
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Converting natural rubber waste into ring-opening metathesis polymers with oligo-1,4-cis-isoprene sidechains†
Mudassar
Abbas
 ab,
Maximilian
Neubauer
a and
Christian
Slugovc
*a
ab,
Maximilian
Neubauer
a and
Christian
Slugovc
*a
aInstitute for Chemistry and Technology of Materials, Graz University of Technology, NAWI Graz, Stremayrgasse 9, A 8010 Graz, Austria. E-mail: slugovc@tugraz.at
bSchool of Textile and Design, University of Management and Technology, C-II Johar Town, , Lahore 54770, Pakistan
First published on 12th March 2018
Abstract
A chemical recycling of natural rubber waste via a degradation/polymerisation approach is described. The vulcanized rubber waste was degraded by cross metathesis with ethyl acrylate as the key-step yielding enoate end-capped oligo-cis-isoprenes, which were subsequently converted into norbornenes via a cycloaddition reaction with cyclopentadiene. Ring-opening Metathesis Polymerisation (ROMP) then yielded main-chain unsaturated polymers bearing oligo-1,4-cis-isoprene side chains with appealing thermal stability and a glass transition temperature of −60 °C.
Natural rubber (NR) is the most widely used feedstock in elastomer production owing to the unique properties of this agricultural product. NR production amounted to about 13.3 million metric tons in 2017
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1 and is used in countless applications spread across many important industries, such as the chemical, transportation or medical industries. The end-of-life treatment of such crosslinked thermosets is typically incineration. However, efforts to chemically recycle natural rubber products have received ever-increasing importance.2 Cross metathesis with ethene (ethenolysis)3 or other alkenes is thought to be a promising way to convert rubber waste into feedstock for the chemical industry.4 In particular, the commercial availability and the high functional group tolerance of ruthenium based olefin metathesis catalysts enabled the preparation of various chain-end functionalized oligomeric 1,4-cis-isoprenes from NR5 and in further course also from (vulcanized) NR based products. The ethenolysis of end-of-life tire granulates (containing primarily NR) yielding oligomeric 1,4-cis-isoprene6 and the conversion of waste tires into telechelic polyisoprene can be considered as benchmarks in the field.7
1 and is used in countless applications spread across many important industries, such as the chemical, transportation or medical industries. The end-of-life treatment of such crosslinked thermosets is typically incineration. However, efforts to chemically recycle natural rubber products have received ever-increasing importance.2 Cross metathesis with ethene (ethenolysis)3 or other alkenes is thought to be a promising way to convert rubber waste into feedstock for the chemical industry.4 In particular, the commercial availability and the high functional group tolerance of ruthenium based olefin metathesis catalysts enabled the preparation of various chain-end functionalized oligomeric 1,4-cis-isoprenes from NR5 and in further course also from (vulcanized) NR based products. The ethenolysis of end-of-life tire granulates (containing primarily NR) yielding oligomeric 1,4-cis-isoprene6 and the conversion of waste tires into telechelic polyisoprene can be considered as benchmarks in the field.7
Herein we wish to report our results on using electron deficient olefins, namely acrylates, instead of ethene or allylic olefins as the cross metathesis partner in the degradation of NR. Related preceding work is restricted to a single patent by Wagener and Schulz, who disclosed the depolymerisation of poly(butadiene) with ethyl acrylate or acryloyl chloride.8 Accordingly, this degradation variant is astonishingly underdeveloped in face of the high potential established for cross metathesis of electron deficient olefins and electron rich olefins,9 allowing for example for the preparation of alternating copolymers by inserting e.g. diacrylates into polymers containing olefins within the main chain.10 Being successful in this endeavour the degradation products (see 1 in Scheme 1) will contain α,β-unsaturated ester moieties facilitating various further transformations.
