
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface-attached poly(phosphoester)-hydrogels with benzophenone groups†
Greta
Becker
ab,
Zhuoling
Deng
c,
Maria
Zober
c,
Manfred
Wagner
a,
Karen
Lienkamp
*c and
Frederik R.
Wurm
 *a
*a
aMax-Planck-Institut für Polymerforschung, Ackermannweg 10, 55128 Mainz, Germany. E-mail: wurm@mpip-mainz.mpg.de
bGraduate School Materials Science in Mainz, Staudinger Weg 9, 55128 Mainz, Germany
cBioactive Polymer Synthesis and Surface Engineering Group, Department of Microsystems Engineering (IMTEK) and Freiburg Center for Interactive Materials and Bioinspired Technologies (FIT), Georges-Köhler-Allee 105, 79110 Freiburg, Germany. E-mail: lienkamp@imtek.uni-freiburg.de
First published on 19th December 2017
Abstract
Photo-reactive poly(phosphoester)s (PPEs) forming surface-attached PPE-networks and hydrogels are presented. These contain benzophenone groups, which can photochemically cross-link polymer films via a C,H-insertion cross-linking (CHiC) reaction. To make the target polymers, a benzophenone-functionalized cyclic phosphate monomer was synthesized and copolymerized with ethylene ethyl phosphate (EEP). Polymerization with EEP and further cyclic phosphates gave terpolymers with additional chemical functionality (e.g. furfuryl or alkene groups). The resulting PPE copolymers and terpolymers were water-soluble and UV-reactive. Copolymerization kinetics of up to three different comonomers were studied by real-time 31P NMR, indicating a gradient-like structure of the copolymers and terpolymers. Cross-linked, surface-attached PPE-networks were formed by spin-coating these polymers onto pre-functionalized substrates, followed by UV irradiation. The resulting surface-attached polymer networks were smooth and hydrophilic (static contact angles of 20–26°). Since PPEs are biocompatible, these networks might be potential anti-fouling coatings for biomedical devices as implants or catheters. The furan and alkenyl groups in two of the terpolymers present a selectively addressable chemical function to further tune the surface properties of PPE-films.
Introduction
Surfaces of biomedical devices (such as implants, biosensors, catheters, or carrier systems for drug delivery) often fail because of undesired adsorption of bacteria, proteins, and other biomolecules.1 Even the adsorption of only a few such molecules can trigger the formation of biofilms, which can cause severe systemic infections.1 Functional polymeric coatings exhibiting antifouling and/or antimicrobial properties are therefore currently intensively investigated.2–4Examples for substance classes that resist biofilm formation are hydrophobic poly(dimethylsiloxane) (PDMS)-based5 or fluorinated6 polymers, which might be used for coating of ship hulls or pipelines. They are classified as “fouling release” coatings: while they have a high interfacial energy with water, which enables amphiphilic proteins to attach to them, these biofoulants can be easily removed by shear stress, e.g. by stress due to hydrodynamic flow.7
“Non-fouling” coatings, on the other hand are typically hydrophilic polymers and have a low interfacial energy with water, leading to lower protein adsorption.7 Prominent protein-repellent examples include poly(ethylene glycol) (PEG)-based polymers, amphiphilic polymer coatings (including those with heterogeneities on the nanoscale prepared from block copolymers), oligosaccharide-containing polymers mimicking the glycocalyx of blood vessels, and zwitterionic polymers.4,7 PEG is particularly well-known to suppress protein adhesion on nanocarriers (“stealth effect”), which led to several PEGylated nanocarriers already in clinical use, and is also the gold standard for macroscopic surfaces with low protein adsorption.8,9 Polyzwitterionic surface-attached networks are also strongly protein-repellent;10–12 a polyzwitterion surface with protein repellency, cell-compatibility, and antimicrobial activity was also recently reported.13
The aim of this study was to explore the potential of poly(phosphoester) (PPE)-based surface-attached networks as potentially protein-repellent coatings. These form hydrogels when immersed into aqueous media. Many PPEs have been reported to be biocompatible and to have low cytotoxicity.14 In particular, poly(ethyl ethylene phosphate) (PEEP), which is hydrophilic15 and protein-repellent, can be considered as a degradable analog of PEG. For example, PPEylated nanoparticles or proteins that were obtained by grafting PPE chains exhibited reduced protein adsorption and low interaction with human immune cells and were thus very similar to PEGylated nanocarriers.9,16,17
To study the bioactivity of PPEs on macroscopic planar surfaces, we designed PPE-copolymers and terpolymers that can form surface-attached polymer networks by simultaneous photo-induced inter-chain crosslinking and surface attachment (Scheme 1). The advantage of this method of surface functionalization is that it is straightforward, efficient and that the thickness of the coating can be tuned over a wider range than when using other methods (e.g. grafting-from or grafting-onto methods, where the coating thickness depends on the molecular weight of the polymer used).
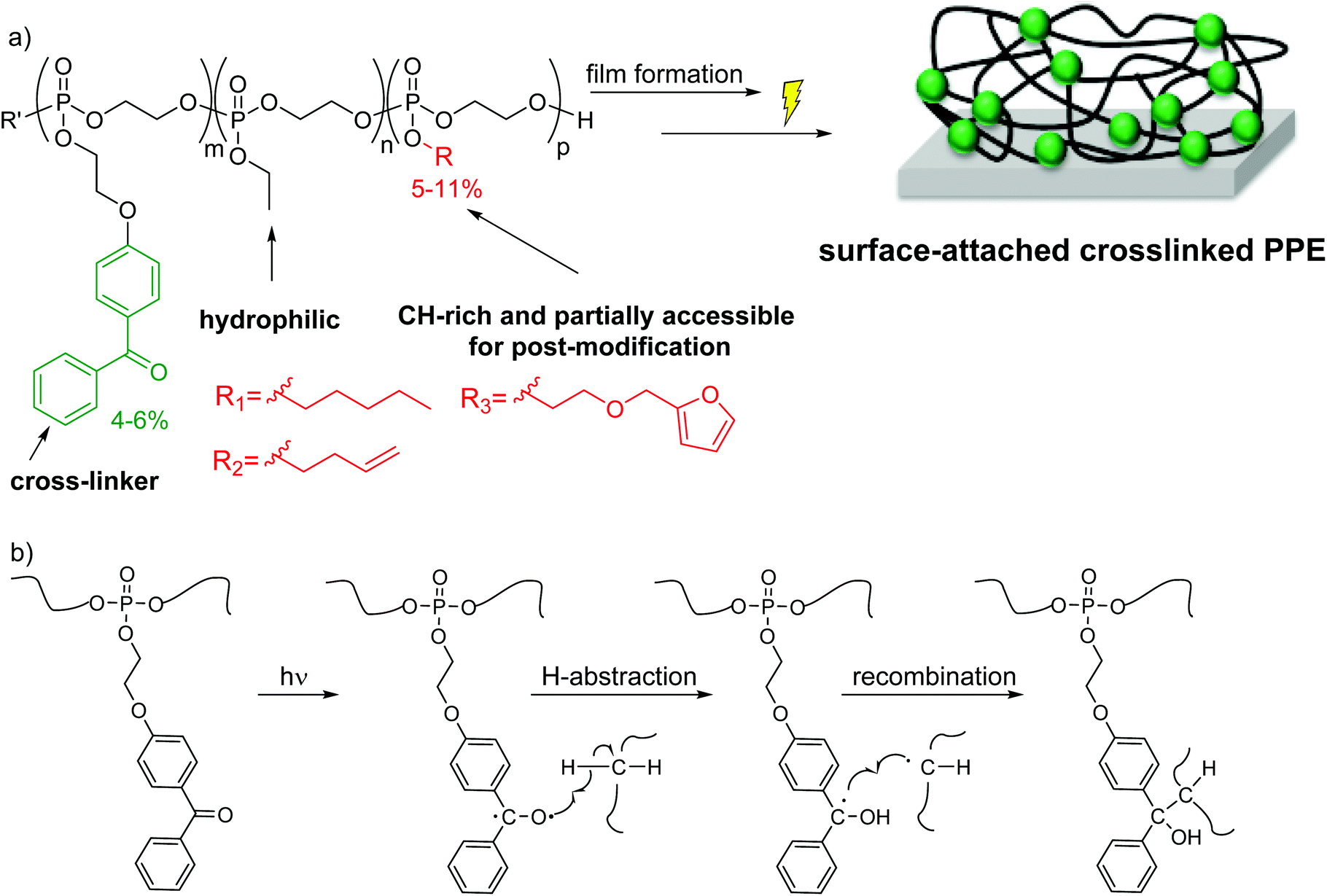 | ||
| Scheme 1 (a) Terpolymers used in this study to form surface-attached PPE-networks; (b) CHiC process for UV-activated cross-linking and surface-attachment of PPEs using benzophenone. | ||
The target polymers consist of the EEP repeat units, repeat units carrying a UV-cross-linkable benzophenone group, higher homologous of EEP, and optionally functional repeat units (Scheme 1a). To be able to make these polymers, we synthesized a novel monomer consisting of a cyclic phosphate and a benzophenone side chain (BeEP, Scheme 2). The BeEP monomer (1) was copolymerized with the protein-repellent EEP monomer (2), its homologs (3 and 4) and optionally other cyclic phosphates carrying functional groups (5 and 6) by ring-opening polymerization (ROP). The resulting copolymers are water-soluble and can be cross-linked by a UV-activated C,H-insertion cross-linking reaction (the so-called CHiC process, Scheme 1b and Scheme S1†).18–20
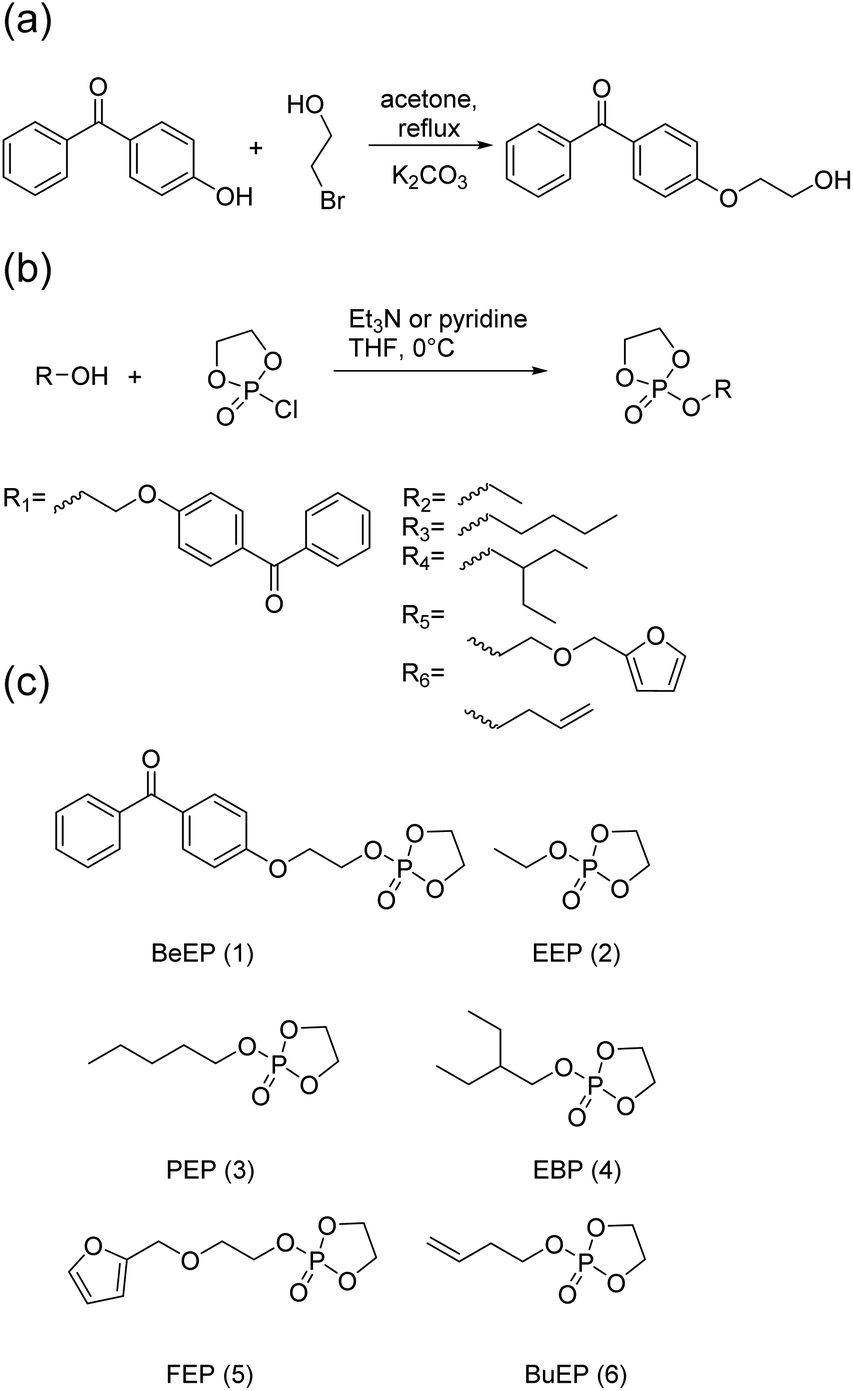 | ||
| Scheme 2 (a) Synthesis of 4-(2-hydroxyethoxy) benzophenone; (b) synthesis of cyclic phosphate monomers; (c) cyclic phosphate monomers used in this study. | ||
As has been described previously for other polymeric systems,18–20 the benzophenone groups in these polymers form a biradical upon UV irradiation, leading to the abstraction of a proton from a nearby alkyl group. When this radical on the former carbonyl C atom of benzophenone collapses with the C radical on the alkyl chain, a covalent bond and thus an inter-chain cross-link is formed (Scheme 1b). Advantageously, the benzophenone group does not need a specific reaction partner but can react with most aliphatic CH groups, so that a polymeric network is formed.20–23 This concept was introduced by Rühe and coworkers into polymer chemisty; they used copolymers containing covalently attached benzophenone to prepare surface-attached polymer networks and hydrogels, e.g. made from dimethylacrylamide and methacryloyloxybenzophenone.23
We here report the synthesis, copolymerization kinetics, and characterization of several benzophenone-containing PPE-copolymers with various pendant groups with different overall hydrophilicity and number of reactive C–H-bonds for the ChiC process. We further present films made from these polymers on silicon surfaces, as well as their characterization. First results of surface modification of these films by Diels–Alder reactions, give an idea how to build up more complex surface architectures from these materials.
