
Water-adaptive and repeatable self-healing polymers bearing bulky urea bonds†
J. I.
Park
a,
A.
Choe
b,
M. P.
Kim
*b,
H.
Ko
 b,
T. H.
Lee
c,
S. M.
Noh
c,
J. C.
Kim
*c and
I. W.
Cheong
*a
b,
T. H.
Lee
c,
S. M.
Noh
c,
J. C.
Kim
*c and
I. W.
Cheong
*a
aDepartment of Applied Chemistry, Kyungpook National University, Buk-gu, Daehak-ro 80, Daegu 41566, Republic of Korea. E-mail: inwoo@knu.ac.kr
bSchool of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology, Ulsan 44919, Korea
cResearch Center for Green Fine Chemicals, Korea Research Institute of Chemical Technology, Ulsan, 44412, Republic of Korea
First published on 27th October 2017
Abstract
Self-healing polymers bearing reversible and bulky urea bonds were prepared by free-radical copolymerization followed by crosslinking with diisocyanates. Linear prepolymers (PtB) were first synthesized from 2-(tert-butylamino)ethyl methacrylate (tBAEMA), methyl methacrylate (MMA), and n-butyl acrylate (BA), in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10, and then crosslinked with 1,6-diisocyanatohexane (HDI) or isophorone diisocyanate (IPDI) to fabricate self-healing polymers. The reversible bonding–debonding between the tBAEMA of the polymer backbone and the isocyanate units of the crosslinker in the self-healing polymer facilitates rapid, repeatable, and water-adaptive self-healing performance, which has been extensively investigated using 1H-NMR, DSC, FT-IR, AFM, optical microscopy, and rheometric analyses. Percentage recovery (%R) and self-healing efficiency (%SHE) were also studied using tensile and single-scratch tests, respectively, and compared with a control sample. It was revealed through extensive analyses that self-healing against a single scratch can be accelerated and repeated up to a certain number of times even in water, although the self-healing polymers are not water-resistant. We also demonstrated that a self-healable tube capable of containing flowing water could be fabricated from a self-healing polymer sheet by using reversible bonding–debonding characteristics of a reversible bulky urea bond.
:10:10, and then crosslinked with 1,6-diisocyanatohexane (HDI) or isophorone diisocyanate (IPDI) to fabricate self-healing polymers. The reversible bonding–debonding between the tBAEMA of the polymer backbone and the isocyanate units of the crosslinker in the self-healing polymer facilitates rapid, repeatable, and water-adaptive self-healing performance, which has been extensively investigated using 1H-NMR, DSC, FT-IR, AFM, optical microscopy, and rheometric analyses. Percentage recovery (%R) and self-healing efficiency (%SHE) were also studied using tensile and single-scratch tests, respectively, and compared with a control sample. It was revealed through extensive analyses that self-healing against a single scratch can be accelerated and repeated up to a certain number of times even in water, although the self-healing polymers are not water-resistant. We also demonstrated that a self-healable tube capable of containing flowing water could be fabricated from a self-healing polymer sheet by using reversible bonding–debonding characteristics of a reversible bulky urea bond.
Introduction
When a polymer is damaged by external forces such as fracturing or scratching, its mechanical strength, surface gloss, toughness, and barrier properties, among others, deteriorate or become permanently lost, leading to maintenance costs for repair or replacement. However, self-healing polymers are able to repair themselves, thereby increasing the lifespan of polymeric products. Self-healing polymers can be categorized into extrinsic and intrinsic systems.1 A representative extrinsic self-healing material is a microcapsule/polymer matrix composite developed by the White group.2 Similarly, microvascular systems that mimic blood vessels have also been developed.3 Although both systems show good performance in healing cracks, their healing mechanisms cannot provide multiple healing capabilities. Very recently, the Chung group developed a capsule system that can avoid secondary damage;4 however, its properties are insufficient for commercial use because the crack-filling healing agent used is too soft to resist external forces other than vibration. On the other hand, intrinsic systems offer multiple healing capabilities through the formation of non-covalent or covalent bonds after bond breakage at the molecular level through hydrogen bonding,5 π–π stacking,6 host–guest interactions,7 reversible cycloaddition reactions,8 exchangeable covalent bonds,9 and stable free-radical-mediated reshuffling.10Regarding exchangeable or reversible covalent bonds, the Cheng group introduced a hindered urea bond (HUB) as a new dynamic covalent bond,11 and developed self-healing polyurethane-urea polymers using an amine with a bulky substituent.12 The urea bond undergoes reversible bonding and debonding, which imparts bond reorganisation and self-repair capabilities after polymer-chain breakage under external forces. Therefore, this bulky reversible urea bond offers multiple self-healing capabilities under ambient conditions without stimuli or catalysis.13 During reversible bonding and debonding, however, free isocyanates produced in the cleaved bond may turn into primary amines through unstable carbamic acids when they react with moisture present in the atmosphere. The resulting primary amines have a high reactivity and they can form irreversible crosslinks when they react with the other free isocyanates in the polymer matrix or wound interface.14 To the best of our knowledge, few systematic studies have been reported on the bonding–debonding behaviour of reversible bulky urea bonds in the presence of water and their impact on self-healing properties in spite of the great concern about the reactivity and toxicity of free isocyanates.15
To address this issue, self-healable acrylic copolymers bearing reversible bulky urea bonds were synthesized by conventional free-radical copolymerisation, and their chemical structures were confirmed by using 1H-NMR, FT-IR, DSC, and size exclusion chromatography (SEC). After the crosslinking of the self-healable acrylic copolymers with two different diisocyanates, their self-healing properties were investigated and compared by using 1H-NMR, rheometry, tensile testing, optical microscopy, and atomic force microscopy (AFM). A single scratch test was conducted to study the effects of an aqueous environment on the self-healing performance and stability, and some experiments were performed under accelerated conditions of elevated temperature and aqueous medium. Furthermore, a tube was fabricated from a polymer sheet by reshaping and annealing to demonstrate that the polymer synthesized in this study could be used as a self-healable tube capable of flowing water as a preliminary test for artificial tissue applications.
Experimental section
Materials
Methyl methacrylate (MMA, 99%, Daejung, Korea), 2-hydroxyethyl methacrylate (HEMA, 97%, Aldrich, USA), and 2-(tert-butylamino)ethyl methacrylate (tBAEMA, 97%, Aldrich, USA) were used as received. n-Butyl acrylate (BA, 99.5%, Junsei, Japan) was used after filtering through an inhibitor removing column (Aldrich, USA). Isophorone diisocyanate (IPDI, a mixture of isomers, Alfa Aesar, USA) and 1,6-diisocyanatohexane (HDI, 98+%, Alfa Aesar, USA) were used as crosslinkers without further treatment. α,α′-Azobis(isobutyronitrile) (AIBN) was recrystallized once from methanol (99.8%, Daejung) before use. N,N-Dimethylformamide (DMF, anhydrous, 99.8%, Aldrich) was used as a polymerisation solvent. Chloroform (CHCl3, >99%, Duksan, Korea) and dibutyltin dilaurate (DBTDL, 95%, Aldrich, USA) were used as received. Hexyl alcohol (HeOH, reagent grade, Aldrich, USA), benzyl isocyanate (BI, 99%, TCI, Japan), and chloroform-d (CDCl3, 99.8 atom%, Aldrich, USA) were also used as received.Prepolymer and self-healing polymer synthesis
To prepare self-healing polymers, linear prepolymers were first prepared using free-radical copolymerisation, followed by crosslinking with diisocyanates. To prepare the prepolymer PtB, BA (150 mmol), MMA (150 mmol), tBAEMA (15 mmol), AIBN (9 mmol), and DMF (526 mmol) were placed into a 250 mL round-bottom flask with a magnetic stirrer bar. The mixture was then stirred and immersed in a thermostatically controlled oil bath at 70 °C. After 4 h, the mixture was poured into an excess amount of water to precipitate and purify the prepolymer. The precipitate was collected and placed in a vacuum oven at 80 °C for 3 days to evaporate water and other residues; the linear copolymer was obtained in a yield of 88%. Self-healing polymers (designated as PtB-HDI and PtB-IPDI) were prepared by crosslinking the previously prepared prepolymer. The prepolymer (PtB, 1 g) diluted by a factor of six in CHCl3 and the crosslinker (32.5 μL HDI or 42.4 μL IPDI) by a factor of ten in CHCl3 were prepared separately and then mixed together for crosslinking. The reaction was maintained at room temperature (RT) under ambient conditions. The molar ratio of the crosslinker to tBAEMA in the prepolymer was fixed at 1:2. A similar prepolymer (PH, the control sample) was also prepared using HEMA instead of tBAEMA for comparison. To this end, BA (150 mmol), MMA (150 mmol), HEMA (15 mmol), AIBN (9 mmol), and DMF (515 mmol) were used, and the polymerisation was carried out under the same conditions described above (the yield of PH was 86%), with the exception that 1 wt% DBTDL, based on the prepolymer, was added to accelerate the crosslinking reaction. The control polymer sample, designated as “PH-HDI”, was prepared using HDI. To this end, the prepolymer (PH, 1 g) dissolved in CHCl3 and the crosslinker (HDI, 33.3 μL) were reacted following the crosslinking procedure described above. The synthesis scheme of prepolymers and self-healing polymers via crosslinking is illustrated in Scheme 1a.