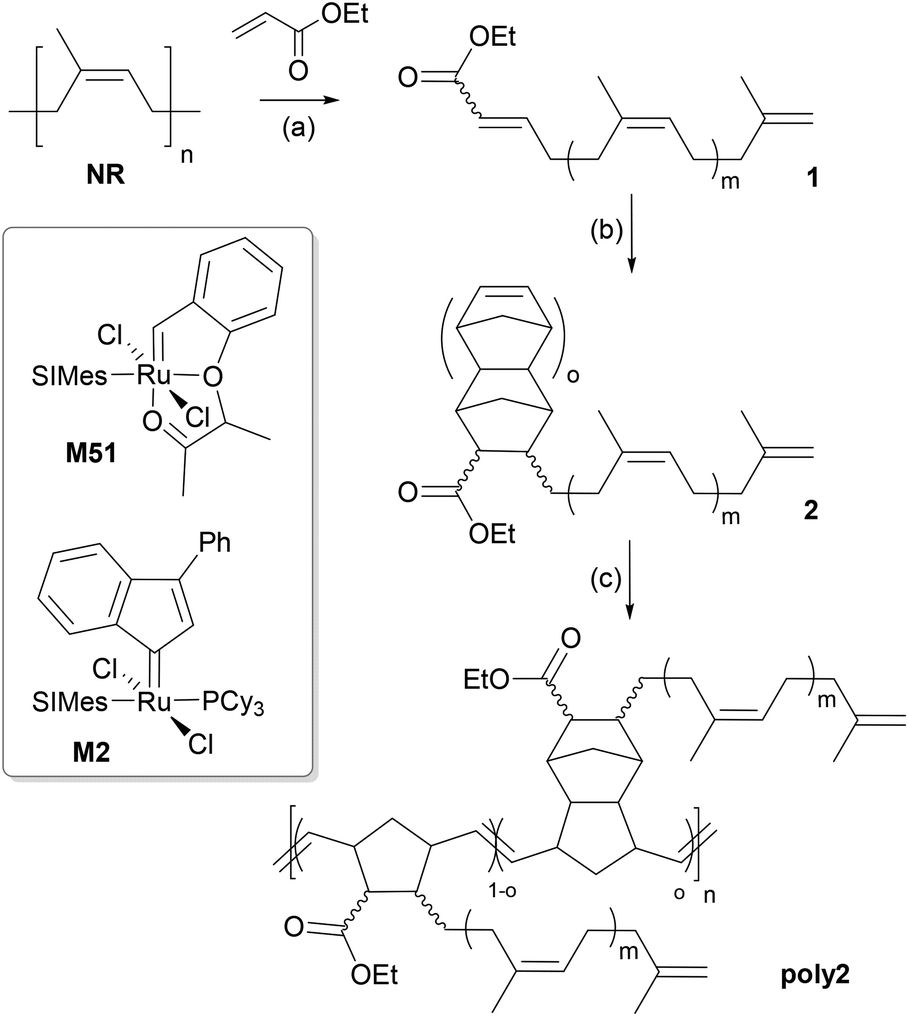 | ||
| Scheme 1 Synthetic procedure for poly2. Reagents and conditions: (a) Ethyl acrylate, M51, toluene, 80 °C, 16 h; (b) dicyclopentadiene, 200 °C, 1 h; (c) M2, 80 °C, 2 h. Typical values for the repeating units are: m ≈ 3; o ≈ 0.4 ± 0.1; n = n.d. | ||
Herein it is aimed at polymerising the degradation products again. For this purpose, the reactivity of enoates will be exploited by performing a Diels–Alder reaction with cyclopentadiene releasing the corresponding norbornene derivatives (2), which will then be polymerised via ring-opening metathesis polymerisation (ROMP) yielding main-chain unsaturated polymers bearing oligo-1,4-cis-isoprene side chains (poly2).
