DOI:
10.1039/C8PY00411K
(Paper)
Polym. Chem., 2018,
9, 3232-3237
UV-triggered shape-controllable PP fabric†
Received
14th March 2018
, Accepted 13th May 2018
First published on 14th May 2018
Abstract
A light-driven polypropylene (PP) fabric as an actuator was fabricated in which a light-responsive polymeric film acts as an active layer and a PP fabric acts as a passive layer. For this, poly[di(ethylene glycol)methyl ether methacrylate-co-pentafluorophenyl acrylate] P(DEGMA-co-PFPA) containing reactive pentafluorophenyl esters was synthesized as a precursor polymer. Owing to the highly reactive esters on P(DEGMA-co-PFPA), photoactive azobenzene moieties were introduced onto the polymer backbone via a post-modification polymerization strategy to form a light-responsive polymer, poly[di(ethylene glycol)methyl ether methacrylate-co-4-(4-methoxy-phenylazo) acrylate] (P(DEGMA-co-MOPAzo)). Interestingly, this copolymer can be attached in a simple and stable manner to the surface of a PP fabric via physical adsorption to build a light-responsive PP fabric. On account of its light responsiveness, this functional polymer PP fabric can immediately respond to a UV stimulus and show a reversible shape transition upon alternating exposure to UV irradiation and the addition of heat.
Introduction
Stimuli-responsive polymers, as a class of “smart” polymers, have been widely investigated owing to their sensitive ability to be triggered by external stimuli, such as light, temperature, humidity, pH, gas etc.1–8 Among these external stimuli, light as a trigger has numerous advantages in terms of its “clean” character, its environmental friendliness, its low cost, and its precise spatial localization. Moreover, its wavelength and irradiation time can be easily controlled. Hence, plenty of light-responsive polymers have been investigated to develop smart materials, such as artificial muscles,9 photorheological fluids,10 actuators,11 and switching devices.12 It is noteworthy that an azobenzene moiety or its derivatives are the most common compounds to build various light-responsive polymers due to their known photo-isomerization, which can undergo reversible switching between an extended trans form and a shortened cis form with alternating exposure to UV and visible light, respectively. On account of this trans to cis transition, azobenzene or its derivatives can switch from unipolar and hydrophobic properties to polar, hydrophobic and nonplanar geometric properties.13 Based on this unique feature, numerous light-responsive polymeric materials driven by an azobenzene moiety have been explored, such as light-responsive self-assembled aggregates, hydrogels and fibers, surfaces and actuators.14–18 Among all of them, light-responsive actuators have recently attracted considerable attention due to their light-remote-controllable character.19,20 For example, Kim et al. programmed polynorbornenes with azobenzene-based dendron side chains to demonstrate the remote-controllable actuator.19 Upon irradiation with UV light, the actuator expanded and bent away from the light source. Liu et al. prepared light-responsive azotolane films and fibers based on two types of reactive linear liquid crystal polymers (LLCPs) with azobenzene moieties. These fibers curled toward the incident direction of visible light at 405 or 445 nm and reverted to their initial state through heating or 530 nm light irradiation.21 Until now, the majority of light-responsive actuators consist of aligning liquid-crystalline azobenzene moieties with one orientation in flexible matrices to obtain a controllable bending direction.22–25 However, cumbersome synthetic routes are required to prepare these complex liquid-crystalline azobenzene moieties. To address this problem, using a bilayer actuator is an effective method to realize a controllable bending direction via facilely casting the light-responsive polymer as an active layer on a passive layer. Based on this strategy, Wen et al. fabricated a photo-driven actuator via casting a linear azobenzene polymer on a silk fibroin film.26 Nevertheless, in their case, the azobenzene moiety with its complex structure was still required to endow the resultant copolymer with good flexibility. Fortunately, the post-polymerization modification is an efficient method to prepare smart polymers with complex structures.27 Hence, poly(pentafluorophenyl acrylate) (PPFPA) has been extensively utilized to prepare various stimuli-responsive polymeric materials with complex structures. Inspired by the advantages of PPFPA, in the current work, the light-responsive polymer is prepared by post-polymerization modification. Considering the flexibility and adsorbability of the targeted light-responsive polymers, di(ethylene glycol)methyl ether methacrylate having a highly flexible chain was copolymerized with PFPA to yield copolymers with suitable glass transition temperatures (Tg). Then, the resultant light-responsive copolymer can be utilized to fabricate a light-responsive PP fabric as an actuator.
