
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInversion of crystallization rates in miscible block copolymers of poly(lactide)-block-poly(2-isopropyl-2-oxazoline)†
Fabian
Pooch
 a,
Marjolein
Sliepen
a,
Kirsi J.
Svedström
b,
Antti
Korpi
c,
Françoise M.
Winnik
ade and
Heikki
Tenhu
*a
a,
Marjolein
Sliepen
a,
Kirsi J.
Svedström
b,
Antti
Korpi
c,
Françoise M.
Winnik
ade and
Heikki
Tenhu
*a
aDepartment of Chemistry, University of Helsinki, P.O. Box 55, Helsinki 00014, Finland. E-mail: heikki.tenhu@helsinki.fi
bDepartment of Physics, University of Helsinki, P.O. Box 64, Helsinki 00014, Finland
cDepartment of Bioproducts and Biosystems, Aalto University, P.O. Box 16100, Aalto 00076, Finland
dWPI International Centre for Nanoarchitectonics (MANA), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba 305-0044, Japan
eDepartement de Chimie, Universite de Montreal, CP 6128 Succursale Centre Ville, Montreal QC H3C 3J7, Canada
First published on 5th March 2018
Abstract
Miscible block copolymers (BCPs) are rarely studied. When one or both components of such BCPs are semi-crystalline polymers, strong effects on the crystallization behavior can be expected. We present a study of 18 miscible BCPs comprised of poly(lactide) (PLLA, semi-crystalline and PDLLA, amorphous) and poly(2-isopropyl-2-oxazoline) (PiPOx, semi-crystalline) with PiPOx volume fractions of 0.14 < ϕPiPOx < 0.82. All BCPs exhibit a single glass transition and form a homogeneous melt. Mixing has a plasticizing effect on PiPOx and increases its crystallization rates (DSC). In contrast, the crystallization rates of PLLA are dramatically reduced, or in most cases entirely prevented. During isothermal crystallization at 130 °C, the crystallization rates of the BCPs were inverted in comparison with those of the parent homopolymers. Crystallization drives the BCPs to phase separate and the formed crystalline structure is that of the parent homopolymers. The fast crystallization of PiPOx confines the observed superstructure. The BCPs were studied on multiple length scales – from the atomic level (WAXS, IR spectroscopy) to the meso level (AFM, SAXS) and the macroscopic superstructure (polarized optical microscopy). A mechanism of the structure evolution is presented.
Introduction
The majority of polymer–polymer mixtures, as blends, linear block copolymers (BCPs) or of more complex architectures are characterized by (micro)phase separation. The two components form a two-phase material. Considerably less prominent are cases in which the two components form a one-phase material, i.e. they are miscible.1Thermodynamics demands a negative Gibbs free energy of mixing ΔGmix for spontaneous mixing of two components.
| ΔGmix = ΔHmix − TΔSmix < 0 | (1) |
In eqn (1), ΔHmix and ΔSmix are the enthalpy and entropy of mixing, respectively, and T is the absolute temperature. For polymer blends, the Gibbs free energy of mixing is given by the famous Flory–Huggins equation:2
 | (2) |
Here, R is the universal gas constant, ν is the total volume, ϕA,B, nA,B, and υA,B are the volume fractions, moles and molar volume of polymers A and B, respectively, nc and υc are the moles and volume of the reference units, respectively, and χ is the Flory–Huggins interaction parameter. χ involves the excess energy of the nearest neighbor interactions and is negative for specific attractive interactions. The logarithmic terms in eqn (2) are the combinatorial entropies. They are always negative (ϕA + ϕB = 1) but for polymers with large molar volumes these terms are negligible. Thus, polymer blends are miscible (ΔGmix < 0) with sufficiently low molar volumes (degree of polymerization) and/or a vanishing (athermal) or negative (attractive) χ-parameter. The thermodynamic criterion for the phase separation of BCPs is the product χN of the interaction parameter and the degree of polymerization.3 If this product exceeds a critical value, phase separation occurs via spinodal decomposition. BCPs require a higher critical χN to (micro)phase separate compared to polymer blends.
In practice, miscibility depends on the length scale of investigation. Different methods have a different resolution on the level of molecular mixing. The most often applied criterion – the appearance of a single glass transition temperature (Tg) – indicates homogeneity on the level of segmental movements (i.e. no domains larger than 15 nm).4 However, intermolecular forces might lead to homogeneity below that limit and are probed with spectroscopic techniques. Such attractions can be caused by H-bonding,5 dipole–dipole6 or acid–base7 interactions.
From a material point of view, miscibility of polymers provides the opportunity to create properties unavailable in the individual one-component systems.8 Through mixing, the thermal, mechanical, rheological, crystallization and degradation properties can be tuned according to the requirements.9–14 Understanding and controlling the structural evolution through a variable temperature range is essential for the application of the materials.
