
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nickel and palladium complexes with fluorinated alkyl substituted α-diimine ligands for living/controlled olefin polymerization†
Robert
Mundil
 a,
Anatolij
Sokolohorskyj
a,
Jan
Hošek
b,
Josef
Cvačka
c,
Ivana
Císařová
d,
Jaroslav
Kvíčala
b and
Jan
Merna
*a
a,
Anatolij
Sokolohorskyj
a,
Jan
Hošek
b,
Josef
Cvačka
c,
Ivana
Císařová
d,
Jaroslav
Kvíčala
b and
Jan
Merna
*a
aDepartment of Polymers, University of Chemistry and Technology, Prague, Technická 5, 166 28 Prague 6, Czech Republic. E-mail: merna@vscht.cz
bDepartment of Organic Chemistry, University of Chemistry and Technology, Prague, Technická 5, 166 28 Prague 6, Czech Republic
cInstitute of Organic Chemistry and Biochemistry v.v.i, Academy of Sciences of the Czech Republic, Prague 6, Czech Republic
dDepartment of Inorganic Chemistry, Faculty of Science, Charles University in Prague, Hlavova 2030, 128 40 Prague 2, Czech Republic
First published on 16th February 2018
Abstract
A series of six nickel and two palladium novel complexes bearing α-diimine ligands with fluorinated alkyl substituents in para-aryl positions was prepared. Two nickel complexes were structurally characterized by X-ray diffraction. Upon activation by methylaluminoxane or borate, the complexes were tested in ethene, propene, and hex-1-ene polymerization using toluene and chlorobenzene. Propene and hex-1-ene polymerization initiated by nickel complexes and ethene polymerization initiated by palladium complexes showed living/controlled character and yielded high molar mass polyolefins with narrow molar mass dispersity (Đ < 1.2). The effect of fluoroalkyl substituents on catalyst activity, thermal stability and chain-walking is only minor. The branching density of polyolefins can be regulated more efficiently by the variation of ortho-aryl and backbone substituents of α-diimine ligands. The topology of polyethylenes prepared by palladium complexes at low ethene pressure is dendritic as shown by Mark–Houwink plots and g′ values obtained from SEC with viscometric detection. The solubility of most of the complexes in fluorinated solvents is limited with the exception of palladium methyl chloride derivatives which suggest the promising method for further design of catalysts suitable for interphase polymerization.
Introduction
In 1995, Johnson et al.1 reported the synthesis and olefin polymerization activity of α-diimine complexes of nickel and palladium. These systems were unique among late metal catalysts in their ability to produce high molar mass materials, rather than oligomers, from both ethene and higher α-olefins. They showed low oxophilicity and therefore higher tolerance to polar functional groups.2,3 A characteristic feature of these catalysts is the chain-walking reaction in which the growing center migrates along the growing polymer chain through a series of β-hydride elimination and reinsertion steps. Chain-walking enables the formation of various types of polyethylenes ranging from almost linear to hyperbranched ones.1,4 In contrast, higher α-olefins are straightened after their insertion and form more linear polymers.5,6 α-Diimine nickel and palladium complexes also belong among the few catalytic systems suitable for living olefin polymerization allowing the preparation of block olefin copolymers including stereoblock copolymers.7–14 Since Brookhart's original discoveries,1,7 an enormous number of scientific reports have been published in the field of olefin polymerization catalyzed by α-diimine complexes.2,3 The catalytic activity and polymerization behavior of α-diimine complexes were tuned by changing ligand steric bulkiness,12,15–18 disrupting symmetry,10,19–22 reducing the imine group23–25 or by changing the ligand electronic structure via different substituents.26,27Relatively low attention was paid to substituents containing fluorine atoms. Fig. 1 gives a generalized overview of already synthesized fluorinated α-diimine ligands that were used for the synthesis of nickel or palladium olefin polymerization catalysts. The effect of the CF3 group in a para-aryl position of palladium α-diimine complexes (I, Fig. 1) and its influence on the catalytic behavior in ethene polymerization were studied by Guan.26,27 It was shown that using catalysts with an electron-withdrawing CF3 group resulted in a lower activity and lower molar mass of the synthesized polyethylenes. On the other hand, the study of polyethylene topology showed more dendritic structures of polyethylenes from CF3 substituted complexes than for those that were produced by catalysts bearing electron-donating ligands. Nickel and palladium α-diimine complexes bearing fluorinated cyclophane ligands (II, Fig. 1) were synthesized by Guan et al.28 and studied in ethene polymerization. Nickel derivatives showed higher thermal stability and Pd complexes provided polyethylenes with higher molar masses than their non-fluorinated analogs. Both, Ni and Pd, complexes produced polyethylenes with a significant decrease in branching density due to the direct interaction between the fluorine atom of the cyclophane ligand and the central metal which suppressed chain-walking. The same influence of fluorine was observed for another series of α-diimine Ni complexes bearing the fluorine atom in ortho-aryl positions (III, Fig. 1).29,30 A series of unsymmetrical α-diimine nickel complexes with an acenaphthene backbone and with one fluorinated N-aryl ring was synthesized (IV, Fig. 1).31 These fluorinated nickel complexes exhibited higher activities toward ethene polymerization than related non-fluorinated systems. Different series of unsymmetrical α-diimine nickel complexes bearing the acenaphthene backbone and CF3 or C6F5 substituents in ortho-aryl positions (V, Fig. 1) were published by Brookhart and co-workers.32 The presence of the CF3 group was found to increase the activity towards ethene polymerization.
 | ||
| Fig. 1 Different fluorinated ligands applied in olefin polymerization catalysts. | ||
Another reason for the incorporation of fluorinated substituents into the structure of catalytic complexes is increasing of fluorophilicity. Highly fluorinated compounds could be used for interphase catalysis utilizing a mixture of immiscible fluorophilic and aprotic non-polar solvents. Such a reaction setup allows an easy recycling of the catalyst or, in the case of polymers, preparation of a product with controlled morphology. Salicylaldiminato nickel complexes with fluorinated substituents were synthesized especially for studying dispersion polymerization behavior in supercritical carbon dioxide (scCO2).33,34 Bis(imino)pyridyl complexes of iron and cobalt with perfluorinated substituents for ethene polymerization were also synthesized.35
Despite numerous reports describing polymerization behavior of fluorinated α-diimine complexes, there have not been published reports on any complexes bearing longer fluorinated alkyl chains. According to the promising impact of fluorinated substituents (F, CF3) on the catalytic behavior of α-diimine complexes toward olefin polymerization, we decided to synthesize novel α-diimine nickel and palladium complexes with fluorinated tails in para-aryl positions (Fig. 2). Another motivation to synthesize these complexes was their potential to be utilized for interphase polymerization or for polymerization in scCO2. The obtained complexes were studied for polymerizations of basic olefins (ethene, propene, and hex-1-ene). Catalytic activity and influence on the chain-walking mechanism were investigated and compared to non-fluorinated analogs.
 | ||
| Fig. 2 Prepared nickel and palladium α-diimine catalyst precursors used in this study. | ||
Experimental section
Materials
All manipulations with air-sensitive compounds were done using standard Schlenk techniques. Nitrogen (SIAD, 99.999%), ethene (SIAD, 99.9%) and propene (SIAD, 99.5%) were purified by passing through a column packed with a Cu-catalyst and molecular sieves to remove traces of oxygen and water. Toluene (p.a., Penta) was dried over sodium and distilled under nitrogen. Chlorobenzene (p.a., Penta) was dried over CaH2 and distilled under nitrogen. Dichloromethane (p.a., Penta) was dried over CaH2 and cryodistilled under nitrogen. Hex-1-ene (99%, Aldrich) was dried over sodium/potassium alloy and distilled under nitrogen. Methylaluminoxane (MAO, 10 wt% solution in toluene), 2,6-diisopropylaniline, 2,6-dimethylaniline, acenaphthenequinone, 2 M solution of trimethylaluminium in hexane, 2 M solution of lithium aluminium hydride in Et2O, 2,3-butanedione, thionyl chloride, palladium on carbon (10 wt%), 40% aqueous solution of glyoxal and Novec™ 7100 (methoxy-nonafluorobutanes) were purchased from Sigma-Aldrich and used as received. Perfluorohexyliodide was purchased from Apollo Scientific, 1H,1H,2H-perfluorooctene was kindly donated by Atochem and HFE 7100 liquid by 3M. 2,6-Dimethyl-4-(1H,1H,2H,2H-perfluorooctyl)aniline (A1) and 2,6-dimethyl-4-perfluorohexylaniline (A2) were obtained according to the method described in the literature.36,37 Methanol and ethanol for the reaction were purchased from Penta s.r.o. and used without any other purification or drying. Diethyl ether, DMF, and THF for reaction were used from a solvent purification system (PureSolv™). Nickel complexes 7, 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1 and palladium complexes 11, 121,26 were synthesized according to the reported procedures. Catalysts were dosed as a solid directly to the polymerization vessel or in the form of dichloromethane solution that was stored at 5 °C.
1 and palladium complexes 11, 121,26 were synthesized according to the reported procedures. Catalysts were dosed as a solid directly to the polymerization vessel or in the form of dichloromethane solution that was stored at 5 °C.
Characterization
Elemental analyses (C, H, N) of new complexes were performed on an Elementar Vario EL III Analyzer. Elemental analysis of highly fluorinated compounds, including perfluoroalkylated ponytail derivatives, was not performed due to the poor combustion of such species,38–40 which can be overcome only with specialized analytical instrumentation.41 The unsatisfactory analysis was found even for single perfluoroalkyl chains, which (as expected)38 worsened with an increasing fluorine content.42,43 Consistent with standard practice in heavy fluorous chemistry, compound identity and purity were gauged from HRMS data and detailed NMR analysis.44–51Fourier transform infrared spectroscopy (FT-IR) measurements were performed on a NICOLET iS50R spectrometer (Thermo Scientific, USA). A Diamond ATR crystal and a DTGS detector were used for the measurements in the range of 4000–400 cm−1.
MALDI-TOF mass spectrometry measurements were performed on an UltrafleXtreme instrument equipped with a 1 kHz Smartbeam II Laser (Bruker Daltonik GmbH, Bremen, Germany) operated in the reflectron mode with an acceleration voltage of 25 kV and 2,5-dihydroxybenzoic acid as a matrix. APCI mass spectra were recorded using an LTQ Orbitrap XL hybrid mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) operated at a resolution of 100000. The APCI source vaporizer and heated capillary temperatures were set to 450 °C and 340 °C, respectively. The samples dissolved in acetonitrile were directly infused into the ion source.
1H NMR spectra of anilines, ligands, complexes, branched polyethylenes from Pd catalysis, polypropylenes, and poly(hex-1-enes) were recorded at 500 MHz in CDCl3 at 25 °C. Linear polyethylenes were measured in C2D2Cl4 at 120 °C. A total number of branches per 1000 carbon atoms (N) was determined by integrating methyl proton signals with respect to signals of all protons in the 1H NMR spectrum and calculated using the formula:

19F NMR spectra were recorded at 500 MHz using CCl3F as the internal standard. 13C NMR spectra of branched polyethylenes and polypropylenes were recorded at 500 MHz in CDCl3 at 25 °C using the INVGATE pulse sequence, 10 s relaxation delay, and 5000 scans to ensure quantitative spectra. All spectra were recorded on a Bruker Avance DRX 500 spectrometer.