The key step, degradation of natural rubber with ethyl acrylate, was designed with the aim to use an as low as reasonable catalyst loading. In order to minimize substrate induced catalyst degradation11 a phosphine free catalyst, namely M51, which is commercialized and showed particularly good performance in the cross metathesis of acrylates and internal olefins was chosen.12
In a typical experiment, finely chopped natural rubber gloves were suspended in a mixture of toluene and an excess of ethyl acrylate (4–5 equiv. in respect to double bonds) under an inert atmosphere of nitrogen. The reaction mixture was heated to 80 °C and after approx. 2 h, the catalyst M51 (0.5 mol% with respect to double bonds) was added. After stirring at 80 °C for 16 h, ethyl vinyl ether was added, insoluble parts were removed by filtration and volatiles were removed under vacuum. Typically, the crude yield amounted to about 160% in relation to the mass of the NR gloves. The reaction mixture was investigated with size exclusion chromatography (SEC) resulting in a molecular mass not higher than 500 g mol−1 (degradation of an unvulcanised NR rubber sample of a molecular mass of about 1 million gave the same result). 1H-NMR investigation of the reaction mixture revealed the presence of signals characteristic of the expected enoate moiety as well as of diethyl fumarate and incompletely degraded natural rubber parts (see the ESI†). Column chromatographic purification allowed for isolation of three fractions (cf. the ESI† for details). While a first minor fraction (approx. 4% of the recovered material) containing oligomeric isoprene species was not further characterized, the second fraction (approx. 84%) consisted of the desired enoates 1 with m = 3.2 (m is the number of NR repeating units, cf.Scheme 1), meaning that on average roughly every fourth double bond of natural rubber was cleaved by ethyl acrylate. The yield of the reaction of vulcanized NR towards 1 with m = 3.2 is 69% (for comparison: the yield of the degradation of native NR is 75% of 1 with m = 3.0). The third fraction (about 12% of the recovered material) was identified as diethyl fumarate.
Increasing the catalyst loading from 0.5 mol% to 1 mol% with respect to NR double bonds leads to enoates 1 with m = 1.6 and a slightly better yield of 80%. A further increase to 2 mol% yielded 1 with m = 0.8 in 79%. Decreasing the catalyst loading to 0.2 mol% caused a distinct drop in yield to less than 50% and higher m-values of about 15 (SEC: Mn = 1800 g mol−1; Đ = 2.2). While prolongation of the reaction time to 32 h did not result in further degradation, a shortening of the reaction time gave 1 with higher m and lower yields so that 16 h reaction time was regarded as an optimum. Furthermore, the reaction was carried out in neat ethyl acrylate since it has been shown that under such conditions particularly low catalyst loadings could be achieved.13 However, in the case of NR as the substrate, only sluggish degradation but pronounced dimerization of ethyl acrylate forming diethyl fumarate14 was noted. The use of methyl acrylate instead of ethyl acrylate resulted in similar m values but distinctly higher fumarate ratios as well as lower yields. Performing the NR degradation without adding ethyl acrylate under otherwise similar reaction conditions led to a reduction of the molecular mass from 1 million to Mn = 48000 g mol−1 (Đ = 1.9).
The compound mixture 1 (with m = 1.6) was characterised in detail using 1H- and 13C-NMR-spectroscopy and GC-MS investigations (see the ESI†). Compounds 1 with m = 0–3 were observed in the gas chromatogram and identified by mass spectrometry. Characteristic peaks in the 1H-NMR spectra comprise a doublet of triplet at 6.98 ppm and a doublet at 5.83 representing the protons of the electron deficient double bond in the E configuration and another doublet of triplet at 6.19 ppm assigned to the corresponding Z-isomer (a E/Z ratio of 92.5 ± 0.5/7.5 ± 0.5 was observed in all cases). An apparently broadened multiplet centred at 5.15 ppm represents the olefinic protons of the oligo(isoprene) chain and two signals at 4.70 and 4.66 ppm are assigned to the protons of the methylene group (see Fig. 1).
 | ||
| Fig. 1 Details of the 1H-NMR spectra of 1 (bottom), 2 (middle) and poly2 (above) showing the assignment of characteristic peaks (* marks residual CH2Cl2). | ||
For further transformations, 1 with an m-value of approx. 3 was selected and reacted with excess dicyclopentadiene (DCPD) at 200 °C for 1 h in a Monowave reactor from Anton Paar.15 At this temperature DCPD undergoes a retro-Diels Alder reaction16 releasing cyclopentadiene, which subsequently reacts with the enoate moieties to give the corresponding norbornene derivatives 2. Moreover, oligomerization of 2via further 4 + 2 cycloadditions is most probably feasible under these conditions.17 The reaction furnished, after removal of hydrocarbons (DCPD and its higher oligomers), the desired product-mixture 2 in typically 95% yield as a low viscous oil. A minor amount of 5 ± 2% of 1 could not be converted into 2, although various reaction conditions were tested. A separation of residual 1 was not attempted. Characteristic peaks of the compound mixture 2 in the corresponding 1H-NMR spectrum comprise four multiplets in the range of 6.27–5.89 ppm assigned to the protons of newly formed double bonds of endo- and exo-substituted norbornenes18 as well as approx. 35% of the corresponding tetracyclo[6.2.1.13,6.02,7]dodec-9-ene-4-carboxylate derivatives (the presence and amount of such derivatives are indicated by the index o in Scheme 1. Typically o ranged from 0.35 to 0.4 as assessed by integrating 13C-NMR spectra; see the ESI†). The occurrence of higher oligomers resulting from further cyclopentadiene additions could not be ruled out.17 Thermogravimetric investigations revealed a thermal stability of 174 °C (defined as the temperature at which 5% mass loss was detected).
