
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemoenzymatic synthesis of polypeptides consisting of periodic di- and tri-peptide motifs similar to elastin†
Prashant G.
Gudeangadi
a,
Kousuke
Tsuchiya
 *a,
Takamasa
Sakai
b and
Keiji
Numata
*a
*a,
Takamasa
Sakai
b and
Keiji
Numata
*a
aEnzyme Research Team, RIKEN Center for Sustainable Resource Science, 2-1 Hirosawa, Wako-shi, Saitama 351-0198, Japan. E-mail: keiji.numata@riken.jp; kosuke.tsuchiya@riken.jp
bDepartment of Bioengineering, School of Engineering, University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
First published on 17th April 2018
Abstract
Proline and valine exist in repetitive motifs in various structural proteins and play an important role in their physiological functions. Herein, we synthesized polypeptides that consist of di- and tri-peptide motifs containing proline and valine via chemoenzymatic polymerization. Di- and tri-peptide ethyl esters (ValGly-OEt, GlyProGly-OEt, and ValProGly-OEt) could be polymerized by papain, whereas proline and valine ethyl esters (Pro-OEt, Val-OEt) were inactive in chemoenzymatic polymerization because of their poor affinity for papain. The copolymer of ValProGly-OEt and ValGly-OEt being poly(ValProGly-co-ValGly), which is composed of a repetitive sequence of elastin, exhibited a temperature-dependent structural transition similar to tropoelastin. The post-polycondensation product of poly(ValProGly-co-ValGly) showed higher molecular weight and elastin-like thermal behaviors.
Introduction
Polypeptides are building blocks in various natural proteins. The chemical and biological properties of proteins depend on their amino acid sequence, referred to as their primary structure. According to the primary structure of proteins, their specific higher order structures are folded and formed to function. Due to their specific structures and functionality, they have been employed as biomaterials in various fields, including tissue engineering and biomedicine.1–7 Structural proteins such as silk, collagen, and elastin are mainly composed of a highly repetitive sequence in which short polypeptide motifs periodically replicate. These repetitive sequences assemble into higher order structures with excellent mechanical properties.6,7 Structural proteins obtain their mechanical properties from certain important amino acids, such as proline in spider silk, collagen, and elastin. Proline exists in repetitive short peptide motifs in these structural proteins, such as glycine-proline-glycine (GlyProGly), in the major ampullate spidroin (MaSp) protein of spider silk8 and glycine-proline-hydroxyproline (GlyProHyp) in collagen.9 On the other hand, elastin contains valine-proline-glycine-valine-glycine (ValProGlyValGly) in its hydrophobic region, and its high elastic properties arise from the tandem sequence of ValProGlyValGly.10 Both proline and valine are essential to realize such elasticity.11Even though conventional solid phase peptide synthesis can be used for constructing precise target sequences, the tedious multistep reactions with several protection and deprotection steps restrict their large-scale production.12,13 Ring-opening polymerization of amino acid N-carboxyanhydrides (NCAs) can also be widely employed for the synthesis of polypeptides with precise control of molecular weight. This method is beneficial for the preparation of di- or tri-block polypeptides but has a poor sequence control.14
On the other hand, chemoenzymatic reactions have emerged as an advantageous method over other solution-based syntheses because of their environmental benignity. These reactions are an atom-economical and facile approach that uses mild aqueous media and is scalable to industrial production.15 Using chemoenzymatic polymerization, we have developed various polypeptides, including not only homopolymers but also random copolymers containing several amino acid residues.16–19 Previously, we demonstrated that synthetic multiblock polypeptides mimicking spider dragline silk were successfully prepared by the post-polymerization of poly(L-alanine) (polyAla) and poly(glycine-r-L-leucine) (poly(Gly-r-Leu)), which have primary structures similar to crystalline and amorphous motifs in the sequence of native spider silk proteins, respectively.20
Di- or tri-peptide esters are also available for use as monomers in chemoenzymatic polymerization, resulting in periodic primary structures. The protease-catalyzed polymerization of AlaGly or LysLeu ethyl ester was performed by Gross et al., and the resulting polypeptides possessed alternating sequences that are hardly accessible by conventional polypeptide synthesis in solution.21,22 Recently, we also demonstrated that even an unnatural amino acid such as 2-aminoisobutyric acid (Aib) can be polymerized using a tri-peptide ester in which Aib is incorporated with L-alanine.23 The resulting polypeptide possessed repetitive primary structures with AlaAibAla units. The periodic insertion of Aib units in the polyAla backbone could induce a substantial transition in the secondary structure from a β-strand to a helical conformation.
Results and discussion
Chemoenzymatic copolymerization