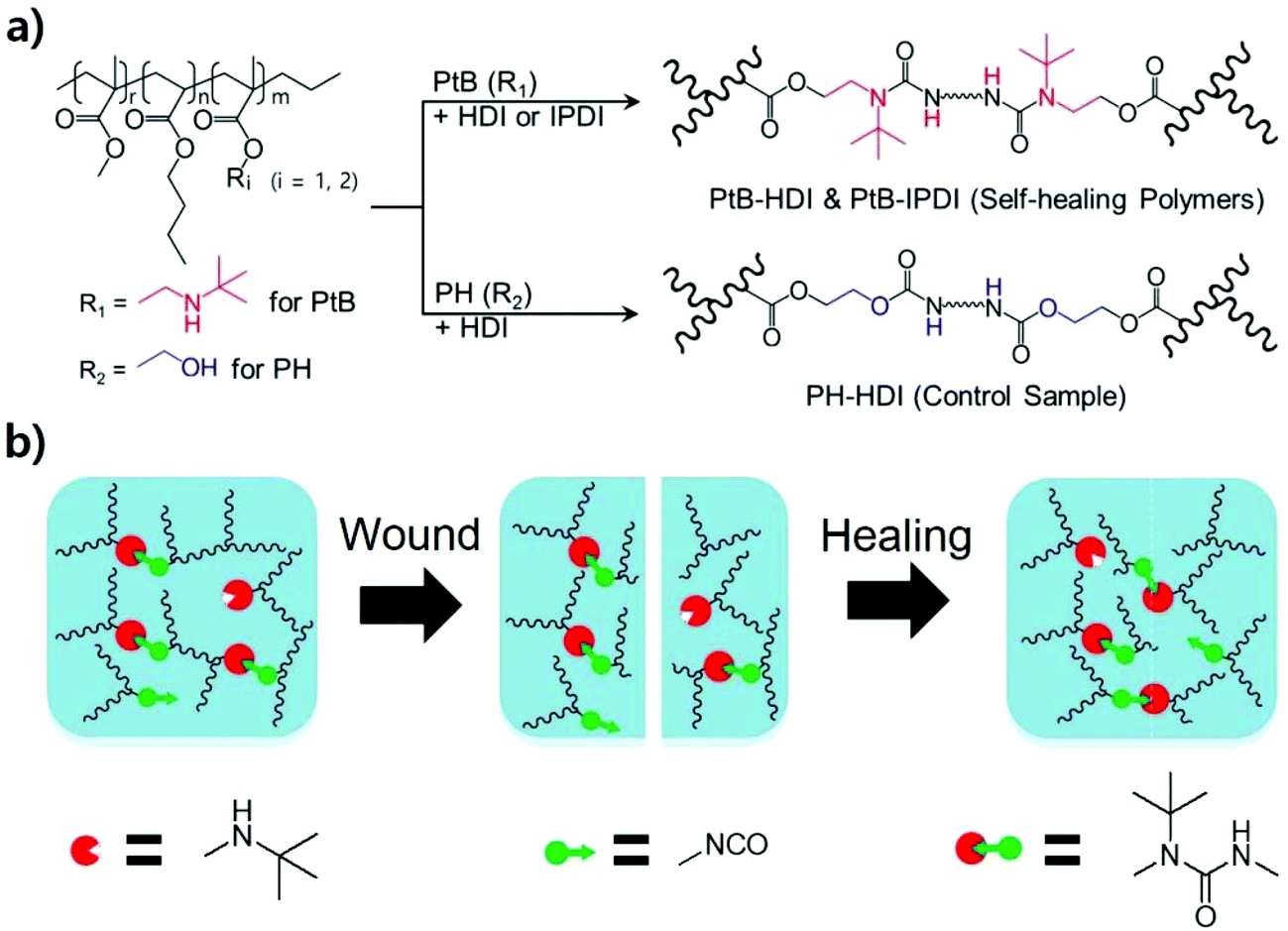 | ||
| Scheme 1 (a) Synthetic scheme and chemical structures of the self-healing polymers (PtB-HDI and PtB-IPDI) and the control sample (PH-HDI). (b) Illustration of the self-healing mechanism between sharply scratched or blade-cut surfaces. | ||
Characterization of prepolymers and self-healing polymers
NMR (AVANCE III 500, Bruker, Germany) and SEC (Alliance e2695, Waters, USA) were used to characterize the prepolymer. SEC analyses were carried out using THF (HPLC grade) as the eluent, at a rate of 1 mL min−1 at 35 °C. A PMMA standard (2.9–955 kg mol−1, Shodex, Japan) was used for calibration. The completion of the crosslinking reaction was confirmed by FT-IR spectroscopy (8400S, Shimadzu, Japan). The Tg values before and after crosslinking were measured from the 2nd heating cycle in a DSC (Q20, TA Instruments, USA). The transparency of the polymer films was measured using UV-Vis spectroscopy (S-3100, SCINCO, Korea).Observation of reversible urea bonds
The tBAEMA comonomer in the self-healing polymer forms a reversible urea bond with an isocyanate, which facilitates self-healing via an exchangeable covalent bonding mechanism.11,12 The cleavage reaction of the urea bond and the subsequent side reaction of the free isocyanate were confirmed by 1H-NMR of the solution sample under accelerated conditions. To examine this, tBAEMA (1 mmol) and BI (1 mmol) were reacted in CDCl3 (5 mL) for 30 min at RT. HeOH (1 mmol) and DBTDL (1 μL) were then injected into the reaction mixture followed by heating at 45 °C. The samples for 1H-NMR analyses were taken immediately (0 h) and after heating for 12 h.During self-healing, two important events must be involved: (1) the physical flow of the molecular segments at or near the wound interface, and (2) the reconnection of the cleavage bond after mechanical damage.16 In this study, rheometric analysis was performed on an oscillatory rheometer (Thermo Scientific Inc., Mars III) to confirm the 2nd event that tBAEMA units are reversibly separated and reconnected. The measurement temperature ranged from 30 to 150 °C at a heating rate of 10 °C min−1, and the temperature was held at both ends for 10 min. The angular frequency was 1 Hz, with a strain of 0.1%, and an 8 mm parallel plate, and an axial force of 5 N were used. Complex viscosities (|η*|s) were calculated from the measurements of storage and loss moduli (G′ and G′′, respectively) using eqn (1):
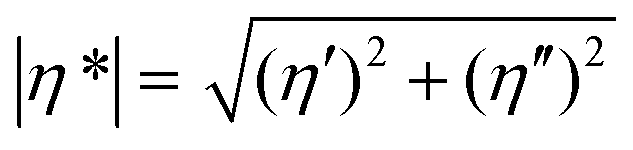 | (1) |
FT-IR spectroscopy was also performed to confirm the presence of free isocyanates due to the debonding of the urea bonds. In order to control the temperature of the polymer specimen from RT to 150 °C, a Peltier device (a thermoelectric module, TEC1-07105T125, Coolerbank, China) connected with a DC power supply (RDP-303, Smart, Korea) was used.
