
Acrylate-based poly-high internal phase emulsions for effective enzyme immobilization and activity retention: from computationally-assisted synthesis to pharmaceutical applications†
G.
Tripodo
 a,
G.
Marrubini‡
a,
M.
Corti‡
a,
G.
Brusotti
a,
C.
Milanese
b,
M.
Sorrenti
a,
L.
Catenacci
a,
G.
Massolini
a and
E.
Calleri
*a
a,
G.
Marrubini‡
a,
M.
Corti‡
a,
G.
Brusotti
a,
C.
Milanese
b,
M.
Sorrenti
a,
L.
Catenacci
a,
G.
Massolini
a and
E.
Calleri
*a
aDepartment of Drug Sciences, University of Pavia, Viale Taramelli 12, Pavia, 70131, Italy. E-mail: enrica.calleri@unipv.it; Fax: +39 (0)382987383; Tel: +39 (0)382987383
bC.S.G.I. - Department of Chemistry, Physical-Chemistry Section, University of Pavia, Viale Taramelli 16, Pavia, 70131, Italy
First published on 29th November 2017
Abstract
This paper focuses on the synthesis of innovative functional materials prepared by the polymerization of high internal phase emulsions (HIPEs) for the bioconjugation of active molecules. By applying a computational method, this paper demonstrates how to produce materials with tailored features, such as pore dimensions (on the nano–micro scale), swelling in water and organic solvents and bulk densities. To this aim, starting from different emulsion compositions, ten materials are synthesised and fully characterized. Six different outputs (i.e. swelling in water and THF, mass loss in water and THF, throat and void diameters) are modeled using D-optimal mixture experimental designs. A clear correlation between the applied synthetic conditions and the final properties of the materials is demonstrated. The responses allow the selection of a single material for the development of a bioreactor prototype. Thus, the selected material is loaded into a glass column and polymerized in situ. An in-column procedure is also used for the covalent binding of horseradish peroxidase (HRP) as a model enzyme. A standard substrate is selected to test the activity of the immobilized enzyme. To conclude, the experimental design models allow one to obtain different materials with different features that could find applications in the fields such as biocatalysis/biochromatography, drug delivery or diagnostics. To the best of our knowledge, for the first time, an acrylate based polyHIPE material is used for the binding of enzymes in the active form and successfully applied under flow conditions.
Introduction
The aim of this study is to produce new materials and to establish their potential applications. In particular, these materials should be able to immobilize proteins or enzymes of pharmaceutical interest maintaining their biological activity for different applications including biocatalysis. Ideally, these materials should be characterized by: (i) an interconnected structure, to ensure solvent and substance exchanges within the material and the surrounding environment; (ii) mechanical stability, to support flow, pressure and friction; (iii) a predominant hydrophobic nature with partial water affinity, to avoid water over-swelling while ensuring an effective wettability to allow water spreading within the material and (iv) the presence of functionalities for biomolecule linkages.In the literature, there are several methods to produce porous materials, not necessarily showing interconnected pores. Porous materials have been obtained by vapour phase polymerization of metal–organic frameworks,1 by spray-drying,2 after post-production solubilisation of water soluble polymers blended into another polymer network,3 through a layer-by-layer process using a uniform polystyrene (PS) latex fabricated by dispersion polymerization,4 by phase separation of cellulose acetate and subsequent hydrolysis,5 or by common salt leaching of NaCl.6 These methods are effective but it is not clear how to obtain interconnected pores within the matrix or tailorable dimensions of the micro-structures. It is also true that the shape and morphology of the final macro-structure could not be accurately controlled by applying some of these methods.
Based on these premises, we focused our attention on the preparation of materials using the high internal phase emulsion (HIPE) technique to obtain polymerized materials [poly(HIPEs)] with easy to control shape and functions.
HIPEs are water in oil emulsions with higher than 70% (w/w or v/v) content in the internal phase. With this technique by polymerizing the external phase, the thin and stretched film surrounding the internal phase droplets polymerizes and, eventually, crosslink to form monoliths with an internally interconnected structure with voids which are the results of internal phase droplets. The porosity of these materials is given by throats which are the result of polymer contraction upon polymerization and crosslinking.7 Obviously, the polymerization and crosslinking take place at the interface of W/O.
PolyHIPEs can be molded, 3D printed and obtained in any shape. Furthermore, differently from other foam-like synthetic materials, they can be obtained as porous material with highly flexible topological characteristics with different porosities (74–99%), pore sizes (1–300 μm), interconnecting pores (0.1–20 μm) and compressive moduli up to 60 MPa.8 These properties can be simply modulated by varying the process parameters such as the stirring rate, mixing time and temperature or changing the composition parameters such as different monomer rates, concentration of surfactant and nature of the monomers.
Recently, different polyHIPEs have been proposed for different applications: chromatography,9,10 scaffolds for regenerative medicine,11 catalysis,12 biocatalysis,13 or 3D printing.14 This versatility justifies and demonstrates that polyHIPEs are effective tools that can be used for different applications.
Among the most used monomers in polyHIPE formation, styrene and divinylbenzene are found in several examples. However, this composition could not lead to materials with effective biocompatibility, biodegradation and post-production functionalization. Other approaches exploit the use of pre-formed polymers, biodegradable polyesters,15,16 polyurethane17 or polysaccharides.18,19 In this field, several complete and exhaustive reviews have been published.7,20,21 The strategy of using pre-formed polymers could be of interest but the possibility of choosing different structural/functional monomers in our scope, is fundamental.
These premises led us to propose an acrylate system.22 As a starting backbone monomer, the first choice was butyl acrylate because of its hydrophobicity. As the crosslinker, a trifunctional crosslinker such as trimethylolpropane triacrylate was evaluated since it would form a strictly interconnected polymeric structure ensuring a good resistance of the final material to mechanical stresses such as flow and friction. Last but not least, the selected functionalized monomer was the epoxy bearing glycidyl methacrylate because the epoxy groups can be further exploited for post-production immobilization of macromolecules such as enzymes, proteins or antibodies. Moreover, after hydrolysis, the epoxy groups lead to the consequent formation of hydroxyl groups (i.e., spontaneous hydrolysis or upon functionalization), that allows the material to gain a certain affinity for water. The hydrophobic nature of the main components is taken into specific account to reduce the swelling of the material in water.