Herein, we synthesized tri-peptide ethyl esters where proline was sandwiched between amino acids with high affinity for papain. Valine was also polymerized using di-peptide or tri-peptide ethyl esters (Scheme 1 and S1†). These di- and tri-peptide motifs are frequently observed in the repetitive motifs of structural proteins, especially elastin. Therefore, the chemoenzymatic copolymerization of these monomers can afford polypeptide materials with elastin-mimicking amino acid sequences. The chemoenzymatic polymerization was performed using papain as a catalyst in 1 M phosphate buffer solution (PBS) at pH 8.0 at 40 °C; the results are tabulated in Table 1. In the case of L-proline methyl (Pro-OMe) and ethyl (Pro-OEt) esters, no precipitation was observed after 2 h, unlike the polymerization of their hydrophobic analogues such as L-leucine and L-alanine esters (Run 1, 2).24,25 The inactivity of proline esters to papain-catalyzed polymerization may be related to the inability of papain to recognize proline residues, which possess a bulky cyclic structure. Because it was found that modification of Aib with protease-recognizable residues can mitigate a mismatch with the substrate pocket in papain,23 the di-peptides Gly-L-Pro ethyl ester (GlyPro-OEt) and benzyl ester (GlyPro-OBn) were synthesized. The homopolymerization of GlyPro-OEt (Run 3) and its copolymerization using Gly-OEt (Run 4) were evaluated in the papain-catalyzed polymerization, but no precipitation was observed in either case. Prolonging the polymerization time (Run 5) and performing the copolymerization at a lower concentration (Run 6) for GlyPro-OBn provided the same results. Hence, we designed and synthesized novel tri-peptide ester monomers in which proline was sandwiched with natural amino acids, namely, Gly-L-Pro-Gly ethyl ester (GlyProGly-OEt) and L-Val-L-Pro-Gly ethyl ester (ValProGly-OEt), as HCl salts by the traditional solution method.26 The chemoenzymatic polymerization of GlyProGly-OEt afforded a precipitate at 31% yield at a feed concentration of 1.0 M (Run 7), which was significantly different from previous results (Run 1–6). Thus, we concluded that the Gly units at both sides of proline is a key factor in papain recognition in this chemoenzymatic polymerization with proline residues.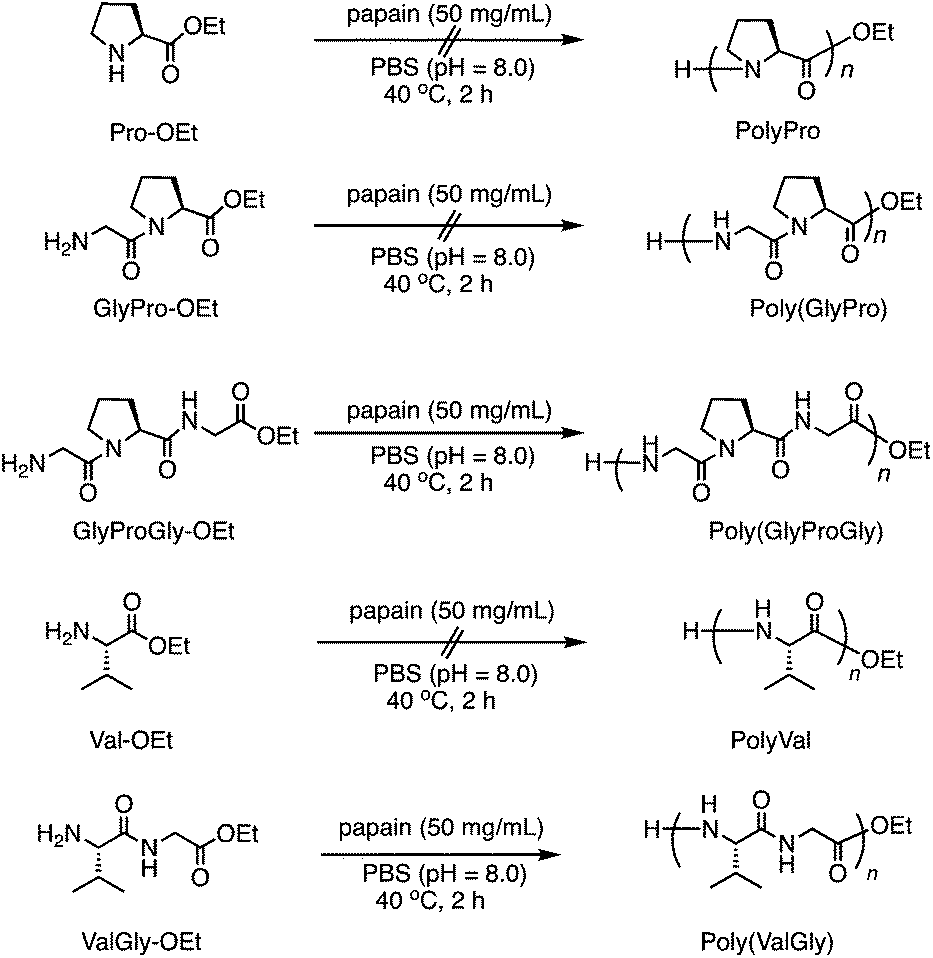 | ||
| Scheme 1 Chemoenzymatic polymerization of Pro- and Val-containing monomers using papain. | ||
| Run | Monomer | Conc. (M) | Time (h) | Yieldb (%) |
|---|---|---|---|---|
| a Polymerization was carried out using monomer (HCl salt) and papain (50 mg mL−1) in phosphate buffer (1 M, pH 8.0) at 40 °C for 2–24 h. b Precipitation was collected by centrifugation, washed with water, and lyophilized. c Carbonate buffer (1 M, pH 10) was used for the reaction. d Molar ratio was 1/1. | ||||
| 1 | Pro-OMe | 1.0 | 2 | 0 |
| 2 | Pro-OEt | 1.0 | 2 | 0 |
| 3 | GlyPro-OEt | 1.0 | 2 | 0 |
| 4 | GlyPro-OEt/Gly-OEt | 1.0 | 2 | 0 |
| 5 | GlyPro-OBn | 1.0 | 24 | 0 |
| 6 | GlyPro-OBn/Gly-OBn | 0.001 | 2 | 0 |
| 7 | GlyProGly-OEt | 1.0 | 2 | 31 |
| 8 | Val-OEt | 1.0 | 2 | 0 |
| 9 | Val-OEt | 0.5 | 12 | 0 |
| 10 | Val-OEt | 0.001 | 12 | 0 |
| 11c | Val-OEt | 1.0 | 2 | 0 |
| 12 | ValGly-OEt | 1.0 | 2 | 31 |
| 13 | ValProGly-OEt | 1.0 | 2 | 1 |
| 14d | GlyProGly-OEt/ValGly-OEt | 1.0 | 2 | 30 |
| 15d | ValProGly-OEt/ValGly-OEt | 1.0 | 2 | 28 |
As another key amino acid residue in elastin, Val-OEt was also tested in the papain-catalyzed polymerization at different feed concentrations (Run 8–10). Even at a lower concentration with prolonged reaction time (Run 10), the polypeptide could not be obtained as a precipitate. Carbonate buffer solution (pH 10) was also employed to promote the polymerization, which also did not afford a precipitate (Run 11). In contrast, the di-peptide L-Val-Gly ethyl ester (ValGly-OEt) could be polymerized at a feed concentration of 1.0 M to afford a precipitate (Run 12). These results also indicated that the Gly unit is a key factor in papain recognition in this chemoenzymatic polymerization. The homopolymerization of ValProGly-OEt, which was highly hygroscopic, afforded very little precipitation under similar conditions to ValGly-OEt (Run 13). This was due to the higher solubilities of monomer and polymer in the buffer.
Furthermore, we carried out the copolymerization of GlyProGly-OEt/ValGly-OEt (Run 14) and ValProGly-OEt/ValGly-OEt (Run 15) (Scheme 2a). The polymerization of GlyProGly-OEt and ValGly-OEt afforded a precipitate in 30% yield. The copolymerization of ValProGly-OEt with ValGly-OEt also afforded a precipitate in 28% yield.
 | ||
| Scheme 2 (a) Chemoenzymatic copolymerization of Pro- and Val-containing di- and tri-peptides using papain and (b) post-polycondensation of poly(ValProGly-co-ValGly). | ||
To confirm the role of enzyme in the polymerization reaction, blank reactions without enzymes were performed (Table S1†), according to the previous literature.27–29 The blank reactions for GlyProGly-OEt and ValGly-OEt were carried out in the absence of papain. The polymerization reaction was performed using 1 M PBS at pH 8.0 at 40 °C for 2 h. No polymerized product was detected by 1H nuclear magnetic resonance (1H NMR) spectroscopy (Fig. S1†), meaning that the reaction didn't proceed without papain. The copolymerization of GlyProGly-OEt/ValGly-OEt and ValProGly-OEt/ValGly-OEt was also carried out in the absence of papain, that resulted in no polymerized products based on 1H NMR measurements (Fig. S2†). Thus, we confirmed that papain acts as a catalyst and drives the polymerization reaction.