Self-healing performance
A wound and self-healing process of a dynamic urea bond is illustrated in Scheme 1b. The healing process is accompanied by the rearrangement of the polymer chain and the new reversible crosslinks are formed through the reaction between tertiary butyl amines and free isocyanates of another adjacent bulky urea group.To explore the self-healing performance, tensile strength and single-scratch analyses were performed. For tensile testing, the prepolymer and the crosslinker were mixed together and poured into a dog-bone shaped mold for curing. After curing for 4 h at RT, the specimen was dried in a vacuum oven at 80 °C for 3 days. After drying, the middle of the specimen was completely cut using a razor blade (Dorco, Korea). The cross-cut surfaces were then gently pressed together for 1 min, and aged in a convection oven at 60 °C for 4 days. Tensile testing was carried out on specimens aged at RT for 1, 2, 3, and 4 days using a force tester (MCT-2150, AND, Japan). The percentage recoveries (%Rs) were calculated in terms of the mechanical properties (stress, strain, and toughness data) measured from the tensile testing experiments and expressed as follows:
 | (2) |
For the single-scratch test, the prepolymer and the crosslinker were mixed together, poured onto a polymer coated-metal plate (7 cm × 15 cm, thickness = 0.8 mm), and cured in a convection oven at 60 °C for 2 days. The film thickness was ∼75 μm. A microstage (Kwonsys, Korea) equipped with a razor blade (ST300, Dorco, Korea) was used for making single scratches. After scratching, the specimen was kept in air or water (a water droplet). The surface was examined at selected time intervals by AFM (Dl-3100, Veeco, USA), in tapping mode, to determine the scratch width (for an in-depth description, see the text accompanying Fig. S1†). From the AFM data, the self-healing efficiency (%SHE) can be obtained and defined as:
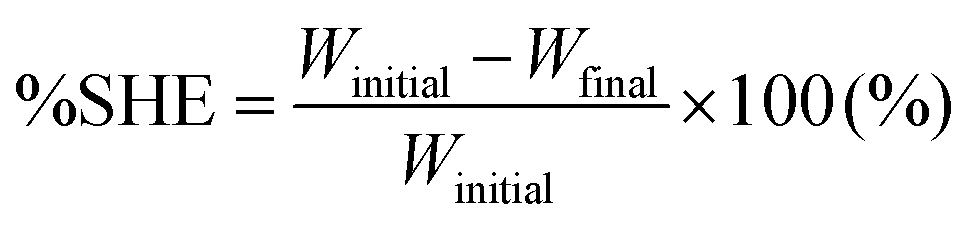 | (3) |
Stability of self-healing polymers in water
As mentioned earlier, the free isocyanates dissociated from the dynamic equilibrium between tBAEMA and the crosslinker can be converted to amines upon coming into contact with water for long periods of time. To study this, self-healing polymer specimens were treated under accelerated conditions and analyzed by DSC, FT-IR, and SEC. The self-healing polymer (PtB-HDI or PtB-IPDI, 3.0 g) was placed in a 20 mL glass vial. The vial was then filled with pure water and kept in an oven at 60 °C. After three weeks, the hydrated self-healing polymer was removed from the vial and dried in the same oven for 24 h. DSC curves were obtained from the 2nd heating cycle over the −50 to 150 °C temperature range. The FT-IR spectrum of the polymer was recorded in ATR mode. For SEC analysis, the dried polymer sample was dissolved in HPLC grade THF and filtered using a 0.2 μm PTFE syringe filter (Advantec, USA). The molecular weights of the THF-soluble portions of the polymer were measured by SEC.Fabrication of self-healable polymer tubes
A polymer tube was fabricated from a self-healing polymer to demonstrate the self-healing performance under water-flowing conditions. First, a polymer sheet was prepared by pouring the mixture of the prepolymer and crosslinker into a rectangular-shaped mold. After curing for 4 h at RT, the sheet was rolled and annealed in an annulus-shaped cylindrical Teflon mold for three days at 60 °C. It had 2 mm-thick walls, a 9 mm inner diameter, and was 70 mm long. Both edges of the tube were then connected to 5 mL single-use syringes (HSW Norm-Ject®, Germany). Tap water was circulated through the tube using an immersion bath circulator (LCB-6D, Labtech, Korea) with a circulation rate of 7 L min−1 at 37 °C. A pierced wound was made using a surgical blade (HFE-SB11, Feather Safety Razor Co., Japan). After covering the wound with a piece of tissue, the tube was gently tied with a rubber band for 5 h. The wound formation and self-healing were filmed using a cell phone (Galaxy S4-LTEA, Samsung, Korea).Results and discussion
Characterization of prepolymers and self-healing polymers
The chemical structures of PH and PtB prepolymers illustrated in Scheme 1 were confirmed by 1H-NMR spectroscopy and the spectra with assignments are shown in Fig. 1. It was observed from the peak-area integration that the ratios of MMA:BA:HEMA in PH and MMA:BA:tBAEMA in PtB were 1.00:0.98:0.11 and 1.00:0.93:0.09, respectively. These ratios are reasonable considering the molar amounts of the monomers used in the formulation (MMA:BA:(HEMA or tBAEMA) = 1:1:0.1).