Poly(lactide) (PLA) is a biodegradable, linear polyester derived from renewable resources. It is a promising alternative to many petroleum-based commodity plastics and entered the market of medical applications.8,15–17 Despite its many positive features, its poor mechanical properties as a pure homopolymer have to be overcome by blending with additives and other polymers.18 Copolymerization, either randomly or in the form of BCPs, is another option to modify the properties of PLA.8,19,20 PLA is produced by ring-opening polymerization (ROP) of lactide (LA), the cyclic diester of lactic acid, which contains two chiral centers and thus exists as three stereo-isomers (L-LA, D-LA and DL-LA). Polymers of DL-LA are amorphous. L- and D-lactide yield semi-crystalline polymers with a melting point Tm from 150 to 160 °C.21
Poly(2-alkyl-2-oxazolines) (POx) are synthetic pseudopeptides with a –(CH2–CH2–N)n– backbone (Scheme 1).22–25 The backbone is non-chiral and the amide-group provides coplanar side-groups. In an extended chain conformation, subsequent side-groups are alternating laterally to either side of the backbone.26 Amide-dipole orientation facilitates chain packing and crystallization.27 The crystallization behaviour of POx was studied with systematic variation of the alkyl-chain length,23,28 but little is known on the effect of the backbone chain length, i.e. the degree of polymerization. POx have been viewed as polymeric analogues of universal solvents such as dimethyl formamide and dimethyl acetamide and found miscible with various commodity plastics (poly(vinyl chloride), polystyrene and poly(vinylidene fluoride)).29,30
 | ||
| Scheme 1 Synthesis of azide-terminated PiPOx, propargyl-terminated PLA and PLA-PiPOx BCPs by CuAAC. | ||
We report here the crystallization behaviour of miscible A-B-type BCPs of PLA (PDLLA and PLLA) and poly(2-isopropyl-2-oxazoline) (PiPOx). PiPOx can crystallize in bulk, but only upon heating to temperatures of around 130 °C to overcome kinetic barriers and provide enough chain mobility.31 We focus first on the theoretical considerations of PLA and PiPOx miscibility and its experimental verification. Then the implications of mixing on the crystallization behavior of the BCPs and the structural evolution are discussed.
Experimental
Materials
L-Lactide (98%, ABCR) and DL-lactide (98%, ABCR) were recrystallized from dry toluene twice prior to use. Sn(Oct)2 (95%, Sigma Aldrich) was kept over molecular sieves (4 Å). Propargyl alcohol (99%, Sigma Aldrich), methyl triflate (98%, Alfa Aesar) and N,N,N′,N″,N″-pentamethyldiethylenetriamine (PMDETA, 99%, Sigma Aldrich) were distilled over CaH2. 2-Isopropyl-2-oxazoline (iPOx) was prepared from isobutyronitrile (99%, Alfa Aesar) and 2-aminoethanol (>99%, Sigma Aldrich) using zinc acetate (anhydrous, 99.9%, Alfa Aesar) as a catalyst as described previously.32 A deuterated solvent for NMR spectroscopy (Euriso-Top, chloroform-d, CDCl3, 99.8% D + 0.03% tetramethylsilane, TMS) was used as received. All other chemicals were used as received.Synthesis
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–), 2.66 (s, 1H per chain, –CH–OH) 4.35 (q, 1H per chain, –CH–OH), 4.72 (m, 1H per chain, –CH2–), 5.16 (q, 1H per lactide unit, –CH–) ppm.
C–), 2.70 (d, 1H per chain, –CH–OH) 4.35 (m, 1H per chain, –CH–OH), 4.72 (m, 1H per chain, –CH2–), 5.18 (m, 1H per lactide unit, –CH–) ppm.
C–), 2.66 (s, 1H per chain, –CH–OH) 4.35 (q, 1H per chain, –CH–OH), 4.72 (m, 1H per chain, –CH2–), 5.16 (q, 1H per lactide unit, –CH–) ppm.
C–), 2.70 (d, 1H per chain, –CH–OH) 4.35 (m, 1H per chain, –CH–OH), 4.72 (m, 1H per chain, –CH2–), 5.18 (m, 1H per lactide unit, –CH–) ppm.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 680 rpm, 30 min). 1H-NMR (500 MHz, CDCl3, δ): 1.11 (s, 6H per oxazoline unit, –CH–(CH3)2), 1.57 (d, 3H per lactide unit, –CH–CH3), 2.52–3.00 (broad, 1H per oxazoline unit, –CH–(CH3)2), 3.06 (s, 3H per chain, CH3–N–), 3.16 (broad, 2H per chain, –CH2–CH2–triazole) 3.49 (broad, 4H per oxazoline unit, –CH2–CH2–), 3.87 (broad, 2H per chain, –CH2–CH2–triazole), 4.38 (q, 1H per chain, –CH–OH), 4.62 (s, 2H per chain,
680 rpm, 30 min). 1H-NMR (500 MHz, CDCl3, δ): 1.11 (s, 6H per oxazoline unit, –CH–(CH3)2), 1.57 (d, 3H per lactide unit, –CH–CH3), 2.52–3.00 (broad, 1H per oxazoline unit, –CH–(CH3)2), 3.06 (s, 3H per chain, CH3–N–), 3.16 (broad, 2H per chain, –CH2–CH2–triazole) 3.49 (broad, 4H per oxazoline unit, –CH2–CH2–), 3.87 (broad, 2H per chain, –CH2–CH2–triazole), 4.38 (q, 1H per chain, –CH–OH), 4.62 (s, 2H per chain, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–CH2–O–), 5.19 (q, 1H per lactide unit, –CH–), 7.60 (m, 1H per chain, –triazole–CH) ppm.