Chemical structure analyses by MALDI-TOF mass and NMR spectroscopy
The chemical structures of these products were confirmed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) and 1H NMR spectroscopy. In its MALDI-TOF MS spectrum (Fig. 1a), poly(GlyProGly) exhibited two series of products up to the monomer with an ethyl ester or carboxylic acid at the C-terminal end. The series of peaks with an interval of m/z 211, which corresponds to the GlyProGly tri-peptide unit, indicates the successful polymerization of GlyProGly-OEt. The polymer showed a relatively higher degree of polymerization than the values of previously reported polymer obtained by chemoenzymatic polymerization.30,31 The molar mass of poly(GlyProGly) was in the range from 400 to 2000 Da. In the MALDI-TOF MS spectrum of ValGly-OEt (Fig. 1a), two series of peaks derived from poly(ValGly) up to the heptamer with a C-terminal ethyl ester or carboxylic acid were detected. The copolymer synthesized from ValProGly-OEt and ValGly-OEt, poly(ValProGly-co-ValGly), afforded peaks with various combinations of di- and tri-peptide units. Molar masses assignable to ValProGly/ValGly units with different ratios were observed, indicating the successful incorporation of both units in the polypeptide chain (Fig. 1b). The copolymer from GlyProGly-OEt and ValGly-OEt, poly(GlyProGly-co-ValGly), also afforded several peaks corresponding to the molar mass of GlyProGly/ValGly units with different ratios (Fig. 1b). In addition to the peaks of poly(GlyProGly-co-ValGly), the peaks derived from poly(ValGly) with ethyl ester and carboxylic acid groups at the C-terminal end were also detected in both cases. This could be due to the high reactivity of the di-peptide ValGly-OEt compared to the tri-peptide GlyProGly-OEt. | ||
| Fig. 1 MALDI-TOF mass spectra of (a) poly(GlyProGly) and poly(ValGly) and (b) poly(ValProGly-co-ValGly) and poly(GlyProGly-co-ValGly) prepared by papain-catalyzed polymerization. | ||
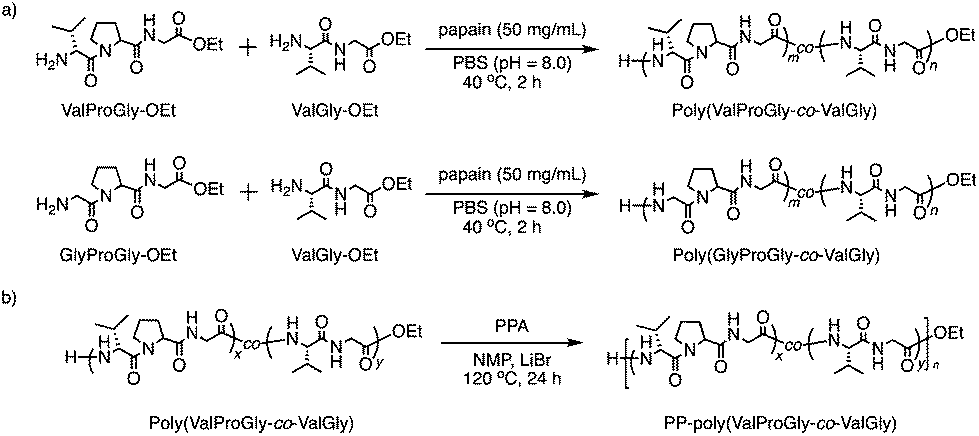
The tri-peptide esters GlyProGly-OEt and ValProGly-OEt and di-peptide ester ValGly-OEt were characterized by both 1H and 13C NMR spectroscopy. The proline peaks at approximately 1.75–2.25 ppm indicate the successful incorporation of proline in the tri-peptide. The –CH3 peak at approximately 1.25 ppm, along with the –CH2 peak at approximately 4.1 ppm of the ester group of the glycine unit, indicates the formation of the tri-peptide. The 1H NMR spectrum of ValProGly-OEt has a relatively similar spectral pattern to that of GlyProGly-OEt. The peaks at approximately 1 ppm are assigned to the two –CH3 groups of valine, along with the peak for the methine proton at approximately 2.4 ppm. The 1H NMR spectrum of ValGly-OEt shows peaks derived from both valine and glycine. The peak at 2.2 ppm of the methine proton and doublet of doublets for two –CH3 groups indicate the presence of valine. The triplet peak at approximately 1.2 ppm and quartet at approximately 4.2 ppm for the ester of glycine, along with the glycine peaks, indicate the formation of the di- and tri-peptides containing both Val and Gly (Fig. S3, S5, and S7†). In the 13C NMR spectrum, the 3 carbonyl signals for the tri-peptide unit and 2 carbonyl signals for the di-peptide unit were observed, indicating the successful formation of both the tri- and di-peptide esters (Fig. S4, S6, and S8†). Fig. 2 shows the 1H NMR spectra of poly(GlyProGly), poly(ValGly) (Fig. 2a), poly(ValProGly-co-ValGly) and poly(GlyProGly-co-ValGly) (Fig. 2b). The 1H NMR spectrum of poly(GlyProGly) shows the characteristic signals of the proline unit at 1.75–2.25 ppm, indicating the formation of the tri-peptide and its polymers. The 1H NMR spectrum of poly(ValGly-OEt) exhibits the characteristic signals derived from the valine unit at 0.75–1.00 ppm. The 1H NMR spectrum of poly(ValProGly-co-ValGly) shows the peaks derived from both the proline and valine units, indicating successful incorporation of both ValProGly and ValGly units. Finally, the 1H NMR spectrum of poly(GlyProGly-co-ValGly) also shows protons derived from both monomers, indicating the incorporation of both units in the polypeptide chain.
