
Ultra-fast aqueous polymerisation of acrylamides by high power visible light direct photoactivation RAFT polymerisation†
Reece W.
Lewis
a,
Richard A.
Evans
b,
Nino
Malic
b,
Kei
Saito
 *c and
Neil R.
Cameron
*ad
*c and
Neil R.
Cameron
*ad
aDepartment of Materials Science and Engineering, Monash University, 22 Alliance Lane, Clayton, Victoria 3800, Australia. E-mail: neil.cameron@monash.edu
bCSIRO Manufacturing Flagship, Clayton, 3168, Australia
cSchool of Chemistry, Monash University, Clayton, 3800, Australia. E-mail: kei.saito@monash.edu
dSchool of Engineering, University of Warwick, Coventry, CV4 7AL, UK
First published on 27th November 2017
Abstract
The effect of visible LED power (λmax = 402 nm, 451 nm) on kinetics and control of direct photoactivation RAFT polymerisations of acrylamide and dimethylacrylamide are investigated. By increasing power supplied to the LEDs from 6 to 208 W, the polymerisation time required to reach >85% conversion is reduced from 12 hours to 11 minutes for acrylamide. Similar conversions are shown to be obtainable in 5 minutes for dimethylacrylamide, all without any exogenous photoinitiator or catalyst. This increase in polymerisation rate is attributed to an increase in both photon flux and a coincident increase in polymerisation temperature at higher light intensities. With both 402 nm and 451 nm LEDs exciting the same n → π* electronic transition, little difference in rate of polymerisation is seen between the two light sources. Minimal reduction in polymerisation control is observed at high irradiation intensity for acrylamide, while an increased production of low molecular weight dead chains is observed for dimethylacrylamide. This is shown to be mitigated by controlling the polymerisation temperature to 17 °C which caused both a reduction in low molecular weight tailing and an increased polymerisation time. Visible light direct photoactivation RAFT is also shown to have application in the synthesis of ultra-high molecular weight acrylamide polymers (Mn > 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 g mol−1).
000000 g mol−1).
Introduction
Various external stimuli have been used to elicit spatial and temporal control during reversible deactivation radical polymerisations (RDRPs).1,2 The use of applied voltage, mechanical force, light and chemical redox triggers have all been investigated.3 Of these triggers, light is one of the most ideal stimuli to afford this control; it is ubiquitous, easily modulated, non-invasive and allows for polymerisations to be conducted at room temperature. Common RDRP techniques such as nitroxide-mediated polymerisation (NMP),4,5 atom transfer radical polymerisation (ATRP)6,7 and reversible addition–fragmentation chain transfer (RAFT) polymerisation8–10 have all seen the development of respective photo-mediated polymerisation approaches.3,11 Photo-mediated RAFT polymerisation is particularly attractive due to the versatile nature of RAFT polymerisation, with tolerance to a range of reaction conditions and functionalities.12 For example, McKenzie et al. utilised visible light irradiation to generate core cross-linked star polymer nanoparticles in a high yielding, one-pot system. Light was then used to reinitiate polymerisation from the trithiocarbonate moiety located in the core, which then formed larger pseudo-miktoarm stars.13In general, there are three different methods to initiate RAFT polymerisations using light. The simplest involves the use of a photo-initiator (PI) to generate radicals independently of the chain transfer agent (CTA). Cai and co-workers extensively used (2,4,6-trimethylbenzoyl)diphenylphosphine oxide (TPO) to successfully initiate RAFT polymerisations under both UV and visible light. Various acrylate, methacrylate,14–16 acrylamide17 and styrenic18 monomers were polymerised with mainly trithiocarbonate CTAs in both organic and aqueous media. This system was particularly effective for poly(ethylene glycol) acrylate monomers, where under mild visible light irradiation in an aqueous solvent well-controlled polymers (Đ < 1.2) were synthesised to above 80% conversion in 10 minutes.19
A second innovative approach using photoinduced electron transfer (PET) was first applied to RAFT by Xu et al.20 This technique (PET-RAFT) involves the addition of a photoredox catalyst (PC), which under targeted irradiation can enter an excited state (PC*) and then reduce thiocarbonylthio compounds (RAFT agents) to a radical anion. This anion can fragment giving an initiating radical which can then propagate and participate in the RAFT process or be deactivated by back electron transfer, producing a dormant polymer chain and returning the PC to its ground state. Both metal complex20,21 and organic PCs22,23 have been found to initiate polymerisation from blue to near infrared (435–850 nm) radiation.24 Since there is no external initiating species, PET-RAFT does not suffer from the production of polymer chains with an initiator-derived moiety at the α-end (which is produced under PI and other conventional initiation systems).25 Another advantage of PET-RAFT is the possibility to conduct polymerisations without prior degassing, since (during an extended induction period) the PC is able to reduce dissolved oxygen into an inactive species.20
Lastly, the initiating species may be generated by photolysis of the weak C–S bond in the thiocarbonylthio group of the CTA, producing an active carbon-centred radical and a more stable thiyl radical (Fig. 1). In this way, the thiocarbonylthio compound acts as a photoiniferter26 (both initiator and CTA). Similar to PET-RAFT, direct photoactivation RAFT (DP-RAFT) avoids the production of initiator-derived moieties at the α-end of the chain, with the advantage that no external PC is required.
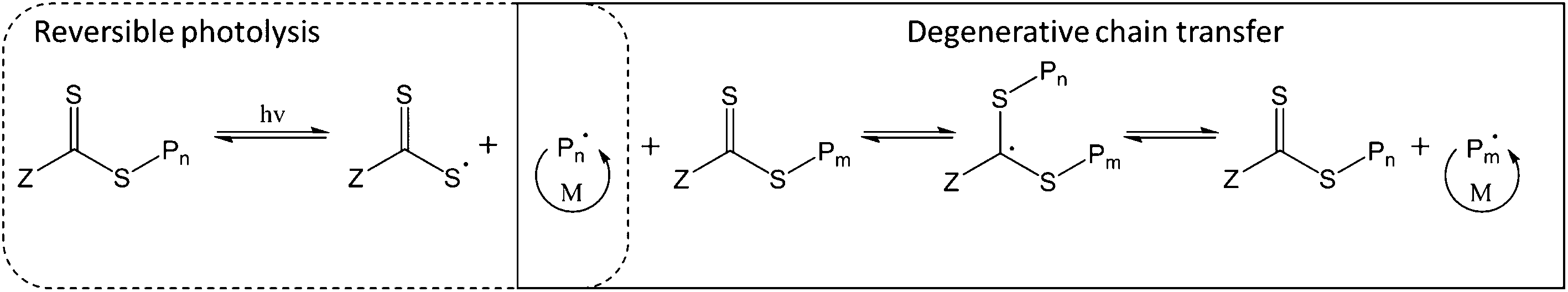 | ||
| Fig. 1 Direct photoactivation RAFT (DP-RAFT) polymerisation mechanism. | ||
Initially, DP-RAFT was investigated using UV (<400 nm) radiation and polymerisations were often characterised by loss of control at moderate to high conversions.27,28 This was likely due to CTA degradation from the high energy UV light, to which the thiocarbonylthio species is highly sensitive.14,29 Recently, there has been a number of publications utilising visible light for DP-RAFT. In this case, the visible light radiation targets the n → π* transition of the thiocarbonyl group (whereas UV radiation targets the stronger π → π* transition).30 Visible light has many advantages over UV radiation; it is safer, more widely available, has a greater depth of penetration and has been reported to result in less CTA degradation.31 In addition, tertiary amine catalysts have been added to DP-RAFT which can accelerate the polymerisation and facilitate removal of dissolved oxygen.32–34 Although utilising an external catalyst, this differs from PET-RAFT in that the CTA is still the photoactive compound, with the tertiary amine interacting after photoexcitation of the CTA. The use of visible light (blue and green LEDs) for catalyst-free DP-RAFT has resulted in the controlled polymerisation of acrylates, methacrylates,35–37 acrylamides,30 styrene,38 acrylonitrile,39 vinyl acetate40 and 2-vinylpyridine.41 Trithiocarbonates, dithioesters, xanthates and dithiocarbamates have all been utilised as both photoinitiator and CTA during these works.