Only a few examples of butyl acrylate based polyHIPEs are shown in the literature.15,23,24 All these examples describe the use of butyl acrylate in combination with other monomers such as divinyl benzene (as a crosslinker) or as acrylate end-capping of poly(ε-caprolactone). On the other hand, butyl acrylate has found applications in the preparation of nanoparticles or biomaterials.25,26
Only few or no examples of polyHIPEs bearing GMA functional groups for post-production functionalization are found in the literature. At the same time, only a few examples of enzymes chemically conjugated to a polyHIPE can be found in the literature and are based on styrene divinylbenzene systems.27,28 In the literature we did not find examples of acrylate-based polyHIPEs which chemically immobilized enzymes retaining the catalytic activity under continuous flow conditions.
For the pharmaceutical industry, biocatalysis is one of the key technologies in the area of “white biotechnology”. From another perspective, flow reactor technology shows many advantages compared with batch methods, including increased safety, high control of reaction parameters, possibility of automation and in-line purifications. The combined use of continuous flow systems in biocatalyzed processes can overcome the main limitations associated with batch biotransformations that, at present, limit a wider use of biocatalysis for industrial applications.29
Biocatalyzed reactions performed in flow chemistry reactors can benefit from improved mass transfer, excellent temperature control and, importantly, continuous substrate feed and product removal, thus limiting the possible substrate/product inhibition of the enzyme activity. The use of continuous flow technologies also offers the possibility to implement enzymatic cascade reactions by confining different enzymatic activities in different environments. Despite these benefits, the potential of biocatalysis in flow reactors is far from being fully exploited.
A key advantage of such a technology is clearly the availability of appropriate supports for enzyme immobilization to be used for different scopes: microscale, lab scale or production scale synthesis.30,31
To accomplish the main aim of obtaining materials able to develop a bioreactor to be used under flow conditions, a property prediction tool, that could guide parameter selection and enable access to polyHIPEs possessing any desired morphology, could be useful.
To this aim, a computational model by applying a design of experiment (DoE) approach was followed. Firstly, a set of HIPEs was prepared within an established range of parameters, then, different outputs from the obtained materials, such as swelling, mass loss etc., were used to validate the model. A correlation between the applied synthetic conditions and the final properties of the materials was found. One of the materials from the computational model was selected and successfully used for the preparation of a bioreactor based on horseradish peroxidase.
Experimental
Materials
Trimethylpropanetriacrylate (TMPT), butyl acrylate (BA), potassium persulfate (KPS), glycidyl methacrylate (GMA), N,N,N′,N′-tetramethylethylenediamine (TEMED), horseradish peroxidase (HRP), O-phenylenediamine (OPD), monobasic potassium phosphate (KH2PO4), anhydrous citric acid, ammonium sulfate, glycine, tetrahydrofuran (THF), absolute ethanol (EtOH), methanol (MeOH) and acetonitrile (ACN) used in this study were obtained from Sigma-Aldrich (Milan, Italy). Hydrogen peroxide solution (30%) was obtained from Belinka Perkemija. Synperonic PE/L 121™ was kindly provided by Croda Italiana Spa. All the materials were used as received. 30 mL Syringe PP/PE without a needle luer lock tip and female luer coupler in polypropylene were obtained from Sigma-Aldrich (Milan, Italy). Water used in this study was double deionized water (DDW), obtained by a Milli-q system from Millipore. An Omnifit® empty glass column (6.6 ID × 100 mm) was used in the bioreactor preparation.Apparatus
Scanning electron microscopy (SEM) analyses were performed on a Zeiss EVO MA10 instrument (Carl Zeiss, Oberkochen, Germany) on gold sputtered samples. Images were acquired from different sides of the samples. ATR-FTIR analyses were carried out using a Spectrum One PerkinElmer FTIR spectrophotometer (resolution: 4 cm−1) (Monza, Italy) equipped with a MIRacle™ ATR device. All the liquid chromatography operations (enzyme immobilization and bioreactor characterization) were carried out using an Agilent Technologies HP-1100 HPLC instrument (Palo Alto, CA, USA) provided with a quaternary pump, a Rheodyne injection valve (20 μL loop), a degasser, a UV-Vis detector and thermostat oven (25 ± 0.5 °C).For HPLC analyses an Agilent Technologies LiChrospher 100 RP-18 5 μm (250 × 4.6 mm ID) column was used. The spectrophotometric analyses were performed by using a Shimadzu UV-1601 spectrophotometer.
Experimental design calculation
All calculations were performed using Microsoft Excel 2010 and R version 3.1.0 (2014-04-10) Copyright(C) 2014 The R Foundation for Statistical Computing. R-based chemometric software routines were used for DoE calculations. The R-based software has been developed by the Group of Chemometrics of the Italian Chemical Society [http://gruppochemiometria.it/gruppo-lavoro-r-in-chemiometria.html]. Simplex graphs and response surface plots were obtained by using Design-Expert® version 7.0.0 software (Stat-Ease Inc., MN, USA).Methods
The general procedure for all the prepared materials was carried out as follows: the oil phase was achieved with a total volume of 8 mL, while the water phase had a total volume of 32 mL. Indeed, a w/o emulsion 80/20 v/v was formed. The oil phase was prepared by mixing an established amounts of two different lipophilic monomers: butyl acrylate as a backbone monomer and glycidyl methacrylate as a functional monomer. The oil phase comprised Synperonic PE/L 121 as the surfactant for the thermodynamic stabilization of the emulsion, and TMPT as a trifunctional crosslinker.