Molar masses of linear polyethylenes were determined by using a high-temperature GPC-IR instrument (Polymer Char) equipped with an infrared and viscometric detector. Separation was performed on two Olexis mixed columns (Polymer Laboratories, 13 μm) at 150 °C in 1,2,4-trichlorobenzene at an elution rate of 1 ml min−1. Molar masses were calculated using the universal calibration approach in GPC One software.
Molar masses of branched polyethylenes obtained by Pd complexes, polypropylenes, and poly(hex-1-enes) were determined using a Waters Breeze chromatographic system equipped with an RI detector operating at 880 nm and multi-angle laser light scattering (MALLS) miniDawn TREOS from Wyatt operating at 658 nm. Separations were performed with two columns (Polymer Laboratories Mixed C) at 35 °C in THF at an elution rate of 1 ml min−1.
DSC measurements were performed on a TA Instrument module Q100 at a rate of 10 °C min−1 for both heating and cooling. Melting temperatures and enthalpies of fusion were obtained from the second heating run. Enthalpy of the fusion 297 J g−1 for hypothetical 100% crystalline polyethylene was used to calculate the degree of crystallinity.52
Crystallographic data for 4 and 6 were collected on a Bruker D8 VENTURE Kappa Duo PHOTON100 by IμS with Mo Kα 0.71073 Å at a temperature of 150(2) K. The absorption corrections were carried out using a numerical method based on crystal shape.
The structures were solved by direct methods (XP)53 and refined by full matrix least squares based on F2 (SHELXL2014).54 The hydrogen atoms on carbons were fixed into idealized positions (riding model) and assigned temperature factors, either Hiso(H) = 1.2Ueq(pivot atom) or Hiso(H) = 1.5Ueq for the methyl moiety. The structure of 6 is complicated by the presence of a large amount of solvent in the unit cells, from which only one molecule of dichloromethane could be discerned. To improve the precision of parameters of the Ni complex the PLATON/SQUEEZE55 procedure was used to correct the data of 6 for the presence of the other disordered solvents.
Crystallographic data for structural analysis of 4 and 6 have been deposited with the Cambridge Crystallographic Data Centre, CCDC no. 1810825 and 1810826,† respectively.
Synthesis of fluorinated anilines
Ligand synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH) ppm. 19F NMR (282.23 MHz, CDCl3): δ −81.3 (t, 4JF–F = 10 Hz, 6F, CF3), −113.8 (m, 4F, CF2CH2), −123.1 (m, 4F, CF2CF2CH2), −123.7 (m, 4F, CF3CF2CF2), −126.8 (m, 4F, CF3CF2) ppm. 13C NMR (100.58 MHz, CDCl3): δ 18.3 (4C, CH3), 36.9 (t, 2JF–C = 22 Hz, 2C, CH2CF2), 105–125 (m, 10C, 8 × CF2, 2 × CF3), 125.4 (2C, Ar–CCH2CF2), 127.0 (4C, Ar–CCH3), 130.9 (4C, Ar–CH), 150 (2C, Ar–CNC), 163 (2C, NCH) ppm. MS (ESI), m/z (%): 829 [M + H]+ (100). HRMS (ESI): calculated for C30H23F22N2 ([M + H]+) 829.1505, found 829.1510. IR (ATR, diamond) 1627 cm−1 (CN).
CH) ppm. 19F NMR (282.23 MHz, CDCl3): δ −81.3 (tt, 4JF–F = 10 Hz, 3JF–F = 3 Hz, 6F, CF3), −115.2 (m, 4F, CF2CH2), −122.4 (m, 4F, CF2CF2CH2), −123.4 (m, 4F, CF3CF2CF2CF2), −124.0 (m, 4F, CF3CF2CF2), −126.7 (m, 4F, CF3CF2) ppm. 13C NMR (75.44 MHz, CDCl3): δ 18.4 (4C, CH3), 26.0 (t, 3JF–C = 4 Hz, 2C, CH2CH2CF2), 33.3 (t, 2JF–C = 22 Hz, 2C, CH2CH2CF2), 105–125 (m, 12C, 10 × CF2, 2 × CF3), 127.2 (4C, Ar–CCH3), 128.4 (4C, Ar–CH), 135.7 (2C, Ar–C), 148.6 (2C, Ar–C), 163.7 (2C, NCH) ppm. MS (ESI), m/z (%): 957 [M + H]+ (100), 611 [M − C8F13H2]+ (30). HRMS (ESI): calculated for C34H27N2F26 ([M + H]+) 957.1754, found 957.1756. IR (ATR, diamond) 1626 cm−1 (CN).
C), 168.4 (2C, NC) ppm. MS (MALDI), m/z (%): 857 [M + H]+ (100). HRMS (MALDI): calculated for C32H27F22N2 ([M + H]+) 857.1817, found 857.1848. IR (ATR, diamond) 1645 cm−1 (CN).
C), 168.5 (2C, NC) ppm. MS (MALDI), m/z (%): 969 [M + H]+ (100). HRMS (MALDI): calculated for C40H43F22N2 ([M + H]+) 969.3069, found 969.3057. IR (ATR, diamond) 1651 cm−1 (CN).
C), 161.3 (2C, CN) ppm. MS (ESI), m/z (%): 953 [M + H]+ (100). HRMS (ESI): calculated for C40H27F22N2 ([M + H]+) 953.1818, found 953.1820 or C40H26F22N2Na ([M + Na]+) 975.1637, found 975.1638. IR (ATR, diamond) 1666 cm−1 (CN).
C), 161.4 (2C, CN) ppm. MS (MALDI), m/z (%): 1065 [M + H]+ (100). HRMS (MALDI): calculated for C48H43F22N2 ([M + H]+) 1065.3069, found 1065.3062. IR (ATR, diamond) 1671 cm−1 (CN).
CH) ppm. 19F NMR (282.23 MHz, CDCl3): δ −81.3 (t, 4JF–F = 10 Hz, 6F, CF3), −113.8 (m, 4F, CF2CH2), −123.1 (m, 4F, CF2CF2CH2), −123.7 (m, 4F, CF3CF2CF2), −126.8 (m, 4F, CF3CF2) ppm. 13C NMR (100.58 MHz, CDCl3): δ 18.3 (4C, CH3), 36.9 (t, 2JF–C = 22 Hz, 2C, CH2CF2), 105–125 (m, 10C, 8 × CF2, 2 × CF3), 125.4 (2C, Ar–CCH2CF2), 127.0 (4C, Ar–CCH3), 130.9 (4C, Ar–CH), 150 (2C, Ar–CNC), 163 (2C, NCH) ppm. MS (ESI), m/z (%): 829 [M + H]+ (100). HRMS (ESI): calculated for C30H23F22N2 ([M + H]+) 829.1505, found 829.1510. IR (ATR, diamond) 1627 cm−1 (CN).
CH) ppm. 19F NMR (282.23 MHz, CDCl3): δ −81.3 (tt, 4JF–F = 10 Hz, 3JF–F = 3 Hz, 6F, CF3), −115.2 (m, 4F, CF2CH2), −122.4 (m, 4F, CF2CF2CH2), −123.4 (m, 4F, CF3CF2CF2CF2), −124.0 (m, 4F, CF3CF2CF2), −126.7 (m, 4F, CF3CF2) ppm. 13C NMR (75.44 MHz, CDCl3): δ 18.4 (4C, CH3), 26.0 (t, 3JF–C = 4 Hz, 2C, CH2CH2CF2), 33.3 (t, 2JF–C = 22 Hz, 2C, CH2CH2CF2), 105–125 (m, 12C, 10 × CF2, 2 × CF3), 127.2 (4C, Ar–CCH3), 128.4 (4C, Ar–CH), 135.7 (2C, Ar–C), 148.6 (2C, Ar–C), 163.7 (2C, NCH) ppm. MS (ESI), m/z (%): 957 [M + H]+ (100), 611 [M − C8F13H2]+ (30). HRMS (ESI): calculated for C34H27N2F26 ([M + H]+) 957.1754, found 957.1756. IR (ATR, diamond) 1626 cm−1 (CN).
C), 168.4 (2C, NC) ppm. MS (MALDI), m/z (%): 857 [M + H]+ (100). HRMS (MALDI): calculated for C32H27F22N2 ([M + H]+) 857.1817, found 857.1848. IR (ATR, diamond) 1645 cm−1 (CN).
C), 168.5 (2C, NC) ppm. MS (MALDI), m/z (%): 969 [M + H]+ (100). HRMS (MALDI): calculated for C40H43F22N2 ([M + H]+) 969.3069, found 969.3057. IR (ATR, diamond) 1651 cm−1 (CN).
C), 161.3 (2C, CN) ppm. MS (ESI), m/z (%): 953 [M + H]+ (100). HRMS (ESI): calculated for C40H27F22N2 ([M + H]+) 953.1818, found 953.1820 or C40H26F22N2Na ([M + Na]+) 975.1637, found 975.1638. IR (ATR, diamond) 1666 cm−1 (CN).
C), 161.4 (2C, CN) ppm. MS (MALDI), m/z (%): 1065 [M + H]+ (100). HRMS (MALDI): calculated for C48H43F22N2 ([M + H]+) 1065.3069, found 1065.3062. IR (ATR, diamond) 1671 cm−1 (CN).
Synthesis of nickel complexes
Nickel complexes were synthesized analogous to the procedure reported for 7 and 81 by the reaction of the corresponding ligand and (dimethoxyethane)nickel(II)bromide ((DME)NiBr2) in dry CH2Cl2.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CH–CHN–Ar)NiBr2 (Ar = 2,6-dimethyl-4-(perfluoropentyl)methylphenyl) (1).
Ligand L1 (0.17 g, 0.20 mmol) and (DME)NiBr2 (0.06 g, 0.19 mmol) were reacted in 10 ml of CH2Cl2 at room temperature overnight, evaporated and extracted with dry n-hexane repeatedly until the n-hexane stopped turning yellow. Evaporation afforded 2 as yellow-brown powder in 91% yield. Anal. calc. (C30H22Br2F22N2Ni) C, 34.42; H, 2.12; N, 2.68. Found: C, 30.27; H, 2.26; N, 2.61. MALDI-TOF m/z: 1039.0940 (C37H27F22N2NiO4 [M + H]+, requires 1039.0968). IR (ATR, diamond) 1632 cm−1 (CN).
CH–CHN–Ar)NiBr2 (Ar = 2,6-dimethyl-4-(2-(perfluorohexyl)ethyl)phenyl) (2).
Using the same procedure as that for 1, ligand L2 (0.15 g, 0.15 mmol) was reacted with (DME)NiBr2 (0.04 g, 0.14 mmol) resulting in a brown powder of 2 in 95% yield. Anal. calc. (C34H26Br2F26N2Ni) C, 34.75; H, 2.23; N, 2.38. Found: C, 32.94; H, 2.66; N, 2.14. MALDI-TOF m/z: 1167.1240 (C41H31F26N2NiO4 [M + H]+, requires 1167.1267). IR (ATR, diamond) 1633 cm−1 (CN).
C(Me)–C(Me)N–Ar)NiBr2 (Ar = 2,6-dimethyl-4-(perfluoropentyl)methylphenyl) (3).