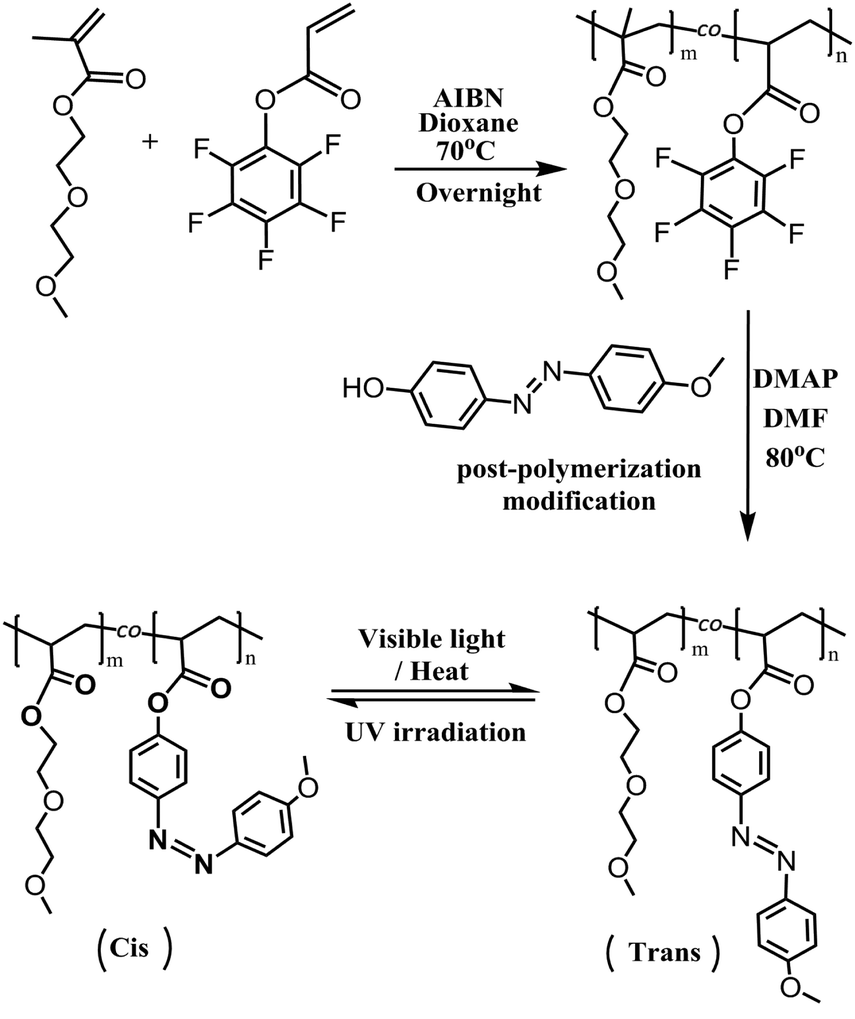
Hence, this work aims at fabricating a light-driven PP fabric as an actuator. First, poly[di(ethylene glycol)methyl ether methacrylate-co-pentafluorophenyl acrylate] P(DEGMA-co-PFPA), containing 70 mol% PFPA, was prepared as a precursor polymer. Then, 4-(4-methoxy-phenylazo)phenyl was introduced onto the polymeric backbone via post-polymerization modification to form the light-responsive copolymer poly[di(ethylene glycol)methyl ether methacrylate-co-4-(4-methoxy-phenylazo) acrylate] P(DEGMA-co-MOPAzo) (see Scheme 1). Due to the features of flexibility and adsorbability of P(DEGMA-co-MOPAzo), it can be durably coated on the surface of the PP fabric to form a light-responsive composite. Finally, the light-responsiveness and the shape-controllable capacity upon light irradiation of this composite are investigated.
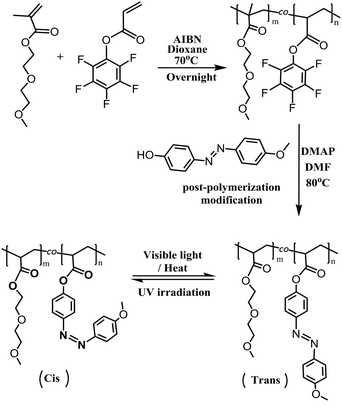 |
| | Scheme 1 The synthetic route of P(DEGMA-co-PFPA) and P(DEGMA-co-MOPAzo). | |
Experimental section
Materials
Di(ethylene glycol)methyl ether methacrylate (DEGMA, Sigma Aldrich, 95%) was purified via passing through a basic alumina column. 2,2′-Azobisisobutyronitrile (AIBN, Sigma Aldrich, 98%) was recrystallized twice from methanol. Pentafluorophenol (PFPA), acryloyl chloride, 4-methoxyaniline, sodium nitrite phenol, 2,6-lutidine, N,N-dimethylpyridin-4-amine (DMPA) and other dried solvents were purchased from Sigma-Aldrich and used as received without further purification. Membrane tubes for dialysis were received from Roth with a 3500 molecular weight cut-off membrane.