The water phase was prepared by dissolving 272 mg of potassium persulfate in nitrogen degassed water. One of the necks of the round bottom flask was provided with a 50 mL dropping funnel with a PTFE stopcock filled with the water phase. The water phase was added drop by drop to the oil phase by manually regulating the dropping funnel stopcock under stirring at 300 rpm by an overhead stirrer supplied with a PTFE D-shaped paddle. The water phase was added over almost 20 min and the system was maintained under stirring and under nitrogen. When all the water phase was added to the oil phase, the stirring speed was increased to 400 rpm and the system was maintained under nitrogen during the complete stirring period. The material obtained at the end of the established stirring time (1 hour) is a highly viscous cream-like emulsion. After the mixing time, the obtained emulsion was added with 272 μL of TEMED and the mixture was stirred for further 2 min at 400 rpm. The material obtained was quickly poured into a Petri dish and allowed to polymerize for 24 h at room temperature. After the 24 h polymerization reaction, a white and light weight solid was achieved. The polyHIPE obtained was transferred into a 200 mL crystallizer and washed as follows: two times in water (2 × 100 mL), one time in EtOH (1 × 100 mL), two times in MeOH (2 × 100 mL) and, eventually, in THF (2 × 100 mL). After washing, the polyHIPEs were dried in an oven at 40 °C for 24 h. Finally, the materials were characterized.
The three factors considered to build up the computational model and, subsequently, to assess the output after the polymerization reaction, were the amount of BA + GMA (factor A ranging from 0.7–0.8), the surfactant amount (factor B ranging from 0.05–0.2), and the crosslinker TMPT amount (factor C ranging from 0.09–0.15). The amounts of the three considered factors were constrained and are given in Table SI1.†
The experimental domain defined by the constrained concentrations of the three factors is the irregular polygon shown in Fig. 1.
 | ||
| Fig. 1 Experimental domain and compositions of the mixtures used in the polymerization reactions. The actual (real compositions), expressed as proportions, are shown together with the compositions coded as upper limit-bound pseudocomponents (compositions in brackets). | ||
Therefore, a D-optimal design for the mixture ought to be used for studying the reaction.2,3 Under the hypothesis that all the responses could be modeled by special cubic mixture models, the plan of the experiments was computed as illustrated in Fig. 1. Ten experiments were selected and carried out; the compositions of the studied mixtures are summarized in Table 1 and Fig. 1.
| Experiment # | A | B | C | u 1 | u 2 | u 3 |
|---|---|---|---|---|---|---|
| BA + GMA | Surfactant | TMPT | ||||
| 1 | 0.800 | 0.110 | 0.090 | 0.000 | 0.600 | 0.400 |
| 2 | 0.700 | 0.150 | 0.150 | 0.667 | 0.333 | 0.000 |
| 3 | 0.755 | 0.155 | 0.090 | 0.300 | 0.300 | 0.400 |
| 4 | 0.800 | 0.080 | 0.120 | 0.000 | 0.800 | 0.200 |
| 5 | 0.750 | 0.100 | 0.150 | 0.333 | 0.667 | 0.000 |
| 6 | 0.752 | 0.128 | 0.120 | 0.317 | 0.483 | 0.200 |
| 7 | 0.710 | 0.200 | 0.090 | 0.600 | 0.000 | 0.400 |
| 8 | 0.800 | 0.050 | 0.150 | 0.000 | 1.000 | 0.000 |
| 9 | 0.700 | 0.175 | 0.125 | 0.667 | 0.167 | 0.167 |
| 10 | 0.700 | 0.200 | 0.100 | 0.667 | 0.000 | 0.333 |
The ten experiments were those that provided the best compromise between the selection of a limited number of experiments and minimized log(normalized determinant) of the dispersion matrix.32,33 As can be seen in Fig. 1, the ten experiments have been selected at the vertices of the polygon, at the midpoints of four of the five sides, and in the centroid of the polygon.
The choice of performing ten experiments and postulate special cubic models a priori instead of simpler models such as the linear or the quadratic model, was made by considering that the simpler models could be computed and validated anyway, since they have a greater number of degrees of freedom (e.g. 4 degrees of freedom for the quadratic model and 7 for the linear model, respectively) and the estimate of the coefficients would be of better quality.32 Actually, one of the encountered problems was that of collinearity between the factors which is expected for mixture designs, especially in highly constrained experimental domains,34 such as the one studied here. Therefore, in order to reduce the level of collinearity, all model computations were carried out using upper level-bound pseudocomponents (ui, i = 1, 2, 3) of the factors (A, B, and C). Once the pseudocomponent model predictive capacity had been defined and found acceptable, the equations in terms of the real values for the factors could be computed by substituting the relationships between A, B, and C and the corresponding ui, for a clearer understanding of the model results.
The upper-bound pseudocomponents were computed using eqn (1):
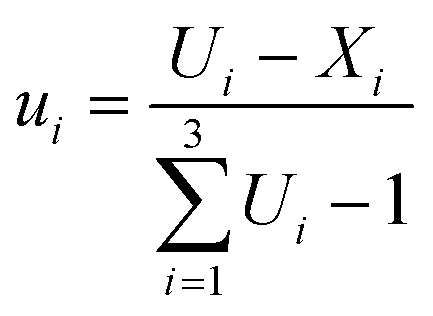 | (1) |
The outputs from the characterization of the polymers under examination used for the validation of the model were the polymer swelling in water (SWaq, Y1) and in THF (SWthf, Y2), the weight loss after wetting with water (WLaq, Y3) and THF (WLthf, Y4), and the material porosity (measured by the throat internal diameters, PID, Y5, and by the void diameters, VD, Y6).
In order to validate this model, a single experiment, with 4 replicates, was conducted. Therefore, an eleventh HIPE (11.a) was achieved to confirm the obtainment of a material with the expected properties as: BA + GMA = 0.788; TMPT = 0.150 and surfactant = 0.062.
 | (2) |
 | (3) |
The semi-quantitative analysis of the epoxy groups in the polyHIPEs was performed by using the software provided with the ATR-FTIR equipment. It allows us to calculate the area of the selected bands. In particular, the bands at 908 and 847 cm−1 and at 1720 cm−1, due to the vibrational bands attributed to the epoxy group ring and to the stretching of the carbonyl group, respectively, were used. The band at 1720 cm−1 was considered invariable with respect to the epoxy signals.