For each of these visible light DP-RAFT polymerisations – which have included monomers with high (acrylamides) and low (methacrylates) inherent rates of polymerisation – achieving conversions greater than 80% has required 5 to 10 hours. These relatively long polymerisation times may be reduced by switching from batch to flow processing, as has recently been demonstrated by Rubens et al.37 Methacrylate monomers were polymerised to quantitative conversion in a microflow reactor in 60 minutes under mild visible light irradiation (LED power of 14.4 W). Polymerisation times could possibly be further reduced by increasing the power output of the visible light source, however, investigations into the effect of irradiation intensity on polymerisation rate and control are limited. Increasing UV irradiation intensity from 3 to 48 mW cm−2 has been shown to reduce the polymerisation time to 10% conversion of methyl acrylate monomer from approximately 200 to 35 minutes. In addition, very little reduction in polymerisation control was observed.42 Other works investigating a smaller range of intensities have also reported increased polymerisation rate at higher UV intensity, with little43 to moderate,44 reduction in polymerisation control. Matyjaszewski et al. looked at increasing green light (530 nm) intensity (by increasing LED power from 1 to 5 W) for the polymerisation of methyl methacrylate with a trithiocarboante CTA. This resulted in an approximate doubling of polymerisation rate with minimal increase in dispersity or alteration of the molecular weight distribution (MWD).35 Kinetic modelling on the effect of irradiation intensity has also been conducted, which highlighted the importance of quantum yield and intensity for the formation of dissociated species.38 The impact of this on polymerisation control was, however, not investigated. Despite the lack of evidence for loss of polymerisation control under increased visible light intensity, the use of high powered LEDs for DP-RAFT has until now not been reported.
In this work, we demonstrate that by increasing LED power output (up to 208 W), polymerisation times to >85% conversion for dimethylacrylamide (DMA) and acrylamide (AM) can be reduced to 5 and 11 minutes respectively. This reduction in polymerisation time is accompanied with a moderate reduction in polymerisation control for DMA polymers (Đ = 1.20 at 208 W), which is shown to be largely due to associated thermal effects.
Experimental
Materials
N,N-Dimethylacrylamide (DMA, 99%) was obtained from Sigma Aldrich and passed through basic alumina prior to use to remove inhibitor. Acrylamide (AM, 98% Sigma) and 1,4 dioxane (99.9%, Merck) were used as received. The chain transfer agent S,S′-bis(α,α′-dimethyl-α′′-acetic acid)trithiocarbonate (DMAT) was synthesised according to literature procedures.45 Analytical data were in agreement with published values. Deuterium oxide (D2O, 99.99% D atom) was obtained from Cambridge Isotope Laboratories and was used for 1H NMR analysis and as a solvent for experiments conducted in NMR tubes.Methods
Mpvs. time calibration curve for both systems, which was near linear across the molar mass ranges.
:[DMAT] = 140:1. The reaction mixture was then degassed by bubbling with argon for 15 minutes and then placed into the desired LED reactor. The reaction was started by turning on the LEDs at the desired power which was set by adjusting the current with a potentiometer. The current was measured with a multimeter connected in series. During the polymerisation, aliquots for 1H NMR and GPC analysis were taken with a degassed syringe. The reaction was quenched by removing the glass tube from the light source and opening to air. The polymer was then purified by precipitation into acetone, followed by freeze drying.
:[DMAT] = 140:1. This stock was then stored in the dark under refrigeration, with 1 mL samples taken into an NMR tube with septum for each induvial experiment. These were then degassed with argon bubbling for 15 minutes, before placing inside a glass jacket connected to a water reservoir and circulating system. Depending on the experiment the temperature in the reservoir was then maintained at either 30 °C by heating or 17 °C by periodic addition of ice. Polymerisation was started by turning on the LEDs at the desired power, with conversion monitored during polymerisation by transferring the NMR tube to an appropriate spectrometer. Purification was as described above.
:[DMAT] = 7000:1. The reaction mixture was then degassed by bubbling with argon for 15 minutes and then placed into the desired LED reactor. The reaction was started by turning on the LEDs at the desired power. The reaction was quenched by removing the glass tube from the light source and opening to air before taking a sample for 1H NMR analysis. The polymer sample was then freeze dried and analysed by GPC.
:[MacroCTA] = 160:1. The reaction mixture was then degassed by bubbling with argon for 15 minutes and then placed into the 402 nm reactor at 108 W. The reaction was quenched by removing the glass tube from the light source and opening to air. The polymer was then purified by precipitation into acetone, followed by freeze drying.
All conversions were calculated by comparing the 1H-NMR integrals of the monomer unsaturated protons to the polymer backbone.
A similar protocol was followed for all DMA polymerisations, except DMA was added in neat. Purification was performed by first freeze drying (to remove water), followed by precipitation from acetone into diethyl ether.
