
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Temperature responsive poly(phosphonate) copolymers: from single chains to macroscopic coacervates†
Thomas
Wolf
a,
Johannes
Hunold
b,
Johanna
Simon
a,
Christine
Rosenauer
a,
Dariush
Hinderberger
 b and
Frederik R.
Wurm
*a
b and
Frederik R.
Wurm
*a
aMax-Planck-Institut für Polymerforschung, Ackermannweg 10, 55128 Mainz, Germany. E-mail: wurm@mpip-mainz.mpg.de
bMartin-Luther-Universität Halle-Wittenberg, Institut für Chemie, Von-Danckelmann-Platz 4, 06120 Halle (Saale), Germany
First published on 19th December 2017
Abstract
We present the first series of random poly(ethylene alkyl phosphonate) copolymers with either high solubility in water or a finely tunable hydrophilic-to-hydrophobic phase transition upon heating (“LCST”). Polymerization via the organocatalytic anionic ring-opening polymerization provided high control over molecular weight (up to 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) and copolymer composition, and resulted in narrow molecular weight distribution (Đ < 1.3). Polymers of molecular weights up to 23000 g mol−1 were obtained. The phase separation temperature was precisely adjusted in a range from 55 °C to 6 °C in water, depending on the copolymer composition. The phase transition mechanism was thoroughly investigated at different length scales via electron paramagnetic resonance spectroscopy (EPR), dynamic light scattering (DLS), UV-Vis spectroscopy and confocal laser scanning microscopy (cLSM), proving the step-wise formation of aggregates close to the cloud point temperature up to macroscopic coacervates.
000 g mol−1) and copolymer composition, and resulted in narrow molecular weight distribution (Đ < 1.3). Polymers of molecular weights up to 23000 g mol−1 were obtained. The phase separation temperature was precisely adjusted in a range from 55 °C to 6 °C in water, depending on the copolymer composition. The phase transition mechanism was thoroughly investigated at different length scales via electron paramagnetic resonance spectroscopy (EPR), dynamic light scattering (DLS), UV-Vis spectroscopy and confocal laser scanning microscopy (cLSM), proving the step-wise formation of aggregates close to the cloud point temperature up to macroscopic coacervates.
Introduction
Responsive materials, that are substances showing a property change upon an external stimulus, are of interest for a variety of applications. They are especially well-studied in the field of “smart” drug carrier development.1–5 Here, polymers have to be water-soluble, biocompatible, non-toxic, and preferably degradable and in addition they should be responsive towards certain triggers. Thermoresponsive polymers are promising materials for the development of drug carriers with the temperature-controlled release of the payload or other “smart” devices. Such polymers undergo a change in their hydrophilicity upon heating and phase separate from an aqueous solution. Especially in sophisticated biomedical applications, a detailed understanding of the different stages of the phase separation is essential to prevent, e.g., premature release of cargo or the formation of large aggregates.Most temperature responsive polymers exhibit a hydrophilic–hydrophobic phase transition upon heating (lower critical solution temperature, LCST) in a defined solvent (water in most cases). The phase separation occurs due to a loss of solvent–polymer hydrogen bonding at elevated temperatures.6–8 As a result, the inter- and intra-chain hydrophobic–hydrophobic interactions dominate and the polymer chain collapses. This is accompanied by an increase of entropy of liberated water molecules previously bound to the polymer.9,10 In addition to the abovementioned criteria, a sharp, reversible and hysteresis-free phase transition is desirable to guarantee a fast and precise response of the material. To prevent premature reactions upon a wrong or insufficient stimulus, a precise adjustment of the phase separation temperature and detailed understanding of the underlying mechanism are essential. To this end elaborate analytical techniques (e.g. UV-Vis, EPR spectroscopy, DLS) can be used to gain a better understanding of these mechanisms on different length scales.11,12
One class of polymers receiving increased attention as candidates in biomedical applications are poly(phosphoester)s (PPEs).13–17 The phosphoric acid ester bonds can be cleaved hydrolytically, eliminating the threat of accumulation of the polymeric material in the body.18–22 Polymers of adjustable hydrophilicity are accessible via different chemistries, and due to the pentavalent phosphorus an additional chemical functionality can be installed into every repeat unit.16,17,23 The anionic ring-opening polymerization (AROP) of cyclic phosphates gives fast access to water-soluble PPEs with high control over the molecular weight; however, transesterification reactions need to be considered.24,25 To date, a variety of PPEs exhibiting LCST behavior have been reported.26–28 Recently, we developed poly(ethylene alkyl phosphonate)s (PPns) that are aliphatic poly(phosphoester)s with a phosphorus–carbon bond in the side-chain, which might be of interest for biomedical applications. These polymers are accessible via the organocatalytic ROP of 2-alkyl-2-oxo-1,3,2-dioxaphospholanes providing excellent control over molecular weight and copolymer composition, and narrow molecular weight distributions.20,21,29
First studies have shown that the polymers are water-soluble, non-toxic and their properties can be finely tuned by variation of the pendant side-group.20,21 In contrast to poly(phosphate) synthesis, transesterification side-reactions during the synthesis of poly(phosphonate)s are less pronounced, allowing living polymerization up to high conversions.15,20,21 Copolymerization of 2-alkyl-2-oxo-1,3,2-dioxaphospholanes was shown to produce random copolymers with material properties (solubility, Tg) dependent on the copolymer composition.29,30
In this work, we study the LCST behavior of a series of novel copolymers of poly(ethylene alkyl phosphonate)s. Polymers with molecular weights between 7700 and 23000 g mol−1, narrow molecular weight distributions and adjustable copolymer compositions were prepared. The phase separation temperature of the resulting copolymers was dependent on the copolymer composition. Copolymers with less than 50 mol% or 30 mol% of the two hydrophobic co-monomers, respectively, were water-soluble and non-thermoresponsive. Above these values phase transitions upon heating an aqueous solution was observed with adjustable macroscopic cloud points between 6 °C and 55 °C in the current set of polymers.
To get a deeper understanding of the phase transition of these polymers, a combination of electron paramagnetic resonance spectroscopy (EPR), dynamic light scattering (DLS), UV-Vis spectroscopy, and confocal laser scanning microscopy (cLSM) was used to analyze the phase transition of these copolymers (Scheme 1). The results of this study will allow the future design of fully degradable nano- or micro-carrier devices based on PPEs.