Experimental Section
Further experimental details can be found in the ESI.†2-(2-(Benzophenone-4-oxy)ethoxy)-2-oxo-1,3,2-dioxaphospholane (BeEP, 1)
4-(2-Hydroxyethoxy)benzophenone (8.64 g, 35.7 mmol, 1 eq.) was dissolved in 60 mL dry THF in a flame-dried 3-necked round-bottom flask. Dry triethylamine (3.97 g, 39.2 mmol, 1.1 eq.) was added and cooled to 0 °C. 2-Chloro-2-oxo-1,3,2-dioxaphospholane (7.62 g, 53.5 mmol, 1.5 eq.) in 40 mL dry THF was added dropwise, stirred for 4 h and stored at −20 °C overnight. During the reaction, hydrogen chloride was formed and precipitated as triethylammonium hydrochloride. The reaction was filtered under inert gas and concentrated at reduced pressure. Column chromatography with an RP-1 column (silica gel deactivated with 5 v% hexamethyldisiloxane, dichloromethane/ethyl acetate 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, Rf = 0.71) gave the pure product BeEP, colorless crystals. Yield: 9.32 g, 78%. 1H NMR (300 MHz, CDCl3): δ [ppm] 7.85–7.82 (d, 2H,
:1, Rf = 0.71) gave the pure product BeEP, colorless crystals. Yield: 9.32 g, 78%. 1H NMR (300 MHz, CDCl3): δ [ppm] 7.85–7.82 (d, 2H, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CHC(CH)–C(O)–Ar–O–CH2–), 7.77–7.74 (d, 2H, Ar–C(O)–C(–CH)CH–CHC–O–CH2–), 7.61–7.55 (t, 1H, –CHCH–CHC(CH)–C(O)–Ar–O–CH2–), 7.51–7.45 (t, 2H, –CHCH–CHC(CH)–C(O)–Ar–O–CH2–), 6.99 (d, 2H, Ar–C(O)–C(–CH)CH–CHC–O–CH2–), 4.57–4.50 (m, 2H, Ar–O–CH2–CH2–O–P(O)–), 4.49–4.35 (m, 4H, –O–CH2–CH2–O–P(O–)–O–CH2–CH2–), 4.28 (t, 2H, Ar–O–CH2–CH2–O–P(O)–). 13C{H} NMR (76 MHz, CDCl3): δ [ppm] 132.54 (Ar–C(O)–Ar–O–), 132.00 (Ar–C(O)–Ar–O–), 129.74 (Ar–C(O)–Ar–O–), 128.22 (Ar–C(O)–Ar–O–), 114.16 (Ar–C(O)–Ar–O–), 67.00 (d, –Ar–O–CH2–), 66.72 (d, –Ar–O–CH2–CH2–O–P–), 66.005 (d, –O–CH2–CH2–O–P).31P{H} NMR (121 MHz, CDCl3): δ [ppm] 17.90. FTIR (cm−1): 3068–2919 (–CH2– and –CH stretching), 1643 (CC stretching), 1601 (diaryl-CO), 1576 (CH– stretching), 1505 (CH– stretching), 1457 (–CH2– deformation), 1444 (O–CH2– deformation), 1418, 1363, 1308, 1284 (PO stretching), 1249, 1174, 1148, 1115, 1085, 1058, 1025 (P–O–C stretching), 998 (P–O–C stretching), 930, 920, 868, 844 (P–O–C stretching), 794, 774, 739 (monosubst. aryl), 703, 694 (–CH2– rocking).
CH–CHC(CH)–C(O)–Ar–O–CH2–), 7.77–7.74 (d, 2H, Ar–C(O)–C(–CH)CH–CHC–O–CH2–), 7.61–7.55 (t, 1H, –CHCH–CHC(CH)–C(O)–Ar–O–CH2–), 7.51–7.45 (t, 2H, –CHCH–CHC(CH)–C(O)–Ar–O–CH2–), 6.99 (d, 2H, Ar–C(O)–C(–CH)CH–CHC–O–CH2–), 4.57–4.50 (m, 2H, Ar–O–CH2–CH2–O–P(O)–), 4.49–4.35 (m, 4H, –O–CH2–CH2–O–P(O–)–O–CH2–CH2–), 4.28 (t, 2H, Ar–O–CH2–CH2–O–P(O)–). 13C{H} NMR (76 MHz, CDCl3): δ [ppm] 132.54 (Ar–C(O)–Ar–O–), 132.00 (Ar–C(O)–Ar–O–), 129.74 (Ar–C(O)–Ar–O–), 128.22 (Ar–C(O)–Ar–O–), 114.16 (Ar–C(O)–Ar–O–), 67.00 (d, –Ar–O–CH2–), 66.72 (d, –Ar–O–CH2–CH2–O–P–), 66.005 (d, –O–CH2–CH2–O–P).31P{H} NMR (121 MHz, CDCl3): δ [ppm] 17.90. FTIR (cm−1): 3068–2919 (–CH2– and –CH stretching), 1643 (CC stretching), 1601 (diaryl-CO), 1576 (CH– stretching), 1505 (CH– stretching), 1457 (–CH2– deformation), 1444 (O–CH2– deformation), 1418, 1363, 1308, 1284 (PO stretching), 1249, 1174, 1148, 1115, 1085, 1058, 1025 (P–O–C stretching), 998 (P–O–C stretching), 930, 920, 868, 844 (P–O–C stretching), 794, 774, 739 (monosubst. aryl), 703, 694 (–CH2– rocking).
2-Pentyloxy-2-oxo-1,3,2-dioxaphospholane (PEP, 3)
A flame-dried 500 mL three-neck flask, equipped with a dropping funnel, was charged with a solution of dry 1-pentanol (12.37 g, 0.14 mol, 1 eq.) and dry triethylamine (14.21 g, 0.14 mol, 1 eq.) in dry THF (100 mL). 2-Chloro-2-oxo-1,3,2-dioxaphospholane (20.00 g, 0.14 mol, 1 eq.) dissolved in dry THF (50 mL) was added dropwise to the stirring solution at 0 °C under argon atmosphere. During the reaction, hydrogen chloride was formed and precipitated as triethylammonium hydrochloride. The reaction was stirred at 0 °C for 4 h and stored at −20 °C overnight. The salt was removed by filtration under inert gas atmosphere and the filtrate concentrated under reduced pressure. The residue was purified by distillation under reduced pressure to give a fraction at 110–117 °C/0.13–0.17 mbar, obtaining the clear, colorless, liquid product PEP. Yield: 20.39 g, 75%. 1H-NMR (300 MHz, CDCl3): δ [ppm] 4.48–4.29 (m, 4H, O–CH2–CH2–O), 4.19–4.08 (m, 2H, CH3–CH2–CH2–CH2–CH2–O–), 1.68 (m, 2H, CH3–CH2–CH2–CH2–CH2–O–), 1.43–1.27 (m, 4H, CH3–CH2–CH2–CH2–CH2–O–), 0.89 (t, 3H, CH3–CH2–CH2–CH2–CH2–O–).13C{H}-NMR (76 MHz, CDCl3): δ [ppm] 69.14 (d, –P–O–CH2–CH2–CH2–CH2–CH3), 65.93 (d, –O–CH2–CH2–O–P–), 29.98 (d, –P–O–CH2–CH2–CH2–CH2–CH3), 27.44 (–P–O–CH2–CH2–CH2–CH2–CH3), 22.14 (–P–O–CH2–CH2–CH2–CH2–CH3), 13.89 (–P–O–CH2–CH2–CH2–CH2–CH3). 31P{H} NMR (121 MHz, CDCl3): δ [ppm] 17.59. FTIR (cm−1): 2958 (–CH2– and –CH3 stretching), 2932 (–CH2– and –CH3 stretching), 2873 (–CH2– and –CH3 stretching), 1469 (O–CH2– deformation), 1367 (–CH3 deformation), 1285 (PO stretching), 1222, 1151 (–CH3 rocking), 1121, 1019 (P–O–C stretching), 1005 (P–O–C stretching), 927, 834 (P–O–C stretching), 754 (–CH2– rocking), 725.