Results and discussion
Investigation into effect of LED power (without temperature control)
Two different LED reactors were constructed with peak emissions (λmax) at 402 nm and 451 nm in order to investigate the effect of LED power and wavelength on DP-RAFT polymerisations (Fig. S1†). Each reactor was designed such that the same glass tube could be used in all polymerisations, minimising variance due to geometrical differences. By modulating the current, total electrical power through the system could be varied from 26 to 208 W (402 nm) and 6 to 26 W (451 nm). In the first set of experiments, AM ([M]0 = 2 M) was polymerised to a targeted degree of polymerisation (DP) 140 in H2O/dioxane (80:20) under various wavelengths and intensities (reported as total electrical power through the LED) of light. DMAT was chosen as the CTA for this investigation (Fig. 2) since trithiocarbonates have been widely and successfully used in DP-RAFT, with DMAT shown to be effective in polymerisations of acrylamide.47,48Fig. 3 displays the overlap between the spectral output of both LEDs and the n → π* transition of DMAT. It can be seen that DMAT undergoes a blue shift from its initial (non-monomer inserted) state to the monomer adduct, in agreement with literature reports for DMAT and other trithiocarbonate CTAs.31 This monomer adduct forms almost immediately and so will dictate the spectral absorbance for the majority of polymerisation. In this case the overlap is found to be similar for both LEDs.
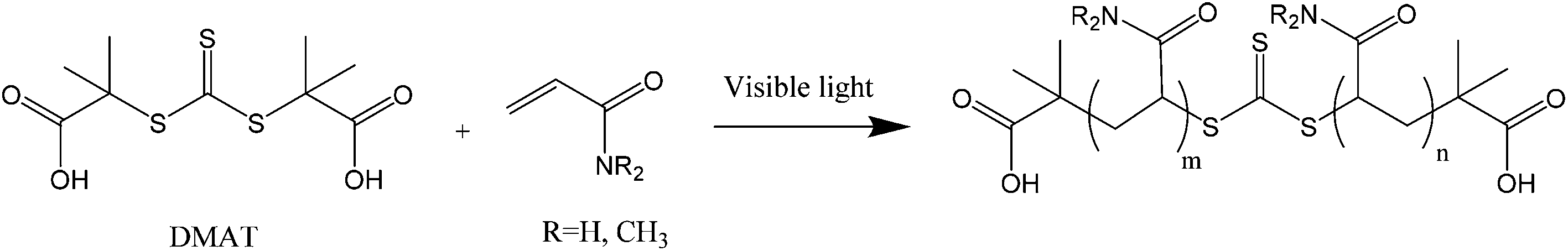 | ||
| Fig. 2 Chemical structure of CTA DMAT used in this study and resulting polyacrylamides. | ||
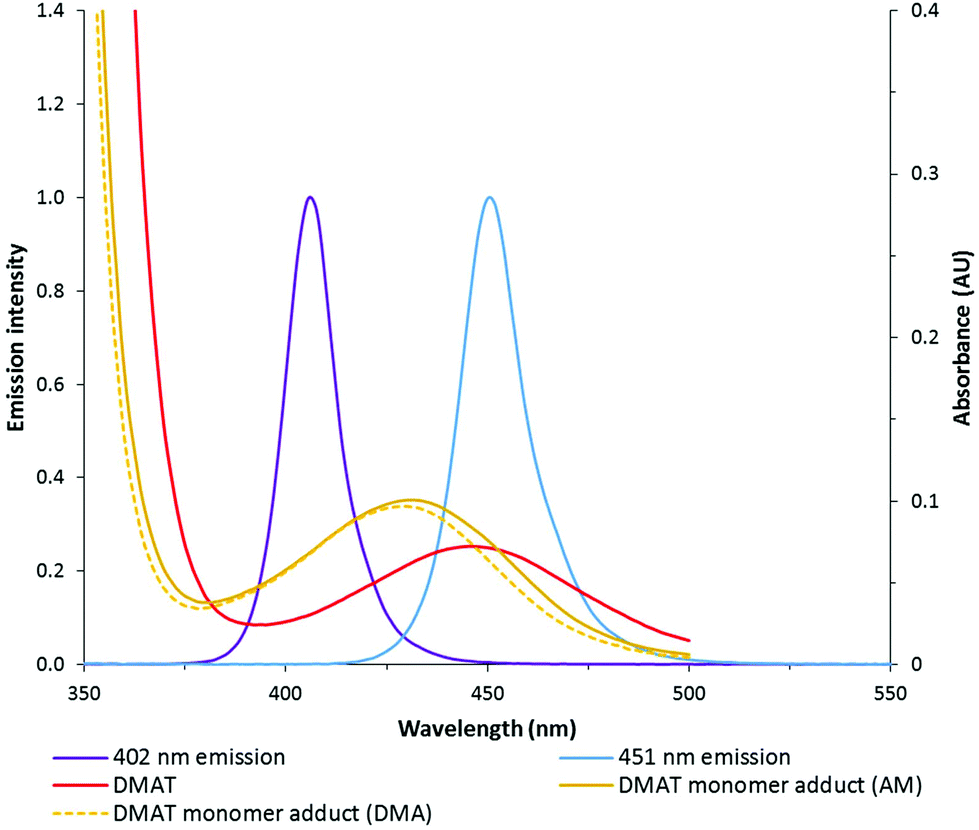 | ||
| Fig. 3 Spectral output of 402 and 451 nm LEDs overlayed with absorption of DMAT (2.14 mM) in both initial and monomer adduct state. | ||
The DP-RAFT polymerisations of AM proceeded with linear pseudo-first order kinetics after a short induction period which was inversely proportional to the irradiation power. A linear increase in number average molecular weight (Mn(GPC)) with conversion was found for all polymerisations, along with narrow MWDs throughout (Đ ≤ 1.20) (Fig. 4). The MWDs for the final sample point of all experiments (85–90% conversion) were found to be symmetrical and precisely overlap with each other (Fig. S2†). Combined, these data indicate that all polymerisations were well controlled. Neither wavelength (402 and 451 nm) nor LED power (6 to 208 W) had an observed detrimental effect on polymerisation control. Polymerising AM at the lowest power (6 W, 451 nm) required 12 hours to reach 85% conversion. Increasing irradiation power was found to directly increase the rate of polymerisation, with the highest irradiation power (208 W, 402 nm) reaching the same conversion in 11 minutes. Wavelength (402 and 451 nm) did not have a noticeable effect on polymerisation rate, perhaps due to the similar overlap in spectral emission of the two LEDs with the absorption of the DMAT monomer adduct (Fig. 3).