 | ||
| Fig. 2 1H NMR spectra of (a) poly(GlyProGly) and poly(ValGly) and (b) poly(ValProGly-co-ValGly) and poly(GlyProGly-co-ValGly), in DMSO-d6/TFA-d (5/1). | ||
Secondary structure characterization by CD spectroscopy
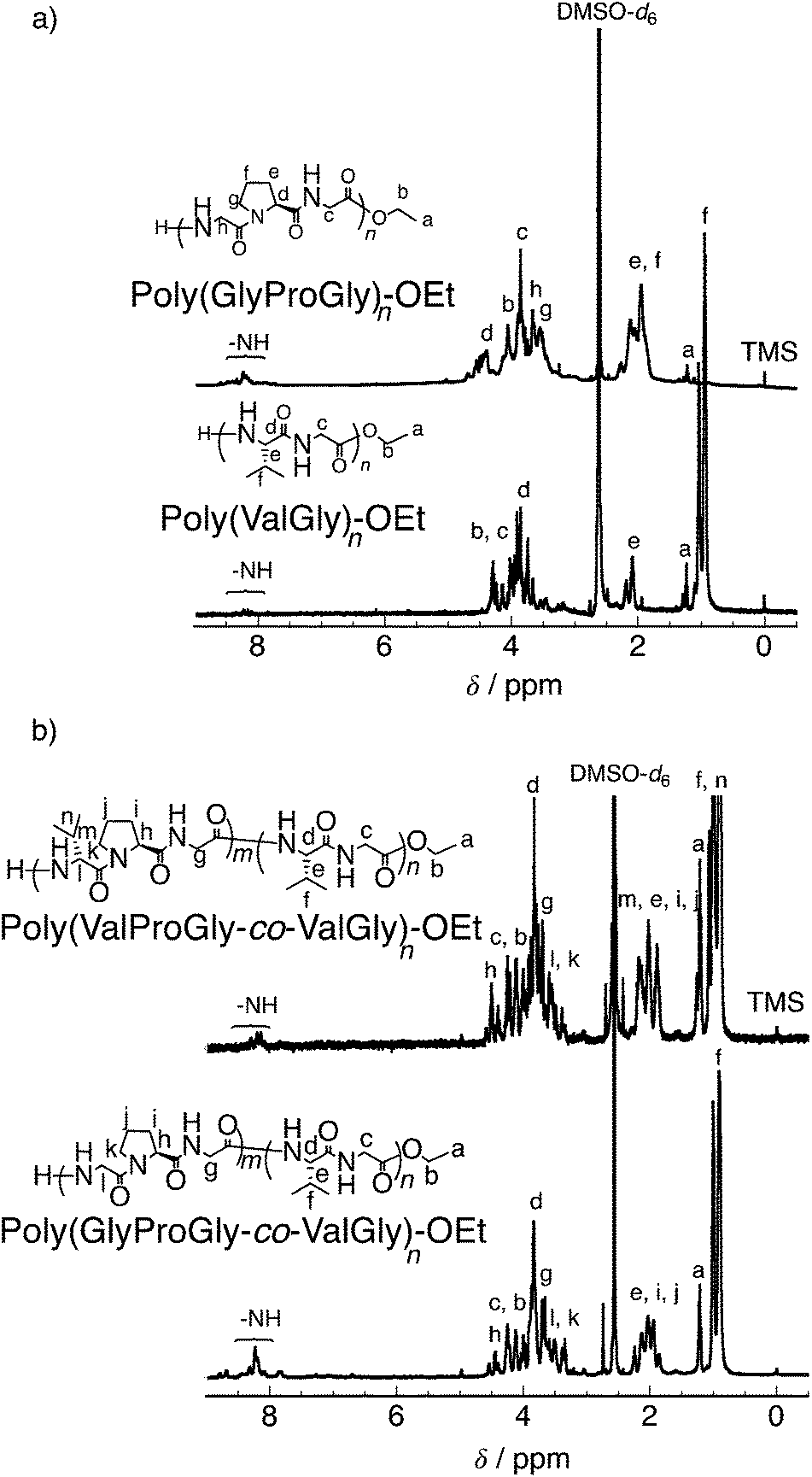
It is well known that the ValProGly triad is responsible for the β-turn confirmation.11 To examine the effect of ValGly on the resulting copolymers, we evaluated the secondary structures of the polypeptides in solution by circular dichroism (CD) spectroscopic analysis. All polypeptides were soluble in water except poly(GlyProGly), which was found to be insoluble. Thus, the CD spectra of the polypeptides were analyzed in 2,2,2-trifluoroethanol (1 mM, containing 0.1% trifluoroacetic acid) and are displayed in Fig. S9.† The contents of the secondary structures were calculated using the DichroWeb online CD analysis server and are tabulated in Table S2.† The CD spectrum of poly(GlyProGly) showed a positive peak centered at approximately 205 nm with a monotonic decrease at lower wavelengths, whereas the CD spectrum of poly(ValGly) showed a negative Cotton effect with negative and positive peaks at approximately 215 and 197 nm, respectively. Poly(GlyProGly) was likely to adopt a more disordered structure than poly(ValGly), which predominantly showed β-strand propensity. This was due to the bent structure of the proline units in poly(GlyProGly), as seen in the amorphous sequence of spider silk proteins showing an elastic nature.8 The CD spectra of poly(GlyProGly-co-ValGly) and poly(ValProGly-co-ValGly) also showed a negative Cotton effect. Poly(GlyProGly-co-ValGly) adopted more β-strand structure than poly(GlyProGly). This result emphasizes that the ValGly unit is essential for inducing β-strands in the polypeptide chain. On the other hand, poly(ValProGly-co-ValGly) adopted a rather unordered structure in 2,2,2-trifluoroethanol.The secondary structure of tropoelastin (a soluble precursor of elastin) is known to vary in response to hydrophobic interactions at different temperatures, which, above a lower critical solution temperature (LCST), cause insoluble aggregation (coacervation).32 The CD spectra of poly(ValProGly-co-ValGly), which possesses a sequence similar to the repetitive motif of elastin, were analyzed in water (1 mM) at different temperatures between 10 and 80 °C to examine the thermal behavior of the polypeptide. Fig. 3 shows CD profiles in a second heating and cooling cycle to eliminate the thermal history of the sample. The secondary structure calculated using DichroWeb is summarized in Table S3.† The initial secondary structure at 20 °C was dominated by a β-strand structure (6.0% helix, 36.5% β-strand, 21.5% β-turn, and 36.0% unordered), which is largely consistent with the reported secondary structure of tropoelastin (3% helix, 41% β-strand, 21% β-turn, and 33% unordered).32 As the temperature increased from 10 to 80 °C, the CD spectra showed a remarkable change. Both positive and negative bands increased in intensity. The helical structure was more preferred at higher temperature (9.3% helix, 37% β-strand, 22% β-turn, and 31% unordered at 80 °C), and the percentage of unordered structure decreased. This behavior was also observed for artificial elastin analogues.32–34 When the sample was cooled from 80 to 10 °C, the unordered content increased with reversible decrease in the helical content. This clearly shows that poly(ValProGly-co-ValGly) undergoes a structural transition similar to natural elastin at elevated temperatures. Unfortunately, a clouding point was not observed for poly(ValProGly-co-ValGly) in water. Because the molecular weight of the obtained poly(ValProGly-co-ValGly) is low (below 1500 Da), the structural change is assumed to be so weak that aggregation cannot be induced by hydrophobic interactions. On the other hand, the CD spectra of poly(GlyProGly-co-ValGly) in water exhibit only a slight change at different temperatures (Fig. S10 and S11†). Therefore, both ValProGly and ValGly sequence units are essential for forming an elastin-like secondary structure.