General procedure for copolymerizations
The polymerization was conducted according to literature procedures for other cyclic phosphate monomers.24 Exemplarily for P1: EEP (993 mg, 6.53 mmol), BeEP (128 mg, 368 μmol) and TU (136 mg, 368 μmol, 5.3 mol% to monomer) were introduced into a tube. A stock solution of DBU in dry DCM (56 mg/0.2 mL, 368 μmol, 5.3 mol% to monomer) and a stock solution of the initiator (2-(benzyloxy) ethanol) in dry DCM (7 mg/0.5 mL, 46 μmol) were prepared and 1 mL dry DCM was added to EEP, BeEP, and TU to give a total concentration of ca. 4 mol L−1. All solutions were cooled down to 0 °C. 0.5 mL of the stock solution of the initiator was added to the stirred solution of EEP, BeEP, and TU. The polymerization was started by rapid addition of 0.2 mL stock solution of DBU to the reaction mixture. The polymerization was terminated after 120 min by the addition of an excess of acetic acid in DCM. DCM was added and washed with 1 M HCl. The organic phase was then extracted with water (3 times) until the aqueous solution exhibited a neutral pH. The organic phase was concentrated, 2 mL DCM added and the polymer precipitated into 40 mL ice-cold diethyl ether. The suspension was centrifuged for 15 min (4000 rpm, 4 °C) and after that, the supernatant was decanted. The polymer was dried under reduced pressure to give the final product.
P1: EEP (993 mg, 6.53 mmol, 142 eq.), BeEP (128 mg, 368 μmol, 8 eq.), DBU (56 mg, 368 μmol, 8 eq.), TU (136 mg, 368 μmol, 8 eq.), initiator (7 mg, 46 μmol, 1 eq.). Yield: 921 mg, 80%. Mn(NMR): 38600 g mol−1. 1H-NMR (300 MHz, DMSO-d6): δ [ppm] 7.76–7.53 (m, 86H Ar–C(O)–Ar–O–CH2–CH2–O–P–), 7.33 (m, 5H, Ar–, initiator), 7.14–7.08 (m, 22H, Ar–C(O)–Ar–O–CH2–CH2–O–P–), 4.47–3.84 (m, 1450H, Ar–C(O)–Ar–O–CH2–CH2–O–P–, –P–O–CH2–CH3, –O–CH2–CH2–O–, backbone), 1.26 (t, 676, –CH3).31P{H} NMR (121 MHz, DMSO-d6): δ [ppm] −1.24, −0.99.
P2: EEP (881 mg, 5.80 mmol, 126 eq.), BeEP (128 mg, 368 μmol, 8 eq.), PEP (143 mg, 736 μmol, 16 eq.), DBU (56 mg, 368 μmol, 8 eq.), TU (136 mg, 368 μmol, 8 eq.), initiator (7 mg, 46 μmol, 1 eq.). Yield: 1002 mg, 83%. Mn(NMR): 21000 g mol−1. 1H-NMR (300 MHz, DMSO-d6): δ [ppm] 7.76–7.52 (m, 58H Ar–C(O)–Ar–O–CH2–CH2–O–P–), 7.33 (m, 5H, Ar–, initiator), 7.14–7.05 (m, 16H, Ar–C(O)–Ar–O–CH2–CH2–O–P–), 4.47–3.87 (m, 847H, Ar–C(O)–Ar–O–CH2–CH2–O–P–, –P–O–CH2–CH3, –P–O–CH2–CH2–CH2–CH2–CH3, –O–CH2–CH2–O–, backbone), 1.70–1.51 (m, 30H, –P–O–CH2–CH2–CH2–CH2–CH3), 1.35–1.16 (m, 404H, –P–O–CH2–CH2–CH2–CH2–CH3, –P–O–CH2–CH3), 0.87 (t, 42H, –P–O–CH2–CH2–CH2–CH2–CH3). 31P{H} NMR (121 MHz, DMSO-d6): δ [ppm] −1.24, −1.14, −0.98, −0.89.
P3: EEP (268 mg, 1.76 mmol, 134 eq.), BeEP (37 mg, 105 μmol, 8 eq.), EBP (22 mg, 105 μmol, 8 eq.), DBU (16 mg, 105 μmol, 8 eq.), TU (39 mg, 105 μmol, 8 eq.), initiator (2 mg, 13 μmol, 1 eq.). Yield: 269 mg, 82%. Mn(NMR): 28600 g mol−1. 1H-NMR (500 MHz, DMSO-d6): δ [ppm] 7.78–7.51 (m, 48H Ar–C(O)–Ar–O–CH2–CH2–O–P–), 7.33 (m, 5H, Ar–, initiator), 7.16–7.06 (m, 13H, Ar–C(O)–Ar–O–CH2–CH2–O–P–), 4.42–3.99 (m, 1051H, Ar–C(O)–Ar–O–CH2–CH2–O–P–, –P–O–CH2–CH3, –O–CH2–CH2–O–, backbone), 3.99–3.87 (m, 34H, –P–O–CH2–CH–(CH2–CH3)2), 1.36–1.29 (m, 17H, –P–O–CH2–CH–(CH2–CH3)2), 1.26 (m, 479H, –P–O–CH2–CH–(CH2–CH3)2, –P–O–CH2–CH3), 0.86 (t, 72H, –P–O–CH2–CH–(CH2–CH3)). 31P{H} NMR (202 MHz, DMSO-d6): δ [ppm] −1.25, −1.01, −0.99.
P4: EEP (881 mg, 5.80 mmol, 126 eq.), BeEP (128 mg, 368 μmol, 8 eq.), FEP (183 mg, 736 μmol, 16 eq.), DBU (56 mg, 368 μmol, 8 eq.), TU (136 mg, 368 μmol, 8 eq.), initiator (7 mg, 46 μmol, 1 eq.). Yield: 907 mg, 76%. Mn(NMR): 34500 g mol−1. 1H-NMR (300 MHz, DMSO-d6): δ [ppm] 7.76–7.52 (m, 108H Ar–C(O)–Ar–O–CH2–CH2–O–P–, –P–CH2–CH2–O–CH2–C–O–CHCH–CH), 7.33 (m, 5H, Ar–, initiator), 7.14–7.05 (m, 22H, Ar–C(O)–Ar–O–CH2–CH2–O–P–), 6.43 (s, 44H, –P–CH2–CH2–O–CH2–C–O–CHCH–CH), 4.45 (s, 53H, –P–CH2–CH2–O–CH2–C–O–CHCH–CH), 4.27–3.88 (m, 1167H, Ar–C(O)–Ar–O–CH2–CH2–O–P–, –P–O–CH2–CH3, –P–CH2–CH2–O–CH2–C–O–CHCH–CH, –O–CH2–CH2–O–, backbone), 3.59 (s, 107H, –P–CH2–CH2–O–CH2–C–O–CHCH–CH), 1.26 (t, 494H, –P–O–CH2–CH3). 31P{H} NMR (121 MHz, DMSO-d6): δ [ppm] −1.29 to −1.18, −0.98, −0.89.
P5: EEP (881 mg, 5.80 mmol, 126 eq.), BeEP (128 mg, 368 μmol, 8 eq.), BuEP (131 mg, 736 μmol, 16 eq.), DBU (56 mg, 368 μmol, 8 eq.), TU (136 mg, 368 μmol, 8 eq.), initiator (7 mg, 46 μmol, 1 eq.). Yield: 1048 mg, 84%. Mn(NMR): 46300 g mol−1. 1H-NMR (300 MHz, DMSO-d6): δ [ppm] 7.79–7.49 (m, 95H Ar–C(O)–Ar–O–CH2–CH2–O–P–), 7.33 (m, 5H, Ar–, initiator), 7.16–7.05 (m, 25H, Ar–C(O)–Ar–O–CH2–CH2–O–P–), 5.87–5.72 (m, 20H, –P–O–CH2–CH2–CHCH2), 5.17–5.06 (m, 51H, –P–O–CH2–CH2–CHCH2), 4.46–3.88 (m, 1720H, Ar–C(O)–Ar–O–CH2–CH2–O–P–, –P–O–CH2–CH3, –P–O–CH2–CH2–CHCH2, –O–CH2–CH2–O–, backbone), 2.44–2.32 (m, 61H, –P–O–CH2–CH2–CHCH2), 1.26 (t, 729H, –P–O–CH2–CH3). 31P{H} NMR (121 MHz, DMSO-d6): δ [ppm] −1.28, −1.24, −0.99.
P6: EEP (937 mg, 6.16 mmol, 134 eq.), BuEP (131 mg, 736 μmol, 16 eq.), DBU (56 mg, 368 μmol, 8 eq.), TU (136 mg, 368 μmol, 8 eq.), initiator (7 mg, 46 μmol, 1 eq.). Yield: 942 mg, 85%. Mn(NMR): 28100 g mol−1. 1H-NMR (300 MHz, DMSO-d6): δ [ppm] 7.34 (m, 5H, Ar–, initiator), 5.87–5.72 (m, 24H, –P–O–CH2–CH2–CHCH2), 5.20–5.05 (m, 43H, –P–O–CH2–CH2–CHCH2), 4.51 (s, 2H, Ar–CH2–O–, initiator), 4.26–3.98 (m, 1058H, –P–O–CH2–CH3, –P–O–CH2–CH2–CHCH2, –O–CH2–CH2–O–, backbone), 2.42–2.38 (q, 46H, –P–O–CH2–CH2–CHCH2), 1.26 (t, 468H, –P–O–CH2–CH3). 31P{H} NMR (121 MHz, DMSO-d6): δ [ppm] −1.27, −1.24, −0.98.
P1, P2, P4, P5: FTIR (cm−1): 3660–3358 (O–H stretching), 2983 (–CH2– and –CH3 stretching), 2957 (–CH2– and –CH3 stretching), 2911 (–CH2– and –CH3 stretching), 1652 (diaryl-CO), 1601 (CC stretching), 1508 (CC stretching), 1454 (–CH2– deformation), 1394 (O–CH2– deformation), 1372 (–CH3 deformation), 1267 (PO stretching), 1167 (–CH3 rocking), 1123 (–CH3 rocking), 1015 (P–O–C stretching), 958 (P–O–C stretching), 797 (O–CH2– stretching), 742 (–CH2– rocking), 704, 683.
P6: FTIR (cm−1): 3660–3358 (O–H stretching), 2983 (–CH2– and –CH3 stretching), 2957 (–CH2– and –CH3 stretching), 2911 (–CH2– and –CH3 stretching), 1642 (CC stretching), 1454 (–CH2– deformation), 1394 (O–CH2– deformation), 1372 (–CH3 deformation), 1267 (PO stretching), 1167 (–CH3 rocking), 1123 (–CH3 rocking), 1015 (P–O–C stretching), 958 (P–O–C stretching), 797 (O–CH2– stretching), 738 (–CH2– rocking), 704, 683.