Characterization
FT-IR measurements were recorded on a Nicolet iS10 spectrometer from Thermo Scientific. 1H NMR spectra were recorded on a Bruker 400 MHz FT-NMR spectrometer in deuterated chloroform and signals are quoted in ppm relative to tetramethylsilane as the internal standard. 19F NMR spectra were measured on a Varian Gemini 2000BB spectrometer in deuterated chloroform. GPC measurements were carried out in a DMF system with an MZ-Gel SDplus linear 5 μm column at 60 °C, and their molecular weights and distributions were calculated against polystyrene standards. Scanning electron microscopy (SEM) images were taken using a Zeiss EVO-MA 10 microscope operating at 5 kV. UV/Vis absorption spectra were measured on a JASCO V-630 UV/Vis-photospectrometer from 200 to 800 nm.
Synthesis of 4-(4-methoxy-phenylazo)phenol (MOPAzo)
4-Methoxyaniline (9.85 g, 0.08 mol) was dissolved in 25 mL deionized water and 38 wt% HCl solution mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, by volume) in a 200 mL flask fitted with an overhead stirrer, a thermometer, and an addition funnel. Then, the ice bath was used to cool down this solution to 0–5 °C. Subsequently, NaNO2 (6.10 g, 0.088 mol), which was dissolved in deionized water (30 mL), was poured into the funnel and added dropwise into the flask. After 30 min, NaOH (8.00 g, 0.20 mol) and phenol (11.28 g, 0.14 mol) were added into the reaction flask, and the solution mixture was stirred at 0 °C for 5 h. Finally, the resultant dark yellow solid was filtered and washed several times with deionized water, followed by purification by column chromatography using a mixture of n-hexane and ethyl acetate (10:1, by volume) as the eluent. Finally, (4-methoxy-phenylazo)phenol was obtained as a yellow solid in 80.1% yield. The reaction mechanism is illustrated in Scheme S1.†1H NMR (400 MHz in CDCl3) (Fig. S1†): 7.95–7.78 (m, 4H, aromatic), 7.05–6.87 (m, 4H, aromatic), 5.46 (s, 1H, –OH), 3.90 (s, 3H, –CH3).
:1, by volume) in a 200 mL flask fitted with an overhead stirrer, a thermometer, and an addition funnel. Then, the ice bath was used to cool down this solution to 0–5 °C. Subsequently, NaNO2 (6.10 g, 0.088 mol), which was dissolved in deionized water (30 mL), was poured into the funnel and added dropwise into the flask. After 30 min, NaOH (8.00 g, 0.20 mol) and phenol (11.28 g, 0.14 mol) were added into the reaction flask, and the solution mixture was stirred at 0 °C for 5 h. Finally, the resultant dark yellow solid was filtered and washed several times with deionized water, followed by purification by column chromatography using a mixture of n-hexane and ethyl acetate (10:1, by volume) as the eluent. Finally, (4-methoxy-phenylazo)phenol was obtained as a yellow solid in 80.1% yield. The reaction mechanism is illustrated in Scheme S1.†1H NMR (400 MHz in CDCl3) (Fig. S1†): 7.95–7.78 (m, 4H, aromatic), 7.05–6.87 (m, 4H, aromatic), 5.46 (s, 1H, –OH), 3.90 (s, 3H, –CH3).
Synthesis of pentafluorophenyl acrylate (PFPA)
Pentafluorophenol (80 g, 0.43 mol) and 2,6-lutidine (51.60 ml, 0.48 mol) were first dissolved in methylene chloride (710 mL) in a 1000 mL three-necked round-bottom flask. Then the temperature was maintained at 0–5 °C in an ice-bath, and acryloyl chloride (42.27 ml, 0.48 mol) was dropped slowly into the solution mixture and stirred at 0–5 °C for 3 h. After removing the ice bath, the solution mixture was kept at room temperature overnight. The resultant precipitate was filtered and washed with DCM, and then the residue was washed three times with water and a saturated solution of sodium chloride. A transparent solution was obtained, and the solution was passed through a silica gel column with DCM as the eluent. Finally, 87.67 mg PFPA was obtained with 87.67% yield. 1H-NMR (400 MHz in CDCl3): 6.14–6.20 ppm (m, H, –CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–), 6.29–6.43 ppm (m, H, –CH2CH–) and 6.67–6.76 ppm (m, H, –CH2CH–) (see Fig. S2†). 19F NMR (188 MHz, chloroform-d) δ −152.00 to −154.02 ppm (m, 2F), −158.33 ppm (t, 1F), −161.64 to −163.69 ppm (m, 2F) (see Fig. S3†); FT-IR: 1770 cm−1 (ester from PFPA) (see Fig. S4†).
CH–), 6.29–6.43 ppm (m, H, –CH2CH–) and 6.67–6.76 ppm (m, H, –CH2CH–) (see Fig. S2†). 19F NMR (188 MHz, chloroform-d) δ −152.00 to −154.02 ppm (m, 2F), −158.33 ppm (t, 1F), −161.64 to −163.69 ppm (m, 2F) (see Fig. S3†); FT-IR: 1770 cm−1 (ester from PFPA) (see Fig. S4†).