Ligand L3 (0.15 g, 0.18 mmol) and (DME)NiBr2 (0.05 g, 0.17 mmol) were reacted in 10 ml of CH2Cl2 at room temperature overnight, evaporated and extracted with dry n-hexane repeatedly until the n-hexane stopped turning yellow. The crude product was dissolved in CH2Cl2 and filtered. The residual solvent was evaporated, and 3 was obtained as a brown-red solid in 88% yield. Anal. calc. (C32H26Br2F22N2Ni) C, 35.75; H, 2.44; N, 2.61. Found: C, 35.56; H, 2.38; N, 2.33. MALDI-TOF m/z: 1067.1255 (C39H31F22N2NiO4 [M + H]+, requires 1067.1281). IR (ATR, diamond) 1637 cm−1 (CN).
C(Me)–C(Me)N–Ar)NiBr2 (Ar = 2,6-diisopropyl-4-(perfluoropentyl)methylphenyl) (4).
Using the same procedure as that for 3, ligand L4 (0.16 g, 0.16 mmol) was reacted with (DME)NiBr2 (0.05 g, 0.15 mmol) resulting in a brown-red powder of 2 in 84% yield. Single crystals suitable for X-ray analysis were obtained by crystallization from dichloromethane solution layered by n-hexane. Anal. calc. (C40H42Br2F22N2Ni) C, 40.47; H, 3.57; N, 2.36. Found: C, 40.38; H, 3.44; N, 2.29. MALDI-TOF m/z: 1179.2513 (C47H47F22N2NiO4 [M + H]+, requires 1179.2533). IR (ATR, diamond) 1641 cm−1 (CN).
C(An)–C(An)N–Ar)NiBr2 (Ar = 2,6-dimethyl-4-(perfluoropentyl)methylphenyl) (5).
Using the same procedure as that for 3, ligand L5 (0.12 g, 0.13 mmol) was reacted with (DME)NiBr2 (0.04 g, 0.12 mmol) resulting in a brown-red powder of 2 in 92% yield. Anal. calc. (C40H26Br2F22N2Ni) C, 41.02; H, 2.24; N, 2.39. Found: C, 40.77; H, 2.21; N, 2.16. MALDI-TOF m/z: 1163.1296 (C47H31F22N2NiO4 [M + H]+, requires 1163.1281), APCI-Orbitrap m/z: 1130.0549 (C42H29BrF22N3Ni [M + H]+, requires 1130.0541). IR (ATR, diamond) 1655 cm−1 (CN).
C(An)–C(An)N–Ar)NiBr2 (Ar = 2,6-diisopropyl-4-(perfluoropentyl)methylphenyl) (6).
Using the same procedure as that for 3, ligand L6 (0.17 g, 0.16 mmol) was reacted with (DME)NiBr2 (0.05 g, 0.15 mmol) resulting in a brown-red powder of 2 in 83% yield. Single crystals suitable for X-ray analysis were obtained by crystallization from dichloromethane solution layered by n-hexane. Anal. calc. (C48H42Br2F22N2Ni) C, 44.92; H, 3.30; N, 2.18. Found: C, 44.40; H, 3.22; N, 1.99. MALDI-TOF m/z: 1275.2483 (C55H47F22N2NiO4 [M + H]+, requires 1275.2533), APCI-Orbitrap m/z: 1242.1805 (C50H45BrF22N3Ni [M + H]+, requires 1242.1793). IR (ATR, diamond) 1660 cm−1 (CN).
CH–CHN–Ar)NiBr2 (Ar = 2,6-dimethyl-4-(perfluoropentyl)methylphenyl) (1).
Ligand L1 (0.17 g, 0.20 mmol) and (DME)NiBr2 (0.06 g, 0.19 mmol) were reacted in 10 ml of CH2Cl2 at room temperature overnight, evaporated and extracted with dry n-hexane repeatedly until the n-hexane stopped turning yellow. Evaporation afforded 2 as yellow-brown powder in 91% yield. Anal. calc. (C30H22Br2F22N2Ni) C, 34.42; H, 2.12; N, 2.68. Found: C, 30.27; H, 2.26; N, 2.61. MALDI-TOF m/z: 1039.0940 (C37H27F22N2NiO4 [M + H]+, requires 1039.0968). IR (ATR, diamond) 1632 cm−1 (CN).
CH–CHN–Ar)NiBr2 (Ar = 2,6-dimethyl-4-(2-(perfluorohexyl)ethyl)phenyl) (2).
Using the same procedure as that for 1, ligand L2 (0.15 g, 0.15 mmol) was reacted with (DME)NiBr2 (0.04 g, 0.14 mmol) resulting in a brown powder of 2 in 95% yield. Anal. calc. (C34H26Br2F26N2Ni) C, 34.75; H, 2.23; N, 2.38. Found: C, 32.94; H, 2.66; N, 2.14. MALDI-TOF m/z: 1167.1240 (C41H31F26N2NiO4 [M + H]+, requires 1167.1267). IR (ATR, diamond) 1633 cm−1 (CN).
C(Me)–C(Me)N–Ar)NiBr2 (Ar = 2,6-dimethyl-4-(perfluoropentyl)methylphenyl) (3).
Ligand L3 (0.15 g, 0.18 mmol) and (DME)NiBr2 (0.05 g, 0.17 mmol) were reacted in 10 ml of CH2Cl2 at room temperature overnight, evaporated and extracted with dry n-hexane repeatedly until the n-hexane stopped turning yellow. The crude product was dissolved in CH2Cl2 and filtered. The residual solvent was evaporated, and 3 was obtained as a brown-red solid in 88% yield. Anal. calc. (C32H26Br2F22N2Ni) C, 35.75; H, 2.44; N, 2.61. Found: C, 35.56; H, 2.38; N, 2.33. MALDI-TOF m/z: 1067.1255 (C39H31F22N2NiO4 [M + H]+, requires 1067.1281). IR (ATR, diamond) 1637 cm−1 (CN).
C(Me)–C(Me)N–Ar)NiBr2 (Ar = 2,6-diisopropyl-4-(perfluoropentyl)methylphenyl) (4).
Using the same procedure as that for 3, ligand L4 (0.16 g, 0.16 mmol) was reacted with (DME)NiBr2 (0.05 g, 0.15 mmol) resulting in a brown-red powder of 2 in 84% yield. Single crystals suitable for X-ray analysis were obtained by crystallization from dichloromethane solution layered by n-hexane. Anal. calc. (C40H42Br2F22N2Ni) C, 40.47; H, 3.57; N, 2.36. Found: C, 40.38; H, 3.44; N, 2.29. MALDI-TOF m/z: 1179.2513 (C47H47F22N2NiO4 [M + H]+, requires 1179.2533). IR (ATR, diamond) 1641 cm−1 (CN).
C(An)–C(An)N–Ar)NiBr2 (Ar = 2,6-dimethyl-4-(perfluoropentyl)methylphenyl) (5).
Using the same procedure as that for 3, ligand L5 (0.12 g, 0.13 mmol) was reacted with (DME)NiBr2 (0.04 g, 0.12 mmol) resulting in a brown-red powder of 2 in 92% yield. Anal. calc. (C40H26Br2F22N2Ni) C, 41.02; H, 2.24; N, 2.39. Found: C, 40.77; H, 2.21; N, 2.16. MALDI-TOF m/z: 1163.1296 (C47H31F22N2NiO4 [M + H]+, requires 1163.1281), APCI-Orbitrap m/z: 1130.0549 (C42H29BrF22N3Ni [M + H]+, requires 1130.0541). IR (ATR, diamond) 1655 cm−1 (CN).
C(An)–C(An)N–Ar)NiBr2 (Ar = 2,6-diisopropyl-4-(perfluoropentyl)methylphenyl) (6).
Using the same procedure as that for 3, ligand L6 (0.17 g, 0.16 mmol) was reacted with (DME)NiBr2 (0.05 g, 0.15 mmol) resulting in a brown-red powder of 2 in 83% yield. Single crystals suitable for X-ray analysis were obtained by crystallization from dichloromethane solution layered by n-hexane. Anal. calc. (C48H42Br2F22N2Ni) C, 44.92; H, 3.30; N, 2.18. Found: C, 44.40; H, 3.22; N, 1.99. MALDI-TOF m/z: 1275.2483 (C55H47F22N2NiO4 [M + H]+, requires 1275.2533), APCI-Orbitrap m/z: 1242.1805 (C50H45BrF22N3Ni [M + H]+, requires 1242.1793). IR (ATR, diamond) 1660 cm−1 (CN).
Synthesis of palladium complexes
Palladium complexes were synthesized according to the modified literature procedure for 121 by the reaction of the corresponding ligand and CODPdMeCl in dry diethyl ether.
C(Me)–C(Me)N–Ar)Pd(Me)Cl (Ar = 2,6-diisopropyl-4-(perfluoropentyl)methylphenyl) (9).
Et2O (10 mL) was added to a Schlenk flask containing CODPdMeCl (41 mg, 0.16 mmol) and a slight excess of ligand L4 (159 mg, 0.16 mmol, 1.05 eq.). The suspension was allowed to stir for an additional 20 h at room temperature. The solvent was removed under vacuum. The product was extracted with n-hexane (3 × 10 mL) and then dried overnight in vacuo. A yellow-orange powder was isolated in 90% yield. Anal. calc. (C41H45ClF22N2Pd) C, 43.75; H, 4.03; N, 2.49. Found: C, 43.34; H, 3.81; N, 2.36. 1H NMR (299.97 MHz, CDCl3): δ 7.17 (s, aromatic 2H), 7.12 (s, aromatic 2H), 3.39 (td, 4H, CH2C5F11), 3.14–2.97 (m, 4H, CH(CH3)2), 2.07 (s, 3H, CH3–CN), 2.04 (s, 3H, CH3–CN), 1.43 (d, 6H, (CH3)2CH), 1.35 (d, 6H, (CH3)2CH), 1.18 (t, 12H, (CH3)2CH), 0.52 (s, 3H, CH3–Pd). 13C NMR (100.58 MHz, CDCl3): δ 174.05, 169.40 (CN); 141.55 (aromatic C–N); 138.98, 138.18 (ortho-aromatic C); 128.36, 127.35 (para-aromatic C); 126.64, 126.15 (meta-aromatic C); 37.04 (CH2–C5F11); 29.01, 28.49 (–CH(CH3)2); 23.72, 23.64, 23.38, 23.06 (–CH(CH3)2); 21.17, 19.78 (CH3–CN); 3.08 ppm (Pd–CH3). IR (ATR, diamond) 1626 cm−1 (CN). MS ESI-m/z: 1123.1907 (C41H44ClF22N2Pd [M − H]−, requires 1123.1882).
C(An)–C(An)N–Ar)Pd(Me)Cl (Ar = 2,6-diisopropyl-4-(perfluoropentyl)methylphenyl) (10).