Pre-functionalization of Si Surfaces
To obtain wafers which are exclusively benzophenone-functionalized, a solution of 4-(3-triethoxysilyl) propyloxybenzophenone (50 mM) in toluene was spin coated on a Si wafer at 1000 rpm, 1000 rpm/sec for 120 s. The wafer was cured for 45 min at 120 °C on a hot plate, washed with toluene, and dried under a continuous nitrogen flow. It was cut into 1.5 × 1.5 cm sized substrates with a diamond cutter. Pre-functionalized Si wafer with a benzophenone/amine ratio of 1:1 were obtained in a similar way: 33.4 mg of (3-aminopropyl) triethoxysilane were added to 3 mL of a solution of 4-(3-triethoxysilyl)propyloxybenzophenone (50 mM) in toluene. The solution was spin-coated on the Si wafer at 3000 rpm, 1000 rpm/sec for 120 s and the wafer was then treated as described above.
For pre-functionalization with trimethox(propyl)silane and (3-aminopropyl)triethoxysilane, ethanol solutions with a total concentration of 50 mM were prepared: (a) propyl/amine ratio of 5:1, 20.5 mg (0.125 mmol, 5 eq.) trimethox(propyl)silane and 5.5 mg (0.0249 mmol, 1 eq.) (3-aminopropyl)triethoxysilane in 3 mL ethanol; (b) propyl/amine ratio of 2:1, 16.4 mg (0.099 mmol, 2 eq.) trimethox(propyl)silane and 11.1 mg (0.050 mmol, 1 eq.) (3-aminopropyl)triethoxysilane in 3 mL ethanol; (c) propyl/amine ratio of 1:1, 12.3 mg (0.075 mmol, 1 eq.) trimethox(propyl)silane and 16.6 mg (0.075 mmol, 1 eq.) (3-aminopropyl)triethoxysilane in 3 mL ethanol. The Si wafer was cut in 1.5 × 1.5 cm sized substrates and a solution of (a), (b) or (c) was spin coated on the substrates at 1000 rpm, 1000 rpm/sec for 30 s. The substrates were cured for 45 min at 120 °C on a hot plate, washed with DCM, and dried under a continuous nitrogen flow.
Pre-functionalization with 3-butenyltriethoxysilane and (3-aminopropyl) triethoxysilane was achieved in the same way with a total silane concentration of 50 mM in ethanol. The butenyl/amine ratios were 5:1, 2:1, or 1:1, respectively.
Immobilization of PPE-networks on silicon substrates
The polymers were dissolved in chloroform at a concentration of 50 mg mL−1 (30, 20 or 10 mg mL−1) and filtered through a syringe filter (Chromafil® Xtra H-PTFE-20, pore size 0.2 μm, ∅ 25 mm from Macherey-Nagel, or Millex-LCR filter, hydrophilic PTFE, pore size 0.45 μm, ∅ 13 mm from Merck). Polymer films were obtained by spin-coating this solution onto the pre-functionalized silicon substrates at 3000 rpm, 1000 rpm/sec for 30 s. The thus obtained PPE-films were directly crosslinked at 254 nm for 30 min in a UV-box. They were then washed with DCM to remove unattached polymer chains (for at least 30 min or overnight) and dried under a continuous nitrogen flow.Results and discussion
Monomer syntheses
The cross-linker monomer 2-(2-(benzophenone-4-oxy)ethoxy)-2-oxo-1,3,2-dioxaphospholane (BeEP, 1) was designed to contain two orthogonal chemistries: first, a UV-reactive benzophenone group that enables inter-chain cross-linking, but is inert in ring-opening polymerization (ROP), and second, a cyclic phosphate group that can undergo ROP. The monomer BeEP (1) was prepared in two steps starting from 4-hydroxybenzophenone. This phenol was transformed to the aliphatic alcohol 4-(2-hydroxyethoxy)benzophenone by reaction with 2-bromoethanol and potassium carbonate (Scheme 2a).25 The resulting alcohol was reacted with 2-chloro-2-oxo-1,3,2-dioxaphospholane (COP)26 to give the cyclic monomer 1 by esterification (Scheme 2b). The aliphatic ethylene spacer was needed because a direct reaction of 4-hydroxybenzophenone with COP did not give a stable product. Monomer 1 was purified by column chromatography over deactivated silica and was isolated as an off-white solid. This monomer could be stored at −28 °C under dry conditions and inert gas for at least one year without decomposition. It was characterized by 1H and 31P{H} NMR spectroscopy (Fig. 1 and S1–3†). The benzophenone resonances were detected in the aromatic region at 6.99–7.85 ppm; the signals for the dioxaphospholane ring were observed at 4.35–4.49 ppm as a multiplet. The 31P{H} NMR spectrum showed a singlet at 17.90 ppm, typical for cyclic phosphate monomers.27,28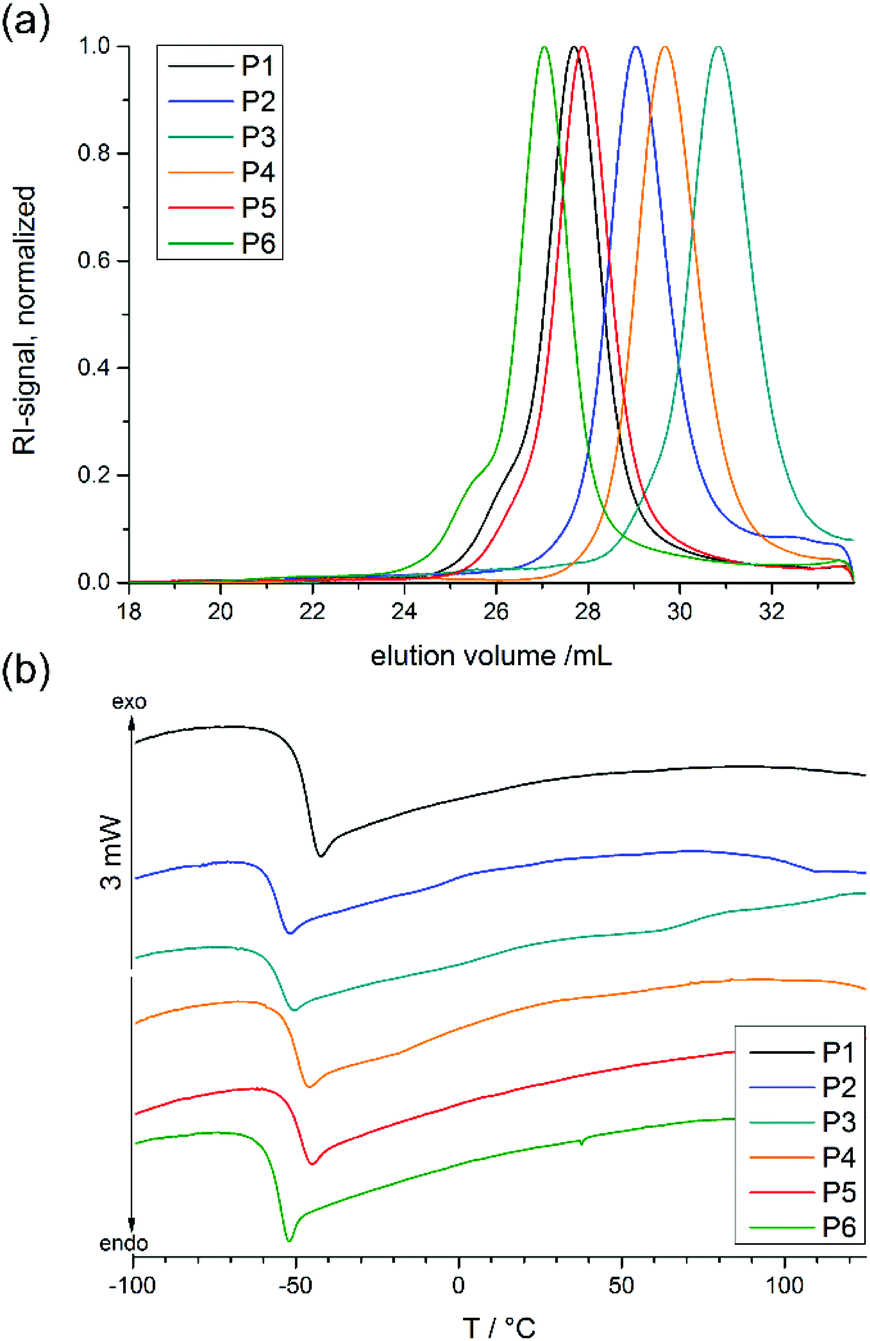 | ||
| Fig. 1 (a) SEC elugrams of all co- and terpolymers (in DMF, RI signal), (b) DSC thermograms of all co- and terpolymers (heating and cooling rate 10 K min−1, 1st run). | ||
2-Ethoxy-2-oxo-1,3,2-dioxaphospholane EEP (2) was synthesized as a second monomer (Scheme 2). EEP forms the water-soluble polymer PEEP, which is usually used as hydrophilic building block in PPE-based drug delivery systems,29,30 and was the major comonomer in the polymers synthesized herein. Further cyclic comonomers used in this study were 2-pentyloxy-2-oxo-1,3,2-dioxaphospholane (PEP, 3), 2-(2-ethyl butoxy)-2-oxo-1,3,2-dioxaphospholane (EBP, 4),31 2-(2-(furan-2-ylmethoxy)ethoxy)-2-oxo-1,3,2-dioxaphospholane (FEP, 5)32 and 2-(but-3-en-1-yloxy)-2-oxo-1,3,2-dioxaphospholane (BuEP, 6).24 Monomers 3 and 4 were used because they contained a larger number of aliphatic CH– groups than EEP and thus served as reaction partner for the benzophenone moieties. FEP (5) and BuEP (6) additionally carry functional groups that are inert under ROP conditions but reactive in Diels–Alder or thiol–ene reactions. These groups can be used for the post-polymerization functionalization of either the polymer or polymer networks.
Polymerization
A series of copolymers containing the cyclic phosphate monomers BeEP and EEP, and optionally the functional monomers 3 to 6, was synthesized by anionic ROP (Scheme 3, Table 1). EEP (2) was used as the main comonomer due to its hydrophilicity, and a low percentage of BeEP (4–6%) was added as to have sufficient cross-linking units. Higher amounts of BeEP were avoided, as BeEP is much more hydrophobic than EEP, and could thus compromise the overall protein-repellency of the copolymers. The target molecular weight was above 20000 g mol−1. Besides the BeEP–EEP copolymer (P1), several terpolymers containing repeat units with longer alkyl chains (PEP with 11 mol% pentyl, or EBP with 5 mol% ethyl butyl, respectively, Table 1) groups were synthesized. These terpolymers had additional methylene groups and could thus react more easily with the BeEP repeat units, thus enabling more efficient network formation. Terpolymers containing furfuryl (11 mol% FEP) or butenyl (9 mol% BuEP), respectively, were also synthesized. These additional functionalities would allow post-polymerization functionalization (Scheme 3b). A copolymer (P6) which contained no benzophenone, but instead 13 mol% butenyl-substituted repeat units, was also produced, which could be potentially cross-linked by photo-chemically activated thiol–ene reactions with multifunctional thiol cross-linkers.