Synthesis of P(DEGMA-co-PFPA)
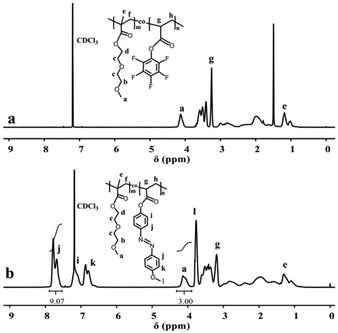
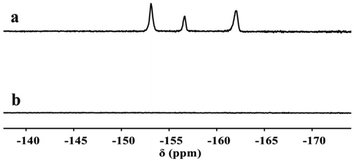
A typical polymerization procedure to obtain P(DEGMA-co-PFPA) with 30 mol% DEGMA was carried out, using for example: PFPA (2.00 g, 1 eq., 8.40 × 10−3 mol), DEGMA (0.68 g, 0.43 eq., 3.60 × 10−3 mol) and AIBN (0.02 g, 0.01 eq., 0.12 × 10−3 mol) dissolved in a 10 mL reaction vial with dioxane. Then, the solution was put into a preheated oil bath at 65 °C overnight after purging with N2 for 30 min. Then, the crude product was cooled down to room temperature, precipitated dropwise into n-hexane three times, and then dried under vacuum at 40 °C for 24 h. The chemical structure of the resulting copolymer was measured via FT-IR, 1H NMR and 19F NMR. IR (neat, cm−1) 1782 cm−1 (ester from PPFPA), 1718 cm−1 (ester from PDEGMA), 1516 cm−1 and 989 cm−1 (pentafluoro phenyl group from PPFPA) (see Fig. 1a and Fig. S5†); 1H NMR (400 MHz, chloroform-d): 1.02 to 3.10 ppm (d, 7H, –CH2–CH– in the backbone of PPFPA and –C(CH3)–CH2– from PDEGMA), 4.13 ppm (m, 3H, –CH3 in the side chain of PDEGMA), 3.41 to 3.59 ppm (m, 8H, –(CH2–CH2–O)2– in the side chain of PDEGMA) (see Fig. 2a); 19F NMR (188 MHz, chloroform-d) δ −153.12, −156.62, −162.02 from PPFPA (see Fig. 3a); GPC: Mn = 4.43 × 104 g mol−1, Mw/Mn = 1.86 (see Fig. S6†). PDEGMA with flexible chains yields P(DEGMA-co-PFPA) with good flexibility and suitable Tg (see Fig. S7 and Table S1†).
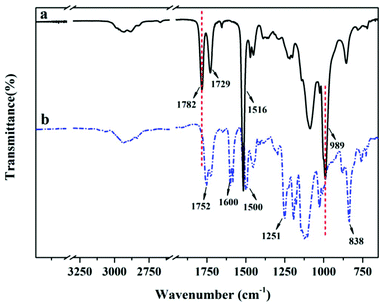 |
| | Fig. 1 FT-IR spectra of P(DEGMA-co-PFPA) (a) and P(DEGMA-co-MOPAzo) (b). | |
 |
| | Fig. 2
1H NMR spectra of P(DEGMA-co-PFPA) (a) and P(DEGMA-co-MOPAzo) (b) in CDCl3. | |
 |
| | Fig. 3
19F NMR spectra of P(DEGMA-co-PFPA) (a) and P(DEGMA-co-MOPAzo) (b). | |
The post-polymerization modification of P(DEGMA-co-PFPA) based on 4-(4-methoxy-phenylazo)phenol (designated as P(DEGMA-co-MOPAzo))
First P(DEGMA-co-PFPA) (1.00 g, 1 eq., 4.48 × 10−3 mol), MOPAzo (0.82 g, 0.8 eq., 3.59 × 10−3 mol) and DMAP (0.10 mg, 0.18 eq., 0.80 × 10−3 mol) were dissolved in 3 mL of DMF in a 10 mL reaction vial, and then nitrogen was degassed in the reaction system for 30 min. Next, the reaction flask was placed in a preheated oil bath at 80 °C with mechanical stirring for 8 h. Then, the crude product was cooled down to room temperature and added dropwise to 50 mL hexane for purification. Re-purification of the modified copolymer was carried out via dialysis against di-water for 72 h using a 3500 molecular weight cut-off membrane, with subsequent freeze-drying (yield: 71%). FT-IR, 1H NMR and 19F NMR were used for the verification of the successful synthesis of P(DEGMA-co-MOPAzo). IR (neat, cm−1) 1752 cm−1 (ester from PMOPAzo), 1600 cm−1, 1500 cm−1, 1251 cm−1 and 838 cm−1 (azobenzene groups from MOPAzo) (see Fig. 1b); 1H NMR (400 MHz, chloroform-d) δ 7.89–6.64 ppm (m, 8H, benzene from PMOPAzo), 4.13 ppm (m, 3H, –CH3 from PDEGMA) (see Fig. 2b). No signal belongs to F in the 19F NMR spectrum (see Fig. 3b).