 | ||
| Fig. 3 CD spectra of poly(ValProGly-co-ValGly) in water in the second (a) heating and (b) cooling cycle at different temperatures between 10 and 80 °C. The concentration of the polypeptide was 1 mM. | ||
Post-polycondensation of poly(ValProGly-co-ValGly)
According to the previous study,20 we performed a post-polycondensation reaction of poly(ValProGly-co-ValGly) in the presence of polyphosphoric acid as a condensing agent at elevated temperature to obtain a higher molecular weight (Scheme 2b). The chain elongation was successfully achieved to afford the post-polycondensation product, PP-poly(ValProGly-co-ValGly), and the obtained polymer was analyzed by 1H NMR spectroscopy and gel permeation chromatography (GPC). The number- and weight-average molecular weights (Mn and Mw) and molecular weight distribution (Mw/Mn) estimated by the GPC profile were 4900, 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, and 1.81, respectively (Fig. S12a†). The molecular weights were determined by using polystyrene standards (Fig. S12b†). In the case when protein molecular weight standards are used to determine the molecular weight (Fig. S12c†), the molecular weight of PP-poly(ValProGly-co-ValGly) is slightly higher. Thus, the polystyrene standards could be applicable to determine the molecular weight of the polypeptide by GPC. Unfortunately, the obtained polymer showed poor solubility in water and common organic solvents, except for N-methyl-2-pyrrolidone (NMP) and TFA. The 1H NMR spectrum of the sample was analyzed in TFA-d and compared with that of the prepolymer, poly(ValProGly-co-ValGly), obtained in the same solvent (Fig. 4). The 1H NMR spectrum showed broad peaks, clearly indicating the higher molecular weight of the polymer. The peaks corresponding to both valine and proline residues are distinguishable. However, the peaks of proline at approximately 2.0–2.6 ppm overlapped with peaks of glycine at approximately 4.0–5.0 ppm. This could be attributed to the longer polypeptide chain and the change in the secondary structure of the obtained PP-poly(ValProGly-co-ValGly).
000, and 1.81, respectively (Fig. S12a†). The molecular weights were determined by using polystyrene standards (Fig. S12b†). In the case when protein molecular weight standards are used to determine the molecular weight (Fig. S12c†), the molecular weight of PP-poly(ValProGly-co-ValGly) is slightly higher. Thus, the polystyrene standards could be applicable to determine the molecular weight of the polypeptide by GPC. Unfortunately, the obtained polymer showed poor solubility in water and common organic solvents, except for N-methyl-2-pyrrolidone (NMP) and TFA. The 1H NMR spectrum of the sample was analyzed in TFA-d and compared with that of the prepolymer, poly(ValProGly-co-ValGly), obtained in the same solvent (Fig. 4). The 1H NMR spectrum showed broad peaks, clearly indicating the higher molecular weight of the polymer. The peaks corresponding to both valine and proline residues are distinguishable. However, the peaks of proline at approximately 2.0–2.6 ppm overlapped with peaks of glycine at approximately 4.0–5.0 ppm. This could be attributed to the longer polypeptide chain and the change in the secondary structure of the obtained PP-poly(ValProGly-co-ValGly).
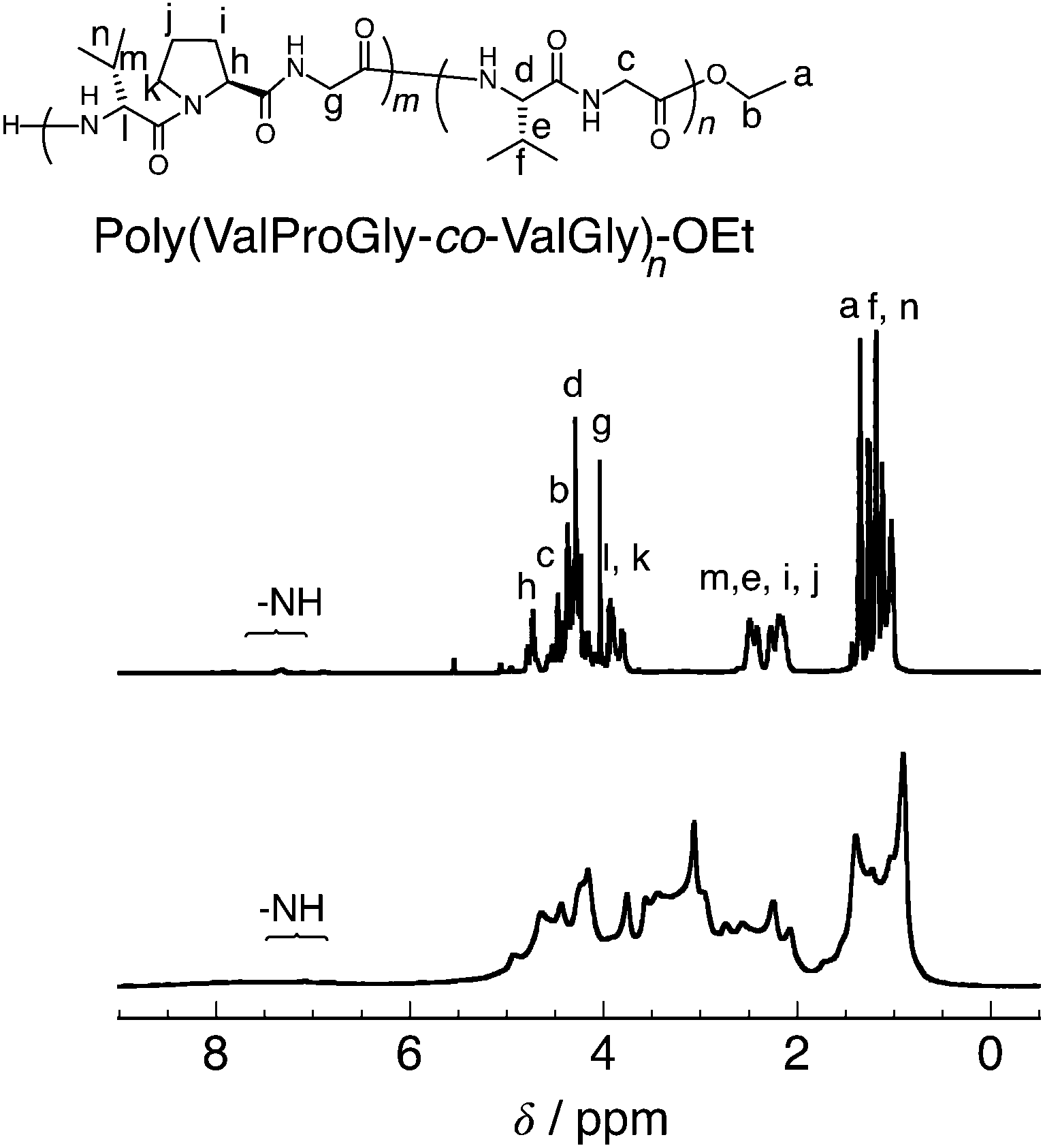 | ||
| Fig. 4 1H NMR of poly(ValProGly-co-ValGly) (top) and PP-poly(ValProGly-co-ValGly) (bottom) in TFA-d. | ||
Thermal properties of PP-poly(ValProGly-co-ValGly)
To characterize the thermal behavior of PP-poly(ValProGly-co-ValGly), we performed thermal gravimetric analysis (TGA) and differential scanning calorimetry (DSC) measurements. The TGA profiles of poly(ValProGly-co-ValGly) before and after post-polycondensation showed a 5% degradation temperature (Td5) at 190 °C regardless of the molecular weight (Fig. 5a). The thermal stability and degradation behavior of PP-poly(ValProGly-co-ValGly) were obviously improved and changed by increasing the molecular weight. The DSC profiles of the first and second heating scans are shown in Fig. 5b. The poly(ValProGly-co-ValGly) prepolymer clearly showed a glass transition temperature (Tg) at approximately 80 °C. This glass transition was reproduced even in the second heating cycle. This revealed that poly(ValProGly-co-ValGly) is dominated by amorphous structures. On the other hand, PP-poly(ValProGly-co-ValGly) showed no Tg in either the first or second heating scans. This indicated that the increase in molecular weight restricts the polymer chain movement, which caused the Tg to shift to a higher temperature. The Tg of PP-poly(ValProGly-co-ValGly) might overlap with thermal degradation, since the Tg of dehydrated elastin ranges from 190 to 200 °C.35,36 To detect the Tg of PP-poly(ValProGly-co-ValGly), we changed the heating and cooling rates at 10 °C min−1 (Fig. S13†). However, the Tg of PP-poly(ValProGly-co-ValGly) was not detected. The storage and loss moduli of PP-poly(ValProGly-co-ValGly) were characterized at different temperatures up to 120 °C (Fig. S14†). The storage and loss moduli of PP-poly(ValProGly-co-ValGly) at 30 °C indicate that the resultant polymer was relatively elastic. In addition, the elastin-like polymer became more elastic and softer at 50 °C.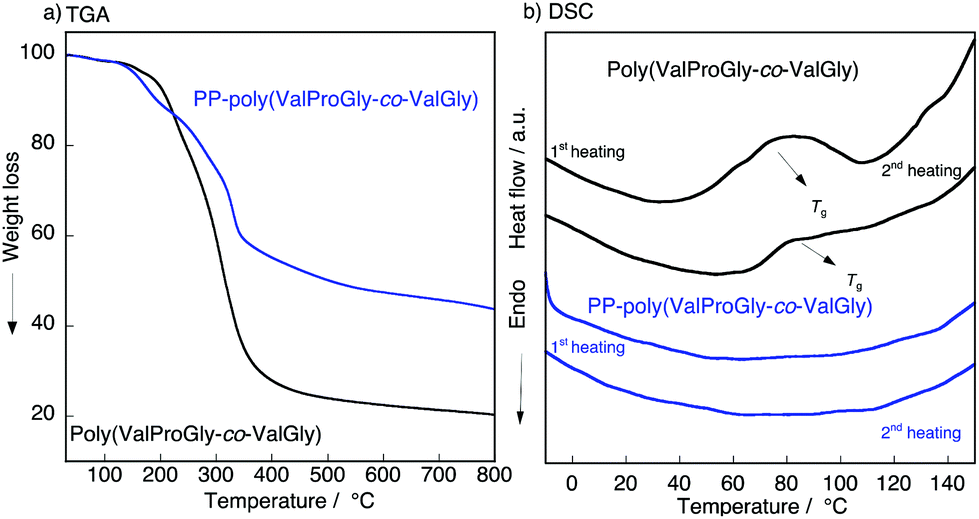 | ||