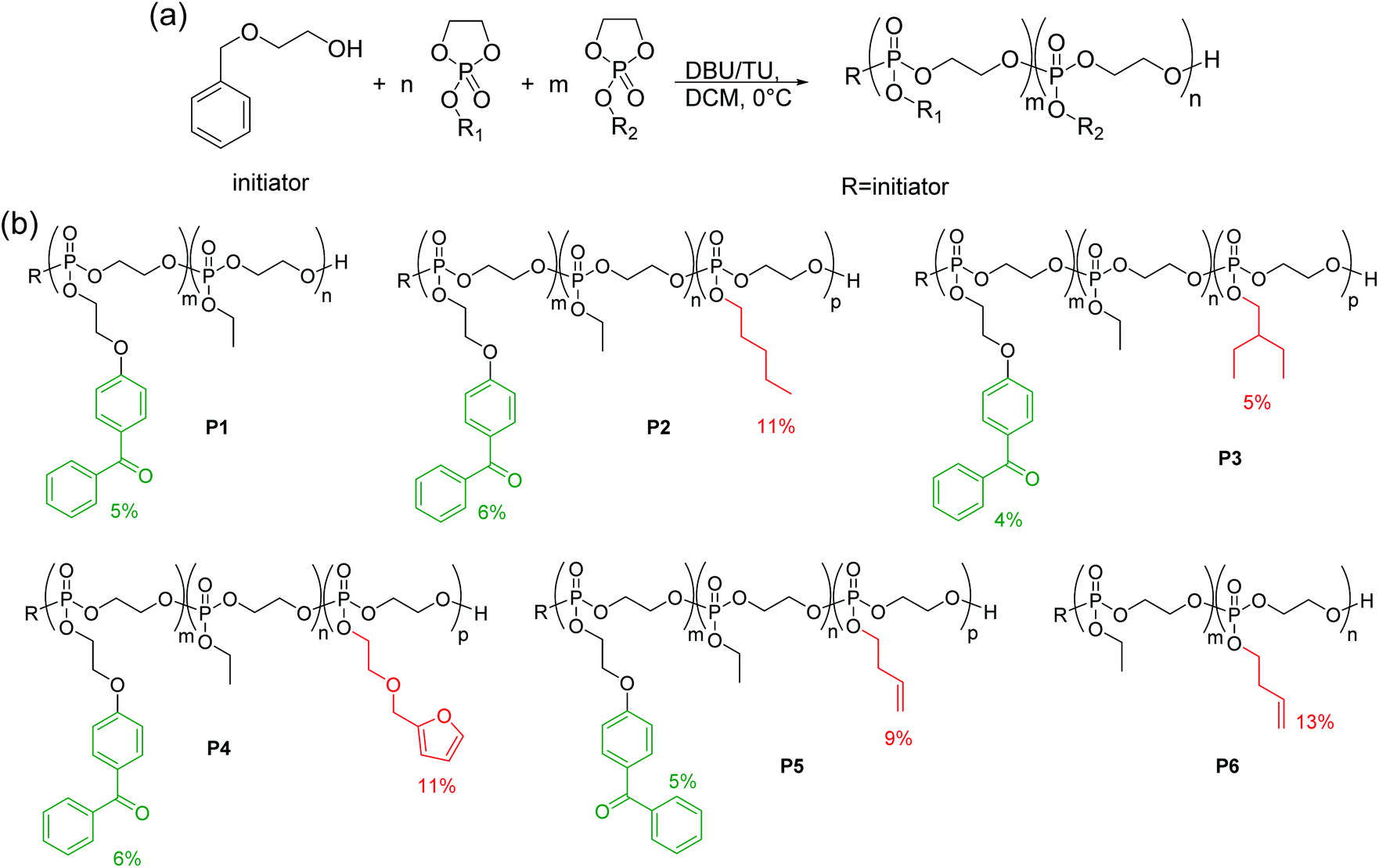 | ||
| Scheme 3 (a) Copolymerization of cyclic phosphates by ROP, (b) co- and terpolymers P1–P6 obtained herein. | ||
| [I]/[1]/[2]/ [3/4/5/6] | DPtha |
1/DPb | 1/%c | 2/DPb | 2/% c | 3/4/5/6/DPb | 3/4/5/6/% |
M
n
b (kg mol−1) |
M
w/Mnd |
T
g
/ °C |
||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a DPth = theoretical degree of polymerization. b DP = degree of polymerization of monomer 1 or 2 determined from the 1H-NMR spectra. c Percentage of comonomer 1 or 2 (determined from NMR). d Determined by SEC in DMF, PEO standard, RI-signal. e Determined by DSC. | ||||||||||||
| P1 | 1-co-2 | 1/8/142/— | 150 | 12 | 5 | 225 | 95 | — | 38.6 | 1.16 | −48 | |
| P2 | 1-co-2-co-3 | 1/8/126/16 | 150 | 8 | 6 | 115 | 83 | 15 | 11 | 21.0 | 1.16 | −58 |
| P3 | 1-co-2-co-4 | 1/8/134/8 | 150 | 7 | 4 | 160 | 91 | 8 | 5 | 28.6 | 1.19 | −56 |
| P4 | 1-co-2-co-5 | 1/8/126/16 | 150 | 11 | 6 | 165 | 83 | 22 | 11 | 34.5 | 1.15 | −51 |
| P5 | 1-co-2-co-6 | 1/8/126/16 | 150 | 13 | 5 | 243 | 86 | 26 | 9 | 46.3 | 1.16 | −50 |
| P6 | 2-co-6 | 1/-/134/16 | 150 | — | — | 156 | 87 | 23 | 13 | 28.1 | 1.10 | −57 |
All polymerizations were carried out with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and N-cyclohexyl-N‘-(3,5-bis(trifluoromethyl)phenyl) thiourea (TU) as catalyst system24 at 0 °C in dry DCM, at a total monomer concentration of 4 mol L−1. The polymers were soluble in non-polar organic solvents such as dichloromethane and chloroform, or polar solvents such as THF, ethanol, methanol, and DMSO. They were insoluble in hexane or diethyl ether. Successful copolymerization and formation of copolymers was proven by size exclusion chromatography (SEC) and 31P{H} NMR measurements. An overview of the analytical data thus obtained is given in Table 1. It demonstrates that the PPE-based polymers had narrow molecular weight distributions (Đ = 1.10–1.19, Fig. 1, Table 1) and relatively high monomer conversions (76–84%). This indicates the controlled nature of the polymerization using the DBU/TU catalyst system. The shift of the phosphorus signals in the 31P{H} NMR spectra (16.83 to 17.90 ppm for the monomers, −0.89 to −1.28 ppm for the polymers) also confirmed that polymer was obtained (see ESI for details†).
Additional 1H DOSY spectra were used to confirm that the polymers obtained were indeed copolymers and not a mixture of different homopolymers (see ESI for details†). Additionally, the aromatic protons of the initiator 2-(benzyloxy)ethanol were used for molecular weight calculations by end group analysis from 1H NMR spectra. End group analysis revealed that the degree of polymerization varied from 138–282 for the polymers, i.e. molecular weights of 21000 to 46300 g mol−1 were obtained under these conditions. 1H NMR spectra of P2 and P4 show the resonances of the aromatic protons in the initiator at 7.33 ppm (signal b in Fig. 2), which were compared with distinct resonances of the three different repeat units in the polymer: the doublet signal at 7.10 ppm (c in Fig. 2) of the BeEP repeat units, the signal at 1.60 ppm (e in Fig. 2) of the PEP units in P2, the singlet at 6.43 ppm (d in Fig. 2) of FEP units in P4, and the triplet signal at 1.26 ppm (f or h in Fig. 2) which belongs to the EEP repeat unit. The signal of the backbone and the methylene groups in the side chains neighboring the P–O bond at 4.50–3.80 ppm (d or f in Fig. 2) gives the degree of polymerization compared to the initiator resonances and coincides with the total number of monomer repeat units (Fig. 2). This 1H NMR analysis shows that the initial feed ratios of the comonomers matched the repeat unit composition of the copolymers. All copolymers exhibited glass transition temperatures (Tg) of −57 to −48 °C, which is typical for PPEs obtained via ROP (Fig. 1).27
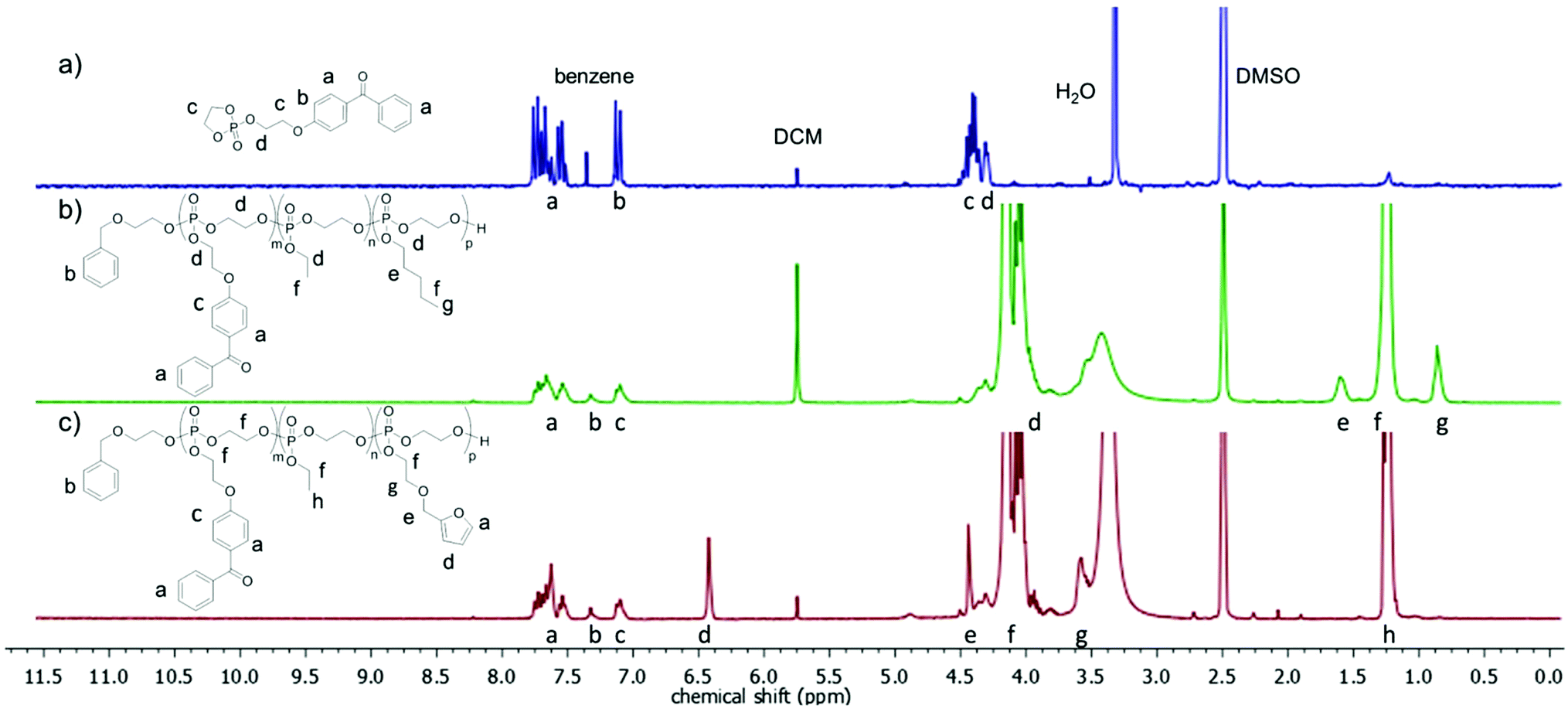 | ||
| Fig. 2 1H NMR spectra (300 MHz, DMSO-d6) of (a) the BeEP monomer (1), (b) terpolymer P2, and (c) terpolymer P4. | ||
Copolymerization kinetics
Since an even distribution of the cross-linker groups in the polymer is important to obtain a homogeneous distribution of cross-links throughout the network, and thus smooth polymer films, the copolymerization kinetics were investigated via real-time 31P{H} NMR. This was done by following the rate of consumption of the monomer BeEP in comparison to EEP and FEP or PEP, respectively. The initial monomer feeds for the kinetics experiments were similar to those used for the synthesis of polymers P1, P2 and P4: for BeEP/EEP, a monomer feed ratio of 0.05/0.95 was used; both the BeEP/PEP/EEP and the BeEP/FEP/EEP system had monomer feed ratios of 0.05/0.1/0.85. The targeted degree of polymerization was 120 for all samples. Reactions were conducted in d2-DCM at −20, 0 or −10 °C, respectively. The monomers were distinguished using the following signals in their 31P{H} NMR spectra (measured at 334 MHz): P1: 17.39 ppm for BeEP/16.91 ppm for EEP at −20 °C (Fig. 3a); P2: 17.52 ppm for BeEP/17.20 ppm for PEP/17.09 ppm for EEP at 0 °C; P4: 17.57 ppm for BeEP/17.41 ppm for FEP/17.11 ppm for EEP at −10 °C. To determine the rate of monomer incorporation, the consumption of each monomer over time was determined from the relative intensities of these signals (Fig. S5.44–S5.46†). All reactions were terminated when no further monomer consumption was detected in the NMR spectra, as increasing viscosity of the solution and lack of stirring in the NMR tube slowed down the polymerization at higher conversions.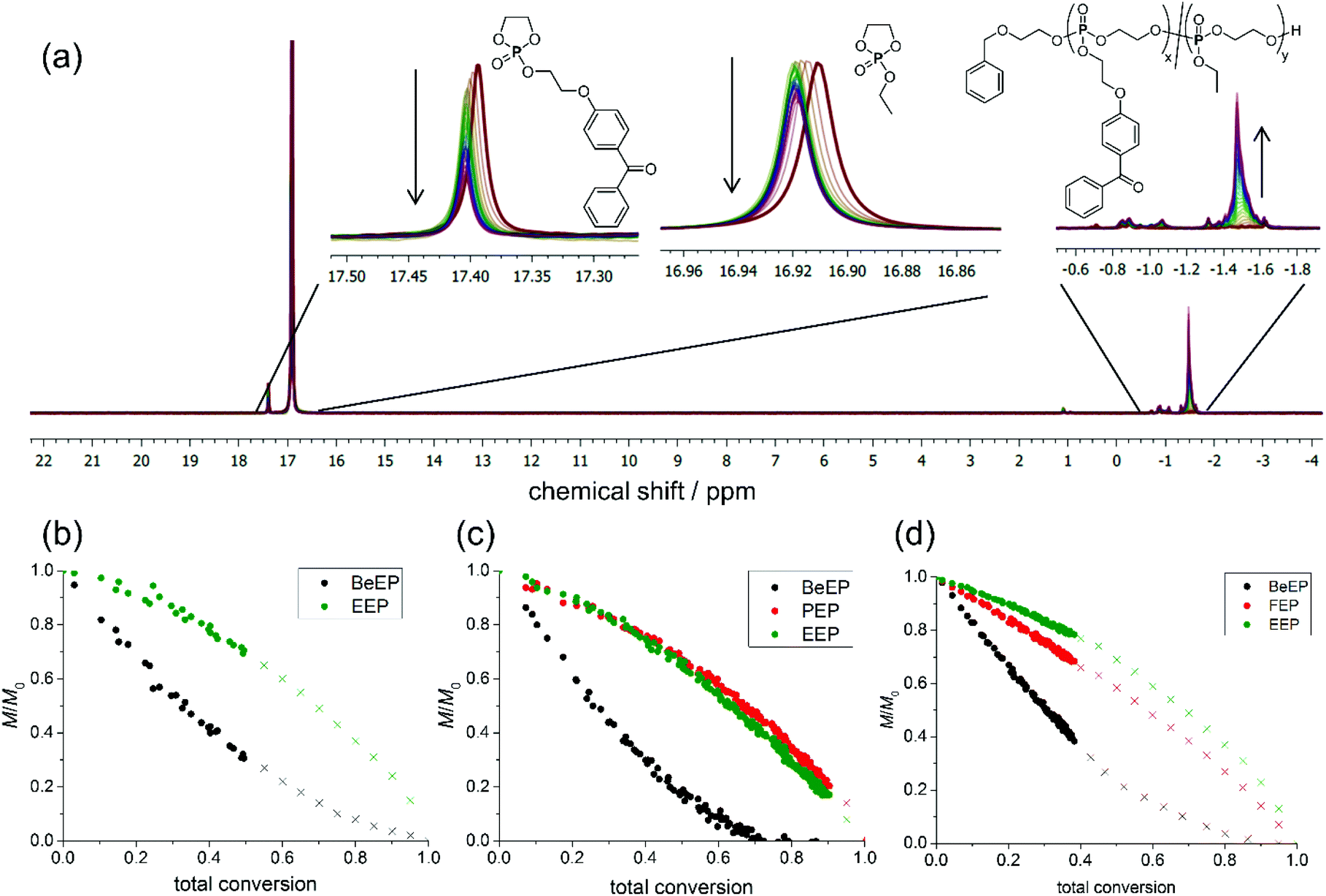 | ||