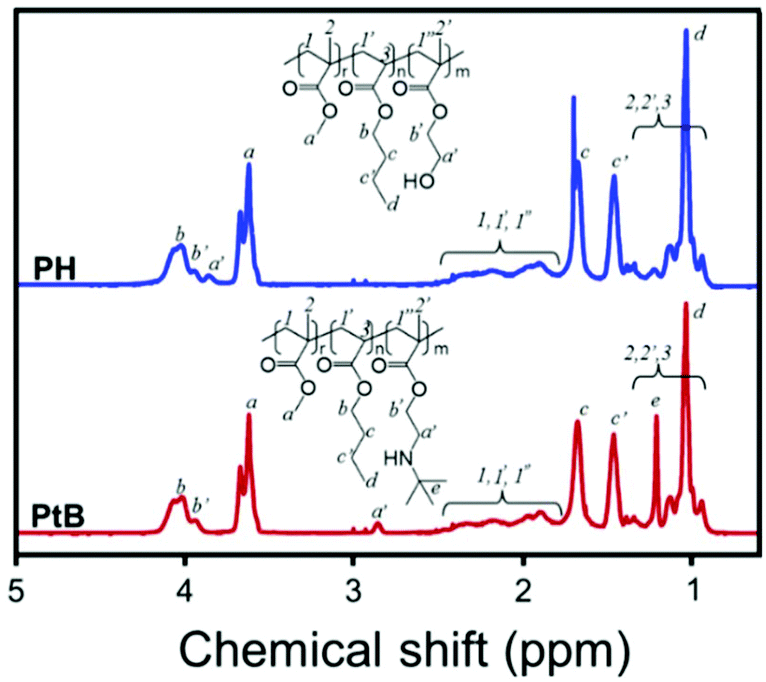 | ||
| Fig. 1 1H-NMR spectra of the control (PH) and self-healing (PtB) prepolymers, and their chemical structures with peak assignments. | ||
To verify the crosslinking reaction of the prepolymers in the presence of HDI or IPDI, FT-IR analyses before and after crosslinking were performed, as shown in Fig. 2a and b. In the case of PH-HDI, an N–H bond from the urethane linkage appears at 1540 cm−1 compared to the prepolymer. For PtB-HDI or PtB-IPDI, a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond from the urea linkage appears at 1650 cm−1. Both control and self-healing polymers show no isocyanate peak at 2270 cm−1, which means that the crosslinking with HDI or IPDI was complete without residual crosslinkers. To determine the Tg changes of these polymers before and after crosslinking, DSC analyses were performed as displayed in Fig. 2c. Single Tgs are observed at −2 °C and 2 °C for the PH and PtB prepolymers, respectively. These values are very close to the Tg values (−2.7 °C and 2.7 °C) calculated from the recipe using the Fox equation (i.e.,
O bond from the urea linkage appears at 1650 cm−1. Both control and self-healing polymers show no isocyanate peak at 2270 cm−1, which means that the crosslinking with HDI or IPDI was complete without residual crosslinkers. To determine the Tg changes of these polymers before and after crosslinking, DSC analyses were performed as displayed in Fig. 2c. Single Tgs are observed at −2 °C and 2 °C for the PH and PtB prepolymers, respectively. These values are very close to the Tg values (−2.7 °C and 2.7 °C) calculated from the recipe using the Fox equation (i.e., 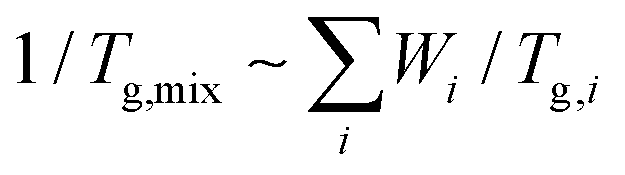 ).17 The DSC data suggest that the mobility of both prepolymers are good because of the low Tg values, which is an important feature for self-healing performance. The degree of polymerisation, molecular weight, and Tg values of the prepolymers and the corresponding crosslinked polymers are listed in Table 1. Furthermore, the transmittance of self-healing and control polymers in the visible light range was measured, as shown in Fig. S2.† All the polymers show above 99% transmittance and are colourless.
).17 The DSC data suggest that the mobility of both prepolymers are good because of the low Tg values, which is an important feature for self-healing performance. The degree of polymerisation, molecular weight, and Tg values of the prepolymers and the corresponding crosslinked polymers are listed in Table 1. Furthermore, the transmittance of self-healing and control polymers in the visible light range was measured, as shown in Fig. S2.† All the polymers show above 99% transmittance and are colourless.
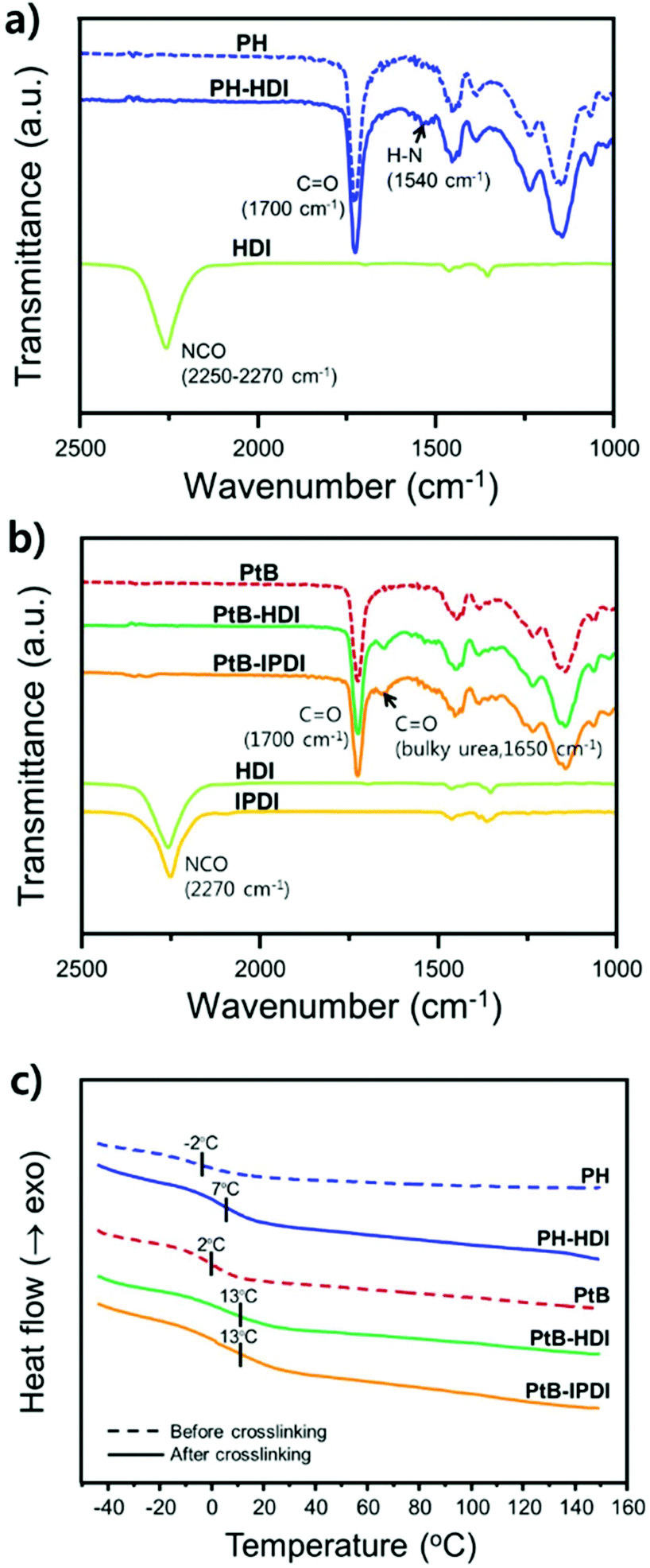 | ||
| Fig. 2 (a) FT-IR spectra of control polymers, PH (prepolymer) and PH-HDI (crosslinked with HDI). (b) FT-IR spectra of self-healing polymers, PtB (prepolymer), PtB-HDI (crosslinked with HDI), and PtB-IPDI (crosslinked with IPDI), respectively. (c) DSC curves of prepolymers (PtB and PH, dashed lines) and the corresponding crosslinked polymers (solid lines) in the 2nd heating cycle. | ||
| Prepolymer code | DPa |
![[M with combining overline]](https://www.rsc.org/images/entities/i_char_004d_0305.gif) n (g mol−1)
n (g mol−1) |
Đ
Mb (—) |
Self-healing polymer code | T g (°C) | |
|---|---|---|---|---|---|---|
| Before crosslinking | After crosslinking | |||||
| a DP: degree of polymerization, estimated from 1H-NMR peak integrations combined with molecular weights obtained from SEC analyses. b Đ M: dispersity index (= Mw/Mn). | ||||||
| PH | Poly(MMA137-r-BA134-r-HEMA16) | 32940 |
1.7 | PH-HDI | −2 | 7 |
| PtB | Poly(MMA70-r-BA65-r-tBAEMA6) | 16372 |
1.5 | PtB-HDI | 2 | 13 |
| PtB-IPDI | 2 | 13 | ||||
Reversible urea bonds
The complex viscosities (|η*|s) of the crosslinked polymers were measured by rheometry as displayed in Fig. 3. For PH-HDI, |η*| is 5.7 × 105 Pa s at 30 °C but gradually decreases to 2.5 × 104 Pa s at 150 °C, which indicates increased polymer mobility owing to thermal relaxation upon heating. Upon cooling, the |η*| increases monotonously. This behaviour implies that the relaxation of the PH-HDI polymer matrix occurs without any topological change during heating, and vice versa. However, the self-healing polymers exhibit completely different behaviour. For example, the |η*| of PtB-HDI slightly decreases with increasing temperature, as observed in PH-HDI; however, the |η*| rapidly drops above the inflection point (red arrows) at 110 °C, indicating that a debonding of the reversible urea bonds has occurred as seen in Diels–Alder