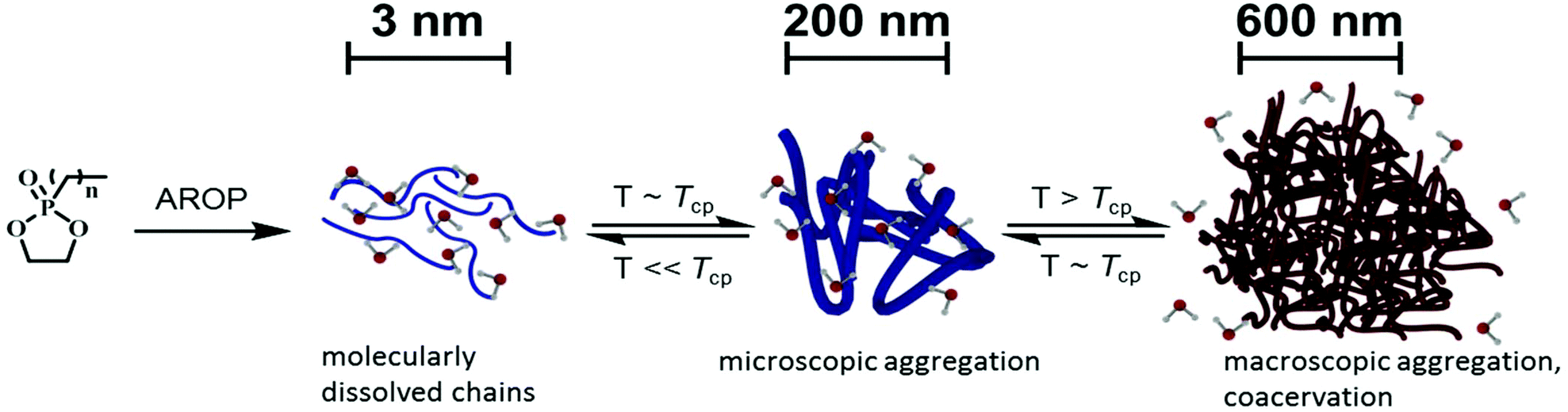 | ||
| Scheme 1 Schematic presentation of this study: anionic ring-opening copolymerization produces well-defined, water-soluble random copolymers. Phase separation behavior of the copolymers in the aqueous medium is investigated through various methods. Formation of nano-inhomogeneities and microscopic aggregates close to the cloud point temperature (Tcp) observed via CW EPR and DLS measurements, respectively. Macroscopic aggregation above the Tcp and the formation of coacervates visualized via turbidity measurements and confocal laser scanning microscopy. | ||
Results and discussion
Monomer synthesis
To generate a library of temperature-responsive poly(ethylene alkyl phosphonate)s, three different five-membered cyclic phosphonates for AROP were synthesized (Scheme 2a). Monomers (1) and (2) were synthesized according to the literature.20 Monomer (3) with a hydrophobic n-hexyl side-chain was newly developed for the current study (Scheme 2b): the Michaelis–Arbuzov reaction of n-hexyl bromide and triethyl phosphite yielded O,O-diethyl n-hexyl phosphonic acid diethyl ester. Conversion to the respective phosphonic acid dichloride was achieved by chlorination with thionyl chloride. Finally, condensation with ethylene glycol provided the cyclic monomer (3). Detailed experimental description, as well as analytical data, can be found in the ESI (Fig. S1–S6†).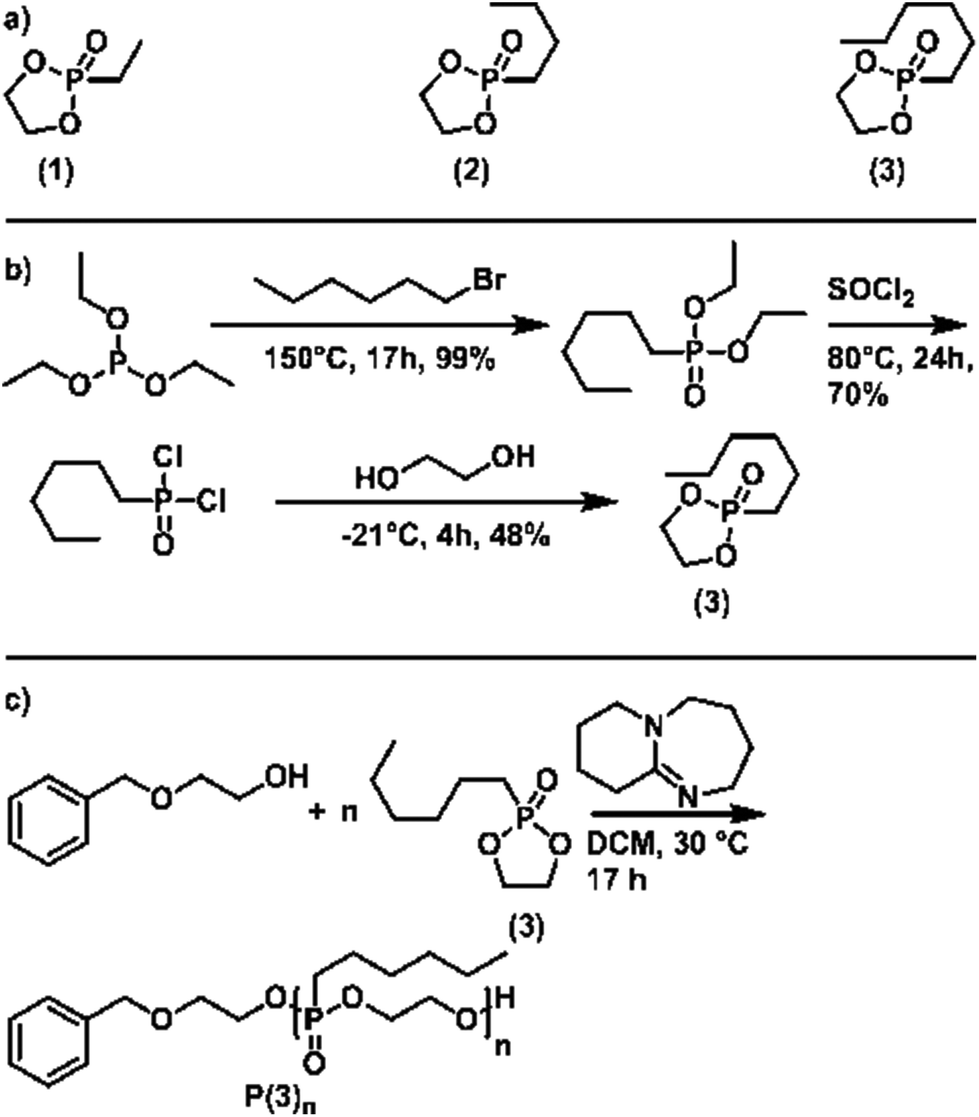 | ||
| Scheme 2 (a) Structure of the 2-alkyl-2-oxo-1,3,2-dioxaphospholane monomers used in this study. (b) Synthesis scheme of 2-n-hexyl-2-oxo-1,3,2-dioxaphospholane (3), starting with the Michaelis–Arbuzov reaction between triethyl phosphite and n-butyl bromide, followed by chlorination with thionyl chloride, and ring-closure with ethylene glycol. (c) Reaction scheme for the 2-(benzyloxy)ethanol-initiated and DBU-catalyzed AROP of (3) to produce P(3)n. | ||
Homopolymerization
First, the polymerization behavior of 2-n-hexyl-2-oxo-1,3,2-dioxaphospholane (3) as well as the respective properties of P(3)n were investigated. Following previously reported results regarding the AROP of dioxaphospholanes, nHexPPn was polymerized in dichloromethane (DCM) with 1,8-diazabicyclo-[5.4.0]undec-7-ene (DBU) as a catalyst at 30 °C with 2-(benzyloxy)ethanol as an initiator, which allowed end-group analysis via1H NMR spectroscopy (Scheme 2c).20Successful polymerization was indicated from the 1H NMR spectra by the broad resonance of the backbone protons ranging from 4.22 to 4.01 ppm after 17 h of polymerization time (Fig. 1a, signal c). Mn was determined via end-group analysis, by a comparison of the initiator signals at 7.26 ppm with the backbone resonances. The accuracy of this method is limited for high molecular weight polymers, but has been proven for PPns with Pn up to 200.21 The 31P{H} spectrum (Fig. 1a, inset) reveals a distinct shift from the monomer (51.4 ppm) to a polymer with two phosphorus signals originating from the backbone (Fig. 1, signal 1, 33.5 ppm) and the terminal phosphorus (Fig. 1, signal 1′, 32.9 ppm).