C–CH2–O–), 5.19 (q, 1H per lactide unit, –CH–), 7.60 (m, 1H per chain, –triazole–CH) ppm.
Characterization
All the recorded 2D scattering patterns of the samples were isotropic. The intensities were integrated radially and averaged azimuthally from a 110°-wide sector. The data were corrected for read-out noise of the detector, background scattering (measured with an empty aluminum ring with two Mylar-foils), polarization, and absorption.
The scattering intensities are given as a function of the scattering angle (2θ), which is related to the length of the scattering vector (q) as:  , where λ is the wavelength of the X-rays. The q-scale was calibrated and the instrumental broadening (Δ) of the reflections was determined using lanthanum hexaboride (LaB6). The d-spacing i.e. the distance between the lattice planes hkl (dhkl) was determined from the position of the corresponding reflection at the q-axis as:
, where λ is the wavelength of the X-rays. The q-scale was calibrated and the instrumental broadening (Δ) of the reflections was determined using lanthanum hexaboride (LaB6). The d-spacing i.e. the distance between the lattice planes hkl (dhkl) was determined from the position of the corresponding reflection at the q-axis as:  .
.
Samples were placed in the sample chamber, which was then depressurized. The slugs were heated to 215 °C by heating the sample holder, which heated the steel combs holding the slugs by metal-to-metal connection, leading the slugs to be in immediate contact with the 215 °C metal from both sides. The samples were left in the elevated temperature for 10 minutes to ensure sufficient heat transfer in the vacuum of the sample chamber, and SAXS data were recorded from the molten samples.
The SAXS measurement for the sample 2L3 at room temperature was conducted using the same set-up as in all the WAXS experiments, except including the following changes: a Bruker HI-Star area detector was used as a detector and a vacuum tube was placed between the sample and the detector. The SAXS data were spherically averaged. The q scale was obtained using the silver behenate calibration sample and background was measured using an empty aluminium ring with Mylar foils.
Results and discussion
A library of 18 PLA-PiPOx BCPs was synthesized by copper catalyzed azide–alkyne cyclo-addition (CuAAC, Scheme 1). Three azide-terminated PiPOx homopolymers were covalently bound to each of the six alkyne-terminated PLA homopolymers (Table 1). This synthetic strategy has the clear advantage that all homopolymers are available for individual characterization and blending. Also, the BCP series with a constant first and variable second block exist with both PiPOx and PLA as the constant block. Thus, the properties of the BCPs can be closely related to their parent homopolymers and systematic variations can be studied. The PiPOx volume fractions of the BCPs ranged from 14 to 82%.| Polymera |
M
n,SECb (disp.) |
M
n,MALDIb |
M
n,BCPc |
ϕ
PiPOxd |
χN |
|
|---|---|---|---|---|---|---|
| a Nomenclature BCPs: “2DL3” = “PiPOx2-block-PDLLA3”. b In kg mol−1, PS calibration, THF as an eluent. c Absolute molecular weight based on MALDI and 1H-NMR in kg mol−1. d Calculated for νPiPOx = 92.3 mL mol and νPLA = 58.1 mL mol and based on the molar ratio of repeating units from 1H-NMR spectra. e N was calculated by relating the molar ratio of repeating units from 1H-NMR to the Mn,MALDI of PiPOx. f In °C, obtained by the Hoffman–Weeks plot (ESI). | ||||||
| PLLA1 | 5.9 (1.11) | — | — | 0 | — | 156 |
| PLLA2 | 10.0 (1.09) | — | — | 0 | — | 178 |