As the final step, the polymerisation of the monomer mixture 2via ROMP using the fast initiating Ruthenium indenylidene complex M31 for solution polymerisation19 or the initiator M2 for solvent-free polymerisation was conducted. While the solution polymerisation in dichloromethane at room temperature with an initiator to monomer ratio of 1:260 yielded poly2 with a number average molecular weight (Mn) of 5000 (Đ = 2.0), bulk polymerisation with M2 (M2:2 = 1:260, reaction temperature 80 °C) furnished 96% poly2 characterized by a Mn of 4500 and a distinctly broader molecular weight distribution (Đ = 4.4). These remarkably low Mn-values are likely caused by cross metathesis reactions competing with the ROMP of the norbornene moieties. This hypothesis is corroborated by the fact that after polymerisation no free 1 (which could not be separated from 2, cf. above) was detected in the reaction mixture (although characteristic signals of the enoate moiety can be seen in the 1H-NMR spectra of poly2, cf.Fig. 1) and that lowering the initiator amount to 1:870 did not significantly affect the Mn value of poly2 (Mn = 5500 g mol−1; Đ = 7.3). The thermal stability of poly2 was at 270 °C, which is approx. 100 °C higher than that of 2, and poly2 exhibited a glass transition temperature (Tg) of −60 °C.
Conclusions
A way to prepare main-chain unsaturated polymers bearing oligo-1,4-cis-isoprene side chains starting from natural rubber waste was presented. The embarked synthetic path comprises first the olefin metathetic degradation of vulcanized natural rubber with the aid of ethyl acrylate as the cross metathesis agent yielding enoate end-capped oligo-cis-isoprenes in high yields at moderate catalyst loadings. The degree of depolymerisation can be tuned by variation of the catalyst loading. These liquid primary degradation products contain a single electron deficient double bond, which distinctly broadens the scope for further transformations compared to degradation products from ethenolysis. The second step exploits the reactivity of the enoate group via transforming it in a solvent-free manner into the corresponding norbornene derivatives in a Diels–Alder reaction with cyclopentadiene released from dicyclopentadiene at high temperatures. In the final third step, these liquid monomers were polymerised via ring-opening metathesis polymerisation. The resulting polymer exhibits a glass transition temperature of about −60 °C and a thermal stability up to 270 °C and should qualify as a component in vulcanizable compounds.Further work on this topic focusing on further simplifying and optimizing the synthesis‡ (repress the fumarate formation, decrease the DCPD amount) using the polymer and 1 as components in rubber formulations and further exploiting the reactivity of the enoate is currently ongoing.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Anton Paar GmbH for allocating a Monowave 50 reactor, Semperit AG for performing low temperature DSC, and Umicore for providing the catalysts; MA is grateful to the Federal Ministry of Science, Research and Economics of Austria for providing an Ernst Mach Follow Up Grant through OEAD-GMBH.Notes and references
- Association of Natural Rubber Producing Countries, Rubber and Plastics News 2018, Jan. 22, 2018.
- P. Sarkar and A. K. Bhowmick, J. Appl. Polym. Sci., 2017 DOI:10.1002/app.45701; S. O. Movahed, A. Ansarifar and S. Estagy, Rubber Chem. Technol., 2016, 89, 54–78 CrossRef CAS; M. Kojima, M. Tosaka and Y. Ikeda, Green Chem., 2004, 6, 84–89 RSC.
- J. Bidange, C. Fischmeister and C. Bruneau, Chem. – Eur. J., 2016, 22, 12226–12244 CrossRef CAS PubMed.
- S. Leimgruber and G. Trimmel, Monatsh. Chem., 2015, 146, 1081–1097 CrossRef CAS.