From this stock solution, different dilutions were prepared (0.25, 0.5, 1.0, 2.0, 2.5, 5.0, and 10.5 mg mL−1). Before the in-flow activity assay, the enzymatic column was equilibrated with 50 mM phosphate buffer solution (pH 7.0). The HPLC analysis was carried out with the injections of 20 μL of the different dilutions. The flow-rate was 0.2 mL min−1, the mobile phase applied was 50 mM phosphate buffer (pH 7.0) and the specific detection was performed at 441 nm. After the in-flow activity assay, the column was washed by the application of 13.5 mL of 50 mM phosphate buffer (pH 7.0). When not in use the column was stored at 2–8 °C in 50 mM phosphate buffer (pH 7.0).
Results and discussion
In this paper, we demonstrate that functionalized polyHIPEs are effective materials for the development of enzyme bioreactors. The main goal of this paper is to prove that by varying the HIPE compositions, it is possible to obtain materials with predictable behaviors in terms of pore dimensions (e.g. to optimize back pressure) and/or water/solvent affinity (e.g. to enhance biocatalyst activity). This prerequisite is fundamental for the application of these materials as functionalized supports for the immobilization of biocatalysts.PolyHIPE experimental design, synthesis and characterization
For the rational design of the materials to be selected for a specific application, a computational approach has been used to: (i) establish the composition range for the HIPE formation; (ii) link the outputs from the characterization to the compositions; and (iii) validate the models by performing a random experiment within the established composition range and evaluate if this composition, within four replicated (different) batches (reproducibility), shows similar outputs within the expected ranges.The monomer used for the polyHIPE synthesis was BA, as the backbone, because of the presence of the C4 chain which can modify the water affinity of the material and improve the separation behaviors of the system in chromatographic systems. The structural stability was also improved by a high crosslinking density achieved by TMPT as the tri-functional crosslinker. GMA was selected as a functional monomer in order to guarantee the conjugation reaction between the nucleophilic groups of the enzyme (mostly –NH2 groups) and the epoxy groups from GMA in the polyHIPE. An important advantage provided by these formulations is that the polymerization reactions were carried out at room temperature since a KPS/TEMED system was used as the radical initiator. As shown in Table 1, ten different polyHIPEs were synthesized and characterized.
To morphologically characterize the obtained polyHIPEs, all the samples were subjected to SEM analysis. The images were taken from different sides of the samples and those from the internal side were used for the calculation of void and throat diameter. In particular, images were acquired from upper, lower and lateral (internal) sides of the samples. As shown in Fig. 2, the typical open-cell structure of the polyHIPE materials was achieved for some of the formulations selected by the computational model.
 | ||
Fig. 2 SEM images of polyHIPEs from 1 to 10, all the shown samples are at a magnification of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×. 000×. | ||
In particular, the polyHIPE open cell typical structure was achieved for 6 materials (1, 2, 4, 5, 6 and 8). The other compositions (3, 7, 9 and 10) did not allow a polyHIPE typical morphology. Actually, the samples with the highest surfactant (3, 7, 9 and 10) did not lead to the typical polyHIPE structure. The effect of the surfactant concentration on the formation of polyHIPEs was previously observed. In particular, it has been shown that by increasing the surfactant concentration, thinner monomer films separating adjacent emulsion droplets could be produced with consequent phase separation.7,38
Throat and void diameters were evaluated from SEM images and are given in Table SI2.†
As from Table SI2,† void and throat diameters can be modulated by varying the composition of the polyHIPE. In particular, throat diameters can be varied between 400–1700 nm while voids can range from 1.5–7.7 μm. This is an important feature because it could lead to the selection of the material based on its behavior.
All the materials were also characterized in terms of weight loss and swelling in water and THF. Moreover, a semiquantitative analysis by ATR-FTIR representative of epoxy group hydrolysis was carried out. The results are shown in Table 2.
| Sample | WL in H2O | WL in THF | SW in H2O | SW in THF | Band area rate A/Ba |
|---|---|---|---|---|---|
| a The ATR-FTIR semiquantitative analysis was performed by calculating the rate of the areas epoxy/ester 908/1720 cm−1 (A) and 847/1720 cm−1 (B). These values are adimensional and to higher values corresponds a lower hydrolysis. | |||||
| 1 | 13.50% ± 8.15% | 3.98% ± 0.93% | 4.16 ± 0.54 | 6.33 ± 0.27 | 0.075/0.153 |
| 2 | 8.90% ± 5.09% | 14.14% ± 0.40% | 2.13 ± 0.19 | 3.95 ± 0.33 | 0.066/0.140 |
| 3 | 6.11% ± 0.59% | 13.87% ± 1.94% | 1.11 ± 0.09 | 1.86 ± 0.19 | 0.094/0.176 |
| 4 | 10.38% ± 2.19% | 9.21% ± 1.80% | 5.56 ± 0.08 | 5.05 ± 0.57 | 0.075/0.143 |
| 5 | 15.11% ± 2.86% | 10.98% ± 1.03% | 4.27 ± 0.10 | 4.89 ± 0.12 | 0.076/0.152 |
| 6 | 10.04% ± 3.77% | 14.86% ± 0.47% | 2.88 ± 0.08 | 4.43 ± 0.24 | 0.074/0.150 |
| 7 | 7.11% ± 0.63% | 18.28% ± 0.25% | 0.55 ± 0.01 | 1.32 ± 0.04 | 0.102/0.161 |
| 8 | 13.17% ± 3.39% | 10.46% ± 0.36% | 4.79 ± 0.98 | 7.10 ± 0.85 | 0.074/0.164 |
| 9 | 4.81% ± 0.32% | 16.70% ± 3.62% | 0.86 ± 0.01 | 1.27 ± 0.03 | 0.084/0.188 |
| 10 | 8.76% ± 0.34% | 29.80% ± 0.05% | 0.80 ± 0.09 | 1.46 ± 0.28 | 0.103/0.169 |