| PLLA3 | 14.6 (1.06) | — | — | 0 | — | 183 |
| PDLLA1 | 4.9 (1.43) | — | — | 0 | — | — |
| PDLLA2 | 9.0 (1.41) | — | — | 0 | — | — |
| PDLLA3 | 17.7 (1.35) | — | — | 0 | — | — |
| PiPOx1 | 2.0 (1.08) | 2.1 | — | 1 | — | 149 |
| 1L1 | 8.1 (1.08) | — | 6.5 | 0.32 | 5.4 | — |
| 1L2 | 12.8 (1.08) | — | 8.2 | 0.26 | 7.1 | — |
| 1L3 | 15.2 (1.18) | — | 10.9 | 0.19 | 9.6 | — |
| 1DL1 | 7.3 (1.21) | — | 7.1 | 0.30 | 6.0 | — |
| 1DL2 | 9.6 (1.37) | — | 8.9 | 0.24 | 7.6 | — |
| 1DL3 | 21.6 (1.37) | — | 15.3 | 0.14 | 13.8 | — |
| PiPOx2 | 9.3 (1.10) | 7.1 | — | 1 | — | 203 |
| 2L1 | 20.5 (1.03) | — | 9.5 | 0.75 | 6.5 | — |
| 2L2 | 23.1 (1.11) | — | 11.6 | 0.62 | 8.5 | — |
| 2L3 | 37.5 (1.08) | — | 14.9 | 0.48 | 11.7 | — |
| 2DL1 | 17.7 (1.11) | — | 11.1 | 0.64 | 8.0 | — |
| 2DL2 | 18.8 (1.21) | — | 14.3 | 0.50 | 11.0 | — |
| 2DL3 | 25.4 (1.29) | — | 19.8 | 0.36 | 16.3 | — |
| PiPOx3 | 15.5 (1.28) | 12.5 | — | 1 | — | 205 |
| 3L1 | 25.9 (1.10) | — | 15.3 | 0.82 | 10.2 | — |
| 3L2 | 34.8 (1.13) | — | 16.6 | 0.75 | 11.4 | — |
| 3L3 | 42.4 (1.08) | — | 20.2 | 0.62 | 14.8 | — |
| 3DL1 | 20.5 (1.34) | — | 16.7 | 0.75 | 11.5 | — |
| 3DL2 | 35.0 (1.33) | — | 19.4 | 0.65 | 14.0 | — |
| 3DL3 | 49.4 (1.39) | — | 25.6 | 0.49 | 19.9 | — |
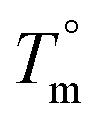
Theoretical considerations of PLA/PiPOx miscibility
The critical value of χN for microphase separation of amorphous BCPs depends on the composition (Fig. 1, dashed line). For a pair of polymers, χ can be calculated from the difference of their solubility parameters δA,B according to eqn (3).33 | (3) |
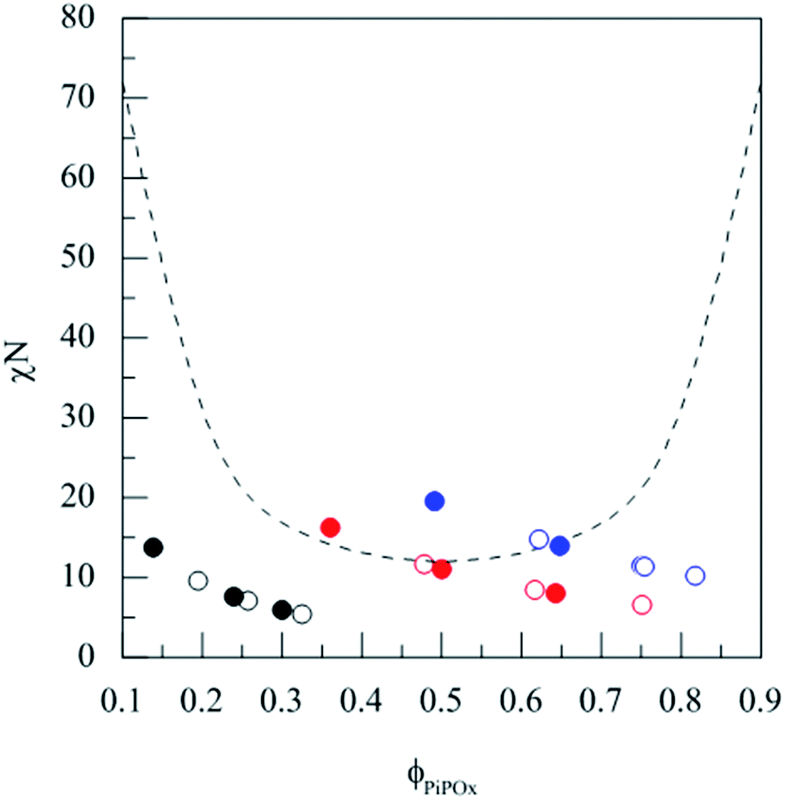 | ||
| Fig. 1 Overview of the 18 BCPs ordered by the segregation strength χN according to eqn (3) and the PiPOx volume fraction ϕPiPOx (color code relates to the parent PiPOx homopolymers: black: PiPOx1, red: PiPOx2, blue: PiPOx3; open and solid symbols correspond to PLLA and PDLLA BCPs, respectively). | ||
With ν as the reference volume (100 cm3 mol−1), δPiPOx = 24.0 J0.5 cm−1.5 and δPLA = 22.7 J0.5 cm−1.5 (ref. 34) (ESI†) this yields χ(298 K) = 0.068. According to this calculation, most of the 18 BCPs are miscible and a few are at the limit to phase separation (Fig. 1).
A shortcoming of eqn (3) is the ignorance of structural features capable of attractive intermolecular interactions. The calculation will exclusively produce positive χ-parameters for any combination of polymers with non-identical solubility parameters. A structural feature of both PLA and PiPOx is the carbonyl group in each repeating unit. In PLLA and PiPOx homopolymers, intermolecular interactions of the carbonyl- and methyl-groups direct the conformational changes and chain packing prior to and during the crystallization process.31,35–37 Furthermore, in poly(ester amides) with a backbone of a defined and periodic sequence of ester- and amide-groups the intermolecular interactions between esters and amides contribute considerably to thermal and mechanical stability.38 It is thus reasonable to assume attractive dipole–dipole interactions between PLA and PiPOx and these interactions favour miscibility. IR-spectroscopy is a sensitive method to probe the molecular environment and changes of the chain conformation due to intermolecular interactions. The IR-spectra of PiPOx1 and its BCPs with PLLA (Fig. 2) show a shift of the carbonyl-stretching band from 1634 to 1643 cm−1 with increasing PLLA molecular weight. As PiPOx1 is amorphous this shift is an indication of attractive intermolecular interaction between PiPOx and PLLA.