- S. Wolf and H. Plenio, Green Chem., 2011, 13, 2008–2012 RSC; X. Michel, S. Fouquay, G. Michaud, F. Simon, J.-M. Brusson, J.-F. Carpentier and S. M. Guillaume, Eur. Polym. J., 2017, 96, 403–413 CrossRef CAS; N. Saetung, I. Campistron, S. Pascual, J.-F. Pilard and L. Fontaine, Macromolecules, 2011, 44, 784–794 CrossRef; A. Martínez, N. Zuniga-Villarreal, S. Gutierrez and M. A. Tlenkopatchev, Curr. Org. Synth., 2016, 13, 876–882 CrossRef.
- S. Wolf and H. Plenio, Green Chem., 2013, 15, 315–319 RSC.
- A. Mouawia, A. Nourry, A.-C. Gaumont, J. F. Pilard and I. Dez, ACS Sustainable Chem. Eng., 2017, 5, 696–700 CrossRef CAS.
- K. B. Wagener and M. Schulz, WO2014/031677A1, 2014 Search PubMed.
- D. J. O'Leary and G. W. O'Neil, Cross Metathesis, in Handbook of Metathesis, ed. R. H. Grubbs and D. J. O'Leary, Wiley-VCH, 2nd edn, 2015, vol. 2, DOI:10.1002/9783527674107.ch16; J. P. Morgan, C. Morrill and R. H. Grubbs, Org. Lett., 2002, 4, 67–70 CrossRef CAS PubMed; A. K. Chatterjee, T.-L. Choi, D. P. Sanders and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 11360–11370 CrossRef PubMed; H. Bilel, N. Hamdi, F. Zagrouba, C. Fischmeister and C. Bruneau, Green Chem., 2011, 13, 1448–1452 RSC; L. M. Pitet and M. A. Hillmyer, Macromolecules, 2011, 44, 2378–2381 CrossRef.
- T.-L. Choi, I. M. Rutenberg and R. H. Grubbs, Angew. Chem., Int. Ed., 2002, 41, 3839–3841 CrossRef CAS PubMed; S. Demel, C. Slugovc, F. Stelzer, K. Fodor-Csorba and G. Galli, Macromol. Rapid Commun., 2003, 24, 636–641 CrossRef.
- G. A. Bailey and D. E. Fogg, J. Am. Chem. Soc., 2015, 137, 7318–7321 CrossRef CAS PubMed.
- P. Vignon, T. Vancompernolle, J.-L. Couturier, J.-L. Dubois, A. Mortreux and R. M. Gauvin, ChemSusChem, 2015, 8, 1143–1146 CrossRef CAS PubMed.
- M. Abbas and C. Slugovc, Tetrahedron Lett., 2010, 52, 2560–2562 CrossRef CAS; M. Abbas and C. Slugovc, Monatsh. Chem., 2012, 143, 669–673 CrossRef.
- T.-L. Choi, C. W. Lee, A. K. Chatterjee and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 10417–10418 CrossRef CAS PubMed.
- D. Huertas, M. Florscher and V. Dragojlovic, Green Chem., 2009, 11, 91–95 RSC.
- A. Leitgeb, J. Wappel, C. A. Urbina-Blanco, S. Strasser, C. Wappl, C. S. J. Cazin and C. Slugovc, Monatsh. Chem., 2014, 145, 1513–1517 CrossRef CAS.
- T. Rager and C. G. Willson, Helv. Chim. Acta, 2000, 83, 2769–2782 CrossRef CAS.
- D. Sarma and A. Kumar, Org. Lett., 2006, 8, 2199–2202 CrossRef CAS PubMed.
- D. Burtscher, C. Lexer, K. Mereiter, R. Winde, R. Karch and C. Slugovc, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4630–4635 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Preparation and characterisation of 1, 2, and poly2. See DOI: 10.1039/c8py00233a |
| ‡ Although herein two chromatographic separations were used with the aim to better define and characterize the obtained products, the overall process could be easily performed without such costly purification steps. In the first step, toluene and surplus ethyl acrylate can be distilled off and subsequently used in another batch; and separation of diethyl fumarate is not necessary, since it will undergo the subsequent Diels–Alder reaction just as the enoates. Surplus DCPD from the second step is also polymerizable so that all those by-products will end up in the final polymer. |
| This journal is © The Royal Society of Chemistry 2018 |