| Fig. 3 Simultaneous co- and terpolymerization of monomer EEP with BeEP and optionally monomers PEP and FEP. (a) Overlay and zoom-in into real-time 31P{H} NMR measurements taken during the copolymerization of EEP with BeEP (P1), (b–d) normalized monomer concentrations versus total monomer conversion for (b) EEP with BeEP (P1), (c) EEP with BeEP and PEP (P2), and (d) EEP with BeEP and FEP (P4) (note: crosses are extrapolated). | ||
In all cases, the statistical copolymerization of the above monomers gave a significantly faster incorporation of BeEP than of EEP, while the consumption of FEP and PEP was, within the limit of the method, comparable to that of EEP (with FEP being only slightly faster incorporated, Fig. 3b–d). For example, for P1 at 40% total conversion, 60% of BeEP was consumed, while 80% of EEP monomer remained in the reaction mixture (Fig. 3b). Likewise, for P2 at 40% conversion, 70% of BeEP was consumed, while 75% of EEP and PEP, respectively, remained in the reaction mixture. In P4, 62% of BeEP was consumed after 40% conversion, while 78% EEP remained. Reactivity ratios were also determined from the thus obtained data. For the copolymerization of BeEP and EEP (P1), the reactivity ratios obtained were r1 = 3.5 and r2 = 0.3 (from the current dataset). These indicate a repeat unit gradient in P1 with BeEP being the preferred comonomer (details and further can be found in the ESI†). Care must be taken not to over-interpret the values. They show a reactivity trend but should not be taken as absolute values, because the reaction rates in ROP, as in most anionic polymerization reactions, are high. Fitting of the curves for the other polymer systems (Fig. 3) also indicated that these were gradient copolymers. They were benzophenone-rich up to about 30% total conversion (50% BeEP conversion), and then rich in non-crosslinkable repeat units. The relative reactivity of the comonomers during the copolymerization was thus BeEP ≫ FEP > PEP ≈ EEP. This means that there was also a polarity gradient in these polymers since BeEP is more hydrophobic than EEP. This fact was not further considered in this study of film formation, but will be analyzed in future work.
Formation of surface-attached PPE-networks
To form surface-attached PPE networks by C,H-insertion reactions (CHiC process),20–23 the here reported polymers were spin-coated onto pre-treated silicon wafers (see Experimental). To be able to do so, several structural parameters of both the polymer and the substrate had to be optimized. For example, we found that the substrates that were silanized with 4-(3-triethoxysilyl)propyloxybenzophenone (Scheme 4) were too hydrophobic and caused dewetting of the hydrophilic PPE polymers. This was solved by using substrates that were pre-functionalized with a mixture of benzophenone and amino groups (3-aminopropyl chains) at a ratio of 1:1 instead.
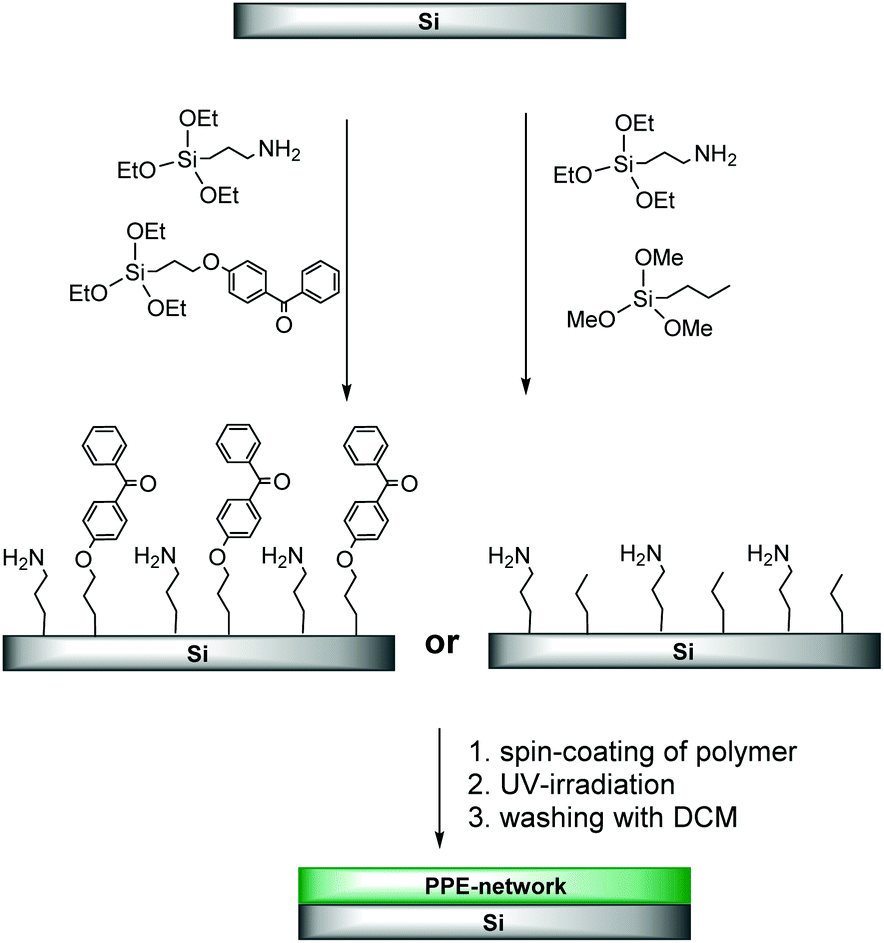 | ||
| Scheme 4 Surface functionalization of Si wafers with PPE: in the first step, Si wafers were functionalized by silanization. PPE copolymers were then spin-coated on the pre-treated wafers, and crosslinked by UV irradiation via the CHiC process. | ||
Additionally, when attempting to prepare networks from polymer P1, the films obtained could be washed off after UV-irradiation, indicating insufficient cross-links inside the network. Apparently, polymer P1, with 95% PEE repeat units, did not contain a sufficient amount of C–H bonds to which productive cross-links could be formed. The terpolymers P2 to P4, on the other hand, with 5% 2-ethyl butyl, 10% pentyl or furfuryl side chains, had significantly more aliphatic C–H groups. P2 and P4 accordingly formed stable surface-attached networks on substrates pre-functionalized with benzophenone and amine (ratio of 1:1) after UV-irradiation. After washing to remove unbound polymer, the layer thicknesses were 164 ± 1 nm for P4, and 78 ± 1 nm for P2 (determined by ellipsometry). P3 (containing EBP units) formed a smooth layer on these pre-functionalized substrates, but after washing the layer was removed completely. This was unexpected because this polymer also contained many aliphatic CH groups and is not yet fully understood. It is possible that the benzophenone cross-linker, which was previously reported to cause polymer main chain cleavage as a side reaction for several polymer backbones,33,34 also had this effect on the poly(phosphoester) backbone. Thus, overall, the functionalization of surfaces with PEE copolymers using benzophenone as cross-linker is far from trivial, and the substrate and polymer structures need to be carefully matched to avoid surface dewetting and/or insufficient cross-linking during network formation.