 | ||
| Fig. 2 IR-spectral region of the carbonyl-stretching band of PiPOx1 and its BCPs with PLLA. | ||
Single glass transition criterion and SAXS of the BCP melt
Fig. 3A compares the Tg region of 3DL3 with its parent homopolymers and a PiPOx3/PDLLA3 blend. This is the BCP with the highest tendency to phase separate according to Fig. 1. The first derivative of a DSC heating curve displays the Tg as a local minimum. | ||
| Fig. 3 T g region of the second DSC heating curves (solid lines) and the first derivative thereof (dashed lines), (A): PiPOx3 (black), PDLLA3 (red), their blend (ϕPiPOx: 0.49, blue) and 3DL3 (green), (B) PiPOx3 (black), PLLA3 (red), their blend (ϕPiPOx: 0.62, blue) and 3L3 (green). | ||
The BCP exhibits a single Tg, whereas two Tg values are observed for the blend with identical compositions. The two Tg values in the blend are shifted from the values of the individual homopolymers. This indicates a two-phase material, where both phases contain each polymer. In each phase, the polymers mix, which causes the transition to shift on the temperature axis according to the local composition. Consequently, in the fully miscible BCP, such composition fluctuations are small compared to the resolution of the analysis method. The result is a single Tg at a temperature between the Tg values of the homopolymers PiPOx3 and PDLLA3.‡ It is expected that the blend has a higher tendency to phase separate compared to a BCP of an identical composition due to the lack of the covalent bond between the blocks. All 18 BCPs exhibited a single and narrow Tg (ESI†). In the case of a blend of PiPOx3 and PLLA3 (Fig. 3B) the Tg region exhibits a broad transition from 45 to 70 °C. This is indicative of a multiphase behaviour of mixing polymers with composition fluctuations in the range of the resolution limits. The comparison of blends of PiPOx3 with either PDLLA3 or PLLA3 shows that the stereochemistry of PLA plays a role in the strength of interaction. The stereo-regular PLLA3 interacts stronger with PiPOx3 and reduces the domain size of the composition fluctuations.
To assess any microphase separated structure in the BCP melt (215 °C) we measured small angle X-ray scattering (SAXS, Fig. 4) of selected samples. No periodic features were found for any of the BCPs.39 Thus, we have confirmed that the amorphous fractions of PiPOx and PLA in the studied BCPs have no tendency to phase separate under ambient conditions (single Tg) and that any crystallization processes start from a disordered melt.
 | ||
| Fig. 4 SAXS curves of selected BCPs in the melt (215 °C). | ||
Crystallization behavior
The crystallization properties of the PiPOx and PLLA homopolymers are discussed in detail in the ESI.† It should be noted that the driving force of crystallization can be expressed by the difference of equilibrium melting temperature and crystallization temperature Tc (degree of undercooling,
and crystallization temperature Tc (degree of undercooling,  ). The values of
). The values of  (Table 1) follow the order PiPOx3 ≈ PiPOx2 > PLLA3 > PLLA2 > PLLA1 > PiPOx1. This means at a given Tc in the mutual crystallization window of all semi-crystalline homopolymers PiPOx3 has the highest and PiPOx1 the lowest driving force of crystallization. In contrast, the maximum crystallization rates of PLLA were dramatically higher than those of PiPOx due to different crystallization mechanisms. The crystallization of PLLA is diffusion limited, whereas PiPOx requires concerted conformational changes and amide dipole orientation prior to crystallization.31,40 In particular, at an isothermal Tc of 130 °C the crystallization rates were in the order PLLA3 > PLLA2 ≫ PiPOx3 ≈ PiPOx2 > PLLA1, while PiPOx1 remained amorphous (Fig. S7†).
(Table 1) follow the order PiPOx3 ≈ PiPOx2 > PLLA3 > PLLA2 > PLLA1 > PiPOx1. This means at a given Tc in the mutual crystallization window of all semi-crystalline homopolymers PiPOx3 has the highest and PiPOx1 the lowest driving force of crystallization. In contrast, the maximum crystallization rates of PLLA were dramatically higher than those of PiPOx due to different crystallization mechanisms. The crystallization of PLLA is diffusion limited, whereas PiPOx requires concerted conformational changes and amide dipole orientation prior to crystallization.31,40 In particular, at an isothermal Tc of 130 °C the crystallization rates were in the order PLLA3 > PLLA2 ≫ PiPOx3 ≈ PiPOx2 > PLLA1, while PiPOx1 remained amorphous (Fig. S7†).
Exemplarily, the isothermal crystallization at 130 °C of PLLA3 and PiPOx3 captured by polarized optical microscopy (POM) is shown in the video (ESI†) and Fig. 7. PLLA3 (Fig. 7A) starts to crystallize in the spherulitic superstructure immediately after reaching the isothermal Tc and the process is completed in 5 min. The radius of the spherulites is 430 ± 100 µm. In contrast, PiPOx3 (Fig. 7B) crystallizes in a granular superstructure (r: 35 ± 7 µm) due to the higher nucleation density (higher ΔT), but the crystallization requires more time. The first granules appear after 12 min and crystallization continues for 45 min after reaching Tc.