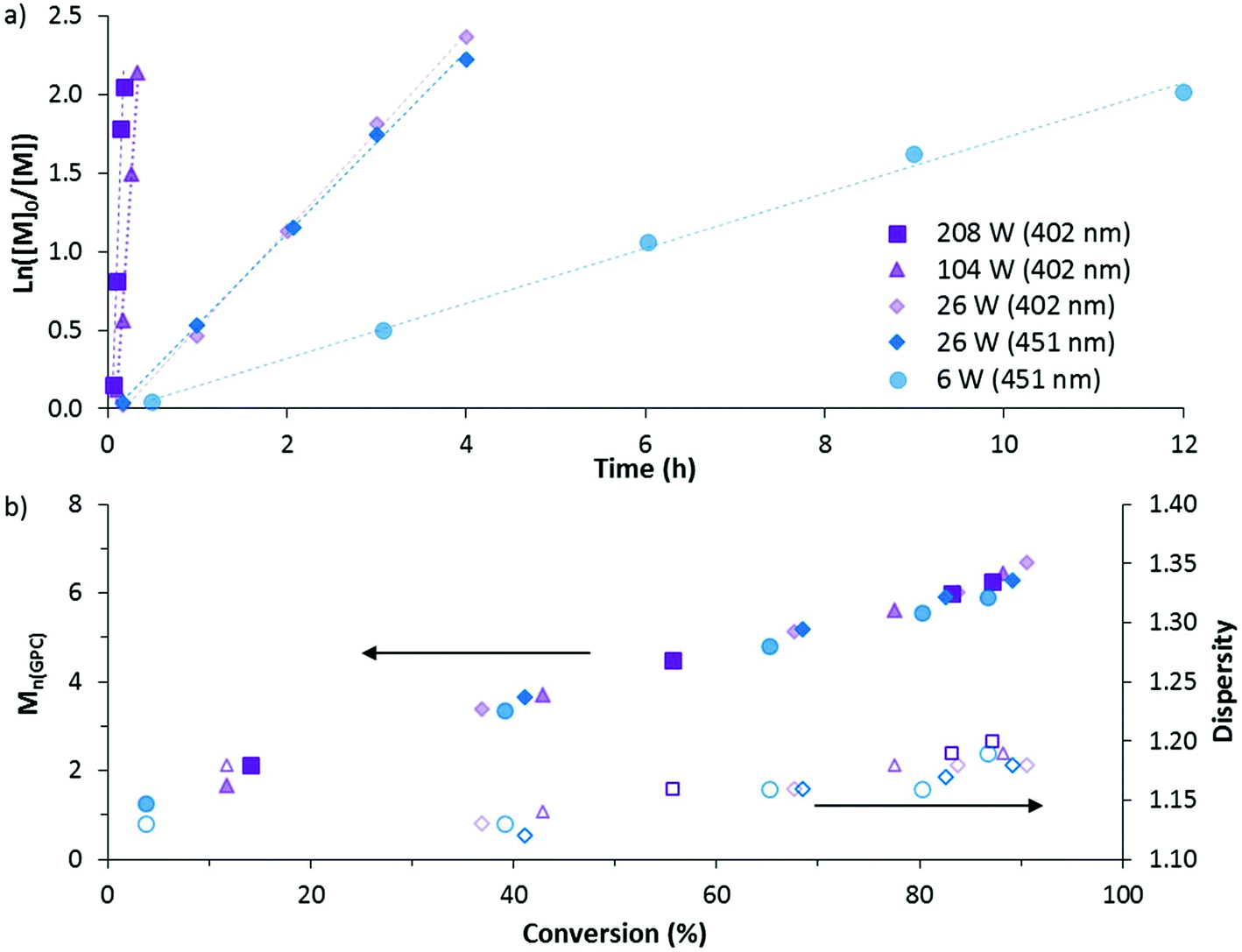 | ||
| Fig. 4 (a) Kinetic plot of DP-RAFT polymerisations ([AM]0 = 2 M, [AM]:[DMAT] = 140:1, H2O/dioxane 80:20) at different wavelengths and intensities. (b) Evolution in number average molecular weight (Mn(GPC) × 10−3) and dispersity (Đ) with increasing conversion for DP-RAFT polymerisation of AM. | ||
The polymers were then purified and chain extended with a further 160 units of AM in order to demonstrate the livingness of each polymerisation. The chain extensions were all done by irradiation with 402 nm light at 104 W for 20 minutes. At this point, conversions were >70% and the polymers were again purified by precipitation into acetone and freeze-dried. All macro-CTAs were successfully chain extended (Fig. S2, ESI†) and – with the exception of the highest irradiation power (208 W) – gave polymers with similar dispersities (1.28–1.29) and Mn(GPC) (10600–11600 g mol−1) (Table 1). The 208 W macro CTA produced a polymer with dispersity 1.33, suggesting that use of such high irradiation power comes with some loss of control.
| Sample | Monomer | Macro CTA Mn,gpc (g mol−1) | Macro CTA Đ | Extension Mn,gpc (g mol−1) | Extension Đ |
|---|---|---|---|---|---|
| a All chain extensions were conducted by adding a further 160 units of the starting monomer under 104 W (402 nm) irradiation for 20 minutes in H2O. | |||||
| 208 W (402 nm) | AM | 6250 | 1.20 | 11300 |
1.33 |
| 104 W (402 nm) | AM | 6440 | 1.19 | 11100 |
1.29 |
| 26 W (402 nm) | AM | 6700 | 1.18 | 11600 |
1.29 |
| 26 W (451 nm) | AM | 6290 | 1.18 | 10900 |
1.28 |
| 6 W (451 nm) | AM | 5290 | 1.19 | 10600 |
1.28 |
| 208 W (402 nm) | DMA | 11800 |
1.20 | 25900 |
1.23 |
Continuing our investigations into the use of high power visible light for DP-RAFT we decided to see if these results could be extended to DMA – another water soluble acrylamide monomer. Utilizing the same polymerisation conditions as was applied to AM (2 M monomer concentration, H2O/dioxane 80:20, [DMA]:[DMAT] = 1:140) a preliminary experiment was conducted at 104 W (402 nm). After 20 minutes of irradiation, near quantitative conversion was reached producing a polymer with dispersity 1.14 and Mn(GPC) 11800 g mol−1. These data indicated that DMA can be polymerised at a faster rate than AM through DP-RAFT and with similar control. Repeating this experiment at the highest irradiation power (208 W), polymerisation time to >85% conversion was reduced to approximately 5 minutes. While kinetic data again demonstrated linear pseudo-first order kinetics, broadening of the MWD had occurred (Fig. 5a and b) with dispersity increasing to 1.20 and clear evidence of low molecular weight dead chains – likely formed from bimolecular termination events.