Using the same procedure as that for 9, CODPdMeCl (30 mg, 0.11 mmol) was reacted with a slight excess of ligand L6 (126 mg, 0.12 mmol, 1.07 eq.) affording 10 as an orange solid in 80% yield. Anal. calc. (C49H45ClF22N2Pd) C, 48.17; H, 3.71; N, 2.29. Found: C, 48.03; H, 3.62; N, 2.16. 1H NMR (299.97 MHz, CDCl3): δ 8.08 (d, aromatic 1 H), 8.04 (d, aromatic 1 H), 7.42 (dt, aromatic 2 H), 7.31 (s, aromatic 2 H), 7.25 (s, aromatic 2 H), 6.58 (d, aromatic 1 H), 6.37 (d, aromatic 1 H), 3.51 (q, 4H, CH2C5F11), 3.45–3.32 (m, 4H, CH(CH3)2), 1.48 (d, 6H, (CH3)2CH), 1.38 (d, 6H, (CH3)2CH), 0.96 (d, 6H, (CH3)2CH), 0.91 (d, 6H, (CH3)2CH), 0.88 (s, 3H, CH3–Pd). 13C NMR (100.58 MHz, CDCl3): δ 172.13, 167.84 (C1); 143.89 (C7); 142.26, 141.36 (aromatic C–N); 140.03, 138.79 (ortho-aromatic C); 131.43 (C5); 131.35 (C6); 130.96 (C5); 128.93 (C4); 128.89 (para-aromatic C); 128.72 (C4); 127.70 (para-aromatic C); 127.13 (meta-aromatic C); 127.11 (C2); 126.51 (meta-aromatic C); 126.35 (C2); 124.79 (C3); 124.66 (C3); 37.51 (CH2–C5F11); 29.13, 28.65 (–CH(CH3)2); 24.08, 23.66, 23.53, 23.23 (–CH(CH3)2); 3.33 ppm (Pd–CH3). IR (ATR, diamond) 1640 cm−1 (C
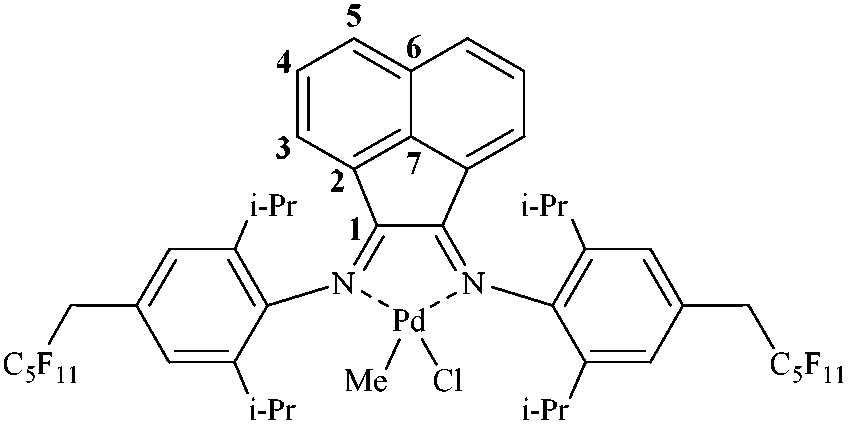 N). MS ESI+ m/z: 1221.2572 (C49H46ClF22N2Pd [M + H]+, requires 1221.2028).
N). MS ESI+ m/z: 1221.2572 (C49H46ClF22N2Pd [M + H]+, requires 1221.2028).
Ethene and propene polymerization with nickel complexes
A 100 ml or 250 ml glass pressure ampoule (Fisher-Porter bottle) with a magnetic stirring bar was evacuated and placed under an ethene/propene atmosphere. Toluene and MAO were added against nitrogen flux and the reactor was then allowed to stir under the desired absolute pressure of ethene/propene for 15 min at the prescribed polymerization temperature. Polymerization was started by the injection of the catalyst solution against nitrogen flux. The reactor was pressurized with ethene/propene, and the pressure was kept constant during the reaction. After the allotted polymerization time, the reaction was terminated by pouring the polymerization mixture into a large excess of ethanol acidified with HCl. Polyethylenes/polypropylenes were separated by filtration, washed with ethanol and dried under vacuum overnight at 50 °C.Ethene polymerization with palladium complexes
A 100 ml glass pressure ampoule (Fisher-Porter bottle) with a magnetic stirring bar was charged with the appropriate amount of solid catalyst. The desired volume of dry chlorobenzene was transferred via a cannula to the reaction ampule and the catalyst was immediately dissolved. Then, the ampule was sealed, cooled to the polymerization temperature and saturated with the appropriate ethene pressure. Polymerization was started by the addition of Na(BArF4) solution into chlorobenzene. An absolute ethene pressure of 8 bars was controlled by using a gas regulator. For the polymerizations at 0.05 bars of ethene, a mixture of ethene and nitrogen was prepared and bubbled through a reaction mixture. The reaction mixture was stirred under constant ethene pressure for the allotted polymerization time. Then, ethene pressure was released and the reaction was quenched by the addition of 0.3 ml of triethylsilane. Chlorobenzene was evaporated on a rotary evaporator. The obtained polymer was dissolved in toluene and passed through a column packed with silica gel and alumina to remove the rest of the catalyst. Then, the solvent was evaporated and the obtained polymer was dried in a vacuum oven at 60 °C to a constant weight.Hex-1-ene polymerization with nickel complexes
Polymerizations were carried out under a nitrogen atmosphere in magnetically stirred 15 ml glass ampoules. The ampoules with toluene, hex-1-ene, and MAO were placed in a bath kept at a desired temperature and tempered for 15 min. The polymerization was initiated by the addition of catalyst solution against nitrogen flux. After the allotted polymerization time the reaction was terminated by pouring the polymerization mixture into a large excess of ethanol acidified with HCl. Polyethylene was separated by filtration, washed with ethanol and dried under vacuum overnight at 50 °C. Reinitiation tests were carried out by the addition of the second portion of the monomer after the consumption of the initial monomer feed.GC method for hex-1-ene polymerization kinetics investigation
The method described in the literature56 was used using cyclohexane in the feed as an internal standard. Polymerization was carried out in the same way as described above with the exception of the addition of 1 ml of cyclohexane after monomer addition. 1 ml samples were withdrawn from the reaction mixture in the course of the polymerization, quenched by pouring into 8 ml ethanol, and decanted to isolate a precipitated polymer. Liquid phases were transferred to 2 ml vials equipped with a septum and measured by gas chromatography immediately. GC analysis was performed on a Varian CP 3800 chromatograph using a capillary column HP Ultra 1 (PDMS, 50 m length, inner diameter 0.32 mm, film thickness 0.52 μm) at 60 °C and 3 ml min−1 nitrogen flow.Results and discussion
Ligand synthesis
Three polyfluorinated anilines, A1, A4 and A5, were synthesized and used for the preparation of diimine ligands L1–L6 (Scheme 1). Aniline A1 with an ethylene spacer between the aromatic ring and a C6F13 moiety was prepared according to the known procedure in a four-step synthesis starting from commercial 2,6-dimethylaniline.36 The same starting aniline was also modified in the para-position with the C6F13 moiety in a two-phase metal free reaction37 to obtain A2. Fluorine atoms in the α-position of the perfluoroalkyl chain attached in the ortho- or para-position of aniline are known to be highly reactive against nucleophilic substitution.57 In our case, anilines A4 and A5 were prepared from A2 and A3, respectively, by substitution of both fluorine atoms in the α-position by hydrogen ones using an ether solution of lithium aluminum hydride. The reaction of anilines A1, A4, and A5 with the corresponding diones in polar solvents afforded a series of new polyfluorinated α-diimine ligands L1–L6 in good yields (Scheme 1). | ||
| Scheme 1 Synthesis of α-diimine ligands with fluorinated alkyl substituents. | ||
Catalyst synthesis
A series of six novel nickel complexes bearing fluorinated alkyl substituents (1–6, Fig. 2) was synthesized by the reaction of the corresponding α-diimine ligands (L1–L6) with (DME)NiBr2 in dichloromethane at room temperature yielding complexes 1–6 in high yields (>80%). Non-fluorinated analogs 7 and 8 were prepared for the comparison with the catalytic behavior of novel complexes in olefin polymerizations. The analysis of nickel complexes by NMR spectroscopy is tangled due to their paramagnetic character. Elemental analysis, IR and mass spectrometry were therefore used for their characterization. Elemental analysis of Ni complexes 1 and 2 bearing only H atoms on the backbone (the highest fluorine content) exhibited the lowest conformity of calculated and experimental values due to the poor combustion of highly fluorinated compounds (see the Experimental section). The rest of the nickel complexes showed very good agreement of theoretical and measured values. Ligand complexation to nickel was confirmed by shifting of CN FTIR signals in ligands (1626–1671 cm−1) by ∼10 cm−1 in the corresponding complexes (1632–1660 cm−1). High-resolution mass spectrometry (MS) of nickel complexes was carried out by APCI (atmospheric pressure chemical ionization) and MALDI-TOF (matrix-assisted laser desorption/ionization with time-of-flight detection) techniques. In all cases, complexes were partially fragmented during the ionization. The formation of the Ni complex was confirmed for all 1–6 studied catalysts. For complexes 5 and 6 bearing naphthalene backbone substituents, both MS techniques were successful. Using APCI, measured in CH3CN, specific masses for (N–N)NiBr·CH3CN (N–N = α-diimine ligand) fragments were found (m/z: 1130.0549 and 1242.1805 for 5 and 6, respectively), whereas by the MALDI technique, using 2,5-dihydroxybenzoic acid as a matrix, (N–N)Ni·DHB (DHB anion) fragments were detected (m/z: 1163.1296 and 1275.2483 for 5 and 6, respectively). For other complexes, only MALDI was successful in their ionization, identifying again (N–N)Ni·DHB adducts (m/z: 1039.0940, 1167.1240, 1067.1255, and 1179.2512 for 1, 2, 3 and 4, respectively).
Single crystals of complexes 4 and 6 suitable for X-ray analysis were obtained by crystallization from dichloromethane solution layered by n-hexane. Crystallographic data are summarized in Table S2 (ESI†). Fig. 3 and 4 show molecular structures of complexes 4 and 6 obtained with X-ray diffraction, while Table S3 (ESI†) gives details of the selected bond distances and angles for the geometry about the nickel atoms in each of the complexes. The geometries of 4 and 6 are similar to non-fluorinated analogs reported in the literature previously,58–62 having aryl rings almost perpendicularly oriented toward the diimine ligand plane and C2v symmetry. Complex 4 possesses pseudo-tetrahedral geometry about the nickel atom. Complex 6 with the acenaphthene backbone ligand forms a dimeric structure in the solid state with two nickel atoms bridged via two bromine atoms, each nickel atom being surrounded by a ligand L6 and three bromides. The geometry of each of the five-coordinated Ni centers can be described as pseudo-hexahedral. Similar dimeric structures were already observed for other Ni complexes.58–61
 | ||
| Fig. 3 View on the molecule of 4 with the atom numbering schema. The displacement ellipsoids are drawn at 30% probability level. The second part of the molecule is generated via mirror perpendicular to the b axis. Symmetry code (i) x, 3/2 − y, z. | ||
 | ||
| Fig. 4 View on the molecule of 6 with the atom numbering schema. The displacement ellipsoids are drawn at 30% probability level. Only quarter of molecule is symmetrically independent, other parts are generated via site symmetry 2/m. Symmetry code (i) x, y, 1 − z; (ii) 1 − x, 1 − y, 1 − z; (iii) 1 − x, 1 − y, z; (iv) 1 − x, 1 − y, z. | ||
Two novel palladium complexes 9 and 10 (Fig. 2) with fluorinated substituents in para-aryl positions and with different diimine backbones were prepared by the reaction of the corresponding ligands (L4 and L6) and CODPdMeCl in dry diethyl ether. The formation of Pd complexes was proved by elemental analysis, NMR and IR spectroscopy and mass spectrometry. Non-fluorinated analogs 11 and 12 were synthesized to compare the catalytic behavior of novel fluorinated complexes and previously published catalysts in ethene polymerizations.