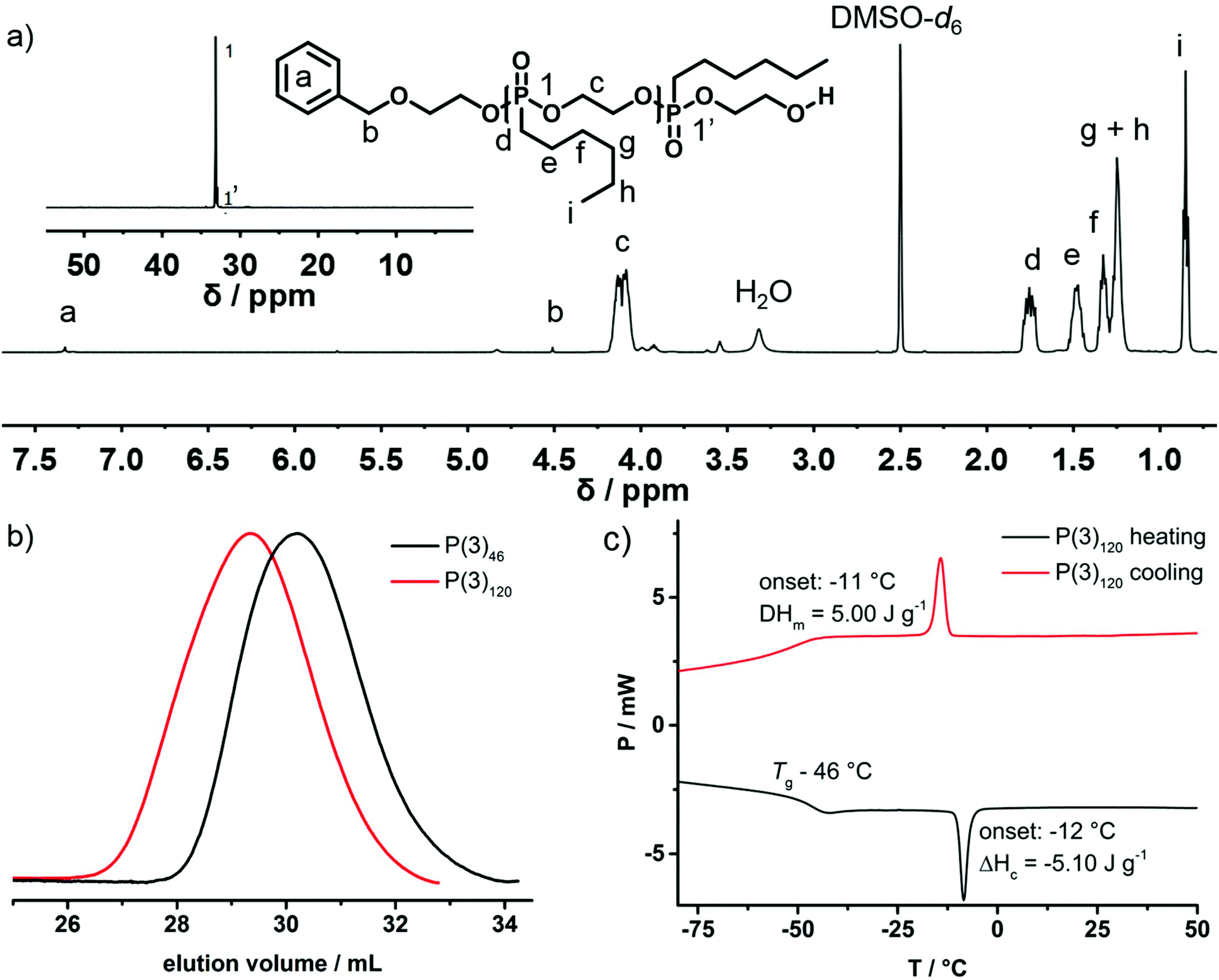 | ||
| Fig. 1 (a) 1H NMR (500 MHz) and 31P{H} NMR (201 MHz) (inset) spectra of P(3)120 in DMSO-d6 at 298 K. (b) SEC elugrams (RI detection) of P(3)120 and P(3)46 in DMF at 333 K. (c) DSC thermogram of P(3)120 at a heating rate of 10 °C min−1 in the temperature range from −80 °C to 50 °C (second heating and cooling curves shown). | ||
SEC analysis (Fig. 1b) shows symmetrical monomodal molecular weight distributions (Đ ∼ 1.20; vs. PEG standards). The molecular weight of the polymer was determined from NMR spectra, but not via SEC, as the solvation behavior and dynamic radii between the PEG standard and PPns are very different. The polymer is insoluble in water but can be readily dissolved in organic solvents such as diethyl ether, dichloromethane, chloroform, dimethyl sulfoxide, or dimethylformamide. Thermal analysis via DSC (Fig. 1c) reveals a low glass transition temperature (Tg = −46 °C) similar to those of previously reported poly(alkylene alkyl phosphonate)s. Additionally, a melting signal with the onset at −10 °C and an enthalpy of fusion of 5 J g−1 is observed, indicating the first side-chain crystallization observed for poly(ethylene alkyl phosphonates)s from AROP.20,21 The polymerizability of monomer (3) ensured that we proceeded with the synthesis of the thermoresponsive poly(ethylene alkyl phosphonate) copolymers P(1x-co-2y) and P(1x-co-3y).