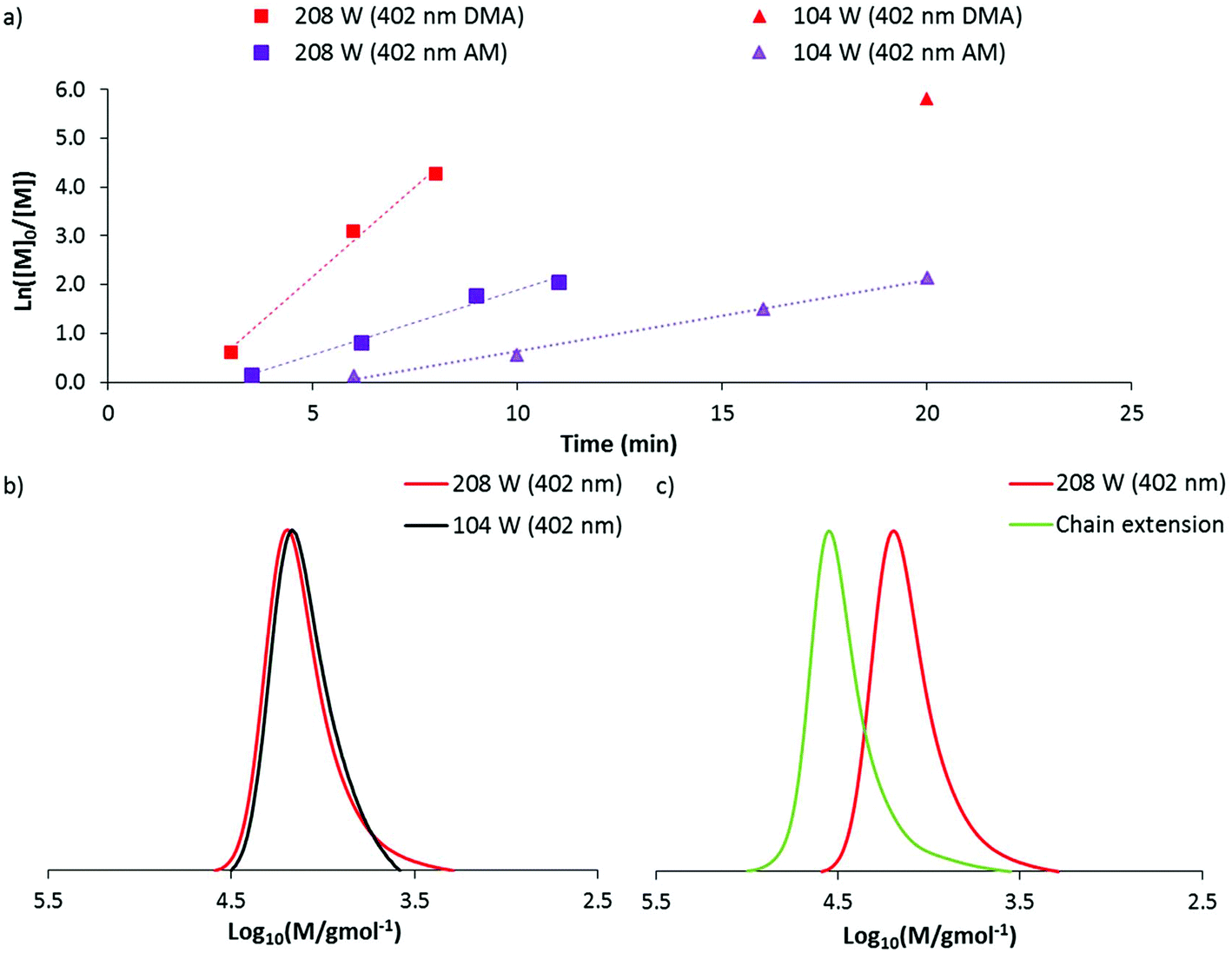 | ||
| Fig. 5 (a) Kinetic plot of DP-RAFT polymerisations of DMA ([DMA]0 = 2 M, [DMA]:[DMAT] = 140:1, H2O/dioxane 80:20) under 402 nm irradiation at different intensities with matching AM polymerisations shown for reference, (b) GPC traces of DP-RAFT DMA polymerisations under 402 nm irradiation at different intensities, (c) GPC traces showing successful chain extension of a DMA polymer synthesised under 208 W (402 nm) irradiation. | ||
The isolated DMA polymer was then successfully chain extended with a further 160 units of DMA under 104 W (402 nm) in H2O for 20 minutes (conversion >99%). The macro CTA is shown as containing a significant proportion of living chains, with a clear shift of MWD to higher molecular weights. Still, it is clear that some of the distribution has not shifted, confirming the presence of low molecular weight dead chains (which were largely non-existent at 104 W) (Fig. 5c). This result again demonstrates the slight trade off in polymerisation control with higher irradiation power.
Until this point, all polymerisations have been conducted without temperature control and at the end of polymerisation under high irradiation powers (104 and 208 W) the product solution was often found to be at a temperature of 65 to 80 °C. These high temperatures are likely the result of both the exothermic nature of polymerisation and the intense irradiation. Thus, increasing irradiation power not only increases the rate of polymerisation through increased photolysis, but also by increasing reaction temperature (since propagation is thermally activated).37 Controlling for this increase in temperature is desirable to develop a mechanistic understanding and is discussed in the next section. However, depending on the degree of polymerisation control required, no temperature control may be advantageous from a materials synthesis point of view due to the experimental simplicity and much reduced polymerisation time.
Investigation into effect of LED power (with temperature control)
In order to control the temperature during polymerisation, an alteration to the experimental set-up was required. Polymerisations were conducted in an NMR tube within a glass jacket with water flowing through at the desired temperature. Selected polymerisations of AM were then repeated at 30 °C using D2O/dioxane 80:20 as the solvent (Fig. 6a). As before, linear pseudo-first order kinetics were observed for all polymerisations, with faster kinetics for experiments run at higher intensities. However, by eliminating the exotherm and controlling the temperature to 30 °C, all polymerisations were significantly slower than without temperature control. For example, at 104 W (402 nm) the polymerisation time to >85% conversion increased from 20 to 150 minutes. Alterations in the geometry (from a glass tube to a smaller NMR tube within a glass jacket) also appear to have an effect on kinetics. This was demonstrated by an experiment run without water in the jacket (NMR tube no temp), which therefore did not afford any direct temperature control. At 104 W (402 nm) slower polymerisation kinetics (45 minutes to >85% conversion) were observed compared to polymerisations conducted in the glass tube as used for previous experiments (20 minutes to >85% conversion).