| Fig. 5 (a) TGA, (b) DSC plot of the poly(ValProGly-co-ValGly) and PP-poly(ValProGly-co-ValGly) at 20 °C min−1. | ||
Secondary structures of PP-poly(ValProGly-co-ValGly)
The structure of poly(ValProGly-co-ValGly) was characterized by FT-IR spectroscopy before and after post-polycondensation, and the amide region of the IR spectra is shown in Fig. 6. After post-polycondensation, a peak derived from the terminal ester group at 1750 cm−1 almost disappeared. This observation indicates that PP-poly(ValProGly-co-ValGly) has fewer terminal ester groups than the poly(ValProGly-co-ValGly) prepolymer as a result of the increase in molecular weight. In the amide I region, the sharp peak derived from the carbonyl group of the β-sheet structures at 1630 cm−1 decreased after the post-polycondensation reaction, and the amide I band was relatively broadened. In addition, PP-poly(ValProGly-co-ValGly) showed a slight shift in the amide II peak and a decrease in the amide III peak. This difference in IR spectra indicated that the post-polycondensation reaction induced disordered secondary structures.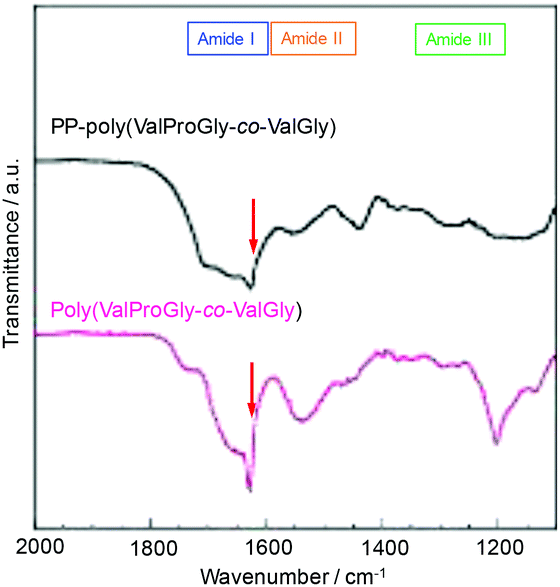 | ||
| Fig. 6 IR spectra of PP-poly(ValProGly-co-ValGly) and poly(ValProGly-co-ValGly) expanded region from 1100 to 2000 cm−1. | ||
Conclusions
In conclusion, we successfully polymerized di- and tri-peptide esters including key amino acids in elastin, namely, proline and valine, via papain-catalyzed polymerization. The modification of proline and valine, which are unfavorable substrates of papain, with a glycine unit allowed us to obtain polypeptides containing repetitive motifs similar to elastin. The copolymer poly(ValProGly-co-ValGly) with a sequence mimicking the highly repetitive sequence of elastin, ValProGlyValGly, was also synthesized. CD spectroscopic analysis revealed that poly(ValProGly-co-ValGly) exhibited a structural transition similar to tropoelastin. The chain elongation of poly(ValProGly-co-ValGly) was achieved by post-polycondensation. The higher-molecular-weight polymer obtained, PP-poly(ValProGly-co-ValGly), showed changes in secondary structure and thermal properties. The Tg of PP-poly(ValProGly-co-ValGly) might be shifted to higher temperature compared to that of poly(ValProGly-co-ValGly), similar to elastin.34 This work mirrors the syntheses of important natural structural proteins such as elastin, which could be used as structural materials in the future. This chemoenzymatic synthesis of polypeptides with periodic sequences will open the door for a new “eco-friendly synthesis” of polypeptide materials mimicking structural proteins.Experimental
Materials
Papain was purchased from Wako Pure Chemical Industries, Ltd (Osaka, Japan) and used as received. The activity was approximately 0.5 U g−1, where one unit is defined as the amount of enzyme needed to hydrolyze 1 mmol of N-benzoyl-DL-arginine p-nitroanilide per minute at pH 7.5 and 25 °C. N-Methyl-2-pyrrolidone (NMP) and N,N-dimethylacetamide (DMAc) were dried over 4 Å molecular sieves. Diethyl ether and triethylamine were dried over 4 Å molecular sieves and stored under a nitrogen atmosphere. All the other reagents were used as received without any purification unless otherwise noted.Measurements
1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on a Varian NMR System 500 instrument (Varian Medical Systems, Palo Alto, CA) at 25 °C and at a frequency of 500 MHz. Dimethylsulfoxide-d6 (DMSO-d6) with trifluoroacetic acid-d (TFA-d) (5/1 in volume) was used as the solvent for the polypeptides with tetramethylsilane as the internal standard. Matrix-assisted laser desorption/ionization time-of-flight (MALDI TOF) mass spectrometry analysis was conducted using an ultrafleXtreme MALDI-TOF spectrophotometer (Bruker Daltonics, Billerica, MA) operating in a reflection mode at an accelerating voltage of 15 kV. The sample was dissolved in water/acetonitrile (0.8 mg mL−1) containing 0.1% TFA, mixed with a solution of α-cyano-4-hydroxycinnamic acid (CHCA) in water/acetonitrile (10 mg mL−1), and deposited on an MTP 384 ground steel BC target plate. Circular dichroism (CD) spectroscopic analysis was conducted using a Jasco J-820 CD spectropolarimeter (JASCO, Tokyo, Japan). Measurements were conducted on polypeptide solutions (1 mM) in 2,2,2-trifluoroethanol (TFE) using a quartz cuvette with a 0.1 cm path length. Each spectrum represents the average of 10 scans from 190 to 290 nm with a resolution of 1 nm obtained at 200 nm min−1 with a bandwidth of 1 nm. The CD analysis at different temperatures was carried out by heating the sample from 10 °C to 80 °C with an increase of 10 °C for each measurement. The sample was cooled from 80 °C to 10 °C by decreasing by 10 °C for each measurement. Gel permeation chromatography (GPC) was performed on a JASCO HPLC system (PU-2086, DG2080-54, AS-2057, CO-2065; JASCO, Tokyo, Japan) with