Copolymerization
The organocatalytic anionic ring-opening copolymerization of monomer (1), resolution in hydrophilic repeat units, with either monomer (2) or (3) (each producing hydrophobic units) was performed at 30 °C with DBU as the respective catalyst in a one-pot reaction for 17 h (Scheme 3).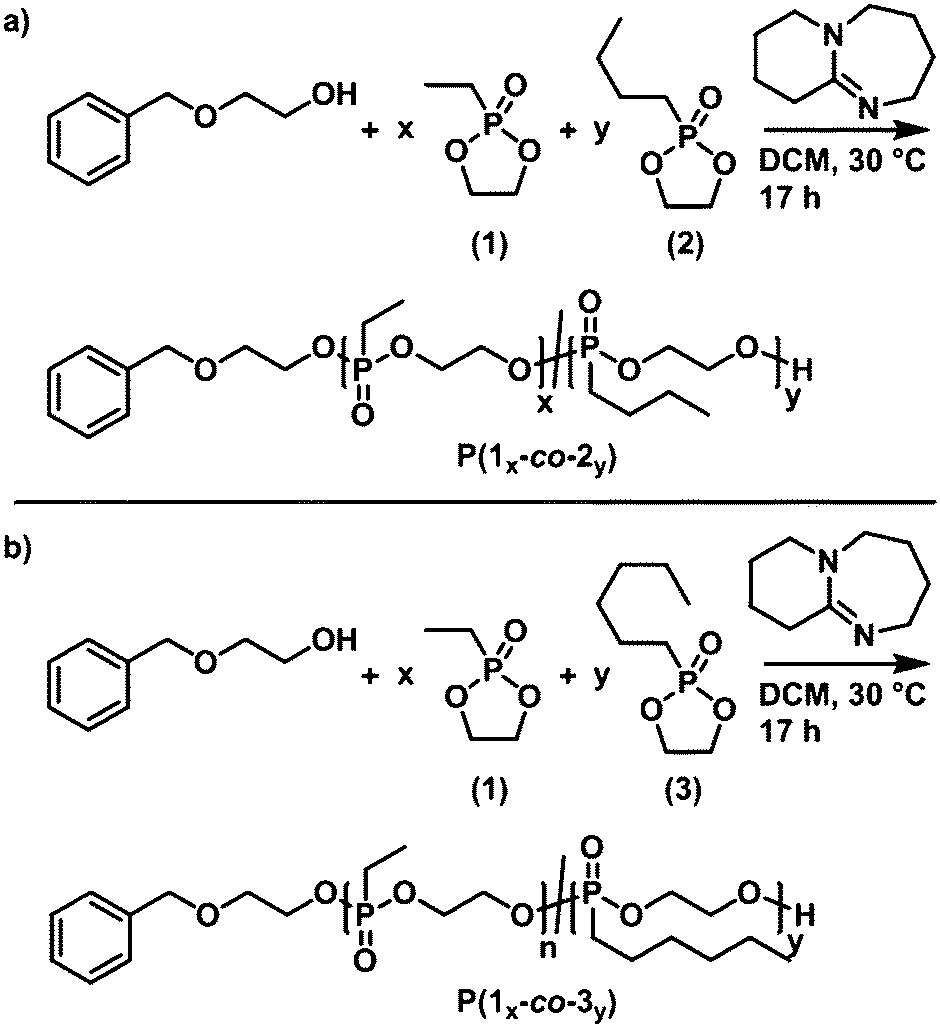 | ||
| Scheme 3 Reaction scheme for the 2-(benzyloxy)ethanol-initiated and DBU catalyzed anionic ring-opening copolymerization of (1) with (a) (2) to form P(1x-co-2y) and with (b) (3) to form P(1x-co-3y). | ||
The representative 1H and 31P{H} NMR spectra of P(1x-co-2y) and P(1x-co-3y) are shown in Fig. 2. Molecular weights can be determined by the end-group analysis of the 1H NMR spectra (vide supra). Polymers with molecular weights ranging from 7700 to 23000 g mol−1 were obtained. 1H NMR spectroscopy further provided information on the copolymer composition in the final material.
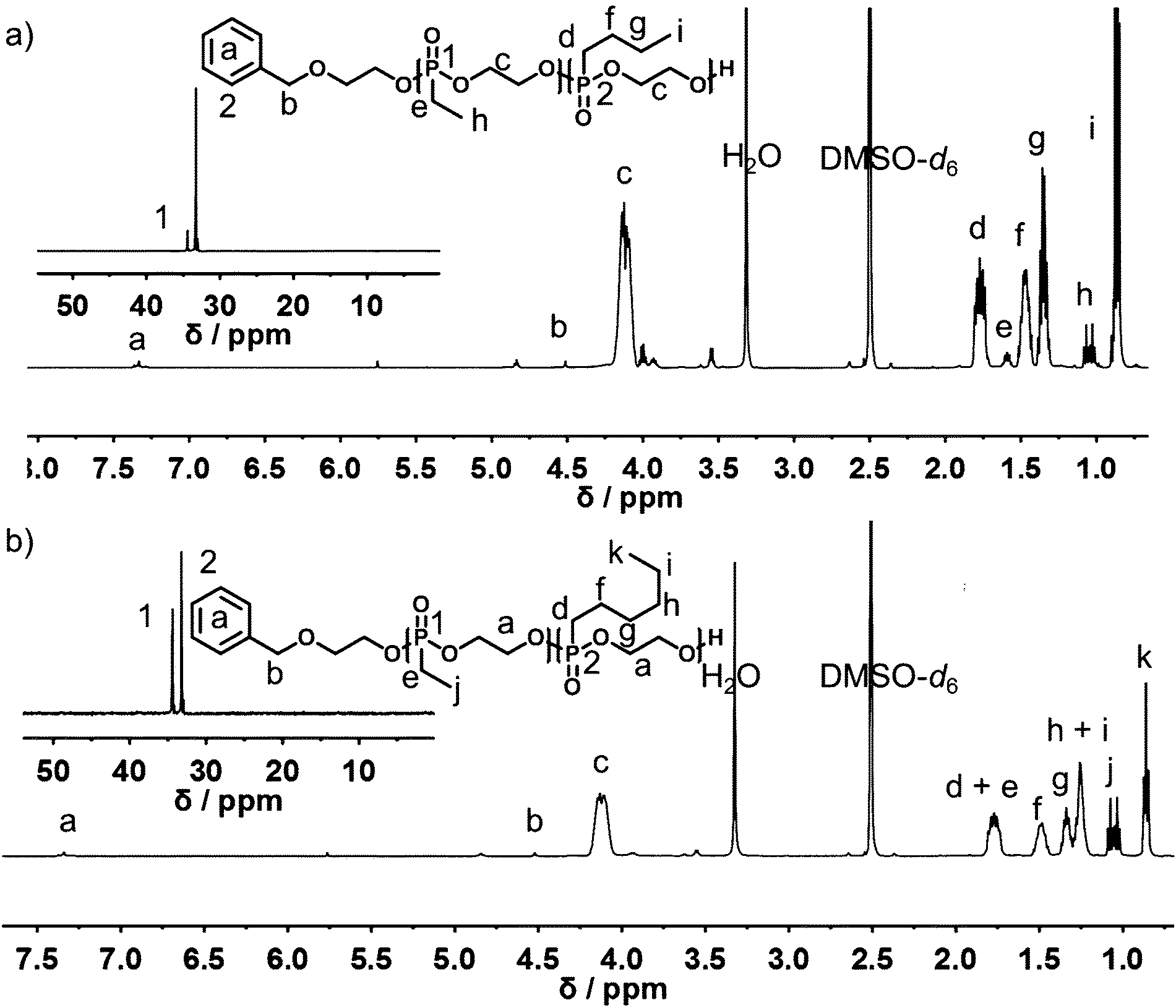 | ||
| Fig. 2 1H NMR (500 MHz) and 31P{H} NMR (201 MHz, inset) spectra of (a) P(115-co-2118) and (b) P(162-co-356) in DMSO-d6 at 298 K. | ||
The resonance of the terminal –CH3 side-chains of (1) at 1.06 ppm is well separated from the respective terminal –CH3 group resonances of (2) and (3) at 0.86 ppm, allowing the calculation of the copolymer composition. In addition to 1H NMR spectroscopy, the comparison of the relative intensities of the backbone resonances in the 31P{H} NMR spectra at 34.4 ppm for (1) and 33.2 ppm for (2) and (3), respectively, also reveals the copolymer composition (Fig. 2, insets). The experimentally found polymer composition matched the monomer feed ratios in all cases, both from 1H or 31P NMR analysis. 1H DOSY NMR spectra (Fig. S10†) further prove successful copolymerization, as all relevant resonances, i.e. -backbone and respective side-chains, have the same diffusion coefficient and are part of the same molecule. All polymers regardless of molecular weight or copolymer composition showed symmetrical monomodal SEC traces with rather narrow molecular weight distribution (1.18 < Đ < 1.30; vs. PEG standard) (Fig. S12 and S14†).