based self-healing polymers.18 The debonding reaction of the urea bond increases polymer mobility and decreases |η*|, which is more influential than the |η*| decrease owing to the simple thermal relaxation above Tg. When the temperature reaches 150 °C, the |η*| curve displays a discontinuity after which it decreases almost linearly with a minimal slope in the 150 °C isothermal region; this might be attributed to a shift in the equilibrium from the covalent bulky urea bond to –NH (tBAEMA) and –NCO (HDI) disconnection (debonding). During the period of temperature decline, the |η*| of PtB-HDI increases, re-exhibits an inflection point, and eventually converges to 5.7 × 105 Pa s, which is the same as the initial value. This oscillating |η*| behaviour supports the reversibility of bonding and debonding of the urea bonds.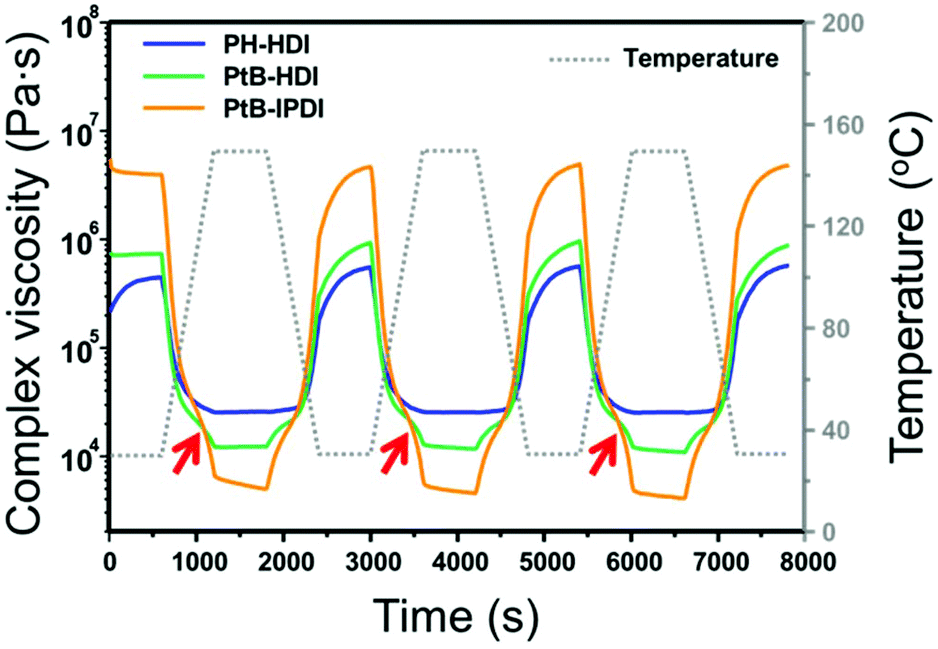 | ||
| Fig. 3 Complex viscosities of the control (PH-HDI) and self-healing polymers (PtB-HDI and PtB-IPDI) at temperature oscillating between 30 and 150 °C. The red arrows indicate the inflection points of the complex viscosities during heating. | ||
It should also be noted that the |η*| of PtB-HDI is higher than that of PH-HDI at 30 °C but lower than that of PH-HDI at 150 °C; i.e., the Δ|η*|s in the self-healing polymers are higher than those in the control sample with oscillating temperature. The decrease in the |η*| of PtB-HDI is not only due to the thermal relaxation of the polymer matrix, but also due to debonding or decrosslinking. In addition, the Δ|η*| of PtB-IPDI is much higher (from 4.8 × 106 to 4.8 × 103 Pa s) than that of PtB-HDI (from 9.0 × 105 to 1.2 × 104 Pa s). This is due to the bulkier and less flexible structure of IPDI than HDI. When PtB-IPDI is decrosslinked at 150 °C, the resulting free IPDI might play the role of a bulkier plasticizer more than HDI in PtB-HDI.
The tBAEMA comonomer has a bulky alkylamino group; therefore, it can form reversible urea bonds with isocyanates to impart self-healing capabilities. This can be confirmed from the 1H-NMR chemical shifts corresponding to the debonding tBAEMA-benzyl isocyanate (BI) and the formation of hexylalcohol (HeOH)-BI (Fig. S4a†). At an elevated temperature of 60 °C, the bond breaking of tBAEMA-BI produces free NCO of BI, and the subsequent irreversible bond forms to yield stable HeOH-BI. These reactions were evidenced by chemical shift changes in the 1H-NMR spectra.
The generation of free NCO from the reversible urea bond can also be monitored by using FT-IR. In Fig. S4b,† the FT-IR spectra of the PtB-HDI self-healing polymer sample at different temperatures (RT and 150 °C) are shown. The free NCO peak from HDI appears at 2270 cm−1 upon heating, which is not visible at RT. In addition, the peak intensity of the CO peak from the urea bond at 1650 cm−1 has been diminished.
Self-healing performance
Tensile tests were carried out using crosscut dog-bone-shaped specimens as shown in Fig. 4. To determine the percentage recovery (%R), toughness, strain, and stress were recorded from stress–strain curves, as summarized in Table S1.† The stress–strain curves of PH-HDI after crosscut show no self-healing ability, although its Tg and moduli are similar to those of PtB-HDI. On the other hand, PtB-HDI and PtB-IPDI show excellent self-healing %Rs. For toughness, strain, and stress after 4 days, PtB-HDI exhibits %Rs of 97, 89, and 95%, respectively, whereas PtB-IPDI shows lower %Rs than PtB-HDI, at 72% (toughness), 87% (strain), and 75% (stress). Recovery from crosscut wounds requires both physical flow and chemical bonding. The self-healing polymers with reversible urea bonds can meet both requirements as compared to the control sample.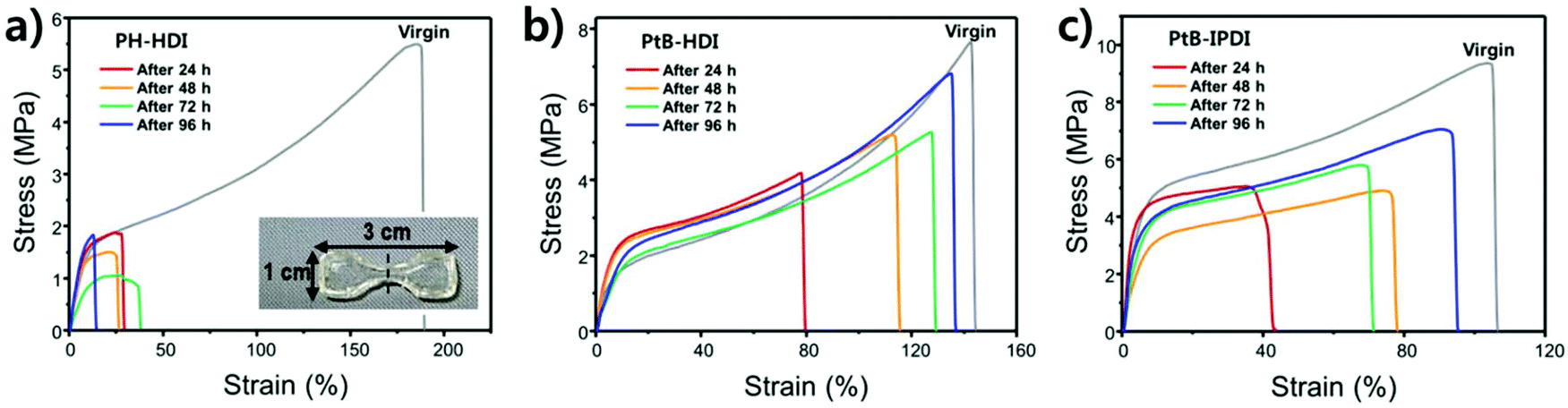 | ||
| Fig. 4 Stress–strain curves of center-cut polymer specimens as a function of self-healing time: (a) PH-HDI (control), (b) PtB-HDI, and (c) PtB-IPDI. The photograph of a typical specimen was inserted in (a) and ‘virgin’ indicates the uncut specimen. | ||
Single-scratch tests were performed using AFM to study the effect of water on the rate and efficiency of self-healing (%SHE), and the results are summarized in Fig. 5. The detailed methods are also provided in the ESI (refer to Fig. S1†). As clearly seen from the optical microscopy images, a single-scratch on the PH-HDI surface cannot be repaired, and both the width and depth of the scratch remain the same in water as well as in air for 24 h, i.e., %SHE values of ∼0%. For PtB-HDI and PtB-IPDI, however, the %SHEs of single-scratches exceed 70% after 2 h of healing in air, and the wounds completely disappear within 24 h.
