198 CrossRef CAS.
- H. Magnusson, E. Malmström, A. Hult and M. Johansson, Polymer, 2002, 43, 301–306 CrossRef CAS.
- X. Zhu, Y. Zhou and D. Yan, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 1277–1286 CrossRef CAS.
- J. A. Alfurhood, P. R. Bachler and B. S. Sumerlin, Polym. Chem., 2016, 7, 3361–3369 RSC.
- R. M. England and S. Rimmer, Polym. Chem., 2010, 1, 1533–1544 RSC.
- S. Rimmer, S. Carter, R. Rutkaite, J. W. Haycock and L. Swanson, Soft Matter, 2007, 3, 971–973 RSC.
- Y. Zheng, S. Li, Z. Weng and C. Gao, Chem. Soc. Rev., 2015, 44, 4091–4130 RSC.
- J. Liu, Y. Wang, Q. Fu, X. Zhu and W. Shi, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1449–1459 CrossRef CAS.
- J. M. J. Fréchet, M. Henmi, I. Gitsov, S. Aoshima, M. R. Leduc and R. B. Grubbs, Science, 1995, 269, 1080–1083 CrossRef PubMed.
- B. Yamada, O. Konosu, K. Tanaka and F. Oku, Polymer, 2000, 41, 5625–5631 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- L. Barner, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2007, 28, 539–559 CrossRef CAS.
- S. R. Carter, R. M. England, B. J. Hunt and S. Rimmer, Macromol. Biosci., 2007, 7, 975–986 CrossRef CAS PubMed.
- A. Gregory and M. H. Stenzel, Prog. Polym. Sci., 2012, 37, 38–105 CrossRef CAS.
- R. T. A. Mayadunne, J. Jeffery, G. Moad and E. Rizzardo, Macromolecules, 2003, 36, 1505–1513 CrossRef CAS.
- J. F. Quinn, R. P. Chaplin and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2956–2966 CrossRef CAS.
- A. E. Smith, X. Xu and C. L. McCormick, Prog. Polym. Sci., 2010, 35, 45–93 CrossRef CAS.
- C. J. Hawker, P. J. Farrington, M. E. MacKay, K. L. Wooley and J. M. J. Fréchet, J. Am. Chem. Soc., 1995, 117, 4409–4410 CrossRef CAS.
- G. Cheng, B. W. Greenland, C. Lampard, N. Williams, M. S. Bahra and W. Hayes, Prog. Org. Coat., 2014, 77, 1516–1522 CrossRef CAS.
- D. Yan, Y. Zhou and J. Hou, Science, 2004, 303, 65–67 CrossRef CAS PubMed.
- Y. Zhou and D. Yan, Chem. Commun., 2009, 1172–1188 RSC.
- H. Hong, Y. Mai, Y. Zhou, D. Yan and J. Cui, Macromol. Rapid Commun., 2007, 28, 591–596 CrossRef CAS.
- Y. Mai, Y. Zhou and D. Yan, Macromolecules, 2005, 38, 8679–8686 CrossRef CAS.
- Y. Liu, C. Yu, H. Jin, B. Jiang, X. Zhu, Y. Zhou, Z. Lu and D. Yan, J. Am. Chem. Soc., 2013, 135, 4765–4770 CrossRef CAS PubMed.
- Y. Zhou and D. Yan, Angew. Chem., Int. Ed., 2004, 43, 4896–4899 CrossRef CAS PubMed.
- A. M. AL-Baradi, S. Rimmer, S. R. Carter, J. P. de Silva, S. M. King, M. Maccarini, B. Farago, L. Noirez and M. Geoghegan, Soft Matter, 2018, 14, 1482–1491 RSC.
- S. Carter, S. Rimmer, A. Sturdy and M. Webb, Macromol. Biosci., 2005, 5, 373–378 CrossRef CAS PubMed.
- S. Ghosh Roy and P. De, Polym. Chem., 2014, 5, 6365–6378 RSC.