The microstructure of the copolymers is expected to have a significant influence on the solubility and phase separation behavior. Therefore, the copolymerization kinetics of (1) with (2) and (3), respectively, were analyzed via31P{H} NMR spectroscopy.
Fig. S15† shows the molar fraction of (1) integrated into the respective copolymer at different times during the copolymerization. Starting from the initial 1:1 ratio of the monomer feed ([A]/([A] + [B]) = 0.5), the copolymer composition remains constant for the whole duration of both copolymerizations. This indicates that all co-monomers are incorporated in the copolymer at the same rate and have the same reactivity, thus resulting in the formation of a random copolymer without any detectable gradients.
DSC analysis shows that the side-chain crystallinity observed for P(3)120 vanishes for the copolymers resulting in completely amorphous materials (Fig. S16 and S17†). Cell-toxicity measurements against the macrophage cell-line RAW 264.7 showed a concentration-dependent cell-toxicity for P(151-co-251) and P(141-co-316). The toxicity was negligible for pharmaceutically suitable concentrations (<100 μg mL−1). Above 250 μg mL−1, a reduction in the viability is observed (Fig. S18†). The effect was more pronounced for the more hydrophobic copolymers P(141-co-316). A similar trend was previously reported for other poly(ethylene alkyl phosphonate)s with hydrophobic side-chains.20 The analytical data of all copolymers are summarized in Tables S1 and S2.†
Thermal behavior in solution
The solubility and thermal behavior of the copolymers in water were thoroughly investigated. First, turbidity measurements were performed to investigate the macroscopic solution behavior. Copolymers with low amounts of the hydrophobic comonomers exhibited high solubility in water (>10 g L−1); however, polymers containing more than 50 mol% of (2) or 30 mol% of (3), respectively undergo a phase separation upon heating. The cloud point temperatures (Tcp) were tailored over a broad temperature range from 6 °C to 55 °C in the current copolymer set (Fig. 3a and b). A linear dependency between the Tcp and a number of hydrophobic side-chains incorporated in the copolymer was found (Fig. 3c). In all cases, little to no hysteresis occurred during the cooling of the samples, indicating a well-reversible phase transition.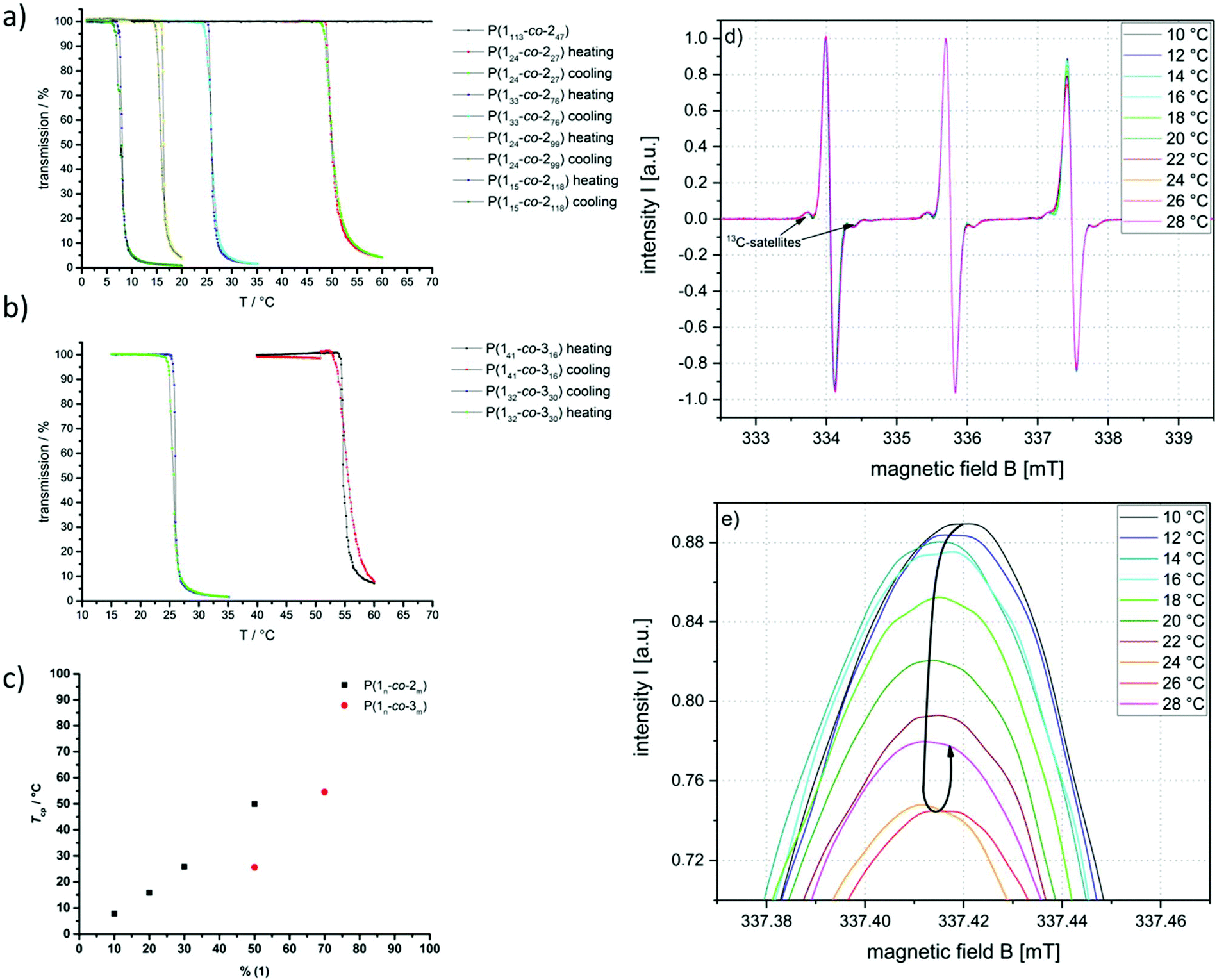 | ||
| Fig. 3 Thermal response of poly(ethylene alkyl phosphonate) copolymers: turbidity measurements (heating and cooling curves) of (a) P(1x-co-2y) and (b) P(1x-co-3y) in ultrapure water at a concentration of 10 g L−1 and a heating/cooling rate of 1 °C min−1. The transmission was measured at 500 nm. (c) Correlation between comonomer ratio and Tcp. CW EPR spectra of an aqueous solution of 15 mg mL−1P(133-co-276) with 0.2 mM TEMPO in DPBS buffer. (d) Full spectra and (e) enlarged high-field region (full high-field peak see Fig. S21†). | ||
In accordance with the literature, the LCST cloud point temperatures were found to be dependent on the ionic strength of the solution.31 The addition of sodium phosphate (0.1 M PBS, pH 7.4) shifted the phase transition by 6 °C to lower temperatures (Fig. S19†). This is in good accordance with the theory of Hofmeister et al. with regard to the antichaotropic effect of sodium ions.31
To gain a more detailed understanding of the phase separation behavior of poly(ethylene alkyl phosphonate)s, three additional methods were used, each using different length scales during the phase separation process: EPR spectroscopy was employed to detect the formation of nano-dimensional structural inhomogeneities. Then, dynamic light scattering was employed to analyze the formation of larger aggregates prior to the macroscopic precipitation of the solution visible by UV-Vis turbidity measurements. Finally, confocal laser scanning microscopy visualized the macroscopic aggregates and analyzed their structures.