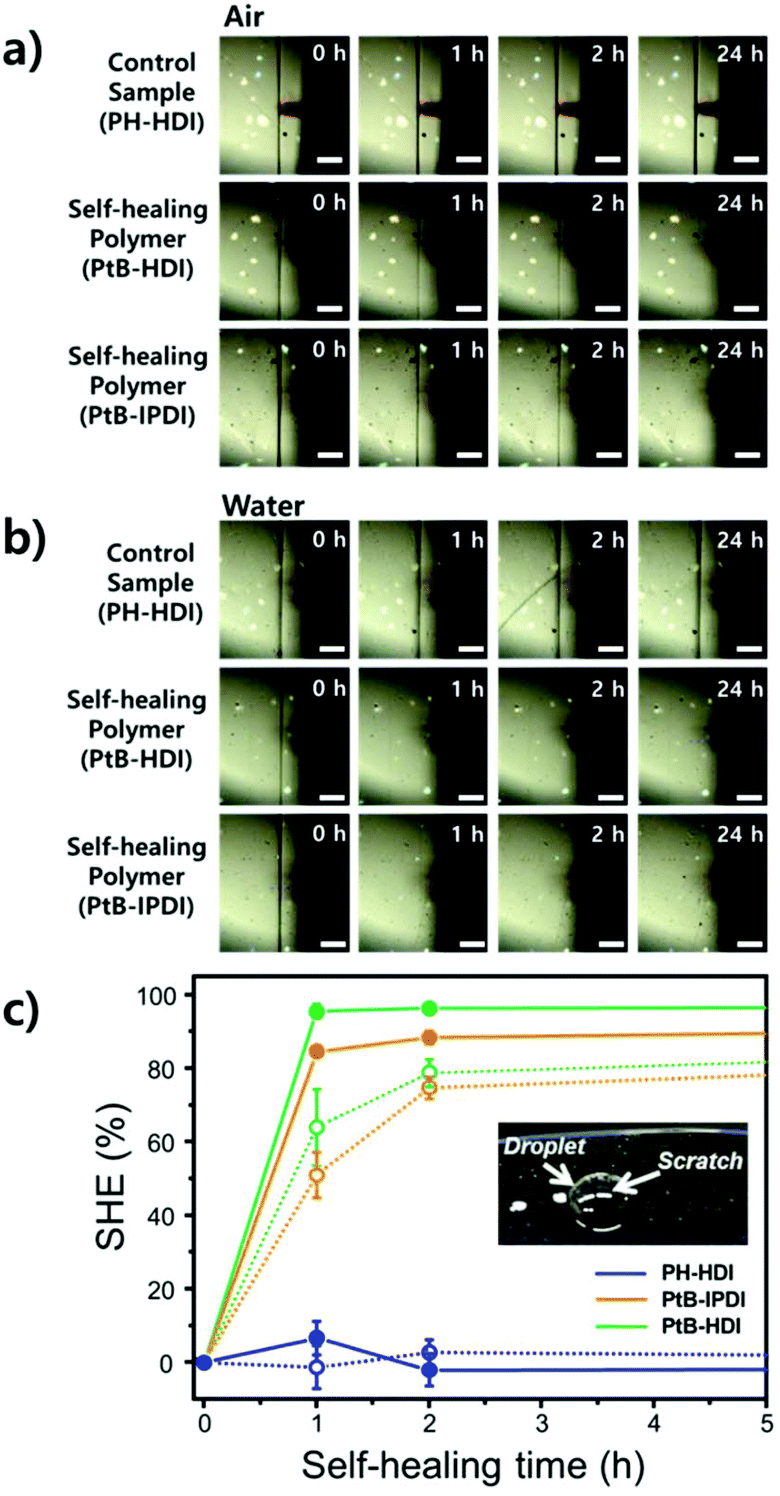 | ||
| Fig. 5 Optical images of single-scratch tests performed on polymer-coated surfaces as a function of self-healing time (a) in air and (b) in water. (c) Self-healing efficiency (%SHE) values obtained from the AFM scratch width data (dashed lines = in air, solid lines = in water). | ||
Upon coming into contact with water, the self-healing performance was enhanced; that is, faster recovery rates and higher %SHEs than those in air (solid lines in Fig. 5c) were observed. When the scratch is covered with a water droplet, the free isocyanate liberated from the scratch can react with water, become a primary amine (–NH2) via unstable carbamic acid (–NHCOOH), and form irreversible urea bonds with other free isocyanates in the surroundings. The primary amine rapidly forms the irreversible urea bond as compared to the bulky amine of tBAEMA. The resulting amines and water molecules in the scratch area may form hydrogen bonds with the self-healing polymer to impart an additional binding force.19 It is also noted that PtB-HDI displays a higher %SHE than PtB-IPDI over the same healing time; these results are consistent with the tensile test results (Fig. 4b and c).
Repetitive scratch-healing and stability in water
The stabilities of the self-healing polymers against water were evaluated by repetitive scratch-healing tests, as shown in Fig. 6a. During these tests, the same position was scratched using a computer-controlled razor blade and covered with a water droplet for 1 h; the droplet was then soaked with paper tissues for observation. This procedure was repeated eight times at RT (see ESI, Fig. S3†). As shown in Fig. 6a, a scratch at a depth of ∼1 μm (the region with a red arrow) was fully recovered during the eight cycles of scratching, but a scratch deeper than 1 μm (∼3 μm, the region with a dotted white arrow) was not recovered after the sixth scratch. Although water can accelerate the healing of scratches (Fig. 5), a repeated contact with water causes the debonding of the urea bond and consequently a persistent side reaction of free isocyanates, eventually making repetitive self-healing difficult. In other words, the consumption of free isocyanates improving the self-healing performance eliminates the reversibility of the self-healing polymer.14 To verify this, DSC and SEC analyses were performed on the self-healing polymers (wPtB-HDI and wPtB-IPDI, the prefix ‘w’ means that the polymer annealed in water) aged for three weeks in 60 °C water, and the results are shown in Fig. 6b and c, respectively. The Tgs of wPtB-HDI and wPtB-IPDI decreased from 13 °C to 5 and 4 °C, respectively. This is attributed to the equilibrium shift of the reversible urea bond. The consumption of isocyanates with water shifts the equilibrium to the reverse direction, i.e., decrosslinking, leading to a decrease in Tg.20 The SEC elution curves of the deteriorated self-healing polymers (THF soluble portion) became very broad, which is completely different from that observed for the PtB prepolymer. It was found from the SEC data that the ĐM increased from 1.5 for pristine PtB to 2.1 and 2.0 for wPtB-HDI and wPtB-IPDI, respectively. Compared to the PtB prepolymer, the SEC elution curves of wPtB-HDI and wPtB-IPDI show small shoulders at a shorter elution time (the high molecular weight region, blue arrows in Fig. 6c), which might be due to the residual less-crosslinked polymers (soluble in THF). Even though the reaction with water seems very slow at RT, the self-healing polymer will eventually be deteriorated and the prepolymer having tBAEMA will gradually be isolated from the crosslinked network structure over time. This phenomenon could also be confirmed by using FT-IR (Fig. S5†). The aged self-healing polymers exhibit NH peaks at ∼3300 cm−1 with minimal CO peaks at 1640 cm−1, which indicates the generation of amines produced by the reaction with water.