- L. Couvreur, C. Lefay, J. Belleney, B. Charleux, O. Guerret and S. Magnet, Macromolecules, 2003, 36, 8260–8267 CrossRef CAS.
- R. K. Heenan, J. Penfold and S. M. King, J. Appl. Crystallogr., 1997, 30, 1140–1147 CrossRef CAS.
- O. Arnold, J. C. Bilheux, J. M. Borreguero, A. Buts, S. I. Campbell, L. Chapon, M. Doucet, N. Draper, R. Ferraz Leal, M. A. Gigg, V. E. Lynch, A. Markvardsen, D. J. Mikkelson, R. L. Mikkelson, R. Miller, K. Palmen, P. Parker, G. Passos, T. G. Perring, P. F. Peterson, S. Ren, M. A. Reuter, A. T. Savici, J. W. Taylor, R. J. Taylor, R. Tolchenov, W. Zhou and J. Zikovsky, Nucl. Instrum. Methods Phys. Res., Sect. A, 2014, 764, 156–166 CrossRef CAS.
- G. D. Wignall and F. S. Bates, J. Appl. Crystallogr., 1987, 20, 28–40 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- S. Carter, B. Hunt and S. Rimmer, Macromolecules, 2005, 38, 4595–4603 CrossRef CAS.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- M. Bergström, J. S. Pedersen, P. Schurtenberger and S. U. Egelhaaf, J. Phys. Chem. B, 1999, 103, 9888–9897 CrossRef.
- C. R. López-Barrón, D. Li, N. J. Wagner and J. L. Caplan, Macromolecules, 2014, 47, 7484–7495 CrossRef.
- U. N. Arua and F. D. Blum, J. Polym. Sci., Part B: Polym. Phys., 2018, 56, 89–96 CrossRef CAS.
- M. M. Mok, S. Pujari, W. R. Burghardt, C. M. Dettmer, S. T. Nguyen, C. J. Ellison and J. M. Torkelson, Macromolecules, 2008, 41, 5818–5829 CrossRef CAS.
- D. F. Anghel, V. Alderson, F. M. Winnik, M. Mizusaki and Y. Morishima, Polymer, 1998, 39, 3035–3044 CrossRef CAS.
- R. Arnold, J. Colloid Sci., 1957, 12, 549–556 CrossRef CAS.
- M. Mandel, Eur. Polym. J., 1970, 6, 807–822 CrossRef CAS.
- T. Swift, L. Swanson, M. Geoghegan and S. Rimmer, Soft Matter, 2016, 12, 2542–2549 RSC.
- Y. Yu and A. Eisenberg, J. Am. Chem. Soc., 1997, 119, 8383–8384 CrossRef CAS.
- A. A. Choucair, A. H. Kycia and A. Eisenberg, Langmuir, 2003, 19, 1001–1008 CrossRef CAS.
- H. Shen and A. Eisenberg, J. Phys. Chem. B, 1999, 103, 9473–9487 CrossRef CAS.
- D. J. Read, D. Auhl, C. Das, J. den Doelder, M. Kapnistos, I. Vittorias and T. C. B. McLeish, Science, 2011, 333, 1871–1874 CrossRef CAS PubMed.
- E. van Ruymbeke, K. Orfanou, M. Kapnistos, H. Iatrou, M. Pitsikalis, N. Hadjichristidis, D. J. Lohse and D. Vlassopoulos, Macromolecules, 2007, 40, 5941–5952 CrossRef CAS.
- I. Hofmeister, K. Landfester and A. Taden, Macromolecules, 2014, 47, 5768–5773 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Assigned 1H NMR spectra; characterization details; investigation into rate of water addition on copolymer self-assembly; UV-vis release data; details of lamellar paracrystal model used to fit SANS data; and additional TEM images. See DOI: 10.1039/c8py00800k |
| This journal is © The Royal Society of Chemistry 2018 |