Fabrication of a photomechanical P(DEGMA-co-MOPAzo)/PP fabric
In this work, a P(DEGMA-co-MOPAzo)/PP fabric bilayer was prepared via spin-coating a 10 wt% P(DEGMA-co-MOPAzo) solution of DMF on a PP fabric at 3000 rpm and then drying in an oven at 80 °C for 12 h. Here, PP acts as a passive layer, which has no responsive behavior upon UV irradiation (see Fig. S8†). P(DEGMA-co-MOPAzo) acts as an active layer, which can be triggered by UV irradiation. Moreover, passive and active layers were combined by physical interaction, benefiting from the flexibility of PDEGMA, which was proved via immersing the functional PP fabric in different solvents (see ESI, Table S2†). The surface morphology of the cross-sectional view and the surface as well as the thickness of the functional PP fabric were analyzed by SEM measurement (see Fig. 5).
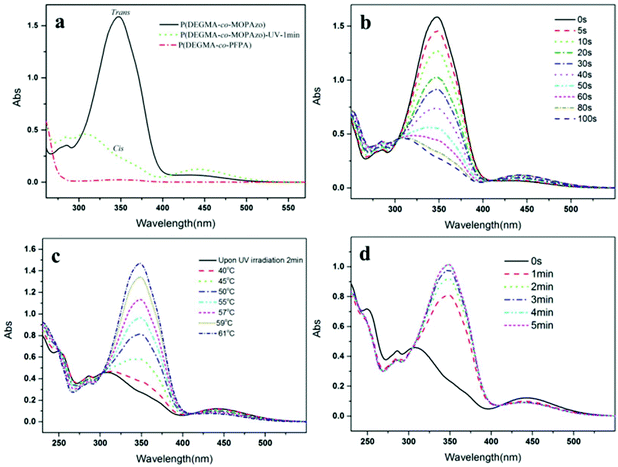 |
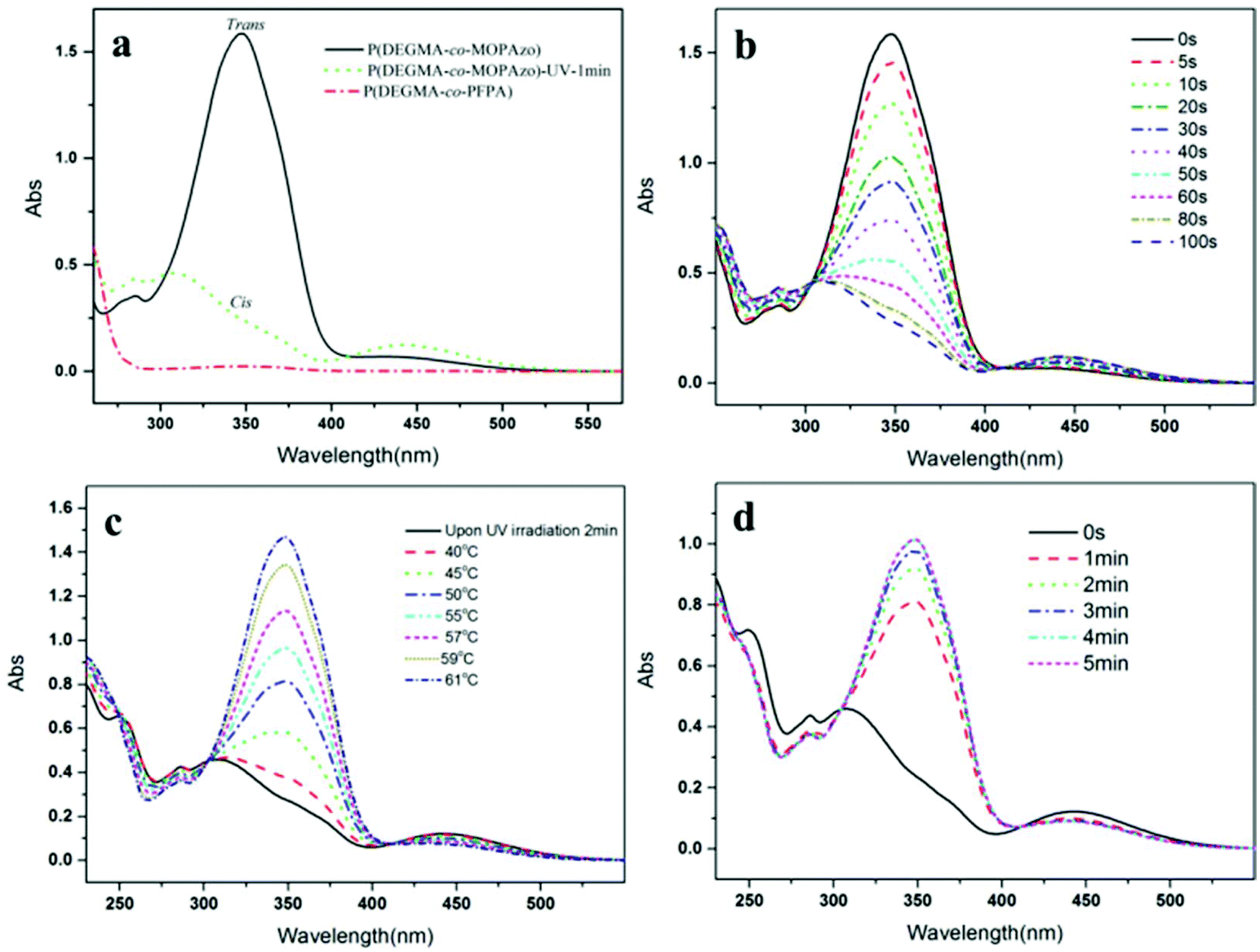
| | Fig. 4 (a) UV-Vis absorption spectra of P(DEGMA-co-PFPA) (short dotted line), upon UV irradiation for 1 min (dotted line) and visible light irradiation (solid line) of P(DEGMA-co-MOPAzo); UV-Vis absorption spectra of P(DEGMA-co-MOPAzo) with time evolution (b) upon UV irradiation, (c) upon heating and (d) when keeping under visible light after irradiation. All of the copolymers were measured in THF with a concentration of 0.03 mg mL−1 at 20 °C. UV irradiation intensity with 365 nm, 0.5 mW cm−2 was used in this measurement. | |
 |
| | Fig. 5 SEM images: Surface (a) and cross-section (b) morphologies of the PP fabric; surface (c) and cross-section (d) morphologies of a P(DEGMA-co-MOPAzo)/PP fabric bilayer. | |
Results and discussion
Structural characterization of synthesized copolymers
After obtaining P(DEGMA-co-PFPA) with 30 mol% PDEGMA, the chemical structure of the copolymer was confirmed by FT-IR, 1H NMR and 19F NMR. In Fig. 1a, multiple characteristic absorption peaks of P(DEGMA-co-PFPA) can be observed in the FT-IR spectrum at 1782 cm−1, 1729 cm−1, 1516 cm−1 and 989 cm−1, of which the peaks at 1782 cm−1 and 1729 cm−1 were ascribed to ester CO of PPFPA and PDEGMA, respectively. 1516 cm−1 and 989 cm−1 were assigned to deformation and stretching vibrations of pentafluorobenzene, indicating the presence of both PDEGMA and PPFPA. 1H NMR was employed to confirm the structure of the synthesized P(DEGMA-co-PFPA) and to further determine the molar contents of DEGMA and PFPA in P(DEGMA-co-PFPA). As shown in Fig. 2a, the integrals of methyl protons at 4.08 ppm from PDEGMA and at 3.29 ppm from PPFPA were calculated with the designed molar ratio of DEGMA:PFPA as 30:70. Moreover, the resulting 19F NMR spectrum (see Fig. 3a) further verified the successful synthesis of P(DEGMA-co-PFPA). After successfully obtaining P(DEGMA-co-PFPA) as a precursor copolymer, post-polymerization modification was applied to fabricate the light-responsive target copolymer. Benefitting from the highly reactive activated PFP ester in the copolymer P(DEGMA-co-PFPA), MOPAzos were introduced into the copolymer to prepare a light-responsive copolymer P(DEGMA-co-MOPAzo) as shown in Scheme 1, following