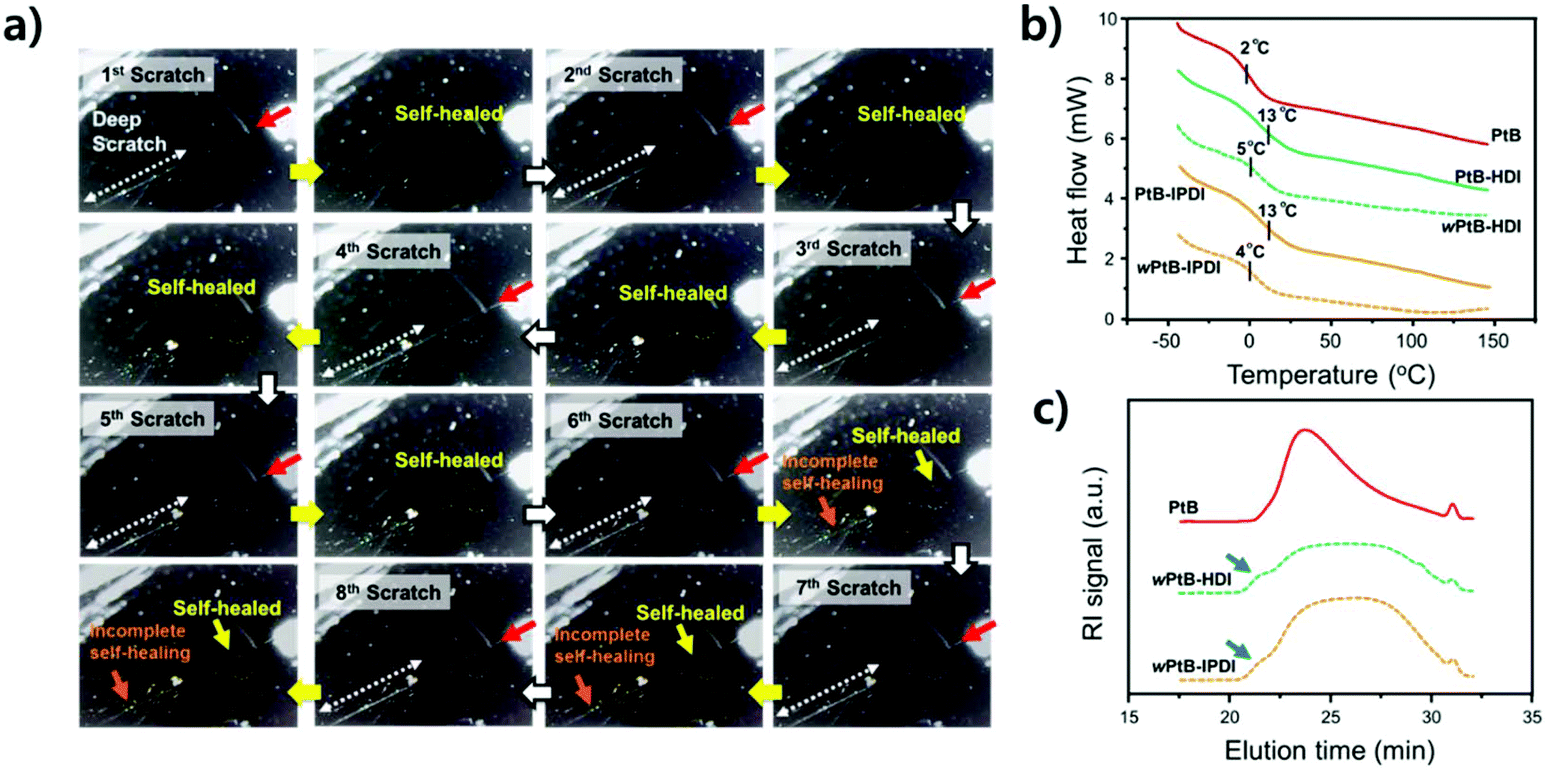 | ||
| Fig. 6 (a) Optical images of repetitive single-scratch tests performed on PtB-HDI self-healing polymer-coating surfaces (the white dotted arrows indicate an ∼3 μm deep region). (b) DSC (2nd heating cycle) curves of pristine and annealed self-healing polymers kept in 60 °C water for three weeks. The annealed samples are designated as wPtB-HDI and wPtB-IPDI, respectively. (c) SEC elution curves of the pristine prepolymer (PtB) and the annealed self-healing polymer extracted from THF. | ||
As described in the tube fabrication, a sheet of PtB-HDI has been turned into a self-healable tube through annealing. Self-healing polymers having dynamic covalent bonds can be re-formed into various shapes owing to the reversible debonding and rebonding mechanism, even though they have crosslinked structures like thermoset polymers. As shown in Fig. 7 or the ESI (Video S1†), the leakage of water had completely ceased after healing for 5 h using a paper tissue with a rubber band around the blade-cut wound. This example can be a preliminary work for preparing artificial blood vessels, even though the thrombosis and durability problems need to be overcome from a practical perspective.21
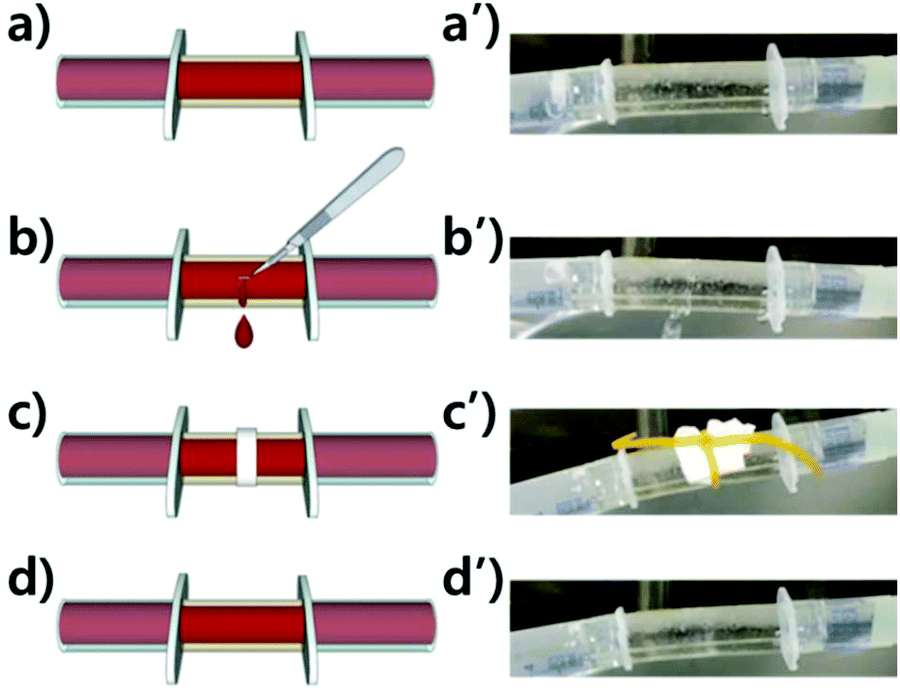 | ||
| Fig. 7 Illustrations and the corresponding photographs for the demonstration of water-flowing self-healable tubes: (a, a′) before blade-cut wound formation, (b, b′) after the wound formation, (c, c′) the wound healing, and (d, d′) after recovery for 5 h. The temperature of flowing water was 37 °C. | ||
Conclusions
The poly(MMA/BA/tBAEMA) prepolymer was synthesized from tBAEMA monomers capable of a dynamic covalent bond and the corresponding self-healing polymers (PtB-HDI and PtB-IPDI) were prepared using diisocyanates (crosslinkers). The reversible bonding and debonding, self-healing performance, and water-adaptability of the resulting self-healing polymers were evaluated and compared with the control sample of PH-HDI. The urea bonds formed between the bulky amine of tBAEMA in the prepolymer and the isocyanate of HDI or IPDI were found to have reversible and repetitive bonding and debonding behaviour through oscillating rheology, NMR, and FT-IR analyses. The tensile testing showed that the self-healing polymer exhibited a mechanical strength of 8–10 MPa, and high percentage recoveries of 75% for PtB-IPDI and 95% for PtB-HDI, respectively, as compared to PH-HDI (33%). The single scratch test showed that the self-healing rate of the self-healing polymers was more than 70% within 2 h, and the rate was improved upon coming into contact with water. Through repetitive scratch healing experiments in the presence of water, it was confirmed that the scratch wound of 3 μm depth was recovered 5 times in spite of the side reaction between free isocyanate and water. Under accelerated conditions, the topological changes of the crosslinked self-healing polymer due to the side reaction could be confirmed by SEC, DSC, and FT-IR. The self-healing polymers in this work have several advantages over the other self-healing systems (e.g., ureido pyrimidinone or ionic bond systems), such as ease of preparation, low cost, high elastic modulus (>106–7 Pa), high transparency and being colourless with no significant water-swelling problem. Besides these, they showed good repetitive self-healing performance despite exposure to water, so they are expected to be used in various fields as long as the water-resistance or the side reactions could be controlled. The prevention of contamination from alcohols or amines would also be one of the most important steps to improve the self-healing performance of the reversible bulky urea system.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are very grateful to the Ministry of Trade, Industry & Energy – South Korea for providing financial support (Grant No. 10067082 and 10070241).References