The CW EPR spectra recorded during the heating of an aqueous solution of 15 mg mL−1 of P(133-co-276) with 0.2 mM 2,2,6,6,-tetramethylpiperidin-1-oxyl (TEMPO) in DPBS buffer are shown in Fig. 3d and e. Under these conditions, which were optimized for the CW EPR measurements, macroscopic phase separation occurred at 25 °C. EPR spectroscopy is a method for studying materials with an unpaired electron, like the spin probe TEMPO. The method is sensitive towards the chemical environment of the probe and minuscule changes in, e.g., hydrophilicity of the surrounding medium can be detected and characterized.32–34 It can be seen in Fig. 3 that the intensity and the line shape of the high-field peak change with increasing temperature. In the range between 10 and 16 °C, the intensity of the high-field line first decreases slightly when the temperature increases, which could e.g. indicate a slight rise of the nanoviscosity around the spin probe or a slight decrease in polarity in the spin probe environment experienced by some of the spin probes.
If, however, the temperature is only increased further by just 2 °C (from 16 to 18 °C), the signal intensity maximum decreases significantly. The reason for the observed intensity decrease of the high-field line is the simultaneous appearance of a second, slightly less polar solvated, probe species at 18 °C. This spin probe species shows a slightly smaller hyperfine splitting constant A (i.e. its high-field line appears at slightly lower magnetic field values) indicating a lower spin probe environmental polarity. Since the hyperfine coupling values of this second, “hydrophobic”, species (∼47 MHz) are still very high compared to those of these species in other, more conventional LCST type polymers (∼44–45 MHz), we can conclude that the less polar polymer-rich regions, probed by the appearance of the second spin probe species, are still water swollen (A in bulk water: ∼48 MHz).11,32–34 As seen in these other LCST-type polymers, CW EPR spectroscopy on TEMPO can detect small, nanoscopically dehydrated regions at temperatures below the cloud point.11 Under the same conditions, no macroscopic aggregation was observed in dynamic light scattering measurements, even at 20 °C (Fig. S20†). This shows that our EPR spectroscopic scheme adds molecular insights that DLS cannot provide for the investigation of the phase separation close to the LCST.
In these LCST polymers, we can distinguish two types of water: one is bulk water with the relatively freely tumbling hydrophilic probe species with comparatively high environmental polarity. The other one is polymer-associated water that forms the hydration shell around the polymer chains where the more hydrophobic probe species with a reduced environmental polarity is located. Thus, at 6–7 °C below the macroscopic cloud point temperature Tcp of about 25 °C a phase separation between still fully solvated and less solvated yet still water-swollen polymer regions on the nanoscale could be observed by EPR spectroscopy. These nanoscopic inhomogeneities continue to grow at a further temperature increase so that the polymer chains are more and more dehydrated (and yet still water swollen in comparison with conventional LCST polymers). As an additional result of this dehydration process, the effective nanoviscosity of the spin probe (i.e. the viscosity, which the probe senses in a radius of about 2–3 nm around it) increases, and also its mobility is reduced. Both effects together lead to a steady signal intensity decrease of the high-field peak, until it reaches its minimum at 24–26 °C, which corresponds quite well to the Tcp. Considering that the TEMPO probes have a van der Waals-radius of about 0.31–0.35 nm (ref. 35) the EPR spectroscopically detected nanoscopic inhomogeneities should not be much larger in diameter than about 5–6 nm. Otherwise, these inhomogeneities would no longer be perceived as such by the relatively small TEMPO probes.11,32
A temperature increase beyond 26 °C leads to a renewed increase in the signal intensity of the high-field peak. This indicates that the polymer chains are further dehydrated (as the phase separation process progresses with the temperature increase), but now some of the hydrophobic probe species are expelled from the inhomogeneities along with polymer-associated water. Consequently, the (spectral) proportion of relatively freely tumbling hydrophilic spin probe species and also the signal intensity of the high field peak increase again.
To further investigate the aggregation during the phase separation process, temperature-dependent DLS measurements were performed. P(124-co-227) with the Tcp at 50 °C (10 g L−1, H2O) was chosen for these experiments, as precise heating of the DLS cell was only viable in a temperature range from 30 to 60 °C in our setup. Fig. 4a shows the hydrodynamic radii measured in the light scattering experiment (left, black) and the relative intensities (right, red) of the observed processes as a function of temperature. At 27 °C, below the Tcp, the majority of the polymers are molecularly dissolved with a small fraction of aggregates only. This can be concluded as there are only two processes observed: one process with a hydrodynamic radius of 1.5 nm corresponding to 70% of the total scattering intensity (molecularly dissolved polymer, circles) and the other process with a hydrodynamic radius of 50 nm corresponding to 30% of the scattering intensity (aggregates, triangles). Due to the strong size dependence of scattering intensity (I ∝ r3) the latter process scatters nearly 40000 times stronger than the 1.5 nm process. Therefore, below Tcp, a negligible fraction of the polymer is already present in an aggregated state while the majority is molecularly dissolved. Upon heating, the relative scattering intensities of these processes change when approaching the phase separation temperature: while keeping a constant radius, the scattering intensity of the 1.5 nm process drops to 12% at 50 °C. The radius and scattering intensity of the second process, however, gradually increase to 120 nm and 88%, respectively at 50 °C. Upon surpassing the Tcp a sharp and fast increase of the radius is observed up to large aggregates of Rh > 250 nm.