a recently developed trans-esterification strategy,28 and its chemical structure was confirmed by IR, UV-vis and 1H NMR measurements. As shown in Fig. 1b, compared with the precursor copolymer, the characteristic absorption bands of PFPA at 1782 cm−1, 1516 cm−1 and 989 cm−1 completely disappeared in P(DEGMA-co-MOPAzo). Moreover, new characteristic absorption bands were observed at 1600 cm−1, 1500 cm−1, 1251 cm−1 and 838 cm−1, which were ascribed to azobenzene groups from MOPAzo. This result demonstrates the successful introduction of azobenzene groups. Moreover, as shown in the 1H NMR spectrum in Fig. 2b, the integral of methyl at 4.08 ppm from PDEGMA and the integral of aromatic protons at 7.85 ppm from MOPAzo corresponded to the molar ratio of DEGMA:MOPAzo (30:70 in molar), thus confirming that complete post-modification had taken place. Furthermore, the characteristic absorption bands of PFPA disappeared in the 19F NMR spectrum (see Fig. 3b). The successful post-modification of P(DEGMA-co-MOPAzo) was further verified by UV-vis spectroscopy. As seen in Fig. 4a, the absorption spectrum showed a strong absorption peak at around 350 nm, which is attributable to the trans-isomerization of azobenzene.25
Azobenzene isomerization behavior of P(DEGMA-co-MOPAzo)
To investigate the specific photo-induced isomerization behavior of P(DEGMA-co-MOPAzo), UV/Vis absorption spectroscopy was used to conduct the isomerization processes. As shown in Fig. 4a, upon UV irradiation for 1 min, the strong absorption peak at around 350 nm, belonging to the π–π* transition of trans-azobenzene absorbance, disappeared compared to that without UV irradiation, which is attributable to the photo-isomerization between the trans- and cis-isomers of the azobenzene groups.29–31 The UV/Vis absorption spectra of P(DEGMA-co-MOPAzo) with time evolution upon UV irradiation are shown in Fig. 4b. The absorbance of P(DEGMA-co-MOPAzo) at 350 nm decreased with increasing time of UV irradiation owing to the fact that during photo-isomerization, the trans-isomer gradually transformed into the cis-isomer. Additionally, the reversibility was investigated using UV-Vis absorption spectroscopy via heating and keeping the polymer solution under visible light. The detailed temperature-evolved absorption spectra of cis isomerized P(DEGMA-co-MOPAzo) are shown in Fig. 4c. On account of the instability of the cis-isomer, P(DEGMA-co-MOPAzo) exhibits good reversibility, and the absorbance of the azobenzene groups at 350 nm completely recovered to the original state at 61 °C. Moreover, the re-isomerization of the copolymer upon exposure to visible light was also investigated, as seen in Fig. 4d.
Bending behaviors of the P(DEGMA-co-MOPAzo)/PP actuator