- B. J. Blaiszik, S. L. B. Kramer, S. C. Olugebefola, J. S. Moore, N. R. Sottos and S. R. White, Annu. Rev. Mater. Res., 2010, 40, 179 CrossRef CAS.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794 CrossRef CAS PubMed; E. N. Brown, S. R. White and N. R. Sottos, J. Mater. Sci., 2004, 39, 1703 CrossRef; J. D. Rule, N. R. Sottos and S. R. White, Polymer, 2007, 48, 3520 CrossRef.
- D. Therriault, S. R. White and J. A. Lewis, Nat. Mater., 2003, 2, 265 CrossRef CAS PubMed; K. S. Toohey, C. J. Hansen, J. A. Lewis, S. R. White and N. R. Sottos, Adv. Funct. Mater., 2009, 19, 1399 CrossRef.
- H. I. Yang, D. M. Kim, H. C. Yu and C. M. Chung, ACS Appl. Mater. Interfaces, 2016, 8, 11070 CAS.
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977 CrossRef CAS PubMed; U. Mansfeld, M. D. Hager, R. Hoogenboom, C. Ott, A. Winter and U. S. Schubert, Chem. Commun., 2009, 3386 RSC.
- R. Fang, Y. Liu, Z. Wang and X. Zhang, Polym. Chem., 2013, 4, 900 RSC; S. Burattini, H. M. Colquhoun, J. D. Fox, D. Friedmann, B. W. Greenland, P. J. F. Harris, W. Hayes, M. E. Mackay and S. J. Rowan, Chem. Commun., 2009, 6717 RSC.
- M. Zhang, D. Xu, X. Yan, J. Chen, S. Dong, B. Zheng and F. Huang, Angew. Chem., 2012, 124, 7117 CrossRef.
- N. Bai, K. Saito and G. P. Simon, Polym. Chem., 2013, 4, 724 RSC; J. S. Park, T. Darlington, A. F. Starr, K. Takahashi, J. Riendeau and H. T. Hahn, Compos. Sci. Technol., 2010, 70, 2154 CrossRef CAS; H. Weizman, C. Nielsen, O. S. Weizman and S. Nemat-Nasser, J. Chem. Educ., 2011, 88, 1137 CrossRef.
- M. Pepels, I. Filot, B. Klumperman and H. Goossens, Polym. Chem., 2013, 4, 4955 RSC; A. Rekondo, R. Martin, A. R. de Luzuriaga, G. Cabañero, H. J. Grande and I. Odriozola, Mater. Horiz., 2014, 1, 237 RSC.
- Y. Amamoto, J. Kamada, H. Otsuka, A. Takahara and K. Matyjaszewski, Angew. Chem., Int. Ed., 2011, 50, 1660 CrossRef CAS PubMed; K. Imato, M. Nishihara, T. Kanehara, Y. Amamoto, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2012, 51, 1138 CrossRef PubMed; C. Yuan, M. Z. Rong, M. Q. Zhang, Z. P. Zhang and Y. C. Yuan, Chem. Mater., 2011, 23, 5076 CrossRef.
- H. Ying and J. Cheng, J. Am. Chem. Soc., 2014, 136, 16974 CrossRef CAS PubMed.
- H. Ying, Y. Zhang and J. Cheng, Nat. Commun., 2014, 5, 3218 Search PubMed.
- Y. Zhang, H. Ying, K. R. Hart, Y. Wu, A. J. Hsu, A. M. Coppola, T. A. Kim, K. Yang, N. R. Sottos, S. R. White and J. Cheng, Adv. Mater., 2016, 28, 7646 CrossRef CAS PubMed.
- Z. Zhang, Y. Hu, Z. Liu and T. Guo, Polymer, 2012, 53, 2979 CrossRef CAS.
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446 RSC.
- Y. Yang, X. Ding and M. W. Urban, Prog. Polym. Sci., 2015, 49–50, 34 CrossRef CAS.
- T. G. Fox, Bull. Am. Phys. Soc., 1956, 1, 123 CAS.
- H. Mallek, C. Jegat, N. Mignard, M. Taha, M. Abid and S. Abid, J. Macromol. Sci., Part A: Pure Appl.Chem., 2013, 50, 728 CrossRef CAS.
- P. Taynton, K. Yu, R. K. Shoemaker, Y. Jin, H. J. Qi and W. Zhang, Adv. Mater., 2014, 26, 3938 CrossRef CAS PubMed; B. K. Ahn, D. W. Lee, J. N. Israelachvili and J. H. Waite, Nat. Mater., 2014, 13, 867 CrossRef PubMed.
- H. Stutz, K. H. Illers and J. Mertes, J. Polym. Sci., Part B: Polym. Phys., 1990, 28, 1483 CrossRef CAS; J. P. Pascault and R. J. J. Williams, J. Polym. Sci., Part B: Polym. Phys., 1990, 28, 85 CrossRef.
- I. H. Jaffer, J. C. Fredenburgh, J. Hirsh and J. I. Weitz, J. Thromb. Haemostasis, 2015, 13, S72 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The table of the percentage recovery of self-healing polymers, 1H-NMR and FT-IR for evidence of an equilibrium shift of the urea bond, and the experimental setup for a single scratch. See DOI: 10.1039/c7py01655g |
| This journal is © The Royal Society of Chemistry 2018 |