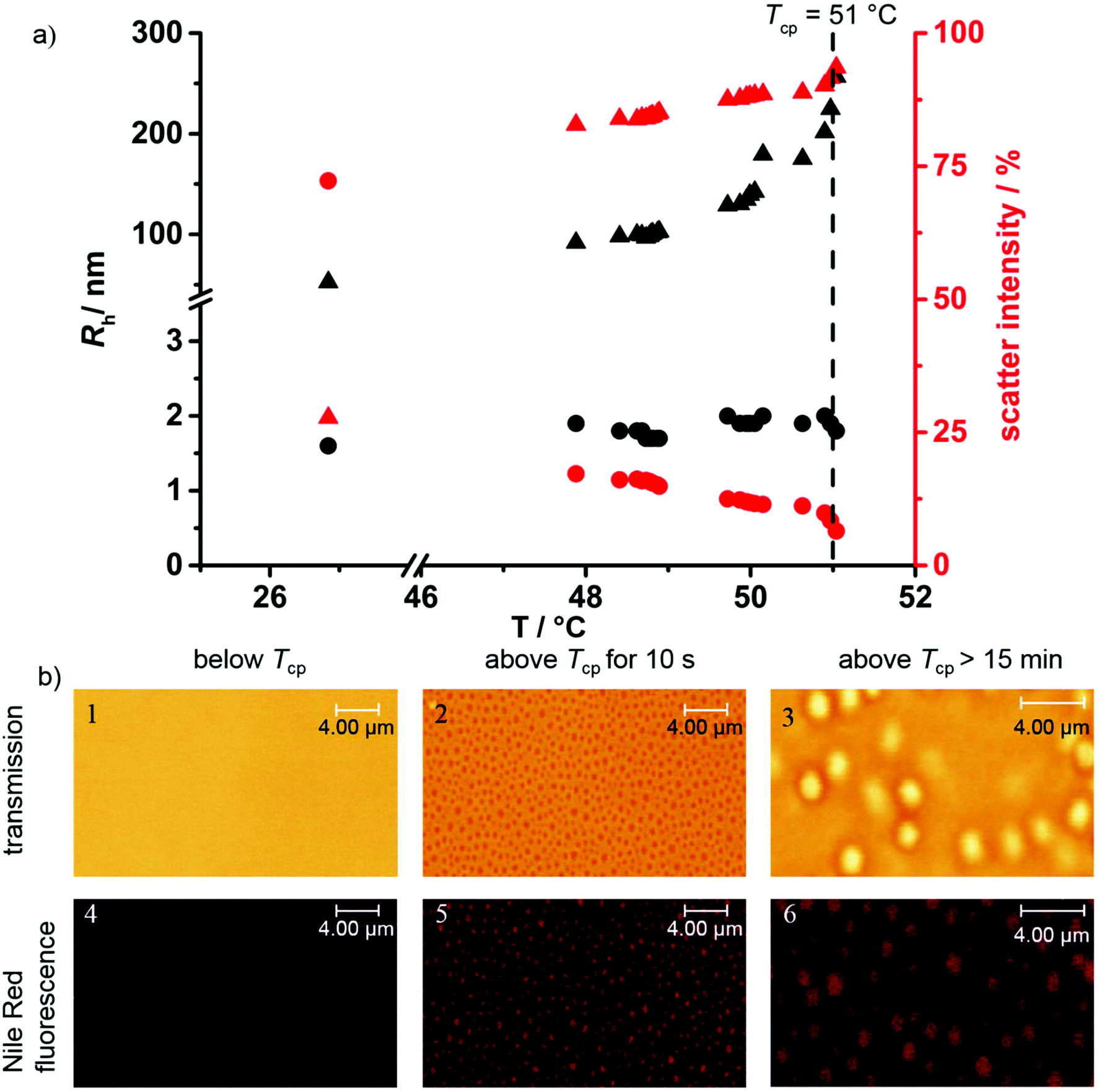 | ||
| Fig. 4 (a) Temperature-dependent DLS measurements of P(124-co-227). (b) Confocal laser scanning microscopy pictures of P(115-co-3118) (Tcp = 8 °C): top row: transmission channel below the Tcp (1), after surpassing the Tcp (2) and above the Tcp (3); bottom row: respective Nile Red fluorescence channel below (4), after surpassing (5) and above the Tcp (6). | ||
Such large structures are easily observed by confocal laser scanning microscopy (cLSM). The respective cLSM pictures of P(115-co-3118) at different temperatures are shown in Fig. 4b. The investigated polymer was switched to a more hydrophobic polymer with a Tcp of 8 °C as the microscope could not be heated and needed to be cooled down during the measurement. In addition to conventional transmission spectroscopy to observe the structures of the aggregates the fluorescence of Nile Red, a solvatochromic dye with weak fluorescence in hydrophilic medium and strong fluorescence in a more hydrophobic environment, was measured. For this purpose, Nile Red was co-dissolved in the aqueous polymer solution below Tcp and heated during the measurement. The concentration of the dye was kept as low as possible (1 μg mL−1) to prevent interactions between the responsive polymer and the dye. However, even at considerably higher concentrations, no change in the solubility behavior of the polymer was observed. Below the Tcp of the copolymer, a clear solution with little fluorescence was observed in the transmission and Nile Red channel, indicating a homogeneous hydrophilic environment under this resolution. This indicates that the subtleties of pre-collapsed dehydration as seen in CW EPR are not detectable with the much larger and more hydrophobic probes. Yet, to characterize the large aggregates formed, this method complements the few nm scale characterization by EPR spectroscopy. If the temperature is raised above the Tcp of the polymer well-defined spherical structures with diameters of ca. 600 nm abruptly become visible. These structures show strong Nile Red fluorescence indicating a less hydrophilic environment compared to the surrounding water phase. This is in good accordance with the observation of Rh = 250 nm aggregates observed from DLS measurements. These aggregates grew in size over time due to coalescence and eventually formed perfectly spherical coacervate droplets with a diameter of ca. 2000 nm, as already observed for a related system.30
With these results, we can propose a mechanism for the LCST phase separation of these poly(ethylene alkyl phosphonate) copolymers.
At low temperatures, the major fraction of the copolymer is molecularly dissolved in water as unimers. Upon increasing the temperature, the steadily decreasing polymer–water hydrogen bond interaction results in the formation of nanoscopic polymer inhomogeneities, as visualized by CW EPR spectroscopy. A further increase in temperature leads to complete dehydration of the polymer and the formation of aggregates (Rh < 100 nm) is detectable by DLS. These slowly increase in size till a threshold temperature is reached upon which a rapid phase separation occurs into a polymer free water-phase and large polymer aggregates (Rh ∼ 250 nm). Macroscopically, this results in the clouding of the solution and the detection of Tcp by transmission spectroscopy. Over time, coalescence of the aggregates results in the formation of large macroscopic droplets and complete phase separation.