Since the PPE-networks were intended as surface-coatings for biomedical devices, it would be advantageous if they could be used on polymer surfaces without a substrate pre-functionalization step. Many polymeric materials (e.g. PMMA, poly(amides)- or -poly(urethanes)) contain a high amount of aliphatic CH groups, and are thus accessible for the CHiC reaction, as shown by Rühe and coworkers.35,36 However, technical polymer substrates cannot be studied with many of the surface analytical techniques used herein (which either need reflective, IR-transparent or very smooth substrates). Therefore, this situation was modeled on silicon wafers by pre-functionalizing them with propyl silanes (to imitate the aliphatic the C–H groups of a polymer substrate) and 3-aminopropyl silanes (to make the surface more hydrophilic). Different ratios of propyl silane to 3-aminopropyl silane (5:1, 2:1 and 1:1) were used.
Polymers P2 and P4 indeed formed surface-attached networks on these substrates, which demonstrated that these PPE polymers can be used to react with substrates that do not carry benzophenones. As is well known, the thickness obtained by spin-coating depends, among other parameters, on the concentration of the polymer solution. The here used polymers complied with this rule, and a “master curve” of layer thickness vs. polymer concentration for P4 is shown in Fig. S50.† Additionally, an influence of hydrophilicity on the thickness of the polymer layers obtained was noticed (Fig. 4): with increasing hydrophilicity of the pre-functionalized substrates, the layer thickness of these coatings increased from 132 ± 2 to 199 ± 2 nm for P4, and from 56 ± 1 to 104 ± 1 nm for P2 (Table S3,†Fig. 4). Thus, PPE-films can spread more easily on the more hydrophilic substrates. This is not only important in the context of spin-coating, which is rarely used to make biomaterials, but also when using these polymers in combination with other coating techniques. It indicates that the surface of the biomaterial must be hydrophilized prior to applying these coatings. A layer thickness of 263 ± 1 nm was observed for P5-networks. When multilayers of two or three films were prepared from P2 and P4, an increase of thicknesses was observed with each layer added (Table S3†). However, these multilayers became visibly less homogeneous and were therefore not further considered in this study.
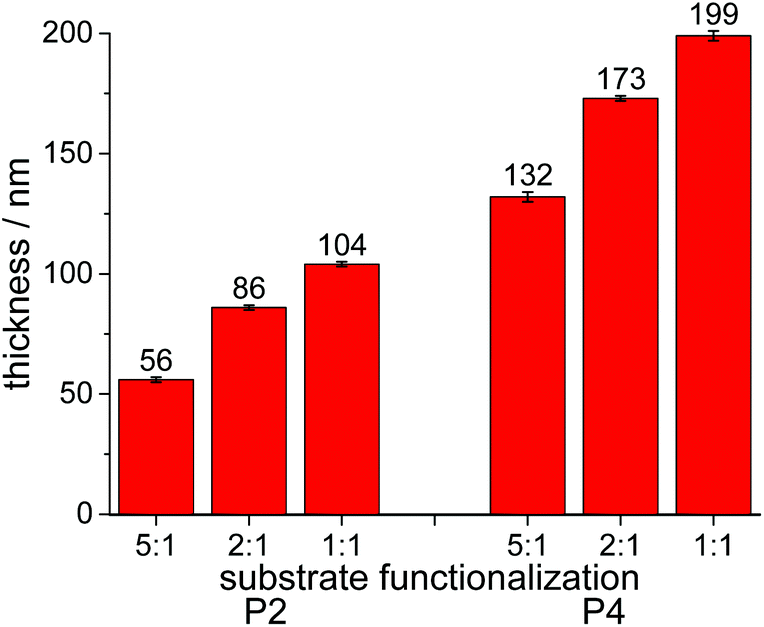 | ||
| Fig. 4 Layer thickness of surface-attached PPE films on substrates pre-functionalized with propyl silane and 3-aminopropyl silane. (a) P2 and P4 on silicon substrates pre-treated with different ratios of propyl silane and 3-aminopropyl silane (from 5:1 to 1:1, respectively). | ||
The physical properties of the surface-attached polymer networks made from P2, P4 and P5 on Si wafers that had been pre-functionalized with a propyl/amine silanes at a ratio of 1:1 were thoroughly characterized. Besides the thickness measured by ellipsometry (Fig. 4 and Table 2), contact angle measurements were performed. These revealed that the materials were highly hydrophilic, with static contact angels of 21, 26 and 26° for P2, P4, and P5, respectively (Fig. 5, Table 2, and Table S2†). This is in line with the expectations since the main comonomer EEP is known to produce hydrophilic PPEs. Atomic force microscopy (AFM) was used to investigate the morphology of the P2 and P4 networks. Height images (Fig. 5) showed a homogeneous morphology of the networks with an average roughness of 0.40 nm for P2, 0.68 nm for P4, and 0.59 nm for P4. Networks of P4 showed nanometer-sized pinholes, probably to minimize the contact area with the comparatively hydrophobic substrate surface. Additional AFM images are shown in the ESI (Fig. S52†). The film morphology was further characterized by scanning electron microscopy (SEM, Fig. S53 of the ESI†). For this, a wafer piece with a P4 coating was frozen in liquid nitrogen and freeze-fractured. The fracture location (i.e. a cross-section perpendicular to the wafer surface) was then investigated by SEM. The images show that the polymer film is very dense. Even at the highest magnification, no internal pores were visible.
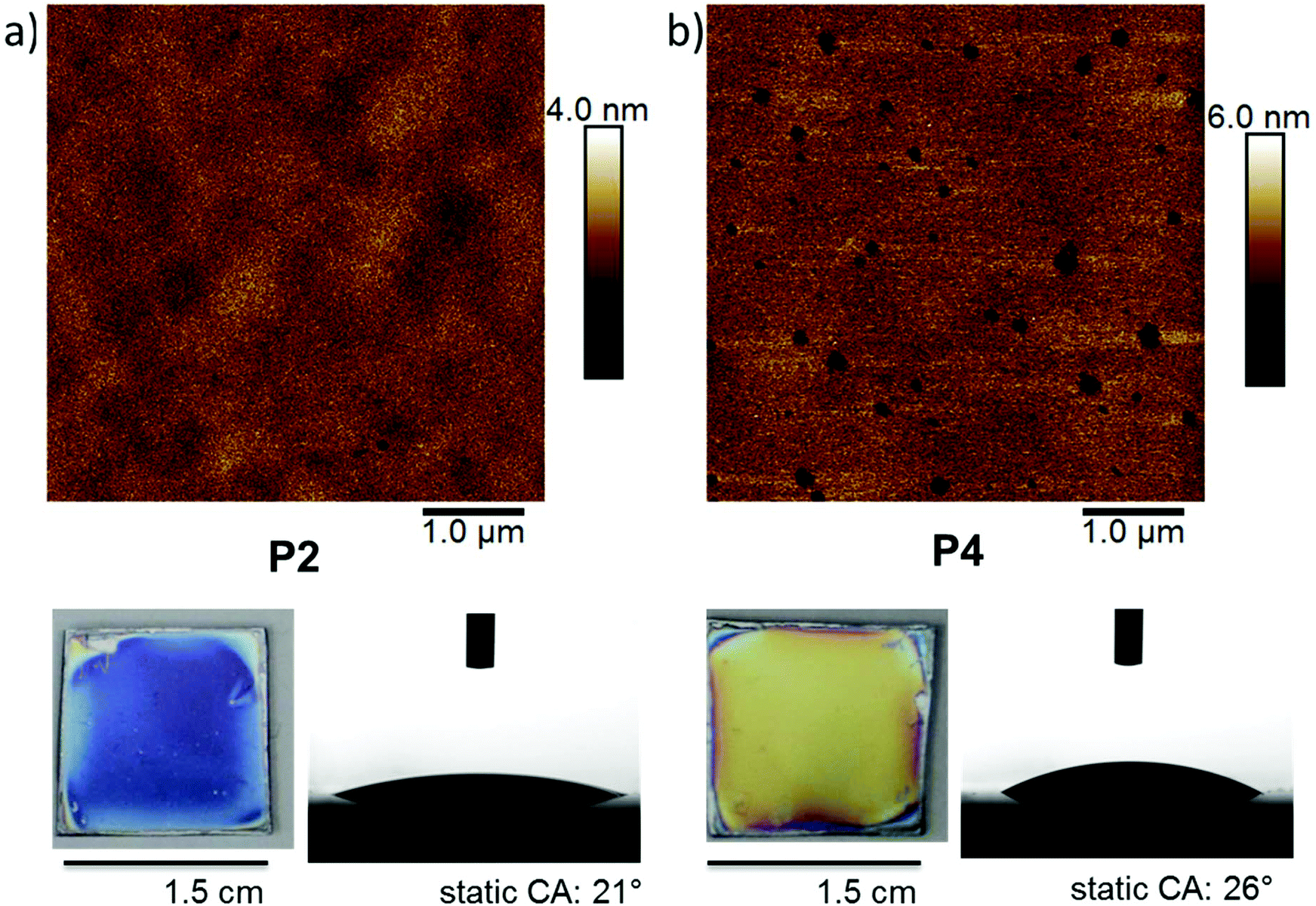 | ||
| Fig. 5 AFM height images of PPE-networks, photographs of the coated silicon substrates and photographs their static contact angle with water: (a) P2 networks and (b) P4 networks. | ||
| Polymer | Dry layer thickness on silicon/nm | Static contact angle/° | Roughness (Rq)/nm |
|---|---|---|---|
| P2 | 104 ± 1 | 21 ± 1 | 0.40 |
| P4 | 199 ± 2 | 26 ± 3 | 0.68 |
| P5 | 263 ± 1 | 26 ± 1 | 0.59 |
The PPE networks were further analyzed by FT-IR spectroscopy and compared with spectra of the monomers and polymers. All spectra are shown in the ESI.† These spectra confirmed that the expected IR bands were present. The dominant bands in the monomer spectra (PO stretching at 1282–1285 cm−1 and P–O–C stretching at 831–884, 980–1005 and 1016–1025 cm−1) could be retrieved in the polymer spectra at 1267, 797, 958 and 1015 cm−1, respectively.37,38 They all exhibited a shift and broadening of the band because of the formation of a linear backbone containing the phosphate groups compared to the strained rings of monomers. Characteristic bands of –CH2, O–CH2 and –CH3 stretching and rocking of EEP monomer at 752, 1100, 1166, 1369, 1393, 1445, 2915 and 2985 cm−1, respectively, could also be observed in all polymer spectra, albeit with slight wavenumber shifts. Further characteristic bands for the pentyl, furfuryl or butenyl groups in the side chains from the comonomers PEP, FEP, and BuEP in P2, P4 and P5 could not be observed because the above-mentioned signals were dominant and all polymers contained the latter repeat units only in small amounts. Characteristic bands of the BeEP monomer could be observed in polymers P1 to P5: a carbonyl band at 1601 cm−1 (at 1601 cm−1 in the monomer spectrum of BeEP) and aromatic CC stretching signals at 704, 1508 and 1652 cm−1 (703, 1505 and 1643 cm−1 in the monomer BeEP). In polymer P6 (only containing EEP and BuEP), a CC stretching band of an alkene group at 1641 cm−1 could be retrieved, in which was also found for the BuEP monomer. In IR spectra of the PPE-networks of P2, P4 and P5 on Si substrates, the dominant signals for PO stretching at 1275 cm−1 and P–O–C stretching at 822, 984 and 1026 cm−1 could be also retrieved, again with a slight shift (around 10 cm−1) to higher wavenumbers. Additionally, stretching bands of –CH2–, –CH3 and CC were visible again at 741, 1398 and 1456 cm−1 (–CH2– stretching) or 1130, 1167, and 1371 cm−1 (–CH3 stretching), as well as 2900–3000 cm−1 or 1506 and 1653 cm−1 again (CC stretching), respectively. Most importantly, the CO stretching band of the benzophenone residue at 1601 cm−1 disappeared because its carbonyl group was transferred into an OH group upon irradiation with UV-light.