The P(DEGMA-co-PFPA)/PP free-standing fabric with a size of 1 mm × 10 mm was treated as an actuator. As shown in Fig. 5, P(DEGMA-co-MOPAzo) can apparently be observed on the top of the PP fabric. The coated P(DEGMA-co-MOPAzo) and P(DEGMA-co-MOPAzo)/PP fabrics had thicknesses of 130 μm and 217 μm, respectively. Subsequently, the bilayer actuator was vertically gripped by a clamp to test its bending behavior in an ambient environment with 30% relative humidity. Here, UV light was used with a 50 mW output-power through a specific wavelength filter with a wavelength of 400 nm. The azobenzene photo-isomerization behavior of the functional PP fabric is illustrated in Fig. 6 (see videos in Movies 1 and 2 in the ESI†). As shown in Fig. 6a and c, the bending behavior of the actuator can be observed in 0.2 s after being irradiated by UV light. Moreover, the corresponding bending angle, which is an included angle between the vertical direction and the moving direction of the actuator, increased with the irradiation time, as shown in Fig. 6b and d. Furthermore, the actuator bent from an initial 30° to 112° in 1.73 s and from 10° to 83° in 1.58 s at the first and the second UV irradiated point, respectively. Upon UV irradiation, the isomerization of azobenzene moieties from rod trans to bent cis in the active layer caused the volume constriction in the active layer, which led to the bending of the functional PP fabric. It is noteworthy that the bent actuator cannot revert to its original state within 1 h upon visible light irradiation. This phenomenon may be ascribed either to the fact that the isomerization of the azobenzene moieties from cis to trans only takes place predominantly located on the surface, or that the converted mechanical energy from visible light isomerization is not sufficient to force the bending of the actuator to its original configuration in a short time. However, the bent actuator reverted to its initial state after being stored at elevated temperatures (e.g. in a 50° C environment for 10 minutes; see Fig. S9†).
 |
| | Fig. 6 Time-profiles of the bending motion of a P(DEGMA-co-MOPAzo)/PP fabric actuator upon UV irradiation at the first point (a) and at the second point (c); (b) and (d) represent the corresponding time-dependent bending angles of (a) and (b), respectively. (Scale bar = 1 cm). | |
Conclusion
In summary, a light-responsive polymer, P(DEGMA-co-MOPAzo), was prepared via post-polymerization modification from the P(DEGMA-co-PFPA) precursor polymer and was successfully used to fabricate a light-driven polypropylene (PP) fabric actuator with light-responsive bending behavior. We found that this copolymer can be easily and stably attached to the surface of a PP fabric via a physical bonding interaction due to the flexible di(ethylene glycol)methyl ether side chains. On account of the light responsiveness of azobenzene moieties, such a functional PP fabric can immediately respond to the UV resource side and display a reversible shape transition upon alternating between exposure to UV irradiation and without exposure to UV irradiation. Hence, these functional PP fabrics having such a photomechanical transition property are expected to be employed in the future as light remotely controllable smart textiles and actuators.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
J. S. and S. L. gratefully acknowledge the China Scholarship Council (CSC grant: 201406240131 and 201306240132) for partial financial support of this work.