The crystallization behaviour changes in a blend of the two polymers (ϕPiPOx: 0.62, Fig. 7C and the video†). As the first crystalline superstructure, granules of PiPOx3 appear after 8.5 min followed by spherulites of PLLA3 after 15 min. The nucleation density of PLLA3 is reduced compared to that of the pure homopolymer resulting in larger spherulites of radius 1200 ± 400 µm. This observation is in accord with the discussed differences in the crystallization mechanism of the two polymers and the multiphase behaviour of mixing. The local molecular mixing of PiPOx and PLLA chains plasticizes PiPOx and this facilitates the required conformational changes prior to crystallization. Subsequently, PLLA is expelled from the regions of crystalline PiPOx, concentrates in PLLA rich domains, and eventually crystallizes. The BCP 3L3 (ϕPiPOx: 0.62, Fig. 7D and the video†) did not form any spherulites as the migration of PLLA was hindered by the covalent bond. Granules of PiPOx appeared after 6.5 min. From WAXS and the DSC heating curve after isothermal crystallization (Fig. 5) it becomes obvious that PLLA3 in 3L3 could not crystallize at all. WAXS further proves that the crystalline structure of PiPOx3 in the samples of the pure homopolymer and 3L3 is identical. The unit cell is unchanged as PLLA is expelled from the crystalline regions. The DSC heating curve of 3L3 exhibits only the melting endotherm of PiPOx3 (Tm: 203 °C).
 | ||
| Fig. 5 (A) WAXS results at room temperature after isothermal crystallization at 130 °C. (B) DSC heating curves (10 °C min−1) after isothermal crystallization at 130 °C for 2 h. | ||
It is interesting to systematically reduce the molecular weight and volume fraction of PiPOx in BCPs with PLLA3 and compare the structural evolution. POM images of 2L3 (ϕPiPOx: 0.48, Fig. 7E) and 3L3 appear similar. Granular structures develop after 3 min. However, WAXS and DSC of 2L3 after 2 h of isothermal crystallization at 130 °C indicate crystallinity of both PiPOx and PLLA (Fig. 5A). That is, only if PLLA3 is the major constituent it can form large enough PLLA rich domains and crystallize. 2L3 is the only BCP in which double crystallinity is observed after isothermal crystallization at 130 °C. The crystallization of both blocks is consecutive. After a short isothermal period of 4 min, DSC confirmed the melting of only PiPOx (ESI†). This verifies the mechanism of a crystallization-driven phase-separation. PLLA is expelled from the crystalline regions of PiPOx and consecutively crystallizes from PLLA rich domains. Consequently, the WAXS pattern of 2L3 consists of the superimposed signals of the pure homopolymers.
The differences of a crystallization-driven phase-separated crystalline-amorphous and double-crystalline structure are shown by AFM (Fig. 6). The two selected BCPs, 2DL2 and 2L3, have similar volume fractions of PiPOx. In the case of 2DL2 stacks, fibrillar structures are observed. The individual fibers are 15 nm wide and consist of the extended chains of crystalline PiPOx.27 The crystalline and amorphous regions are clearly separated in the phase image due to the different mechanic response. In the case of 2L3, double-crystallinity reduces the phase contrast and a lamellar phase-separated structure is observed. The lamellar structure was further confirmed by SAXS (Fig. 6C). The positions of the observed peaks correspond to q-values of 0.16, 0.08 and 0.03 Å−1.
 | ||
| Fig. 6 AFM phase images of (A) 2DL2 (ϕPiPOx: 0.50) and (B) 2L3 (ϕPiPOx: 0.48) after isothermal crystallization at 130 °C (scale bars: 400 nm), and (C) the SAXS pattern of 2L3 after isothermal crystallization at 130 °C. | ||
In the sample of 1L3 (ϕPiPOx: 0.19) PLLA spherulites appear as a sole crystalline superstructure (Fig. 7F). Both WAXS and DSC (Fig. 5) indicate that PiPOx is amorphous in this sample. Its presence as a miscible component clearly disturbs the nucleation and crystallization of PLLA. The spherulites appeared after 1 min and their growths lasted for 90 min, resulting in a radius of 5500 µm. The DSC heating scan exhibited a bimodal melting peak due to the crystallites of different sizes. However, the WAXS patterns of 1L3 and PLLA3 display the same peaks indicating that the same crystal structure is formed in block- and homopolymers.
 | ||
| Fig. 7 Polarized optical micrographs after isothermal crystallization at 130 °C for the indicated time periods of: (A) PLLA3, (B) PiPOx3, (C) PiPOx3/PLLA3 blend (ϕPiPOx: 0.62), (D) 3L3 (ϕPiPOx: 0.62), (E) 2L3 (ϕPiPOx: 0.48) and (F) 1L3 (ϕPiPOx: 0.19); scale bar: 1 mm. | ||
It should be noted that the crystallization properties of PLLA in the BCPs cannot be predicted based on the PLLA volume fraction only. A sample of 1L1 (ϕPLLA: 0.68) remained entirely amorphous (Fig. 5A) even though its ϕPLLA is higher than that of 2L3. This is due to the lower  of PLLA1. At 130 °C, the degree of supercooling is not sufficient to drive the crystallization of 1L1.
of PLLA1. At 130 °C, the degree of supercooling is not sufficient to drive the crystallization of 1L1.