Solubility tests of all novel Ni and Pd complexes with fluorinated tails were performed in different common solvents and also in one fluorinated solvent (Table 1), namely NovecTM 7100 (methoxy-nonafluorobutanes), which was tested concerning the potential application of novel fluorinated complexes in interphase polymerizations.
| Cat. | Novec™ 7100 | n-Hexane | Toluene | Diethyl ether | Dichloromethane |
|---|---|---|---|---|---|
| X (insoluble), ∼ (slightly soluble), √ (soluble). | |||||
| 1 | ∼ | X | X | ∼ | ∼ |
| 2 | ∼ | X | X | ∼ | ∼ |
| 3 | X | X | √ | ∼ | √ |
| 4 | X | X | √ | ∼ | √ |
| 5 | X | X | √ | √ | √ |
| 6 | X | X | √ | √ | √ |
| 9 | ∼ | X | √ | ∼ | √ |
| 10 | √ | ∼ | √ | √ | √ |
The poorest solvent of all tested ones was n-hexane as expected because it is commonly used for the extraction of crude complexes. Other conventional solvents (toluene, diethyl ether, and dichloromethane) were in general evaluated as suitable solvents. Nickel complexes 1 and 2 bearing hydrogen atoms on the ligand backbone were partially soluble in Novec as they have the highest fluorine content. The rest of the Ni complexes (3, 4, 5 and 6) with bulkier backbones were completely insoluble in Novec due to the decreased fluorophilicity. Palladium complexes 9 and 10 showed surprisingly better solubility than their Ni analogs. Complex 9 was slightly soluble in Novec and catalyst 10 bearing the bulkier acenaphthene backbone was dissolved entirely in Novec in 24 h. This means that complex 10 would be the most appropriate candidate for interphase catalysis. However, slower dissolution of complex 10 in Novec than in common solvents suggests that interphase polymerization would not be possible. Generally, the better solubility in fluorinated solvents can be expected for metal–alkyl–halogene complexes (e.g. PdMeCl in 9 and 10) or complexes with a higher content of fluoroalkyl groups.
Ethene polymerization by Ni complexes
Nickel α-diimine complexes 1–6 were activated by 200 eq. of MAO and tested in ethene polymerization. Polymerization experiments were performed in toluene at 25 °C and 2 bars of ethene (Table 2). Complexes 7 and 8, the non-fluorinated analogs of complexes 4 and 6, were used for the comparison with fluorinated catalysts.| Run | Cat. | TOFa 103 [h−1] | M n [kg mol−1] | Đ [−] | T m [°C] | α [%] | N |
|---|---|---|---|---|---|---|---|
| n Ni = 1.2 μmol, Al/Ni = 750, total volume = 100 ml, tp = 30 min, 2 bars of ethene (1 bar on gauge).a Turnover frequency: mol ethene/(molNitp).b Molar mass and dispersity determined by HT-SEC in 1,2,4-trichlorobenzene at 150 °C.c Melting temperature and crystallinity determined by DSC from the 2nd heating run.d Number of branches per 1000 C atoms determined by 1H NMR. | |||||||
| 1 | 1 | 16 | 17 | 2.28 | 132 | 47 | 3 |
| 2 | 2 | 21 | 18 | 2.57 | 126 | 45 | 7 |
| 3 | 3 | 43 | 87 | 2.43 | 117 | 34 | 16 |
| 4 | 4 | 61 | 175 | 1.92 | 59 | 12 | 58 |
| 5 | 5 | 42 | 72 | 2.54 | 125 | 35 | 9 |
| 6 | 6 | 108 | 134 | 2.09 | 83 | 23 | 49 |
| 7 | 7 | 54 | 495 | 1.62 | 34 | 8 | 48 |
| 8 | 8 | 84 | 79 | 1.89 | 35 | 2 | 71 |
Complexes 1 and 2 (Fig. 2) bearing hydrogen atoms as ligand backbone substituents displayed lower activity and provided PEs with lower molar masses than the other derivatives 3–6 bearing bulkier backbone substituents. This observation corresponds with the behavior of non-fluorinated nickel α-diimine complexes published previously.1 All prepared complexes produced polyethylenes with monomodal molar mass distribution and with the dispersity values around 2, which is typical of single-site catalysts.63 Branching numbers of synthesized polyethylenes well corresponded to the ligand bulkiness.1,64 Complexes 1, 2, 3, and 5, substituted with methyl groups in ortho-aryl positions, yielded very linear PEs with 3–16 br. per 1000 C atoms. Complexes 4 and 6, bearing bulkier iPr substituents, provided polyethylenes with ∼50–60 br. per 1000 C.
The comparison of fluorinated complexes 4 and 6 with their non-fluorinated analogs 7 and 8 shows higher polymerization activity for fluorinated derivatives which could be caused by the increase of nickel center electrophilicity due to the substitution of the diimine ligand by electron-withdrawing fluorinated alkyl groups (Table 2, runs 4 and 6 vs. runs 7 and 8, respectively). Molar masses of PEs prepared with all catalysts are several times lower than the theoretical values (obtained from polymer yield and amount of Ni precursors), which together with broad molar mass distribution, shows the high extent of transfer reactions during ethene polymerization.
Concerning the effect of the ligand structure on the chain-walking extent, there is an opposite trend for complexes 4 and 7 with methyl backbone substituents and complexes 6 and 8 with the naphthalene backbone. Thus, the backbone substituent of the diimine ligand plays a more important role in influencing the metal center toward chain-walking reaction than a fluorinated alkyl group.
Propene polymerization by Ni complexes
Propene polymerizations were performed in toluene at −10 °C or 25 °C and at 2 bars of propene (Table 3). Complexes 1 and 2 bearing hydrogen atoms as backbone substituents yielded polypropylenes with the lowest molar masses and with relatively broader dispersities (Đ ≈ 1.70) at −10 °C. Broader molar mass distributions and lower molar masses than theoretical values suggested that small hydrogen atoms at the ligand backbone cannot protect Ni centers from transfer reactions. On the other hand, complexes 3, 4 and 6 with bulkier backbone substituents (Me or An) afforded polypropylenes with very narrow molar mass distribution, suggesting the living character of polymerization at −10 °C. Surprisingly, catalyst 5 was the only complex with bulkier backbone substituents than H atoms which didn't show the living character of propene polymerization. This is due to lower sterical interactions of the α-diimine backbone and ortho-aryl substituents, which is weaker for the acenaphthene backbone of 5 than for the methyl backbone used in 3.65 Thus, the combination of the acenaphthene backbone and small methyl ortho-aryl substituents of complex 5 results in insufficient blocking of axial coordination sites and higher probability of transfer reactions leading to the loss of living behavior. The comparison of theoretical and experimental molar masses for polypropylenes with narrow dispersities showed that nickel complexes are not entirely activated by MAO (Table 3, runs 11, 12, 14, 15 and 16). The highest activities represented by TOF values were observed for complexes with naphthalenediyl backbone substituents. Polypropylenes synthesized by catalysts with less bulky methyl o-aryl substituents (Table 3, runs 9, 10, 11 and 13) showed the highest number of branches due to the suppressed chain-walking mechanism. Complexes 4 and 6 provided polypropylenes with slightly decreased branching in comparison to their non-fluorinated analogs 7 and 8. It is probably caused by the electron-withdrawing effect of para-fluoroalkyl substituents that supports the chain-walking mechanism and leads to chain straightening.26,27| Run | T [°C] | Cat. | TOFa 103 [h−1] | M n [kg mol−1] | Đ [−] |
M
n
thc [kg mol−1] |
T g [°C] | N |
|---|---|---|---|---|---|---|---|---|
| n Ni = 5 μmol, Al/Ni = 200, total volume = 18 ml, tp = 1.5 h, 2 bars of propene.a Turnover frequency: mol propene/(molNitp).b Molar mass and dispersity determined by SEC RI-PS calib.c Theoretical molar mass, Mnth = mp/nNi.d Glass transition temperature determined by DSC from the 2nd heating run.e Number of branches per 1000 C atoms determined by 1H NMR.f tp = 2 h. | ||||||||
| 9 | −10 | 1 | 0.4 | 12 | 1.64 | 23 | −18 | 332 |
| 10 | −10 | 2 | 1.2 | 13 | 1.66 | 74 | −21 | 320 |
| 11 | −10 | 3 | 0.7 | 63 | 1.14 | 43 | −22 | 309 |
| 12 | −10 | 4 | 0.4 | 66 | 1.13 | 22 | −35 | 250 |
| 13 | −10 | 5 | 1.6 | 51 | 1.97 | 102 | −18 | 311 |
| 14 | −10 | 6 | 3.2 | 244 | 1.16 | 201 | −15 | 284 |
| 15 | −10 | 7 | 0.5 | 119 | 1.13 | 33 | −25 | 261 |
| 16 | −10 | 8 | 2.7 | 315 | 1.13 | 173 | −13 | 293 |
| 17f | 25 | 1 | 0.6 | 11 | 2.03 | 51 | −39 | 298 |
| 18f | 25 | 2 | 1.3 | 11 | 3.90 | 109 | −39 | 267 |
| 19f | 25 | 3 | 1.4 | 106 | 1.79 | 115 | −36 | 240 |
| 20f | 25 | 4 | 1.2 | 183 | 1.52 | 100 | −40 | 212 |
| 21f | 25 | 5 | 2.6 | 64 | 1.82 | 219 | −35 | 257 |
| 22f | 25 | 6 | 3.1 | 312 | 2.06 | 197 | −23 | 256 |
| 23f | 25 | 7 | 1.0 | 269 | 1.56 | 80 | −31 | 228 |
| 24 | 25 | 8 | 3.0 | 412 | 1.74 | 189 | −23 | 244 |
Propene polymerization at 25 °C showed only a slight increase of TOF and molar mass values compared to polymerization at −10 °C. Furthermore, molar mass distribution increased significantly, clearly indicating the loss of the living character at 25 °C. The increase of reaction temperature is also reflected in decreased Tg values of polypropylenes as a result of enhanced chain-walking which leads to 1,3-insertion of propene and straightening of the chain.
To further investigate the livingness/controllability of propene polymerization with novel catalysts, we have chosen highly active complex 6, which produced narrowly dispersed polypropylene with a molar mass close to the theoretical value at −10 °C (Table 3, run 14). A linear increase of molar mass up to 250 kg mol−1 with polymer yield, the retention of very low dispersity values and no tailing of SEC peaks demonstrate very well controlled propene polymerization by 6/MAO at −10 °C (Fig. 5 and S4, ESI†).