From the data shown in Table 2 different considerations arise. First, samples not characterized by an open-cell structure show the lowest ability to swell in both water or THF. This is not surprising because the material being less interconnected will not allow a good penetration of the solvent inside the material. On the other side, materials 1, 2, 4, 5, 6 and 8 show swelling values related to the void diameter. This is particularly true for the swelling in THF. In particular, samples 2, 5 and 6 with void diameters of 1.5–2.4 μm had swelling values of 3.95–4.89 in THF. Samples 1, 4 and 8 which show void diameters of 3.9–7.7 μm had higher swelling values of 5.05–7.10 in THF. The swelling in water could not follow the same order as in THF and this is not surprising because of the predominantly hydrophobic nature of the material. So, a “levelling effect” of the material itself on the swelling behavior in water could be predictable as also confirmed by the computational analysis (see below). However, some slight difference (even if not statistically significant) could be noted among the samples so we thought that the reason could be a different hydrolysis yield of the epoxy group during the synthesis (leading to the formation of hydrophilic OH groups) based on the formulation. Even for this reason, we performed a semi-quantitative ATR-FTIR analysis to study the hydrolysis of epoxy groups during the process. In particular, we compared the area of the band at 1720 cm−1 which is related to the stretching vibration of the carbonyl ester group (chosen as a quantitatively “fixed” band) with those from the epoxy group at 908 and 847 cm−1. The semiquantitative analysis was performed by calculating the rate of the areas 908/1720 cm−1 (A) and 847/1720 cm−1 (B). These values are adimensional and are given in Table 2. The higher the value of the found rates, the lower is the hydrolysis of the epoxy groups (being a rate epoxy/ester). It was not possible to find the highest value of the ratio (no hydrolysis) because of problems in handling the liquid monomers (BA + GMA + TMPT) for ATR-FTIR analysis. Obviously, the absence of bands related to the epoxy groups would indicate full hydrolysis; we previously demonstrated that on formulating the polyHIPE without the GMA, the indicated bands were not present.22 The results from the semiquantitative analysis indicate that epoxy group hydrolysis is independent of the polyHIPE composition (i.e. samples 1, 2, 4, 5, 6 and 8) while the hydrolysis yield was lower for samples 3, 7, 9 and 10, which did not show any open-cell morphology, with the epoxy groups being less exposed to the environment. One could also speculate that the “no-hydrolysis values” are those found for samples 3, 7, 9 and 10 (no open-cell morphology) which are close to the values found for the other samples indicating a low general hydrolysis during the polyHIPE formation. Obviously, this is a fundamental result because assures that the integrity of the epoxy groups is independent from the chosen composition.
The last step in the synthesis of the polyHIPEs was the selection of a random composition within the established domain which was prepared in 4 different batches to test reproducibility of the materials and prediction ability (validity) of the model. In this manner, the compositions A: BA + GMA 0.788, B: surfactant 0.062, C: TMPT 0.15 were prepared and the resulting polyHIPEs were characterized as before. Fig. 3 shows the SEM images of the new polyHIPE material.
 | ||
| Fig. 3 SEM images of polyHIPE 11.a; 11.b; 11.c; and 11.d at a 10000× magnification. | ||
The SEM images were acquired for the composition 11 (4 replicates) showing a porous network for all the four materials which consequently were recognized as polyHIPEs. The throat and void diameters were then calculated and expressed as average diameter ± SD, Table SI3.† As is shown, all the diameters for throats or voids are comparable in the four repeated samples, within a range of 0.1 μm, indicating an effective reproducibility of the adopted experimental method for the material preparation.
Table SI4† summarizes the main results from the performed characterization on sample 11. Also in this case, considering the possibly large experimental error for water loss or swelling determination, the found experimental values show comparable values for the four prepared samples.
The results from all the characterized compositions (samples 1–10) were used to compute the model equations and are given in Table 3.
| Models equations | R 2 | Adj. R2 | F (p-value) | RMSECV |
|---|---|---|---|---|
| SWaq is the swelling of the polymer in water, SWthf is the swelling of the polymer in THF, WLaq is the weight loss after polymer wetting with water, and WLthf is the weight loss after polymer wetting with THF measured in %; PID, polymer throat internal diameter, and VD, polymer void diameter measured in μm. C.I.(95%, n = 10) is the confidence interval at the 95% probability level for the mean of n = 10 observations collected in duplicate measurements. NA, not applicable. | ||||
| SWaq = −2.02·u1 + 4.81·u2 − 11.10·u3 + 8.07·u1·u2 + 26.01·u1·u3 + 24.04·u2·u3 − 57.94·u1·u2·u3 | 0.998 | 0.995 | 328.79 (<0.001) | 0.26 |
| SWthf = 1.016·u1 + 7.102·u2 + 1.503·u3 | 0.853 | 0.811 | 20.37 (0.0012) | 1.20 |
| WLaq; no model resulted statistically significant. The data are summarized by descriptive statistics. | NA | NA | NA | NA |
| Mean(WLaq) ± C.I.(95%, n = 10) = 10 ± 3%; median 9.6%; range (min–max) 5.0–17.4% | ||||
| WLthf = 19.62·u1 + 10.50·u2 − 50.85·u3 − 12.06·u1·u2 + 128.71·u1·u3 + 73.27·u2·u3 | 0.890 | 0.752 | 6.34 (0.027) | 4.33 |
| PID = −0.46·u1 + 1.64·u2 + 0.45·u3 | 0.878 | 0.837 | 24.8 (<0.001) | 0.33 |
| VD = −1.01·u1 + 5.94·u2 − 0.37·u3 | 0.834 | 0.787 | 17.6 (0.0018) | 1.47 |
Some selected diagnostic parameters for appraising the quality of the model computed are listed. As far as the fitting of the experimental points is concerned, the R2 and the adjusted R2 are given in Table 3. The model statistical significance computed using the ANOVA of the regression is described by the F parameter together with the model probability level (p-value).