Conclusions
In summary, we studied the hydrophilic-to-hydrophobic phase transition of thermo-responsive aliphatic poly(phosphonate)s on different length scales: the nanometer-sized regime was studied by CW EPR spectroscopy, revealing two types of water: first, bulk water with the freely tumbling hydrophilic probe and second, polymer-associated water that forms the hydration shell around the polymer chains where the more hydrophobic probe species with a reduced environmental polarity is located. Upon reaching/exceeding a critical temperature, fast aggregate growth and the formation of macroscopic droplets were observed (by dynamic light scattering and confocal microscopy) resulting in the clouding of the solution (quantified by turbidity measurements). The LCST behavior was studied based on a series of poly(ethylene alkyl phosphonate) copolymers, which were prepared by the ring-opening polymerization of 2-alkyl-2-oxo-1,3,2-dioxaphospholanes with ethyl, butyl, and hexyl side chains. Random copolymers with molecular weights between 7700 and 23000 g mol−1 and molecular weight distributions between 1.18 and 1.30 were prepared, which exhibited a thermo-responsive behavior over a broad temperature range depending on the copolymer composition. The results of this study give a deeper insight into the LCST phase transition in PPE-copolymers and will allow the future design of fully degradable thermo-responsive materials based on PPEs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Angelika Manhart (MPIP) for synthetic assistance, Manfred Wagner (MPIP) for NMR measurements, Anke Kaltbeitzel (MPIP) for assistance with the cLSM measurements, and Franziska Zeuner and Heike Schimm (both MLU) for technical support. The authors acknowledge the support from the “Deutsche Forschungsgemeinschaft” (DFG WU 750/6-1) and the Fonds der Chemischen Industrie (FCI, JH and DH). Open Access funding provided by the Max Planck Society.References
- B. D. Ratner and S. J. Bryant, Annu. Rev. Biomed. Eng., 2004, 6, 41–75 CrossRef CAS PubMed.
- S. R. Meyers and M. W. Grinstaff, Chem. Rev., 2012, 112, 1615–1632 CrossRef CAS PubMed.
- S. Anastase-Ravion, Z. Ding, A. Pelle, A. S. Hoffman and D. Letourneur, J. Chromatogr. B: Biomed. Sci. Appl., 2001, 761, 247–254 CrossRef CAS.
- H. Kanazawa, K. Yamamoto, Y. Matsushima, N. Takai, A. Kikuchi, Y. Sakurai and T. Okano, Anal. Chem., 1996, 68, 100–105 CrossRef CAS PubMed.
- C. de Las Heras Alarcon, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- S. Nayak, H. Lee, J. Chmielewski and L. A. Lyon, J. Am. Chem. Soc., 2004, 126, 10258–10259 CrossRef CAS PubMed.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2009, 48, 7978–7994 CrossRef CAS PubMed.
- M. Sahn, T. Yildirim, M. Dirauf, C. Weber, P. Sungur, S. Hoeppener and U. S. Schubert, Macromolecules, 2016, 49, 7257–7267 CrossRef CAS.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS PubMed.
- F. D. Jochum and P. Theato, Chem. Soc. Rev., 2013, 42, 7468–7483 RSC.
- D. Kurzbach, M. J. Junk and D. Hinderberger, Macromol. Rapid Commun., 2013, 34, 119–134 CrossRef CAS PubMed.
- J. Herzberger, D. Kurzbach, M. Werre, K. Fischer, D. Hinderberger and H. Frey, Macromolecules, 2014, 47, 7679–7690 CrossRef CAS.
- T. Steinbach and F. R. Wurm, Biomacromolecules, 2016, 17, 3338–3346 CrossRef CAS PubMed.
- F. Zhang, S. Zhang, S. F. Pollack, R. Li, A. M. Gonzalez, J. Fan, J. Zou, S. E. Leininger, A. Pavia-Sanders, R. Johnson, L. D. Nelson, J. E. Raymond, M. Elsabahy, D. M. Hughes, M. W. Lenox, T. P. Gustafson and K. L. Wooley, J. Am. Chem. Soc., 2015, 137, 2056–2066 CrossRef CAS PubMed.
- T. Steinbach and F. R. Wurm, Angew. Chem., Int. Ed., 2015, 54, 6098–6108 CrossRef CAS PubMed.
- G. Becker, T. A. Marquetant, M. Wagner and F. R. Wurm, Macromolecules, 2017, 50(20), 7852–7862 CrossRef CAS.
- K. N. Bauer, H. T. Tee, M. M. Velencoso and F. R. Wurm, Prog. Polym. Sci., 2017, 73, 122–161 CrossRef.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed.
- H. Lin, T. Wolf, F. R. Wurm and M. A. Kelland, Energy Fuels, 2017, 31, 3843–3848 CrossRef CAS.
- T. Wolf, T. Steinbach and F. R. Wurm, Macromolecules, 2015, 48, 3853–3863 CrossRef CAS.
- T. Steinbach, S. Ritz and F. R. Wurm, ACS Macro Lett., 2014, 3, 244–248 CrossRef CAS.
- H. Wang, L. Su, R. Li, S. Zhang, J. Fan, F. Zhang, T. P. Nguyen and K. L. Wooley, ACS Macro Lett., 2017, 6, 219–223 CrossRef CAS.
- G. Becker, L. M. Ackermann, E. Schechtel, M. Klapper, W. Tremel and F. R. Wurm, Biomacromolecules, 2017, 18(3), 767–777 CrossRef CAS PubMed.
- A. P. Dove, ACS Macro Lett., 2012, 1, 1409–1412 CrossRef CAS.
- O. Nuyken and S. D. Pask, Polymers, 2013, 5, 361–403 CrossRef.
- T. Steinbach, R. Schroder, S. Ritz and F. R. Wurm, Polym. Chem., 2013, 4, 4469–4479 RSC.
- Y.-C. Wang, Y. Li, X.-Z. Yang, Y.-Y. Yuan, L.-F. Yan and J. Wang, Tunable Thermosensitivity of Biodegradable Polymer Micelles of Poly(ε-caprolactone) and Polyphosphoester Block Copolymers, Macromolecules, 2009, 42(8), 3026–3032 CrossRef CAS.
- Y. Iwasaki, T. Kawakita and S.-i. Yusa, Chem. Lett., 2009, 38, 1054–1055 CrossRef CAS.
- T. Wolf, J. Nass and F. R. Wurm, Polym. Chem., 2016, 7, 2934–2937 RSC.
- T. Wolf, T. Rheinberger and F. R. Wurm, Eur. Polym. J., 2017, 95, 756–765 CrossRef CAS.
- F. Hofmeister, Arch. Exp. Pathol. Pharmakol., 1888, 24, 247–260 Search PubMed.
- M. J. Junk, W. Li, A. D. Schluter, G. Wegner, H. W. Spiess, A. Zhang and D. Hinderberger, Angew. Chem., Int. Ed., 2010, 49, 5683–5687 CrossRef CAS PubMed.
- D. Kurzbach, M. N. Reh and D. Hinderberger, ChemPhysChem, 2011, 12, 3566–3572 CrossRef CAS PubMed.
- M. J. N. Junk, W. Li, A. D. Schlüter, G. Wegner, H. W. Spiess, A. Zhang and D. Hinderberger, Macromol. Chem. Phys., 2011, 212, 1229–1235 CrossRef CAS.
- V. Bercu, M. Martinelli, C. A. Massa, L. A. Pardi and D. Leporini, J. Chem. Phys., 2005, 123, 174906 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures and additional analytic data. See DOI: 10.1039/c7py01811h |
| This journal is © The Royal Society of Chemistry 2018 |