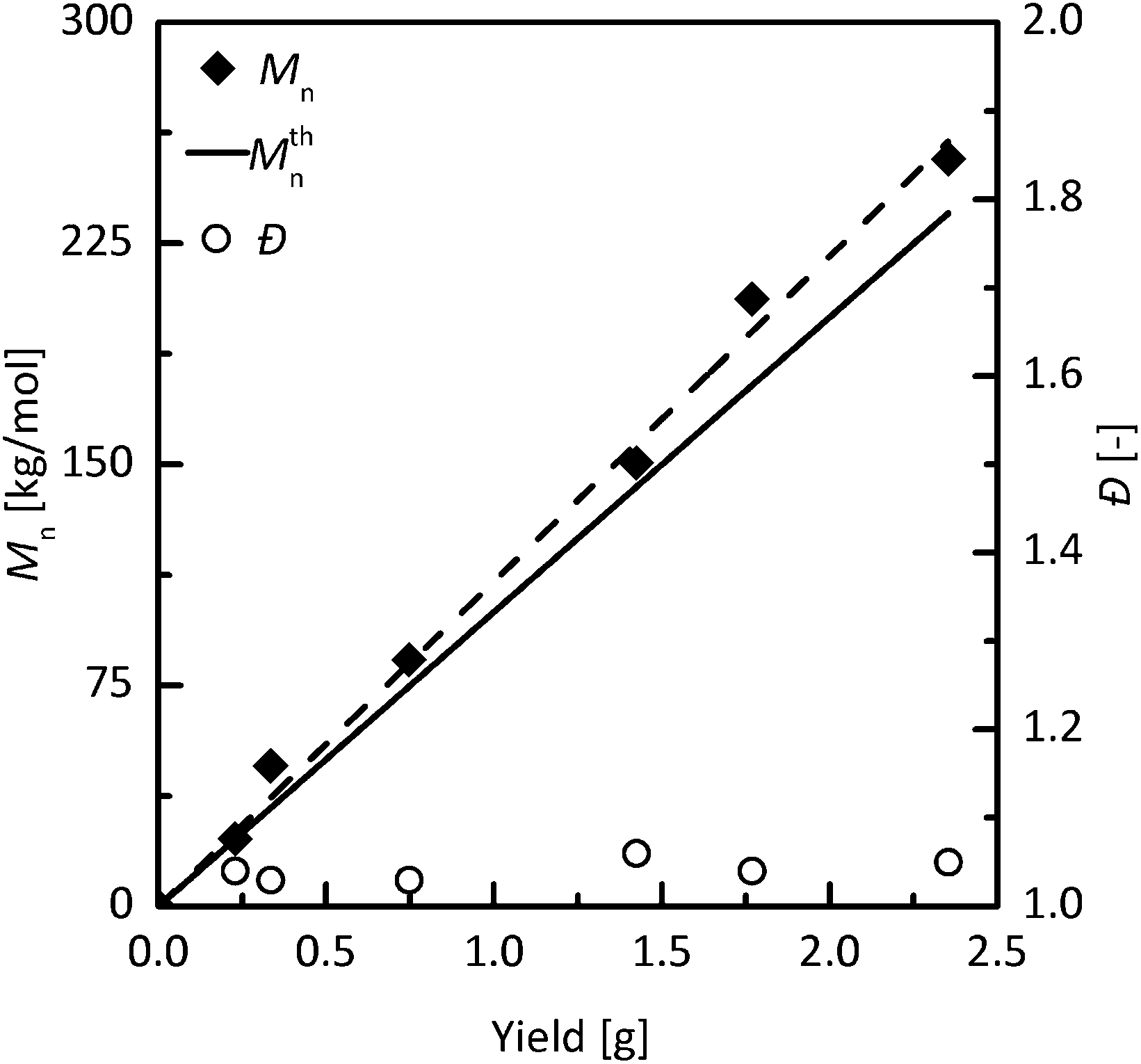 | ||
| Fig. 5 Dependence of polypropylene number average molar mass (diamonds) and the dispersity (circles) on polymer yield obtained in propene polymerization initiated by 6/MAO at −10 °C in toluene. Propene (2 bars), nNi = 10 μmol, [Al]/[Ni] = 200, total volume = 40 ml. | ||
Hex-1-ene polymerization by Ni complexes
Similarly, like for propene polymerization, complexes 3, 4 and 6 afforded poly(hex-1-ene)s with very low molar mass dispersities at −10 °C (Table 4). Molar masses of the prepared poly(hex-1-ene)s are generally higher than the theoretical values as a consequence of the incomplete activation of Ni complexes by MAO as observed earlier.12,18,56,66,67 Complexes 1 and 2 with hydrogen backbone substituents and complex 5 yielded poly(hex-1-ene)s with broadened molar mass distribution indicating that steric hindrance of the axial position of the complex is insufficient for these complexes to protect the metal center from transfer reactions. Branching of polyhexenes can be regulated by ligand bulkiness. Complexes 1 and 2 with the least bulky ligands yielded poly(hex-1-ene) with a number of branches that would be formed if only 1,2-insertion of hex-1-ene takes place, which corresponds well with our previous observation.18 The most rearranged poly(hex-1-ene) was prepared by complex 4. The increase of reaction temperature leads to the loss of livingness of hex-1-ene polymerization as indicated by increased molar mass dispersities as it was observed in the case of propene polymerization (Table 3). Reaction temperature also increased the extent of chain-walking resulting in polyhexenes with more rearranged structures as displayed by lower branching numbers. To investigate the livingness of hex-1-ene polymerization more in detail, we used complex 6. Molar mass of the resulting poly(hex-1-ene) can be easily controlled in a broad range by setting the monomer/Ni ratio at −10 °C (Fig. 6 and Table S1, ESI†). | ||
| Fig. 6 Dependence of number average molar mass (Mn) and the dispersity (Đ) on hex-1-ene/Ni ratio in hex-1-ene polymerization initiated by 6/MAO at −10 °C in toluene. nNi = 10 μmol, Al/Ni = 200, total volume = 10 ml, tp = 5 h. | ||
| Run | Cat. | T [°C] | t p [h] | Yield [%] | TOFa [h−1] | M n [kg mol−1] | Đ [−] |
M
n
thc [kg mol−1] |
T g [°C] | N |
|---|---|---|---|---|---|---|---|---|---|---|
| n Ni = 10 μmol, Al/Ni = 200, total volume = 10 ml, [hex-1-ene] = 0.8 M.a Turnover frequency: mol hex-1-ene/(molNitp).b Molar mass and dispersity determined by SEC RI – PS calib.c Theoretical molar mass, Mnth = mp/nNi.d Glass transition temperature determined by DSC from the 1st heating run.e Number of branches per 1000 C atoms determined by 1H NMR.f nNi = 5 μmol, Al/Ni = 200, total volume = 18 ml, [hex-1-ene] = 0.3 M; n.d. = not determined. | ||||||||||
| 25 | 1 | −10 | 5 | 31 | 45 | 12 | 1.63 | 19 | −60 | 167 |
| 26 | 2 | −10 | 5 | 65 | 100 | 15 | 1.71 | 43 | −58 | 167 |
| 27 | 3 | −10 | 5 | 78 | 140 | 75 | 1.13 | 58 | −57 | 143 |
| 28 | 4 | −10 | 5 | 29 | 50 | 60 | 1.09 | 20 | −54 | 118 |
| 29 | 5 | −10 | 5 | >99 | 160 | 56 | 1.42 | 66 | −56 | 148 |
| 30 | 6 | −10 | 5 | 92 | 240 | 176 | 1.08 | 99 | −51 | 142 |
| 31 | 7 | −10 | 5 | 36 | 60 | 86 | 1.09 | 25 | −54 | 113 |
| 32 | 8 | −10 | 5 | >99 | 170 | 146 | 1.09 | 62 | −50 | 139 |
| 33f | 1 | 25 | 24 | 16 | 10 | 11 | 1.59 | 15 | −64 | 135 |
| 34f | 2 | 25 | 24 | 20 | 10 | 10 | 2.09 | 17 | −61 | 123 |
| 35f | 3 | 25 | 24 | 49 | 20 | 64 | 1.76 | 43 | −52 | 98 |
| 36f | 4 | 25 | 24 | 84 | 40 | 154 | 1.39 | 79 | −50 | 90 |
| 37f | 5 | 25 | 24 | 73 | 30 | 50 | 1.76 | 64 | −55 | 102 |
| 38f | 6 | 25 | 24 | 94 | 45 | 150 | 1.61 | 89 | −52 | 110 |
| 39f | 7 | 25 | 24 | 74 | 35 | 235 | 1.40 | 95 | −53 | 98 |
| 40f | 8 | 25 | 24 | 92 | 40 | 166 | 1.50 | 82 | −53 | 116 |
Another investigation of livingness of hex-1-ene polymerization initiated by 6/MAO was based on a monomer resumption test. As checked by the absence of hex-1-ene in the mixture by GC, an initial feed of hex-1-ene (400 eq. to Ni) was totally consumed in 6 h at −10 °C. After this period, a sample of polymer was taken for SEC analysis, and the same amount of hex-1-ene (400 eq. to Ni) was added to the reaction mixture. After an additional 16 h, the conversion of second monomer feed to polymer was over 99% and the molar mass of the polymer was doubled while dispersity remained low (Fig. 7). Moreover, hex-1-ene polymerization controllability by 6/MAO was further investigated by detailed kinetic studies and compared to the non-fluorinated system 8/MAO. Controlled polymerization behavior was proved by linear molar mass development with monomer conversion for both systems at −10 °C (Fig. 8A). A small increase of dispersity values can be ascribed to the contamination of reaction feed during the withdrawal of samples for conversion and molar mass determination rather than to the imperfection of polymerization as indicated by very low dispersity values achieved after the same reaction time in independent experiments (Table 4, run 30 or 32). The linear first-order plot finally demonstrated polymerization controllability for hex-1-ene polymerization catalyzed by 6 and 8/MAO at −10 °C (Fig. 8B). Both catalytic systems exhibited very similar polymerization behavior at −10 °C, showing that para-aryl fluorinated substituents do not interfere with the livingness of polymerization.