The Root Mean Square Error in Cross-Validation (RMSECV) is an indicator of the model prediction ability. The RMSECV is computed by processing as many models as experiments, and excluding each time one experiment, whose response was then predicted based on the remaining experiments. Ideally, the lower the RMSECV the better is the model in prediction. The equation for computing the RMSECV is:
 | (4) |
| Component change | Effect = Ychange − Ycenter (Effect % = 100 × Effect/Ycenter) | ||||
|---|---|---|---|---|---|
| SWaq | SWthf | WLthf | PID | VD | |
| A: BA + GMA | |||||
| 0.725 → 0.745 | 3.344 − 2.877 = 0.467 | 4.475 − 4.037 = 0.438 | 11.701 − 14.569 = −2.868 | 0.929 − 0.729 = 0.2 | 2.960 − 2.454 = 0.506 |
| (X2/X3 = 1.067) | (+16%) | (+11%) | (−20%) | (+27%) | (+21%) |
| B: Surfactant | |||||
| 0.128 → 0.148 | 1.980 − 2.877 = −0.897 | 3.235 − 4.037 = −0.802 | 17.004 − 14.569 = 2.435 | 0.468 − 0.729 = −0.261 | 1.540 − 2.454 = −0.914 |
| (X1/X3 = 6.267) | (−31%) | (−20%) | (+17%) | (−36%) | (−37%) |
| C: TMPT | |||||
| 0.120 → 0.140 | 3.253 − 2.877 = 0.376 | 4.094 − 4.037 = 0.057 | 13.912 − 14.569 = −0.657 | 0.650 − 0.729 = −0.079 | 2.509 − 2.454 = 0.055 |
| (X1/X2 = 5.875) | (+13%) | (1%) | (−5%) | (−11%) | (+2%) |
The definitive confirmation of the predictive ability of the models was obtained after collecting the data of replicates of the experiments corresponding to the composition of the reaction mixture [A: BA + GMA 0.788, B: surfactant 0.062, C: TMPT 0.15]. The experimental values obtained for SWaq, SWthf, WLaq, WLthf, PID, and VD, together with the predicted values computed by the models for each response, are presented in Table SI5.†
Based on these results, it can be concluded that the models were all reliable, since the agreement between the predicted value and the experimental value was within the limits of the experimental error and none of the results of the validation experiments were significantly different from the predicted values.
Therefore, the models were applicable for predicting the properties of the formulations prepared by using the compositions included in the experimental domain.
In mixture design data analysis, one difficulty is that the relevance of the effects of the components cannot be directly deduced by observing the magnitude of the coefficients computed. The mixture models must be analyzed taking into consideration that a change in one component is accompanied by the simultaneous change in the other two components of the mixture, since the overall composition of the mixture must respect the constrain A + B + C = 1. Several methods for estimating the effects of the components in a mixture have been reported,34 and in the present study the approach described by Cafaggi and coworkers was chosen owing to its simplicity.32 Briefly, the effect of component i is computed as the difference between the responses at two points, centered on the centroid of the experimental domain. The two points have a fixed difference in terms of component i. The other two components instead are kept at a constant ratio which corresponds to the composition of the mixture represented in the centroid of the experimental domain. By applying this method, it has been possible to compute quantitatively the effects given in Table 4 which can be evaluated qualitatively as shown in Fig. SI1a–SI1e.†
Based on the results reported in Table SI4† and on the study of the response surfaces plotted in Fig. SI1a–SI1e,† the following conclusions could be drawn.
The SWaq and SWthf are affected primarily by the surfactant amount in the mixture (Table 4 and Fig. SI16a, SI16b†). By increasing the surfactant by 0.02% actually the models showed a decrease in responses for both SWaq and SWthf of about 31% and 20%, respectively. The effect of BA + GMA is also important and similar on either swelling, but to a lesser extent (+16% and +11% on SWaq and SWthf, respectively). The effect of TMPT is evidenced only on the swelling in THF, where the increase of TMPT causes an increase of about 13% in this polymer feature.
As far as the weight loss in water is concerned, as evidenced by the experimental data, no significant changes are evident as a consequence of the changes in the mixture used for the polymerization. The average WLaq is around 10% (median 9.6%). The fact that no model could be computed is due to the great scatter of the data around the mean with no evident tendency due to changes in the mixture composition, Table 4.
The weight loss in THF appears instead influenced by BA + GMA and by the surfactant amount to the same extent, although in opposite directions. The response decreases drastically as the BA + GMA amount increased in the mixture, and the response increases instead if the surfactant amount increases (Fig. SI1c†).
The throat internal diameters (PID, Fig. SI1d†) and void diameters (VD, Fig. SI1e†) are affected in the same way by the changes in the composition of the polymerization mixture. The major effect is that of the amount of surfactant, followed by that of the amount of BA + GMA. When the amount of surfactant is increased, this brings to throats and voids with smaller diameters. The effect of increasing the amount of BA + GMA instead is to produce throats and voids with larger diameters. The effect of TMPT is measurable only on the throat internal diameters, where the tendency shown is that an increase in the amount of TMPT produced polymers with smaller throats.
Evaluation of polyHIPE as a material for enzyme immobilization
For this experiment, the polyHIPE #6 has been chosen as the model material because of its low swelling in water and with a regular SEM morphology. Filling the glass column with the emulsion before polymerization allows us to achieve the polymerization directly in the column and, consequently, to slightly modify the procedure for the radical induction. To avoid a premature polymerization of the HIPE prior to the column filling, TEMED was not added directly to the emulsion (in the flask) but a “syringe-to-syringe” method was used to quickly transfer the radically initiated emulsion directly to the column (refer to the Experimental part). Approximately a 10 × 6 mm ID polyHIPE bed was obtained. For this experiment we selected HRP since it is well known that this enzyme preserves its catalytic activity after immobilization on epoxy-functionalized materials.39–41The covalent immobilization of the HRP to the polyHIPE was accomplished by following a consolidated in situ procedure (see the Experimental part).
The immobilization yield was determined spectrophotometrically by measuring the absorbance of the enzymatic solution before and after the immobilization procedure. For quantitation, a calibration curve was derived at 280 nm and a linear correlation was found (y = 0.8562x − 0.0009; R2 = 0.9996).