a Shodex KD-804 column (Showa Denko K. K., Tokyo, Japan) and a UV detector (UV-2075). Measurements were carried out on polypeptide samples (2 mg mL−1) eluted with NMP containing 10 mM of lithium bromide at a flow rate of 1.0 mL min−1. The number- (Mn) and weight-average molecular weights (Mw) and the molecular weight distribution (Mw/Mn) were estimated using polystyrene standards of the following molecular weights: 1.32 × 103, 3.25 × 103, 1.01 × 104, 2.85 × 104, 6.60 × 104, and 1.56 × 105. Conalbumin from chicken egg white (7.5 × 104 Da), ovalbumin from hen egg (4.3 × 104 Da), Ribonuclease A from bovine pancreas (1.37 × 104 Da), and aprotinin from bovine lung (6.5 × 103 Da) were used for comparison between proteins and polystyrenes regarding elution time. The protein molecular weight standards were chosen from the Gel filtration calibration kit LMW (GE Healthcare, Chicago, USA). The infrared (IR) spectra of the samples were recorded by an IRPrestige-21 Fourier transform infrared spectrophotometer (Shimadzu Corporation, Kyoto, Japan) with a MIRacle A single reflection ATR unit using a Ge prism. DSC measurements were conducted by using a DSC (PerkinElmer, MA, USA). The polymer sample of 3–5 mg was weighed and sealed in an aluminum pan, which was heated and cooled at 20 °C min−1 from −10 °C to 150 °C under a nitrogen atmosphere. The device was calibrated with an empty cell to establish a baseline and with indium to characterize the heat flow and temperature of the system. For the rheological analysis, powder samples were set in a rheometer (MCR302; Anton Paar, Austria) under a dry nitrogen gas flow. The measurement limit of the rheometer was approximately 0.1 Pa. The storage modulus (G′) and the loss modulus (G′′) were measured by heating the sample from 30 °C to 120 °C with an increase of 10 °C for each data point. The strain and frequency were 1% and 1 Hz, respectively. Measurements were performed at least three times for one data point.Synthetic procedures
Synthesis of poly(GlyProGly) and poly(ValGly) by chemoenzymatic polymerization
A solution of GlyProGly-OEt hydrochloride (1.0 g, 3.89 mmol) in phosphate buffer (2.68 mL, 1.0 M, pH = 8.0) was placed in a glass tube equipped with a stirring bar. To this solution papain (1.94 g) was added. The final concentrations of GlyProGly-OEt and papain were 1.0 M and 50 mg mL−1, respectively. The mixture was stirred at 40 °C at 800 rpm for 2 h using an EYELA ChemiStation PPS-5511 instrument (Tokyo Rikakikai Co. Ltd, Tokyo, Japan). After 2 h, the reaction mixture was cooled to room temperature; the precipitate was collected by centrifuging at 9000 rpm and 4 °C for 30 min. The crude product was washed twice with deionized water, centrifuged, and lyophilized to obtain poly(GlyProGly)-OEt as a white powder. The yield was 0.310 g (31%). Poly(ValGly) was synthesized by chemoenzymatic polymerization using the same experimental procedure.Synthesis of poly(GlyProGly-co-ValGly) and poly(ValProGly-co-ValGly) by chemoenzymatic polymerization
A solution of GlyProGly-OEt hydrochloride (0.3 g, 1.16 mmol) in phosphate buffer (0.5719 mL, 1.0 M, pH = 8.0) was added to ValGly-OEt (0.2357 g, 1.16 mmol), and the resulting solution was placed in a glass tube equipped with a stirring bar. To this solution, papain (58.30 mg) was added. The concentration of papain was 50 mg mL−1. The mixture was stirred at 40 °C and 800 rpm for 2 h using an EYELA ChemiStation PPS-5511 instrument. After 2 h, the reaction mixture was cooled to room temperature; the precipitate was collected by centrifuging at 9000 rpm and 4 °C for 30 min. The crude product was washed twice with deionized water, centrifuged, and lyophilized to obtain poly(GlyProGly-co-ValGly) as a white powder. The yield was 0.09 g (30%). Poly(ValProGly-co-ValGly) was synthesized by chemoenzymatic polymerization using the same experimental procedure.Synthesis of polyPro and polyVal by chemoenzymatic polymerization
A solution of Pro-OEt hydrochloride (0.1 g, 0.56 mmol) in phosphate buffer (0.43 mL, 1.0 M, pH = 8.0) was polymerized by papain (28.00 mg). The final concentrations of Pro-OEt and papain were 1.0 M and 50 mg mL−1, respectively. The other reaction conditions were same as poly(GlyProGly-co-ValGly). In addition to Pro-OEt, Pro-OBn and Val-OEt were polymerized by the same method.Synthesis of poly(GlyPro) and poly(GlyPro-co-Gly) by chemoenzymatic polymerization
A solution of GlyPro-OEt hydrochloride (0.1 g, 0.499 mmol) in phosphate buffer (0.374 mL, 1.0 M, pH = 8.0) was polymerized by using papain (25.00 mg). The final concentrations of GlyPro-OEt and papain were 1.0 M and 50 mg mL−1, respectively. The other reaction conditions and purification method were the same as for polyPro. The same experimental procedure was followed for the homopolymerization of GlyPro-OBn. For poly(GlyPro-co-Gly), a solution of GlyPro-OEt hydrochloride (0.215 g, 1.074 mmol) and Gly-OEt hydrochloride (0.150 g, 1.074 mmol) in phosphate buffer (0.78 mL, 1.0 M, pH = 8.0) was reacted with papain (50.00 mg). The other reaction conditions and purification method were the same as above.Blank control reactions for chemoenzymatic polymerization