 | ||
| Fig. 7 SEC-RI chromatograms of polyhexenes obtained by 6/MAO after 6 h of polymerization (dash) and after reinitiation by the addition of the second portion of monomer after additional 16 h of polymerization (solid). T = −10 °C, [hex-1-ene] = 0.4 M + 0.4 M, nNi = 10 μmol, [Al]/[Ni] = 200, total volume = 10 ml. | ||
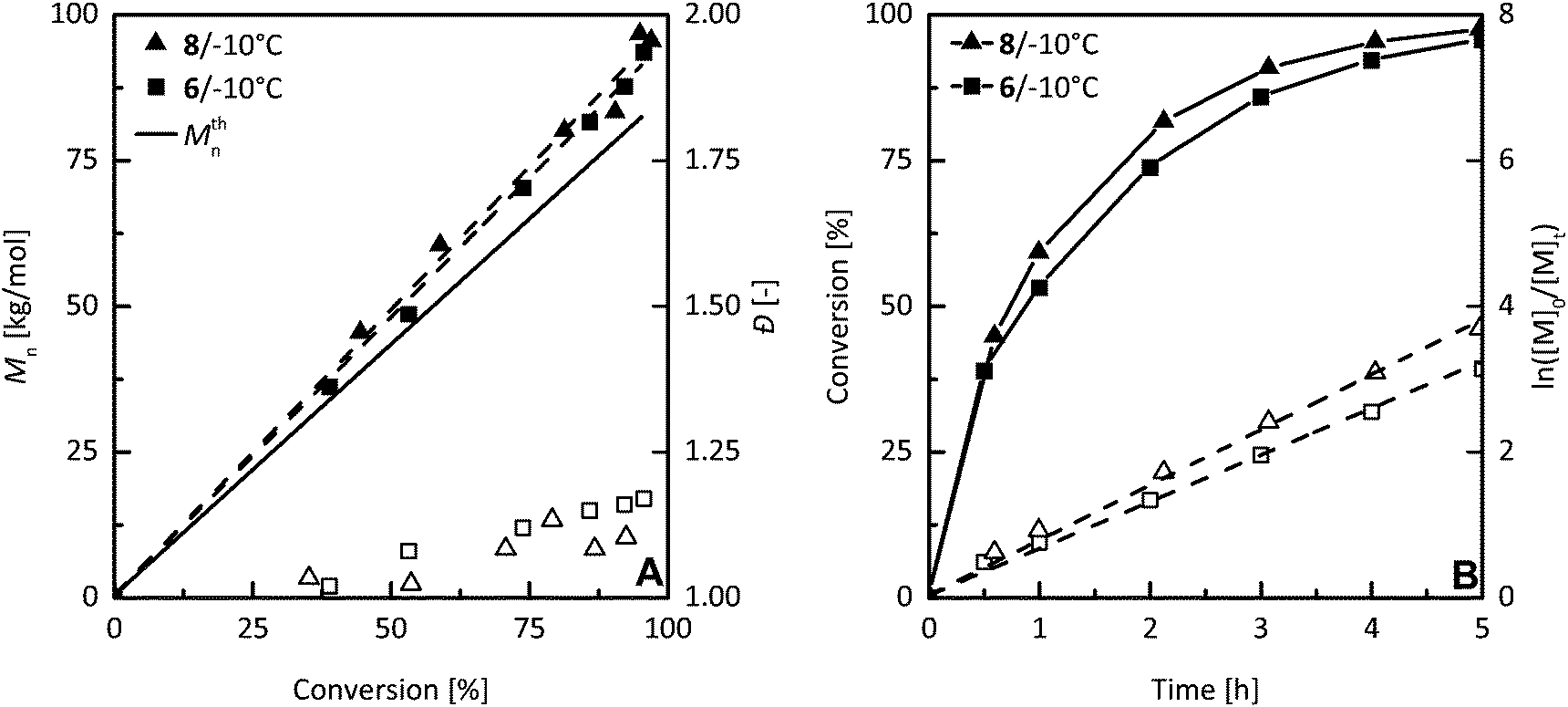 | ||
| Fig. 8 Dependence of number average molar mass (full symbols) and the dispersity (empty symbols) on monomer conversion to polymer (A) and monomer conversion to the polymer (full symbols) and semilogarithmic dependence of ln([M0]/[M]t) (empty symbols) on time (B) in hex-1-ene polymerization initiated by 6/MAO and 8/MAO at −10 °C in toluene. [Hex-1-ene] = 0.8 M, nNi = 10 μmol, [Al]/[Ni] = 200, total volume = 10 ml. | ||
Fluoro substituents were often reported to enhance the thermal stability of the catalyst.28–30 Therefore, we investigated the livingness of the polymerization and lifetimes of fluorinated complex 6 and its non-fluorinated analog 8 at 50 °C (Fig. S7 and S8, ESI†). The increase of molar mass with conversion for 6/MAO displayed only slightly higher slope than that with experiments at −10 °C. In contrast, non-fluorinated complex 8/MAO exhibited a much higher deviation of molar masses from theoretical values. This means that fluorinated complex 6 is efficiently activated by MAO both at −10 and 50 °C whereas the activation efficiency of 8 by MAO at 50 °C is significantly decreased. However, first-order plots of complexes 6 and 8 clearly deviate from linearity, showing the termination of growing centers. Thus, the thermal stability of nickel diimine complexes is not influenced significantly by para fluoroalkyl substituents, which cannot interact directly with the metal center and stabilize it in contrast to fluoro substituents in ortho-aryl positions.28–30
Ethene polymerization by Pd complexes
Two novel palladium complexes 9 and 10 (Fig. 2), bearing fluorinated para-aryl substituents, were tested in ethene polymerization and compared with basic catalyst 12 and previously published complexes 11, bearing an electron-accepting chloro substituent in the para position26 (Fig. 2). Complexes 9–12 were dissolved in chlorobenzene and activated in situ by the addition of 2 equivalents of NaBArF4.Polymerizations were conducted at 0 °C to suppress side reactions and maintain the living character of polymerization (Table 5). Different ethene pressures (0.05 vs. 8 bars) were used to vary the degree of the chain-walking mechanism which is also known to influence the topology of the resulting polyethylenes.4 Ethene polymerizations showed living character with narrow molar mass dispersities (Đ < 1.1) for all tested complexes at both pressures. Polymerizations were also well controlled with fast and efficient catalyst activation which was demonstrated by similar values of experimental and theoretical molar masses. The only exception from controlled behavior was complex 10 (Table 5, run 46) which provided polyethylene with slightly broader dispersity (Đ = 1.4) at low ethene pressure. The reason probably lies in different backbone substituents of 10 (1,8-naphthalenediyl) compared to other complexes bearing methyl backbone substituents. A structural difference of 10 is also pronounced in the lowest molar masses and activities (Table 5, runs 42 and 46) which is in agreement with previously published mechanistic data.65 Polymerizations with complexes 9 and 11, substituted by electron-withdrawing fluoroalkyl and chloro groups, at high ethene pressure showed comparable polymerization activities, PE molar masses, and distributions as the unsubstituted complex 12, which was used as a benchmark. Different polymerization results were observed for low ethene pressure experiments (Table 5, runs 45–48). Whereas complexes 9 and 11 were again comparable, the reference non-fluorinated complex 12 showed approximately 2 times higher values of activity and molar mass (Table 5, compare runs 45 and 47 with run 48). This difference was probably caused by the presence of electron-withdrawing substituents that are known to decrease the activity and molar masses in ethene polymerization catalyzed by Pd diimine catalysts.26 The reversed relative order of activities of 9 and 11versus12 at low and high ethylene pressures are probably caused by difference in the trapping rate of ethylene to metal centers. Ethylene trapping forms alkyl-ethylene complexes which are the resting state species in the catalytic cycle.65 At high pressure, trapping is very fast and does not limit the polymerization rate, which is roughly the same for all complexes as the electronic effect of chloro and fluoroalkyl substituents is not very strong. On the other hand, at very high ethylene dilution, the activity is probably influenced significantly by different abilities of complexes to trap ethylene under these conditions. Thus, slower trapping by 9 and 11 results in their lower polymerization rates compared to 12. Branching numbers N of all polyethylenes synthesized by Pd complexes were consistent and were about the value of 100 branches per 1000 C atoms. It is known that PEs with similarly high branching densities can display different topologies, ranging from linear to dendritic ones.4 To evaluate the topology of the prepared polymers, we used a simple experimental parameter MRIw/MLSw, which can be obtained from a single measurement by size-exclusion chromatography coupled with a refractometric (RI) and light-scattering (LS) detector. Similarly to the values of branching ratios g and g′,68–70 the ratio MRIw/MLSw decreases with an increasing degree of branching and compactness of the polymer coil.71,72 Polymerizations that were conducted at high ethene pressure exhibited MRIw/MLSw values over 1 (Table 5, runs 41–44) which suggested linear topology as was expected concerning the reaction conditions.4 On the other hand, the decrease of ethene pressure resulted in the increase of the chain-walking mechanism4,32 which was demonstrated by the MRIw/MLSw values reaching 0.5 (Table 5, runs 45–48). To further investigate the topology of prepared PEs by a more fundamental approach, we constructed Mark–Houwink (MH) plots and calculated other topological parameters (g′ and viscosity ratio) from data obtained by using a HT SEC-IR-viscometer in trichlorobenzene at 150 °C. PE samples prepared by Pd catalysts 9 and 10 were investigated and compared to PEs prepared by α-diimine Pd catalysts 11 and 12 that were previously studied by Guan.26,27 PEs prepared at high ethene pressure (runs 41–44) displayed MH plots that lie close to the linear HDPE reference (Fig. 9). These observations are in agreement with the values of topological parameters g′ ∼ 0.5 and viscosity ratio ∼2 which suggests moderately branched to linear topology (Table 5, runs 41–44).
 | ||
| Fig. 9 Mark–Houwink plot of PEs prepared by Pd complexes. | ||
| Run | Cat. | p C2 [bar] | t [h] | TOFa [h−1] | M n [kg mol−1] | Đ [−] |
M
n
thc [kg mol−1] |
M
RIw/MLSwd |
N | g′f | Viscosity ratio |
|---|---|---|---|---|---|---|---|---|---|---|---|
| c Pd = 0.5 μmol ml−1, 2 eq. NaBArF4, total volume = 15 ml, T = 0 °C.a Turnover frequency: mol ethene/(molPdtp).b Molar mass and dispersity determined by SEC-MALS in THF at 35 °C.c Theoretical molar mass, Mnth = mp/nPd.d Ratio of weight average molar masses obtained from relative PS calibration and LS detection.e Number of branches per 1000 C atoms determined by 1H NMR.f Obtained from HT SEC-IR-η in 1,2,4-trichlorobenzene at 150 °C. | |||||||||||
| 41 | 9 | 8 | 5 | 400 | 56 | 1.01 | 56 | 1.25 | 95 | 0.53 | 1.8 |
| 42 | 10 | 8 | 5 | 190 | 12 | 1.04 | 19 | 1.34 | 109 | 0.42 | 1.9 |
| 43 | 11 | 8 | 5 | 520 | 71 | 1.04 | 72 | 1.21 | 99 | 0.53 | 1.8 |
| 44 | 12 | 8 | 5 | 380 | 51 | 1.04 | 53 | 1.20 | 101 | 0.49 | 1.8 |
| 45 | 9 | 0.05 | 21 | 120 | 80 | 1.04 | 69 | 0.59 | 107 | 0.12 | 4.3 |
| 46 | 10 | 0.05 | 21 | 70 | 23 | 1.42 | 40 | 0.79 | 111 | 0.16 | 3.3 |
| 47 | 11 | 0.05 | 21 | 100 | 60 | 1.08 | 60 | 0.59 | 113 | 0.10 | 4.7 |
| 48 | 12 | 0.05 | 21 | 260 | 159 | 1.03 | 150 | 0.54 | 96 | 0.16 | 3.7 |
In contrast, PEs prepared at low ethene pressure (0.05 bar) showed a significant shift of their MH plots towards lower values of intrinsic viscosity (runs 45–48, Fig. 9), suggesting more branched topology of these PE samples. Topological parameter g′ as low as 0.10–0.15 as well as viscosity ratio values ∼4 (Table 5, runs 45–48) clearly indicate dendritic topology of PEs prepared by new fluorinated complexes 9 and 10.
Conclusions
Novel α-diimine nickel and palladium complexes with fluorinated substituents were successfully synthesized and tested in olefin polymerization. Nickel complexes with a bulky backbone and ortho-aryl substituents polymerized propene and hex-1-ene in a controlled manner at −10 °C whereas palladium complexes allowed controlled ethylene polymerization even at 0 °C. In propene polymerization chain-walking was supported at −10 °C when fluorinated alkyl Ni derivatives were used, supporting the observation in the literature.26 However, ethylene polymerization catalyzed by Pd complexes showed that fluorinated alkyl substituents and chloro group in a para position of ligand aryl rings don't have an essential impact on PE branching (chain-walking), suggesting that these substituents have an electroneutral character. The degree of chain-walking extent was more significantly influenced by the structure of backbone substituents (R1) decreasing in the order – Me, An and H. The topology of PEs prepared by Pd catalysts can be efficiently tuned by the variation of reaction conditions and catalyst structure from linear to dendritic one.The insolubility of most of the novel Ni and Pd complexes in fluorinated solvents shows their insufficient fluorophilicity needed to perform interphase olefin polymerization, which could provide polyolefins with narrow molar mass and simultaneously controlled particle morphology. Solubility results of PdMeCl complexes suggest which structural features are important for the design of complexes with better solubility in fluorinated solvents. Prepared fluoroalkyl substituted palladium complexes, tolerant for polar groups, might also be of interest for the application in scCO2. Our future intention is to increase catalyst fluorophilicity by additional ligand substitution by perfluoroalkyl groups.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in the frame of a joint project by Czech Science Foundation (No. 15-15887J) and Deutsche Forschungsgemeinschaft (No. LE 1424/7-1) and from specific university research (MSMT No. 20/2017). Albena Lederer is kindly acknowledged for fruitful discussion.Notes and references
- L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414–6415 CrossRef CAS.