To qualitatively test the strength of the surface adhesion of the polymer networks, the so-called tape test was conducted. For this test, regular office tape was glued on top of the surface-attached polymer network (in this case P2), and it was then peeled off at right angles to the surface. The thickness before this treatment and after this treatment was measured by ellipsometry. This data as well as photographs of the wafer before and after the tape test are shown in Fig. S51 in the ESI.† The data shows that only a few nanometers thin layer of polymer was removed. This, in combination with the fact that the polymer did not delaminate from the substrate, demonstrates substantial adhesion to the functionalized silicon substrate.
Summary & conclusions
For the formation of the surface-attached polymer networks and hydrogels made from poly(phosphate esters), a cyclic phosphate monomer suitable for anionic ring-opening polymerization (ROP) and containing a photo-reactive benzophenone group in the pendant chain (BeEP) has been designed and copolymerized with ethylene ethyl phosphate (EEP) as comonomer. Thus, hydrophilic functional polymers were obtained. Terpolymerization with additional comonomers gave functional polymers bearing pentyl (PEP), furfuryl (FEP) or butenyl (BuEP) pendant groups. The obtained terpolymers had 5% benzophenone repeat units and about 10% pentyl, furfuryl or butenyl functionalities, respectively. Molecular weights up to 46300 g mol−1 and molecular weight distributions as low as 1.15–1.19 were obtained. The polymerization kinetics studies by 31P{H} NMR measurements indicated that a gradient-like polymer structure was obtained, and that and the relative reactivity of the monomers was BeEP ≫ FEP > EEP ≈ PEP. This gradient structure is relevant since it indicates that there are zones in the polymer with high cross-linker density and others that are more loosely equipped with cross-linker groups. The failure to obtain surface-attached layers form all polymers described herein might be linked to this gradient, however, this hypothesis needs to be studied further to be verified. Surface-attached PPE-networks were obtained by spin-coating of the polymer solutions onto functionalized silicon substrates, followed by UV-irradiation. The layer thicknesses varied between 56 and 263 nm and depended on the applied polymer and the hydrophilicity of the substrates. Importantly, the polymers spread more easily on the more hydrophilic substrates. AFM images showed a homogeneous and smooth morphology of the P4 and P5 networks, with contact angles between 21 and 26° revealing hydrophilic surfaces. Initial studies to functionalize these substrates after polymerization using the furane-containing PPE-coating showed that modification after crosslinking of the coating can be performed, which might be a promising strategy to build up more complicated surface architectures and will be studied in the future to generate PPEylated medical devices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. B. is recipient of a fellowship through funding of the Excellence Initiative (DFG/GSC 266) in the context of the graduate school of excellence “MAINZ” (Material Sciences in Mainz). We thank Gunnar Glaßer (MPIP) for SEM measurements. Funding of the Deutsche Forschungsgemeinschaft (DFG) to F. R. W. (WU750/6-1) and K. L. (LI1714/5-1) is gratefully acknowledged. Open Access funding provided by the Max Planck Society.References
- L. Hall-Stoodley, J. W. Costerton and P. Stoodley, Nat. Rev. Microbiol., 2004, 2, 95–108 CrossRef CAS PubMed
.
- W. Hartleb, J. S. Saar, P. Zou and K. Lienkamp, Macromol. Chem. Phys., 2016, 217, 225–231 CrossRef CAS
- V. B. Damodaran and N. S. Murthy, Biomater. Res., 2016, 20, 18 CrossRef PubMed
- I. Banerjee, R. C. Pangule and R. S. Kane, Adv. Mater., 2011, 23, 690–718 CrossRef CAS PubMed
- A. Ekin, D. C. Webster, J. W. Daniels, S. J. Stafslien, F. Cassé, J. A. Callow and M. E. Callow, J. Coat. Technol. Res., 2007, 4, 435 CrossRef CAS
- J. C. Yarbrough, J. P. Rolland, J. M. DeSimone, M. E. Callow, J. A. Finlay and J. A. Callow, Macromolecules, 2006, 39, 2521–2528 CrossRef CAS
- S. Krishnan, C. J. Weinman and C. K. Ober, J. Mater. Chem., 2008, 18, 3405–3413 RSC
- J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm, F. R. Wurm and H. Frey, Chem. Rev., 2016, 116, 2170–2243 CrossRef CAS PubMed
- S. Schöttler, G. Becker, S. Winzen, T. Steinbach, K. Mohr, K. Landfester, V. Mailänder and F. R. Wurm, Nat. Nanotechnol., 2016, 11, 372–377 CrossRef PubMed
- G. Cheng, G. Li, H. Xue, S. Chen, J. D. Bryers and S. Jiang, Biomaterials, 2009, 30, 5234–5240 CrossRef CAS PubMed
- W. Yang, L. Zhang, S. Wang, A. D. White and S. Jiang, Biomaterials, 2009, 30, 5617–5621 CrossRef CAS PubMed
- S. Jiang and Z. Cao, Adv. Mater., 2010, 22, 920–932 CrossRef CAS PubMed
- M. Kurowska, A. Eickenscheidt, D.-L. Guevara-Solarte, V. T. Widyaya, F. Marx, A. Al-Ahmad and K. Lienkamp, Biomacromolecules, 2017, 18(4), 1373–1386 CrossRef CAS PubMed
- K. N. Bauer, H. T. Tee, M. M. Velencoso and F. R. Wurm, Prog. Polym. Sci., 2017, 73, 61–122 CrossRef CAS
- T. Steinbach and F. R. Wurm, Angew. Chem., Int. Ed., 2015, 54, 6098–6108 CrossRef CAS PubMed
- T. Steinbach, G. Becker, A. Spiegel, T. Figueiredo, D. Russo and F. R. Wurm, Macromol. Biosci., 2016, 17, 1600377 Search PubMed
- T. Steinbach and F. R. Wurm, Biomacromolecules, 2016, 17, 3338–3346 CrossRef CAS PubMed
- O. Prucker, C. A. Naumann, J. Ruehe, W. Knoll and C. W. Frank, J. Am. Chem. Soc., 1999, 121, 8766–8770 CrossRef CAS
- K. Schuh, O. Prucker and J. Ruehe, Adv. Funct. Mater., 2013, 23, 6019–6023 CrossRef CAS
- M. Körner, O. Prucker and J. Rühe, Macromolecules, 2016, 49, 2438–2447 CrossRef
- C. K. Pandiyarajan, O. Prucker and J. Rühe, Macromolecules, 2016, 49, 8254–8264 CrossRef CAS
- O. Prucker, C. A. Naumann, J. Rühe, W. Knoll and C. W. Frank, J. Am. Chem. Soc., 1999, 121, 8766–8770 CrossRef CAS
- R. Toomey, D. Freidank and J. Rühe, Macromolecules, 2004, 37, 882–887 CrossRef CAS
- B. Clément, B. Grignard, L. Koole, C. Jérôme and P. Lecomte, Macromolecules, 2012, 45, 4476–4486 CrossRef
- A. M. Lehaf, M. D. Moussallem and J. B. Schlenoff, Langmuir, 2011, 27, 4756–4763 CrossRef CAS PubMed
- G. Becker and F. R. Wurm, Tetrahedron, 2017, 73, 3536–3540 CrossRef CAS
- T. Steinbach, R. Schröder, S. Ritz and F. R. Wurm, Polym. Chem., 2013, 4, 4469–4479 RSC
- L. K. Müller, T. Steinbach and F. R. Wurm, RSC Adv., 2015, 5, 42881–42888 RSC
- Y. H. Lim, K. M. Tiemann, G. S. Heo, P. O. Wagers, Y. H. Rezenom, S. Zhang, F. Zhang, W. J. Youngs, D. A. Hunstad and K. L. Wooley, ACS Nano, 2015, 9, 1995–2008 CrossRef CAS PubMed
- Z. Zhao, J. Wang, H.-Q. Mao and K. W. Leong, Adv. Drug Delivery Rev., 2003, 55, 483–499 CrossRef CAS PubMed
- S. Zhang, J. Zou, F. Zhang, M. Elsabahy, S. E. Felder, J. Zhu, D. J. Pochan and K. L. Wooley, J. Am. Chem. Soc., 2012, 134, 18467–18474 CrossRef CAS PubMed
- G. Becker, T. A. Marquetant, M. Wagner and F. R. Wurm, Macromolecules, 2017, 50(20), 7852–7862 CrossRef CAS
- S. K. Christensen, M. C. Chiappelli and R. C. Hayward, Macromolecules, 2012, 45, 5237–5246 CrossRef CAS
- E. K. Riga, J. S. Saar, R. Erath, M. Hechenbichler and K. Lienkamp, Polymers, 2017, 9(12), 686 CrossRef
- J. D. J. S. Samuel, T. Brenner, O. Prucker, M. Grumann, J. Ducree, R. Zengerle and J. Rühe, Macromol. Chem. Phys., 2010, 211, 195–203 CrossRef CAS
- M. Moschallski, J. Baader, O. Prucker and J. Rühe, Anal. Chim. Acta, 2010, 671, 92–98 CrossRef CAS PubMed
-
M. Hesse, H. Meier and B. Zeeh, Spektroskopische Methoden in der organischen Chemie, Georg Thieme Verlag, Stuttgart, 1995 Search PubMed
-
G. Socrates, Infrared and Raman Characteristic Group Frequencies: Tables and Charts, Wiley, 2004 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7py01777d |
| This journal is © The Royal Society of Chemistry 2018 |