References
- F. Liu and M. W. Urban, Prog. Polym. Sci., 2010, 35, 3–23 CrossRef.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 CrossRef.
- M. A. Ward and T. K. Georgiou, Polymers, 2011, 3, 1215–1242 CrossRef.
- J. L. Willett, Macromol. Chem. Phys., 2008, 209, 764–772 CrossRef.
- W. Feng, W. Zhou, S. Zhang, Y. Fan, A. Yasin and H. Yang, RSC Adv., 2015, 5, 81784–81789 RSC.
- L. T. De Haan, J. M. N. Verjans, D. J. Broer, C. W. M. Bastiaansen and A. P. H. J. Schenning, J. Am. Chem. Soc., 2014, 136, 10585–10588 CrossRef PubMed.
- M. Li, L. Lei, Q. Zhang and S. Zhu, Macromol. Rapid Commun., 2016, 37, 957–962 CrossRef PubMed.
- M. P. M. Dicker, A. B. Baker, R. J. Iredale, S. Naficy, I. P. Bond, C. F. J. Faul, J. M. Rossiter, G. M. Spinks and P. M. Weaver, Sci. Rep., 2017, 7, 9197–9205 CrossRef PubMed.
- R. Kumar, A. M. Ketner and S. R. Raghavan, Langmuir, 2010, 26, 5405–5411 CrossRef PubMed.
- Q. Shi, J. Li, C. Hou, Y. Shao, Q. Zhang, Y. Li and H. Wang, Chem. Commun., 2017, 53, 11118–11121 RSC.
- W. R. Browne and B. L. Feringa, Annu. Rev. Phys. Chem., 2009, 60, 407–428 CrossRef PubMed.
- S. Lin, J. Shang, X. Zhang and P. Theato, Macromol. Rapid Commun., 2018, 39, 1700313 CrossRef PubMed.
- F. P. Nicoletta, D. Cupelli, P. Formoso, G. De Filpo, V. Colella and A. Gugliuzza, Membranes, 2012, 2, 134–197 CrossRef PubMed.
- S. Samai, C. Sapsanis, S. P. Patil, A. Ezzeddine, B. A. Moosa, H. Omran, A.-H. Emwas, K. N. Salama and N. M. Khashab, Soft Matter, 2016, 12, 2842–2845 RSC.
- M. Han and M. Hara, J. Am. Chem. Soc., 2005, 127, 10951–10955 CrossRef PubMed.
- S. Peng, Q. Guo, P. G. Hartley and T. C. Hughes, J. Mater. Chem. C, 2014, 2, 8303–8312 RSC.
- M. Hendrikx, A. P. H. J. Schenning, M. G. Debije and D. J. Broer, Crystals, 2017, 7, 231–251 CrossRef.
- D. Y. Kim, S. Shin, W. J. Yoon, Y. J. Choi, J. K. Hwang, J. S. Kim, C. R. Lee, T. L. Choi and K. U. Jeong, Adv. Funct. Mater., 2017, 27, 1606297 Search PubMed.
- G. Han, J. Nie and H. Zhang, Polym. Chem., 2016, 7, 5088–5092 RSC.
- Y. Liu, W. Wu, J. Wei and Y. Yu, ACS Appl. Mater. Interfaces, 2017, 9, 782–789 Search PubMed.
- Z. Liu, R. Tang, D. Xu, J. Liu and H. Yu, Macromol. Rapid Commun., 2015, 36, 1171–1176 CrossRef PubMed.
- D. Liu and D. J. Broer, Langmuir, 2014, 30, 13499–13509 CrossRef PubMed.
- Y. Yi, M. Nakata, A. R. Martin and N. A. Clark, Appl. Phys. Lett., 2007, 90, 163510 CrossRef.
- B. W. Lee and N. A. Clark, Science, 2001, 291, 2576–2580 CrossRef PubMed.
- H. Wen, W. Zhang, Y. Weng and Z. Hu, RSC Adv., 2014, 4, 11776–11781 RSC.
- H. Zhao and P. Theato, Polym. Chem., 2013, 4, 891–894 RSC.
- A. Das and P. Theato, Macromlecules, 2015, 48, 8695–8707 CrossRef.
- N. R. Krekiehn, M. Müller, U. Jung, S. Ulrich, R. Herges and O. M. Magnussen, Langmuir, 2015, 31, 8362–8370 CrossRef PubMed.
- E. Merino and M. Ribagorda, Beilstein J. Org. Chem., 2012, 8, 1071–1090 CrossRef PubMed.
- A. Khan and S. Hecht, Chem. – Eur. J., 2006, 12, 4764–4774 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data, IR, NMR, GPC and SEM. See DOI: 10.1039/c8py00411k |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Shaojian
Lin
a and
Patrick
Theato
a,
Shaojian
Lin
a and
Patrick
Theato