- S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169–1204 CrossRef CAS PubMed.
- V. C. Gibson and S. K. Spitzmesser, Chem. Rev., 2003, 103, 283–316 CrossRef CAS PubMed.
- Z. Guan, P. M. Cotts, E. F. McCord and S. J. McLain, Science, 1999, 283, 2059–2062 CrossRef CAS PubMed.
- E. F. McCord, S. J. McLain, L. T. J. Nelson, S. D. Ittel, D. Tempel, C. M. Killian, L. K. Johnson and M. Brookhart, Macromolecules, 2007, 40, 410–420 CrossRef CAS.
- J. M. Rose, A. E. Cherian and G. W. Coates, J. Am. Chem. Soc., 2006, 128, 4186–4187 CrossRef CAS PubMed.
- C. M. Killian, D. J. Tempel, L. K. Johnson and M. Brookhart, J. Am. Chem. Soc., 1996, 118, 11664–11665 CrossRef CAS.
- A. C. Gottfried and M. Brookhart, Macromolecules, 2001, 34, 1140–1142 CrossRef CAS.
- A. C. Gottfried and M. Brookhart, Macromolecules, 2003, 36, 3085–3100 CrossRef.
- A. E. Cherian, J. M. Rose, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 13770–13771 CrossRef CAS PubMed.
- J. M. Rose, F. Deplace, N. A. Lynd, Z. Wang, A. Hotta, E. B. Lobkovsky, E. J. Kramer and G. W. Coates, Macromolecules, 2008, 41, 9548–9555 CrossRef CAS.
- J. Merna, Z. Hošt’álek, J. Peleška and J. Roda, Polymer, 2009, 50, 5016–5023 CrossRef CAS.
- J. Peleška, Z. Hoštálek, D. Hasalíková and J. Merna, Polymer, 2011, 52, 275–281 CrossRef.
- G. Leone, M. Mauri, F. Bertini, M. Canetti, D. Piovani and G. Ricci, Macromolecules, 2015, 48, 1304–1312 CrossRef CAS.
- D. Zhang, E. T. Nadres, M. Brookhart and O. Daugulis, Organometallics, 2013, 32, 5136–5143 CrossRef CAS.
- D. H. Camacho and Z. Guan, Macromolecules, 2005, 38, 2544–2546 CrossRef CAS.
- D. H. Camacho, E. V. Salo, J. W. Ziller and Z. Guan, Angew. Chem., Int. Ed., 2004, 43, 1821–1825 CrossRef CAS PubMed.
- A. Sokolohorskyj, R. Mundil, A. Lederer and J. Merna, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3193–3202 CrossRef CAS.
- X. Wang, L. Fan, Y. Ma, C.-Y. Guo, G. A. Solan, Y. Sun and W.-H. Sun, Polym. Chem., 2017, 8, 2785–2795 RSC.
- X. Sui, C. Hong, W. Pang and C. Chen, Mater. Chem. Front., 2017, 1, 967–972 RSC.
- D. Meinhard and B. Rieger, Chem. – Asian J., 2007, 2, 386–392 CrossRef CAS PubMed.
- D. Pappalardo, M. Mazzeo, S. Antinucci and C. Pellecchia, Macromolecules, 2000, 33, 9483–9487 CrossRef CAS.
- H. Gao, H. Hu, F. Zhu and Q. Wu, Chem. Commun., 2012, 48, 3312–3314 RSC.
- H. Hu, L. Zhang, H. Gao, F. Zhu and Q. Wu, Chem. – Eur. J., 2014, 20, 3225–3233 CrossRef CAS PubMed.
- H. Hu, D. Chen, H. Gao, L. Zhong and Q. Wu, Polym. Chem., 2016, 7, 529–537 RSC.
- C. Popeney and Z. Guan, Organometallics, 2005, 24, 1145–1155 CrossRef CAS.
- C. S. Popeney and Z. Guan, Macromolecules, 2010, 43, 4091–4097 CrossRef CAS.
- C. S. Popeney, A. L. Rheingold and Z. Guan, Organometallics, 2009, 28, 4452–4463 CrossRef CAS.
- S. Ahmadjo, S. Damavandi, G. H. Zohuri, A. Farhadipour, N. Samadieh and Z. Etemadinia, Polym. Bull., 2017, 74, 3819–3832 CrossRef CAS.
- S. Ahmadjo, S. Damavandi, G. H. Zohuri, A. Farhadipour and Z. Etemadinia, J. Organomet. Chem., 2017, 835, 43–51 CrossRef CAS.
- X. Wang, L. Fan, Y. Yuan, S. Du, Y. Sun, G. A. Solan, C.-Y. Guo and W.-H. Sun, Dalton Trans., 2016, 45, 18313–18323 RSC.
- D. P. Gates, S. A. Svejda, E. Oñate, C. M. Killian, L. K. Johnson, P. S. White and M. Brookhart, Macromolecules, 2000, 33, 2320–2334 CrossRef CAS.
- A. Bastero, G. Franciò, W. Leitner and S. Mecking, Chem. – Eur. J., 2006, 12, 6110–6116 CrossRef CAS PubMed.
- D. Guironnet, T. Friedberger and S. Mecking, Dalton Trans., 2009, 8929–8934, 10.1039/B912883B.
- W. Zhang, S. Wang, S. Du, C.-Y. Guo, X. Hao and W.-H. Sun, Macromol. Chem. Phys., 2014, 215, 1797–1809 CrossRef CAS.
- J. Hošek, M. Rybáčková, J. Čejka, J. Cvačka and J. Kvíčala, Organometallics, 2015, 34, 3327–3334 CrossRef.
- 2002.
- R. Wickbold, Angew. Chem., 1954, 66, 173–174 CrossRef CAS.
- V. P. Fadeeva, V. D. Tikhova and O. N. Nikulicheva, J. Anal. Chem., 2008, 63, 1094–1106 CrossRef CAS.
- P. d. Voogt and M. Sáez, TrAC, Trends Anal. Chem., 2006, 25, 326–342 CrossRef.
- Q. Liu, W. Zhang, D. Jia, X. Hao, C. Redshaw and W.-H. Sun, Appl. Catal., A, 2014, 475, 195–202 CrossRef CAS.
- T. Bříza, J. Kvíčala, O. Paleta and J. Čermák, Tetrahedron, 2002, 58, 3841–3846 CrossRef.
- J. Kvíčala, T. Bříza, O. Paleta, K. Auerová and J. Čermák, Tetrahedron, 2002, 58, 3847–3854 CrossRef.
- P. Dudziński, A. V. Matsnev, J. S. Thrasher and G. Haufe, Org. Lett., 2015, 17, 1078–1081 CrossRef PubMed.
- S. Xie, S. A. Lopez, O. Ramström, M. Yan and K. N. Houk, J. Am. Chem. Soc., 2015, 137, 2958–2966 CrossRef CAS PubMed.
- N. Morlanés, K. S. Joya, K. Takanabe and V. Rodionov, Eur. J. Inorg. Chem., 2015, 2015, 49–52 CrossRef.
- M. Sha, R. Pan, P. Xing and B. Jiang, J. Fluorine Chem., 2015, 169, 61–65 CrossRef CAS.
- B. Zhang and A. Studer, Org. Lett., 2014, 16, 3990–3993 CrossRef CAS PubMed.
- M. Enders, B. Görling, A. B. Braun, J. E. Seltenreich, L. F. Reichenbach, K. Rissanen, M. Nieger, B. Luy, U. Schepers and S. Bräse, Organometallics, 2014, 33, 4027–4034 CrossRef CAS.
- Y. Sun, H. Sun, J. Jia, A. Du and X. Li, Organometallics, 2014, 33, 1079–1081 CrossRef CAS.
- E. M. Sletten and T. M. Swager, J. Am. Chem. Soc., 2014, 136, 13574–13577 CrossRef CAS PubMed.
- B. Wunderlich, Thermal analysis, Elsevier, 1990 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- A. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- U. Subramanyam and S. Sivaram, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1093–1100 CrossRef CAS.
- L. Strekowski, S.-Y. Lin, H. Lee and J. C. Mason, Tetrahedron Lett., 1996, 37, 4655–4658 CrossRef CAS.
- J. Yuan, Y. Mu, J. Zhao, W. Xu, J. Chen and Z. Zhang, J. Organomet. Chem., 2014, 761, 32–41 CrossRef CAS.
- R. J. Maldanis, J. S. Wood, A. Chandrasekaran, M. D. Rausch and J. C. W. Chien, J. Organomet. Chem., 2002, 645, 158–167 CrossRef CAS.
- J. O. Liimatta, B. Löfgren, M. Miettinen, M. Ahlgren, M. Haukka and T. T. Pakkanen, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1426–1434 CrossRef CAS.
- H.-R. Liu, P. T. Gomes, S. I. Costa, M. T. Duarte, R. Branquinho, A. C. Fernandes, J. C. W. Chien, R. P. Singh and M. M. Marques, J. Organomet. Chem., 2005, 690, 1314–1323 CrossRef CAS.
- C. S. B. Gomes, P. T. Gomes and M. T. Duarte, J. Organomet. Chem., 2014, 760, 101–107 CrossRef CAS.
- A. Vaughan, D. S. Davis and J. R. Hagadorn, Polymer Science: A Comprehensive Reference, Elsevier, 2012 Search PubMed.
- L. Deng, T. K. Woo, L. Cavallo, P. M. Margl and T. Ziegler, J. Am. Chem. Soc., 1997, 119, 6177–6186 CrossRef CAS.
- D. J. Tempel, L. K. Johnson, R. L. Huff, P. S. White and M. Brookhart, J. Am. Chem. Soc., 2000, 122, 6686–6700 CrossRef CAS.
- F. Zhai and R. F. Jordan, Organometallics, 2017, 36, 2784–2799 CrossRef CAS.
- F. Peruch, H. Cramail and A. Deffieux, Macromolecules, 1999, 32, 7977–7983 CrossRef CAS.
- W. H. Stockmayer and M. Fixman, Ann. N. Y. Acad. Sci., 1953, 57, 334–352 CrossRef CAS.
- B. H. Zimm and W. H. Stockmayer, J. Chem. Phys., 1949, 17, 1301–1314 CrossRef CAS.
- B. I. Voit and A. Lederer, Chem. Rev., 2009, 109, 5924–5973 CrossRef CAS PubMed.
- S. Podzimek, Light Scattering, Size Exclusion Chromatography and Asymmetric Flow Field Flow Fractionation, Wiley, 2011 Search PubMed.
- A. M. Striegel, W. W. Yau, J. J. Kirkland and D. D. Bly, Modern Size-Exclusion Liquid Chromatography: Practice of Gel Permeation and Gel Filtration Chromatography, Wiley, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Polymer preparation and characterization data and crystallographic data of new complexes. CCDC 1810825 and 1810826. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8py00201k |
| This journal is © The Royal Society of Chemistry 2018 |