Plasticization of PiPOx and inversion of crystallization rates
Plasticization is the property of a compound to increase the chain flexibility of a polymer, expressed by a decreasing Tg.Plasticization is utilized in many cases to increase the crystallinity of polymers and thereby improve the mechanical properties.41 In the case of PiPOx, plasticization facilitates the required conformational changes and increases the crystallization rates. Plasticization of PiPOx3 was observed in the blend with PLLA3 (Fig. 7C and the video†) by an earlier onset of PiPOx crystallization. In the corresponding BCP 3L3, the onset of crystallization is even earlier as in the blend indicating that the level of molecular mixing determines the extent of plasticization. Fig. 8 shows the crystallization rates of all BCPs with an observable exothermal peak during the isothermal crystallization at 130 °C. In comparison, the crystallization rates of the respective pure PiPOx homopolymers are indicated by dashed lines. In all cases the crystallization rates of the BCPs are faster than those of their parent PiPOx homopolymers. With increasing PLA molecular weight and a constant PiPOx block, the crystallization rates increase. The plasticization is stronger for the PDLLA BCPs due to the different strength of interaction between PiPOx and PDLLA/PLLA. The different methyl side-group orientations of PDLLA/PLLA in the molten state cause different chain conformations and adjust the interplay with PiPOx. In contrast, PiPOx disturbs the crystallization of PLLA. The sample of 1L3 was the only BCP with a distinguishable exothermal peak of PLLA crystallization at 130 °C. The crystallization rate compared to PLLA3 was reduced by a factor of 20.
 | ||
| Fig. 8 Inverse crystallization half times of: PDLLA BCPs (black circles), PLLA BCPs (red triangles), and the parent PiPOx homopolymers (dashed lines) at an isothermal Tc of 130 °C. | ||
Conclusions
Miscibility is an important influencing factor in the crystallization of BCPs. We set out with a series of 18 BCPs comprised of PLA and PiPOx with a wide compositional and molecular weight range. Our synthetic approach towards the BCPs proved useful to find clear relationships between the BCPs and their parent homopolymers. Based on the theoretical considerations and experimental results, all 18 BCPs were found to be miscible. Mixing has a plasticizing effect on PiPOx and a detrimental effect on the crystallization of PLLA. The mechanism of structural evolution during isothermal crystallization is shown in Fig. 9. Starting from a mixed melt (A1/B1), PiPOx is plasticized by PLA (A2/B2). The required conformation changes of the PiPOx backbone take place and allow the orientation of amide-dipoles. This process expels PLA and causes its migration. PiPOx crystallizes in its original unit cell (A3/B3). Long range migration of PLA is prevented in the BCP through the covalent bond and tethering to the crystalline phase of PiPOx (B3). Local PLLA rich domains can only crystallize if the volume fraction of PLLA is large enough and the degree of supercooling is sufficient (B4). In the blend PLLA macro-phase separates (A3) and crystallizes (A4) with reduced nucleation density. The crystallization-driven phase-separation of PLA-PiPOx BCPs leads to: (1) a double-crystalline lamellar nanostructure within the confinement of a granular superstructure of PiPOx or (2) a crystalline-amorphous fibrillar nanostructure. | ||
| Fig. 9 Mechanism of the structural evolution of PLA/PiPOx blends and BCPs during isothermal crystallization from the homogeneous melt. | ||
The bulk behaviour of unprecedented PLA-PiPOx BCPs is dominated by the miscibility of the components. Further understanding of the interplay of miscibility, stereochemistry and crystallization could stimulate a variety of potential biomedical applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
FP thanks Tina Borke for discussion of the manuscript. This work was supported by Tekes (Project number: 1921/31/2012) and made use of the instrumentation of Aalto University Nanomicroscopy Center (Aalto-NMC).References
-
O. Olabisi, L. M. Robeson and M. T. Shaw, Polymer-Polymer Miscibility, Academic Press, 1979 Search PubMed
.