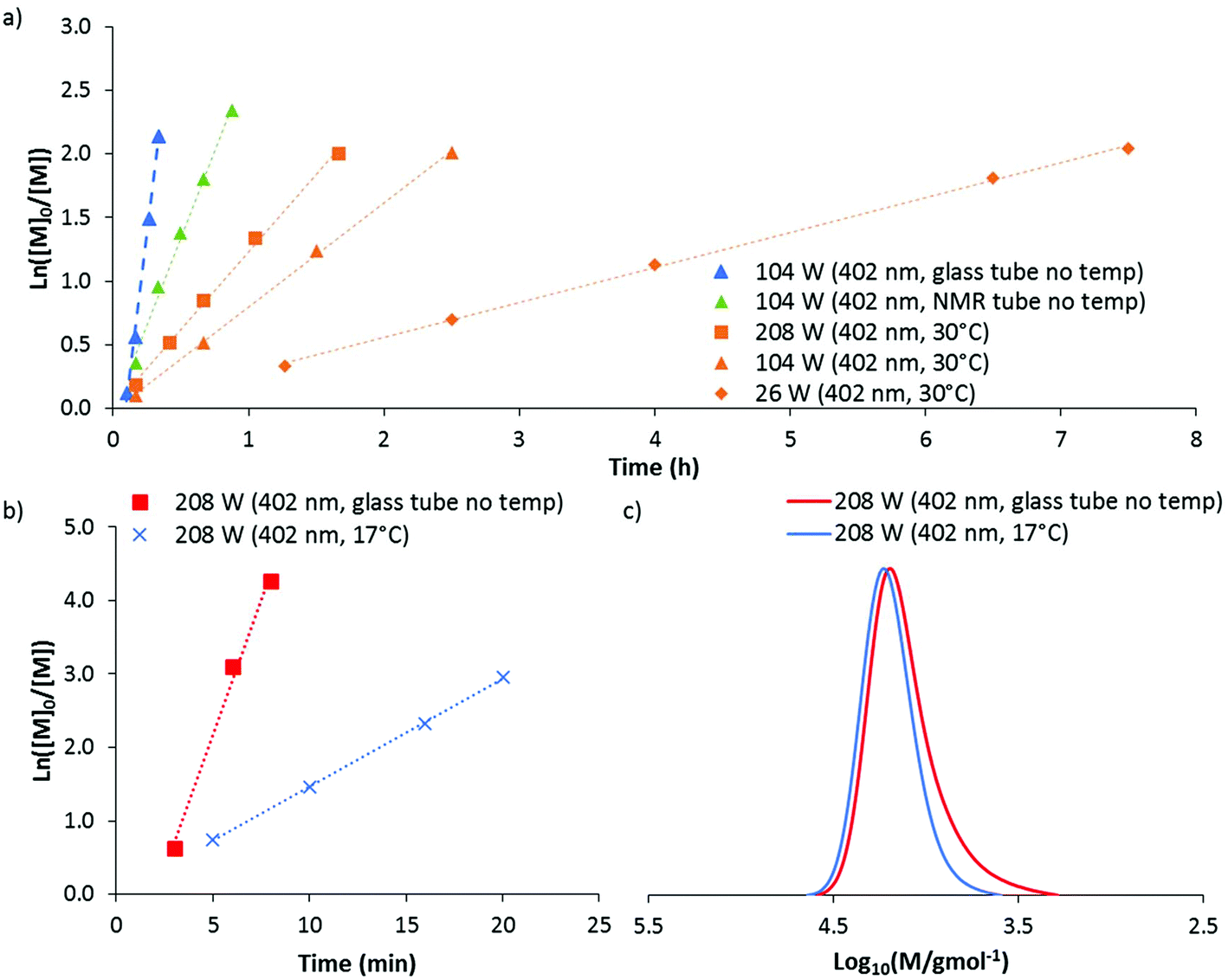 | ||
| Fig. 6 (a) Kinetic plot of DP-RAFT polymerisations of AM in an NMR tube under temperature controlled (30 °C) and non-temperature controlled conditions ([AM]0 = 2 M, [AM]:[DMAT] = 140:1, D2O/dioxane 80:20). Effect of temperature control in DP-RAFT of DMA under 208 W (402 nm) irradiation on (b) kinetics and (c) MWD. | ||
The resultant MWD from each of the polymerisations conducted at 30 °C was found to match very closely with those initially conducted without temperature control. Mn(GPC) was between 6090 and 7870 g mol−1 and dispersities between 1.21 and 1.23 (Table S1†).
Using this jacketed set-up we decided to investigate if the production of low molecular weight dead chains during the polymerisation of DMA at 208 W (402 nm) could be reduced if the polymerisation was cooled to 17 °C. 95% conversion was obtained in 20 minutes, which (like the data obtained for AM polymerisations) followed linear pseudo-first order kinetics but was significantly slower than without temperature control (Fig. 6b). Importantly, the resultant polymer was found to have significantly reduced low molecular weight tailing (Fig. 6c), with a dispersity of 1.12 (compared to 1.20 without temperature control). This suggests that the moderate loss of control observed at higher irradiation power for DMA can be partly attributed to the coincident increase in temperature and not necessarily due to the increased photon flux. The detrimental effect of higher temperatures may be due to hydrolysis of the CTA, which has been demonstrated to occur for DMAT at temperatures above 50 °C in aqueous acrylamido polymerisations.49
If we (as an approximation) relate electrical power through the LED system to irradiation power (IP) we can plot the natural log of IP against the natural log of the first order rate constant for each polymerisation (Fig. 7). This plot can give an indication of the experimental power law relationship between IP and the rate constant. Looking at the data for AM polymerisations at 30 °C under 402 nm irradiation, a power law relationship of 0.73 can be found. This is somewhat larger than the expected dependence on initiator concentration for a RAFT polymerisation of 0.5,50 possibly due to limitations of our set-up to provide exact temperature control at all points within the NMR tube during polymerisation. Overall, this plot demonstrates that the observed increase in rate of polymerisation at higher intensities is not just attributable to higher reaction temperatures, but also increased photon flux. Additionally, experiments performed at 70 °C in the dark confirmed that irradiation is required for initiation. Other control experiments have been conducted confirming the dual role of the CTA in both initiation and control of polymerisation (Table S2†).