A solution of GlyProGly-OEt hydrochloride (1.0 g, 3.89 mmol) in phosphate buffer (2.68 mL, 1.0 M, pH = 8.0) was placed in a glass tube equipped with a stirring bar. The final concentration of GlyProGly-OEt was 1.0 M. The mixture was stirred at 40 °C and 800 rpm for 2 h using an EYELA ChemiStation PPS-5511 instrument. After 2 h, the reaction mixture was cooled to room temperature. The reaction mixture was lyophilized to obtain the crude for the 1H NMR measurements, because no precipitate was observed. In addition to GlyProGly-OEt, ValGly-OEt, GlyProGly-OEt/ValGly-OEt and ValProGly-OEt/ValGly-OEt were used for the blank control reactions.Post-polycondensation of poly(ValProGly-co-ValGly) using polyphosphoric acid (PPA)
A solution of poly(ValProGly-co-ValGly) (50 mg) in NMP (1 mL) containing lithium bromide (0.0868 g, 1 mmol) was added to polyphosphoric acid (PPA) (0.07 g) placed in a 10 mL flask equipped with a stir bar and a stopcock under a nitrogen atmosphere. The solution was stirred at 120 °C for 24 h under nitrogen. After cooling to room temperature, the mixture was poured into water and stirred for 2 h. The precipitate was collected by centrifugation at 9000 rpm and 4 °C for 15 min. The crude product was washed with water, centrifuged twice, and lyophilized to afford a brown powder (35%).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Impulsing Paradigm Change through Disruptive Technologies Program (ImPACT) of the Japan Science and Technology Corporation (JST), the RIKEN Engineering Network, and the JSPS KAKENHI Grant Number JP17K18361.Notes and references
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS PubMed.
- U. Hersel, C. Dahmen and H. Kessler, Biomaterials, 2003, 24, 4385–4415 CrossRef CAS PubMed.
- H. Shin, S. Jo and A. G. Mikos, Biomaterials, 2003, 24, 4353–4364 CrossRef CAS PubMed.
- R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS PubMed.
- K. Numata and D. L. Kaplan, Adv. Drug Delivery Rev., 2010, 62, 1497–1508 CrossRef CAS PubMed.
- K. Numata, Polym. J., 2015, 47, 537–545 CrossRef CAS.
- T. Passioura and H. Suga, Chem. Commun., 2017, 53, 1931–1940 RSC.
- A. D. Malay, K. Arakawa and K. Numata, PLoS One, 2017, 12, e0183397 Search PubMed.
- B. Brodsky and A. V. Persikov, Adv. Protein Chem., 2005, 70, 301–339 CrossRef CAS PubMed.
- S. M. Mithieux and A. S. Weiss, Adv. Protein. Chem., 2005, 70, 437–461 CrossRef CAS PubMed.
- Y. Chen and Z. Guan, J. Am. Chem. Soc., 2010, 132, 4577–4579 CrossRef CAS PubMed.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- I. Coin, M. Beyermann and M. Bienert, Nat. Protoc., 2007, 2, 3247–3325 CrossRef CAS PubMed.
- F. Guzman, S. Barberis and A. Illanes, Electron. J. Biotechnol., 2007, 10, 279–314 CAS.
- K. Tsuchiya and K. Numata, Macromol. Biosci., 2017, 1700177 CrossRef PubMed.
- K. Numata and P. J. Baker, Biomacromolecules, 2014, 15, 3206–3212 CrossRef CAS PubMed.
- K. Yazawa and K. Numata, Molecules, 2014, 19, 13755–13774 CrossRef PubMed.
- J. Fagerland, A. F. Wistrand and K. Numata, Biomacromolecules, 2014, 15, 735–743 CrossRef CAS PubMed.
- Y. Ma, Z. Li and K. Numata, ACS Biomater. Sci. Eng., 2016, 2, 697–706 CrossRef CAS.
- K. Tsuchiya and K. Numata, ACS Macro Lett., 2017, 6, 103–106 CrossRef CAS.
- X. Qin, A. C. Khuong, Z. Yu, W. Du, J. Decatur and R. A. Gross, Chem. Commun., 2013, 49, 385–387 RSC.
- X. Qin, W. Xie, S. Tian, J. Cai, H. Yuan, Z. Yu, G. L. Butterfoss, A. C. Khuong and R. A. Gross, Chem. Commun., 2013, 49, 4839–4841 RSC.
- K. Tsuchiya and K. Numata, Chem. Commun., 2017, 53, 7318–7321 RSC.
- G. Li, V. K. Raman, W. Xie and R. A. Gross, Macromolecules, 2008, 41, 7003–7012 CrossRef CAS.
- P. J. Baker and K. Numata, Biomacromolecules, 2011, 13, 947–951 CrossRef PubMed.
- E. Galoppini and M. A. Fox, J. Am. Chem. Soc., 1996, 118, 2299–2300 CrossRef CAS.
- R. A. Gross, A. Kumar and B. Kalra, Chem. Rev., 2001, 101, 2097–2124 CrossRef CAS PubMed.
- S. Matsumura, Macromol. Biosci., 2002, 2, 105–126 CrossRef CAS.
- S. Shoda, H. Uyama, J. Kadokawa, S. Kimura and S. Kobayashi, Chem. Rev., 2016, 116, 2307–2413 CrossRef CAS PubMed.
- Y. Ma, R. Sato, Z. Li and K. Numata, Macromol. Biosci., 2016, 16, 151–159 CrossRef CAS PubMed.
- K. Tsuchiya and K. Numata, Macromol. Biosci., 2016, 16, 1001–1008 CrossRef CAS PubMed.
- B. Vrhoski, S. Jensen and A. S. Weiss, Eur. J. Biochem., 1997, 250, 92–98 Search PubMed.
- A. M. Tamburro, M. Lorusso, N. Ibris, A. Pepe and B. Bochicchio, Chirality, 2010, 22, E56–E66 CrossRef CAS PubMed.
- D. Weller, J. R. McDaniel, K. Fischer, A. Chilkoti and M. Schmidt, Macromolecules, 2013, 46, 4966–4971 CrossRef CAS.
- V. Samouillan, C. Andre, J. Dandurand and C. Lacabanne, Biomacromolecules, 2004, 5, 958–964 CrossRef CAS PubMed.
- X. Hu, X. Wang, J. Rnjak, A. S. Weiss and D. L. Kaplan, Biomaterials, 2010, 31, 8121–8131 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Scheme S1, Fig. S1–14, Tables S1–S3. See DOI: 10.1039/c8py00034d |
| This journal is © The Royal Society of Chemistry 2018 |