-
G. Strobl, The Physics of Polymers, Springer, Berlin Heidelberg, 3rd edn, 2007 Search PubMed
- L. Leibler, Macromolecules, 1980, 13, 1602–1617 CrossRef CAS
- D. S. Kaplan, J. Appl. Polym. Sci., 1976, 20, 2615–2629 CrossRef CAS
- M. M. Coleman and P. C. Painter, Prog. Polym. Sci., 1995, 20, 1–59 CrossRef CAS
- W.-C. Chen, S.-W. Kuo, U.-S. Jeng and F.-C. Chang, Macromolecules, 2008, 41, 1401–1410 CrossRef CAS
- S. Katayose and K. Kataoka, Bioconjugate Chem., 1997, 8, 702–707 CrossRef CAS PubMed
- P. Saini, M. Arora and M. N. V. R. Kumar, Adv. Drug Delivery Rev., 2016, 107, 47–59 CrossRef CAS PubMed
- P. Supaphol, N. Dangseeyun, P. Thanomkiat and M. Nithitanakul, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 676–686 CrossRef CAS
- P. P. Lizymol and S. Thomas, Polym. Degrad. Stab., 1993, 41, 59–64 CrossRef CAS
- T. Takayama, M. Todo and H. Tsuji, J. Mech. Behav. Biomed. Mater., 2011, 4, 255–260 CrossRef PubMed
- Z. Yang and C. D. Han, Macromolecules, 2008, 41, 2104–2118 CrossRef CAS
- H. Tsuji, Adv. Drug Delivery Rev., 2016, 107, 97–135 CrossRef CAS PubMed
- B. Zhong and Z. Y. Al-Saigh, J. Appl. Polym. Sci., 2012, 123, 2616–2627 CrossRef CAS
- E. Castro-Aguirre, F. Iñiguez-Franco, H. Samsudin, X. Fang and R. Auras, Adv. Drug Delivery Rev., 2016, 107, 333–366 CrossRef CAS PubMed
- R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835–864 CrossRef CAS PubMed
- Y. Ramot, M. H. Zada, A. J. Domb and A. Nyska, Adv. Drug Delivery Rev., 2016, 107, 153–162 CrossRef CAS PubMed
- S. Farah, D. G. Anderson and R. Langer, Adv. Drug Delivery Rev., 2016, 107, 367–392 CrossRef CAS PubMed
- P. Olsén, T. Borke, K. Odelius and A.-C. Albertsson, Biomacromolecules, 2013, 14, 2883–2890 CrossRef PubMed
- T. Isono, Y. Kondo, I. Otsuka, Y. Nishiyama, R. Borsali, T. Kakuchi and T. Satoh, Macromolecules, 2013, 46, 8509–8518 CrossRef CAS
- S. Saeidlou, M. A. Huneault, H. Li and C. B. Park, Prog. Polym. Sci., 2012, 37, 1657–1677 CrossRef CAS
- T. G. Bassiri, A. Levy and M. Litt, J. Polym. Sci., Part B: Polym. Phys., 1967, 5, 871–879 CrossRef
- M. Litt, F. Rahl and L. G. Roldan, J. Polym. Sci., Part A-2, 1969, 7, 463–473 CrossRef CAS
- R. Hoogenboom and H. Schlaad, Polymers, 2011, 3, 467–488 CrossRef CAS
- R. Hoogenboom and H. Schlaad, Polym. Chem., 2017, 8, 24–40 RSC
- R. Hoogenboom, Angew. Chem., Int. Ed., 2009, 48, 7978–7994 CrossRef CAS PubMed
- A. L. Demirel, M. Meyer and H. Schlaad, Angew. Chem., Int. Ed., 2007, 46, 8622–8624 CrossRef CAS PubMed
- A. L. Demirel, P. Tatar Güner, B. Verbraeken, H. Schlaad, U. S. Schubert and R. Hoogenboom, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 721–729 CrossRef CAS
- K. Aoi and M. Okada, Prog. Polym. Sci., 1996, 21, 151–208 CrossRef CAS
- S. Kobayashi, M. Kaku and T. Saegusa, Macromolecules, 1988, 21, 334–338 CrossRef CAS
- S. Sun and P. Wu, Phys. Chem. Chem. Phys., 2015, 17, 31084–31092 RSC
- H. Witte and W. Seeliger, Justus Liebigs Ann. Chem., 1974, 1974, 996–1009 CrossRef
- I. W. Hamley, P. Parras, V. Castelletto, R. V. Castillo, A. J. Müller, E. Pollet, P. Dubois and C. M. Martin, Macromol. Chem. Phys., 2006, 207, 941–953 CrossRef CAS
- R. F. Fedors, Polym. Eng. Sci., 1974, 14, 147–154 CAS
- J. Zhang, H. Tsuji, I. Noda and Y. Ozaki, J. Phys. Chem. B, 2004, 108, 11514–11520 CrossRef CAS
- C. Li, S. Luo, J. Wang, H. Wu, S. Guo and X. Zhang, Biomacromolecules, 2017, 18, 1440–1448 CrossRef CAS PubMed
- D. A. Brant, A. E. Tonelli and P. J. Flory, Macromolecules, 1969, 2, 228–235 CrossRef CAS
- H. Tetsuka, Y. Doi and H. Abe, Macromolecules, 2006, 39, 9071–9079 CrossRef CAS
- I. W. Hamley and V. Castelletto, Prog. Polym. Sci., 2004, 29, 909–948 CAS
- P. Pan, W. Kai, B. Zhu, T. Dong and Y. Inoue, Macromolecules, 2007, 40, 6898–6905 CrossRef CAS
- J. K. Palacios, J. Zhao, N. Hadjichristidis and A. J. Müller, Macromolecules, 2017, 50, 9683–9695 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: POM videos of isothermal crystallization, polymer characterization, calculation of solubility parameters, Tg regions of all BCPs, detailed crystallization properties of the homopolymers, isothermal crystallization of 2L3. See DOI: 10.1039/c8py00198g |
| ‡ The small transition at 65 °C is likely a result of crystallization induced phase-separation during the previous cooling scan. |
| This journal is © The Royal Society of Chemistry 2018 |