The immobilization yield was 24.13%, corresponding to 50 mg of immobilized enzyme indicating the successful immobilization of HRP on the epoxy groups of the polyHIPE-based stationary phase.
To verify that the enzyme does not undergo a reduction in UV absorbance during the immobilization process, the same experiment was performed by fluxing the same HRP solution in a support without epoxy functionalities. After 24 h the enzyme absorbance was equal to 99.5% with respect to the initial absorbance.
Enzyme kinetic evaluation by an in-flow activity assay
Horseradish peroxidase is a typical peroxidase enzyme obtained by the roots of horseradish. Among all peroxidases, HRP is known to catalyze various oxidative transformations of organic compounds using H2O2 as an oxidative agent, such as the conversion of OPD in 2,3-diaminophenazine. In this study, HRP was selected as a protein to be immobilized onto the polyHIPE monolithic column because, as a peroxidase, it is regarded as a highly selective and efficient biocatalyst. Moreover, HRP has some advantageous characteristic properties like low price, a significant resistance to organic solvents and a very wide number of substrates available on the market.42 OPD was selected as a HRP chromogenic substrate because its oxidative conversion leads to the obtainment of the product 2,3-diaminophenazine. 2,3-Diaminophenazine can be easily detected by using a UV-Vis detector at 441 nm.37 Moreover, at 441 nm, OPD does not show any absorbance. For this reason, 441 nm was selected as the wavelength for the HPLC-UV-Vis analysis in the in-flow activity assay, Fig. 4. According to the conditions reported above, different OPD concentrations were injected into a HPLC-UV-Vis system and the monolithic polyHIPE HRP-column was used in the biocatalysis process. Fig. 4 shows the chromatograms obtained by injecting increasing substrate concentrations. As expected, the product peak areas increase with the substrate concentration. | ||
| Fig. 4 Chromatograms of OPD solution at different concentrations (mg mL−1) in phosphate buffer at pH 7 (50 mM) as the mobile phase, detection at 441 nm. | ||
In order to evaluate the enzymatic kinetic, the reaction rate for each substrate concentration was calculated as follows:
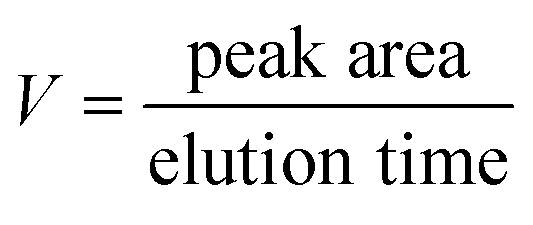 | (5) |
This relation was expressed by the Michaelis–Menten equation that represents the kinetic profile of enzymatic reactions:
 | (6) |
In order to calculate the reaction kinetic values, such as Km and Vmax, this equation was expressed as a double reciprocal plot (eqn (7)):
 | (7) |
Thus, this equation represents the graphic representation of this enzyme kinetic, where the slope was Km/Vmax, the y-intercept was 1/Vmax and the x-intercept was −1/Km. In this case, a curve y = 0.01688x + 0.00035 was achieved as shown in Fig. SI3.† Therefore, kinetic values were calculated as: Vmax = 2857.1 area per min; Km = 48.23 mM.
These results showed a good linearity and a correct outgoing for an enzymatic reaction. To date the bioreactor has been used for approximately 50 bioconversions without loss of the initial catalytic activity confirming the high stability of the supported enzyme. Maintenance of their structural stability with functional efficiency and reproducibility provides an excellent base to further investigate the applicability of these supports in different fields.
Conclusions
The synthesis of the polyHIPEs and method validation were performed by a design of experiment (DoE) approach. The prepared materials were physicochemically characterized and, most of them showed the typical interconnected pore structure of the polyHIPE. Among the obtained polymeric monoliths, one was selected for the chemical immobilization of a model enzyme on a pre-polymerization-filled chromatographic column. HRP, chemically immobilized into the polyHIPE, retained its catalytic activity. In conclusion the special structure of polyHIPEs is well suited for in flow-applications. In fact, differently from particle based supports, polyHIPE monolithic supports offer new possibilities since the diffusive limitations are minimized. Moreover the highly interconnected channel network totally permeable for the eluent flow, allows us to reduce the back pressure of the system. The unique properties of these supports make them very attractive for bioconversion processes with the advantage to be realized with different functionalities and on different scales, e.g. from micro-analytical to industrial one.With the vast array of research on enzyme immobilization this result paves the way for highly efficient and economically biotechnological processes in the field of biotransformation, diagnostics and pharmaceutical and food industries.
Conflicts of interest
There are no conflicts to declare.References
- T. T. Bui, Y. S. Kim, H. Chun, D. D. La and S. V. Bhosale, Mater. Lett., 2017, 192, 80–83 CrossRef CAS.
- S. W. Tan, A. Ebrahimi and T. Langrish, Mater. Des., 2017, 117, 178–184 CrossRef CAS.
- D. Karamessini, G. C. Lainioti, V. Deimede and J. K. Kallitsis, J. Appl. Polym. Sci., 2017, 134 CrossRef CAS , 44539.
- X. B. Wei, C. R. Gong, X. J. Chen, G. L. Fan and X. H. Xu, Front. Mater. Sci., 2017, 11, 33–41 CrossRef.
- Y. R. Xin, Q. C. Xiong, Q. H. Bai, M. Miyamoto, C. Li, Y. H. Shen and H. Uyama, Carbohydr. Polym., 2017, 157, 429–437 CrossRef CAS PubMed.
- K. Schacht, J. Vogt and T. Scheibel, ACS Biomater. Sci. Eng., 2016, 2, 517–525 CrossRef CAS.
- N. R. Cameron, Polymer, 2005, 46, 1439–1449 CrossRef CAS.
- J. Naranda, M. Susec, U. Maver, L. Gradisnik, M. Gorenjak, A. Vukasovic, A. Ivkovic, M. S. Rupnik, M. Vogrin and P. Krajnc, Sci. Rep., 2016, 6, 28695 CrossRef CAS PubMed.