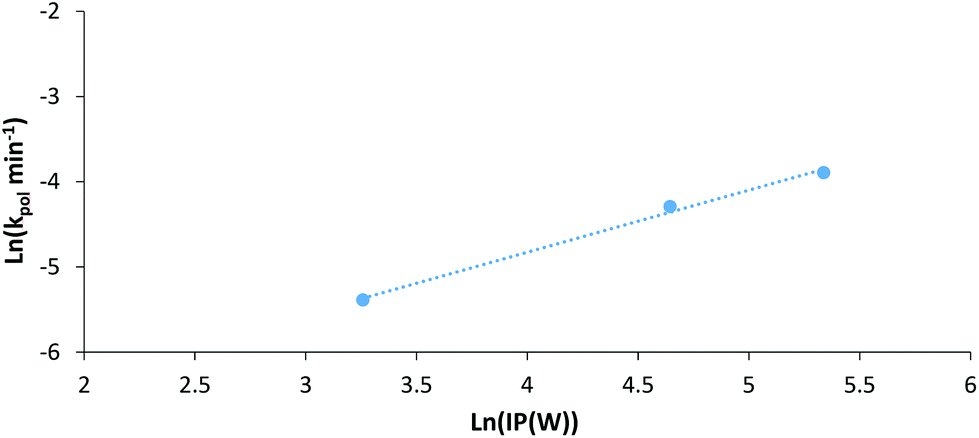 | ||
| Fig. 7 log–log plot of the pseudo-first order polymerisation rate constant (kpol) and irradiation power (IP, approximated from power through LEDs) for AM DP-RAFT polymerisations under 402 nm irradiation controlled to 30 °C. | ||
Ultra-high molecular weight polymers by visible light direct activation RAFT
Inspired by the recent work of Carmean et al.51 which demonstrated the applicability of DP-RAFT polymerisations to generate ultra-high molecular polymers under UV irradiation, we decided to investigate if similar results could be obtained under visible light. Polyacrylamide Mn values of 500000 and 2000000 g mol−1 were targeted by increasing [Monomer]:[DMAT] from 140:1 to 7000:1 and 28000:1 respectively. A range of LED powers and wavelengths were investigated with polymerisation times in the order of hours required to reach conversions of >70% conversion (Table S3†). Selected GPC traces are presented in Fig. 8, demonstrating the formation of monomodal peaks shifting to earlier retention times when higher molecular weights are targeted. Molecular weights were found to be in close agreement with theoretical predictions with moderate dispersities (1.32 ≤ Đ ≤ 1.44). Additionally the retention of the trithiocarbonate moity (λmax = 305 nm) in these high molecular weight chains was also supported by close agreement in RI and UV detector (λ = 305 nm) data for AM polymerisations (Fig. S4†).
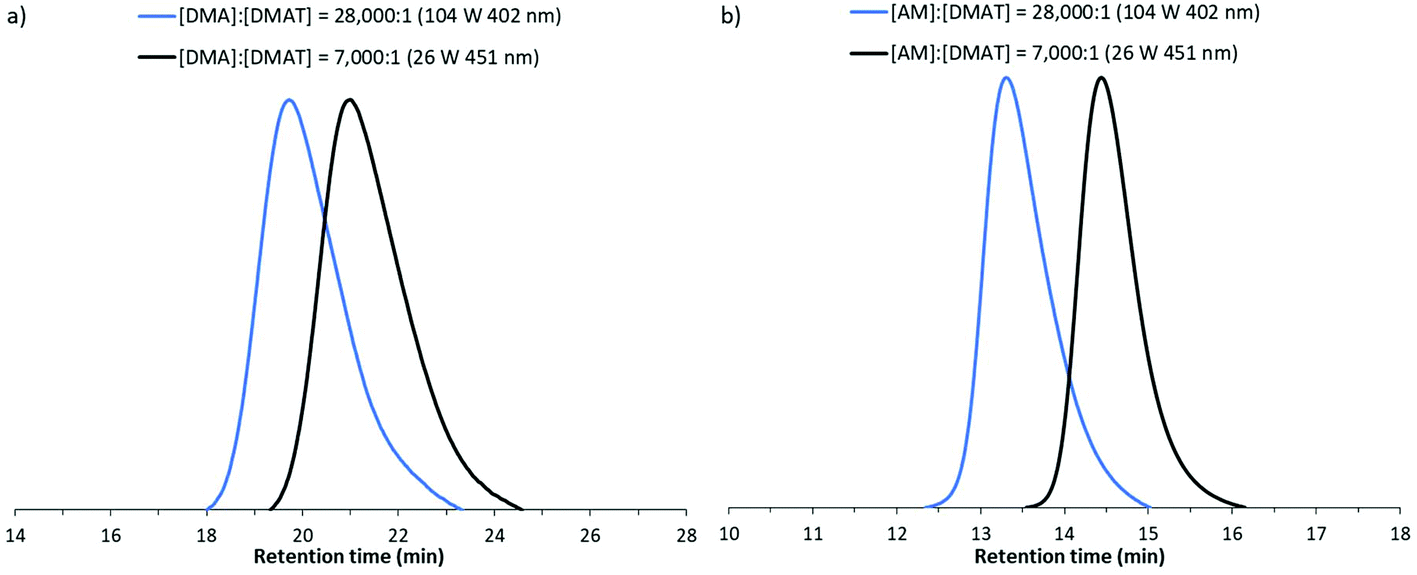 | ||
| Fig. 8 GPC traces for ultra-high molecular weight DP-RAFT polymerisations of (a) DMA (as characterised by Shimadzu DMAc GPC) and (b) AM (as characterised by Waters Alliance aqueous GPC) under visible light irradiation. | ||
Conclusions
By increasing the power supplied to LEDs during direct photoactivation RAFT (DP-RAFT), significant reductions in polymerisation time to high conversion is possible. At 208 W (402 nm) conversions of greater than 85% are obtainable in 11 minutes for acrylamide (AM) and 5 minutes for dimethylacrylamide (DMA). This represents a significant reduction from the 12 hours required to achieve similar conversions for AM under 6 W (451 nm) irradiation. Choice of wavelength (between 402 nm and 451 nm LEDs) did not appear to have a direct effect on polymerisation control or kinetics for the studied CTA – monomer system. The increase in polymerisation rate was attributed to both an increase in photon flux and the coincident increase in reaction temperature from the intense irradiation and polymerisation exotherm. By chain extending each AM homopolymer produced by DP-RAFT, it was found that only at the highest irradiation power (208 W) some loss in polymerisation control was observed. Similar results were found for DMA polymerisations, where increased formation of low molecular weight dead chains was observed in the MWD after polymerisation at 208 W. This loss of control at high irradiation powers can be partly attributed to the simultaneous increase in reaction temperature, as was demonstrated by the synthesis of narrow dispersity (Đ = 1.12) DMA polymers at 17 °C under 208 W (402 nm) irradiation. Lastly, we have demonstrated the applicability of DP-RAFT under visible light irradiation for rapid synthesis (<3 h) of ultra-high molecular weight acrylamide polymers (Mn > 1000000 g mol−1).
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The Ph.D position of RWL was supported by the Chemicals and Plastics Manufacturing Innovation Network and Training Program at Monash University.References
- A. D. Jenkins, R. G. Jones and G. Moad, Pure Appl. Chem., 2010, 82, 483–491 CAS.
- F. A. Leibfarth, K. M. Mattson, B. P. Fors, H. A. Collins and C. J. Hawker, Angew. Chem., Int. Ed., 2013, 52, 199–210 CrossRef CAS PubMed.
- S. Shanmugam, J. T. Xu and C. Boyer, Macromol. Rapid Commun., 2017, 38, 1700143 CrossRef PubMed.
- M. K. Georges, R. P. N. Veregin, P. M. Kazmaier and G. K. Hamer, Macromolecules, 1993, 26, 2987–2988 CrossRef CAS.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
- J. S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, Chem. Rev., 2007, 107, 2270–2299 CrossRef CAS PubMed.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 CrossRef CAS.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Q. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS PubMed.
- X. C. Pan, M. A. Tasdelen, J. Laun, T. Junkers, Y. Yagci and K. Matyjaszewski, Prog. Polym. Sci., 2016, 62, 73–125 CrossRef CAS.
- M. Semsarilar and S. Perrier, Nat. Chem., 2010, 2, 811–820 CrossRef CAS PubMed.
- T. G. McKenzie, E. H. H. Wong, Q. Fu, A. Sulistio, D. E. Dunstan and G. G. Qiao, ACS Macro Lett., 2015, 4, 1012–1016 CrossRef CAS.
- L. C. Lu, N. F. Yang and Y. L. Cai, Chem. Commun., 2005, 5287–5288 RSC.
- W. D. Jiang, L. C. Lu and Y. L. Cai, Macromol. Rapid Commun., 2007, 28, 725–728 CrossRef CAS.
- Y. Shi, G. H. Liu, H. Gao, L. C. Lu and Y. L. Cai, Macromolecules, 2009, 42, 3917–3926 CrossRef CAS.
- G. H. Liu, H. Shi, Y. R. Cui, J. Y. Tong, Y. Zhao, D. J. Wang and Y. L. Cai, Polym. Chem., 2013, 4, 1176–1182 RSC.
- H. J. Zhang, J. J. Deng, L. C. Lu and Y. L. Cai, Macromolecules, 2007, 40, 9252–9261 CrossRef CAS.
- Y. Shi, H. Gao, L. C. Lu and Y. L. Cai, Chem. Commun., 2009, 1368–1370 RSC.
- J. T. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS PubMed.
- J. T. Xu, K. Jung, N. A. Corrigan and C. Boyer, Chem. Sci., 2014, 5, 3568–3575 RSC.
- M. Chen, M. J. MacLeod and J. A. Johnson, ACS Macro Lett., 2015, 4, 566–569 CrossRef CAS.
- J. T. Xu, S. Shanmugam, H. T. Duong and C. Boyer, Polym. Chem., 2015, 6, 5615–5624 RSC.
- S. Shanmugam, J. T. Xu and C. Boyer, Angew. Chem., Int. Ed., 2016, 55, 1036–1040 CrossRef CAS PubMed.
- Y. Wu, W. Zhang, Z. B. Zhang, X. Q. Pan, Z. P. Cheng, J. Zhu and X. L. Zhu, Chem. Commun., 2014, 50, 9722–9724 RSC.
- T. Otsu, M. Yoshida and T. Tazaki, Makromol. Chem., Rapid Commun., 1982, 3, 133–140 CrossRef CAS.
- J. F. Quinn, L. Barner, C. Barner-Kowollik, E. Rizzardo and T. P. Davis, Macromolecules, 2002, 35, 7620–7627 CrossRef CAS.
- Y. Z. You, C. Y. Hong, R. K. Bai, C. Y. Pan and J. Wang, Macromol. Chem. Phys., 2002, 203, 477–483 CrossRef CAS.
- J. F. Quinn, T. P. Davis, L. Barner and C. Barner-Kowollik, Polymer, 2007, 48, 6467–6480 CrossRef CAS.
- T. G. McKenzie, Q. Fu, E. H. H. Wong, D. E. Dunstan and G. G. Qiao, Macromolecules, 2015, 48, 3864–3872 CrossRef CAS.
- L. Lu, H. J. Zhang, N. F. Yang and Y. L. Cai, Macromolecules, 2006, 39, 3770–3776 CrossRef CAS.
- Q. Fu, T. G. McKenzie, S. Tan, E. Nam and G. G. Qiao, Polym. Chem., 2015, 6, 5362–5368 RSC.
- M. L. Allegrezza, Z. M. DeMartini, A. J. Kloster, Z. A. Digby and D. Konkolewicz, Polym. Chem., 2016, 7, 6626–6636 RSC.
- Q. Fu, K. Xie, T. G. McKenzie and G. G. Qiao, Polym. Chem., 2017, 8, 1519–1526 RSC.
- J. T. Xu, S. Shanmugam, N. A. Corrigan and C. Boyer, Controlled Radical Polymerization, Vol 1: Mechanisms, 2015, vol. 1187, pp. 247–267 Search PubMed.
- K. Jung, C. Boyer and P. B. Zetterlund, Polym. Chem., 2017, 8, 3965–3970 RSC.
- M. Rubens, P. Latsrisaeng and T. Junkers, Polym. Chem., 2017, 8, 6496–6505 RSC.
- B. Cabannes-Boue, Q. Z. Yang, J. Lalevee, F. Morlet-Savary and J. Poly, Polym. Chem., 2017, 8, 1760–1770 RSC.
- J. J. Li, C. L. Ding, Z. B. Zhang, J. Zhu and X. L. Zhu, React. Funct. Polym., 2017, 113, 1–5 CrossRef CAS.
- C. L. Ding, C. W. Fan, G. Q. Jiang, X. Q. Pan, Z. B. Zhang, J. Zhu and X. L. Zhu, Macromol. Rapid Commun., 2015, 36, 2181–2185 CrossRef CAS PubMed.
- J. X. Luo, M. C. Li, M. H. Xin, W. F. Sun and W. C. Xiao, Macromol. Chem. Phys., 2016, 217, 1777–1784 CrossRef CAS.
- H. Wang, Q. B. Li, J. W. Dai, F. F. Du, H. T. Zheng and R. K. Bai, Macromolecules, 2013, 46, 2576–2582 CrossRef CAS.
- M. Y. Khan, M. S. Cho and Y. J. Kwark, Macromolecules, 2014, 47, 1929–1934 CrossRef CAS.
- H. X. Zhou and J. A. Johnson, Angew. Chem., Int. Ed., 2013, 52, 2235–2238 CrossRef CAS PubMed.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- R. W. Lewis, R. A. Evans, N. Malic, K. Saito and N. R. Cameron, Polym. Chem., 2017, 8, 3702–3711 RSC.
- D. B. Thomas, A. J. Convertine, L. J. Myrick, C. W. Scales, A. E. Smith, A. B. Lowe, Y. A. Vasilieva, N. Ayres and C. L. McCormick, Macromolecules, 2004, 37, 8941–8950 CrossRef CAS.
- W. Bai, L. Zhang, R. Bai and G. Z. Zhang, Macromol. Rapid Commun., 2008, 29, 562–566 CrossRef CAS.
- A. J. Convertine, B. S. Lokitz, A. B. Lowe, C. W. Scales, L. J. Myrick and C. L. McCormick, Macromol. Rapid Commun., 2005, 26, 791–795 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- R. N. Carmean, T. E. Becker, M. B. Sims and B. S. Sumerlin, Chem, 2017, 2, 93–101 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7py01752a |
| This journal is © The Royal Society of Chemistry 2018 |