- F. Du, L. Sun, X. Zhen, H. Nie, Y. Zheng, G. Ruan and J. Li, Anal. Bioanal. Chem., 2015, 407, 6071–6079 CrossRef CAS PubMed.
- R. Su, G. Ruan, H. Nie, T. Xie, Y. Zheng, F. Du and J. Li, J. Chromatogr., A, 2015, 1405, 23–31 CrossRef CAS PubMed.
- W. Busby, N. R. Cameron and A. B. C. Jahoda, Polym. Int., 2002, 51, 871–881 CrossRef CAS.
- M. Depardieu, N. Kinadjian and R. Backov, Eur. Phys. J.: Spec. Top., 2015, 224, 1655–1668 CrossRef CAS.
- S. J. Pierre, J. C. Thies, A. Dureault, N. R. Cameron, J. C. M. van Hest, N. Carette, T. Michon and R. Weberskirch, Adv. Mater., 2006, 18, 1822–1826 CrossRef CAS.
- N. A. Sears, P. S. Dhavalikar and E. M. Cosgriff-Hernandez, Macromol. Rapid Commun., 2016, 37, 1369–1374 CrossRef CAS PubMed.
- Y. Lumelsky, I. Lalush-Michael, S. Levenberg and M. S. Silverstein, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 7043–7053 CrossRef CAS.
- Y. Hu, H. Gao, Z. Du, Y. Liu, Y. Yang and C. Wang, J. Mater. Chem. B, 2015, 3, 3848–3857 RSC.
- Y. Zhu, R. Zhang, S. Zhang, Y. Chu and J. Chen, Langmuir, 2016, 32, 6083–6088 CrossRef CAS PubMed.
- H. Liu and C. Wang, RSC Adv., 2014, 4, 3864–3872 RSC.
- B. H. L. Oh, A. Bismarck and M. B. Chan-Park, Biomacromolecules, 2014, 15, 1777–1787 CrossRef CAS PubMed.
- S. D. Kimmins and N. R. Cameron, Adv. Funct. Mater., 2011, 21, 211–225 CrossRef CAS.
- M. S. Silverstein, Prog. Polym. Sci., 2014, 39, 199–234 CrossRef CAS.
- G. Brusotti, E. Calleri, C. Milanese, L. Catenacci, G. Marrubini, M. Sorrenti, A. Girella, G. Massolini and G. Tripodo, Polym. Chem., 2016, 7, 7436–7445 RSC.
- D. Yin, Y. Guan, H. Gu, Y. Jia and Q. Zhang, RSC Adv., 2017, 7, 7303–7309 RSC.
- S. Livshin and M. S. Silverstein, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4840–4845 CrossRef CAS.
- A. A. Rane and K. L. Christman, J. Am. Coll. Cardiol., 2011, 58, 2615–2629 CrossRef CAS PubMed.
- J. Pyun, C. B. Tang, T. Kowalewski, J. M. J. Frechet and C. J. Hawker, Macromolecules, 2005, 38, 2674–2685 CrossRef CAS.
- G. Ruan, Z. Wu, Y. Huang, M. Wei, R. Su and F. Du, Biochem. Biophys. Res. Commun., 2016, 473, 54–60 CrossRef CAS PubMed.
- Y. Yücel, C. Demir, N. Dizge and B. Keskinler, Biomass Bioenergy, 2011, 35, 1496–1501 CrossRef.
- D. J. Pollard and J. M. Woodley, Trends Biotechnol., 2007, 25, 66–73 CrossRef CAS PubMed.
- S. Datta, L. R. Christena and Y. R. S. Rajaram, 3 Biotech, 2013, 3, 1–9 CrossRef PubMed.
- E. Calleri, G. Cattaneo, M. Rabuffetti, I. Serra, T. Bavaro, G. Massolini, G. Speranza and D. Ubiali, Adv. Synth. Catal., 2015, 357, 2520–2528 CrossRef CAS.
- S. Cafaggi, R. Leardi, B. Parodi, G. Caviglioli and G. Bignardi, Chemom. Intell. Lab. Syst., 2003, 65, 139–147 CrossRef CAS.
- P. F. De Aguiar, B. Bourguignon, M. S. Khots, D. L. Massart and R. Phan-Than-Luu, Chemom. Intell. Lab. Syst., 1995, 30, 199–210 CrossRef.
- J. Cornell, Experiments with mixtures. Designs, models, and the analysis of mixture data, 2002 Search PubMed.
- E. Calleri, G. Massolini, D. Lubda, C. Temporini, F. Loiodice and G. Caccialanza, J. Chromatogr., A, 2004, 1031, 93–100 CrossRef CAS PubMed.
- E. Calleri, C. Temporini, E. Perani, C. Stella, S. Rudaz, D. Lubda, G. Mellerio, J. L. Veuthey, G. Caccialanza and G. Massolini, J. Chromatogr., A, 2004, 1045, 99–109 CrossRef CAS PubMed.
- M. Iacono and A. Heise, Polymer, 2015, 7, 1427–1443 CAS.
- N. R. Cameron, D. C. Sherrington, L. Albiston and D. P. Gregory, Colloid Polym. Sci., 1996, 274, 592–595 CAS.
- C. Topcular and H. Ayhan, J. Biomater. Sci., Polym. Ed., 2007, 18, 595–607 CrossRef CAS PubMed.
- J. M. Abad, M. Velez, C. Santamaria, J. M. Guisan, P. R. Matheus, L. Vazquez, I. Gazaryan, L. Gorton, T. Gibson and V. M. Fernandez, J. Am. Chem. Soc., 2002, 124, 12845–12853 CrossRef CAS PubMed.
- V. Kumar, N. Misra, N. K. Goel, R. Thakar, J. Gupta and L. Varshney, RSC Adv., 2016, 6, 2974–2981 RSC.
- X. Gao, S. Huang, P. Dong, C. Wang, J. Hou, X. Huo, B. Zhang, T. Ma and X. Ma, Catal.: Sci. Technol., 2016, 6, 3585–3593 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7py01626c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |