
From mono- to tetraacylgermanes: extending the scope of visible light photoinitiators†
Anna
Eibel
 a,
Judith
Radebner
b,
Michael
Haas
b,
David E.
Fast
a,
Hilde
Freißmuth
a,
Eduard
Stadler
a,
Paul
Faschauner
b,
Ana
Torvisco
b,
Iris
Lamparth
c,
Norbert
Moszner
c,
Harald
Stueger
b and
Georg
Gescheidt
*a
a,
Judith
Radebner
b,
Michael
Haas
b,
David E.
Fast
a,
Hilde
Freißmuth
a,
Eduard
Stadler
a,
Paul
Faschauner
b,
Ana
Torvisco
b,
Iris
Lamparth
c,
Norbert
Moszner
c,
Harald
Stueger
b and
Georg
Gescheidt
*a
aInstitute of Physical and Theoretical Chemistry, NAWI Graz, Graz University of Technology, Stremayrgasse 9, 8010 Graz, Austria. E-mail: g.gescheidt-demner@tugraz.at
bInstitute of Inorganic Chemistry, NAWI Graz, Graz University of Technology, Stremayrgasse 9, 8010 Graz, Austria
cIvoclar Vivadent AG, Bendererstrasse 2, 9494 Schaan, Liechtenstein
First published on 20th November 2017
Abstract
We have investigated the inititiaton efficiency of carefully selected germanium-based photointiators for radical polymerization. To establish a systematic relationship between structure and reactivity, we have developed a convenient synthetic protocol for the preparation of a trisacylgermane, closing the gap from mono- to tetraacylgermane photoinitiators. The studied acylgermanes display distinct, wavelength-dependent photobleaching upon irradiation up to 470 nm. In particular, tetraacylgermanes featuring ortho-alkyl substituents reveal red-shifted n–π* bands, in line with excellent photobleaching upon visible light irradiation. Quantum yields of decomposition (determined at 385 nm) have been found to be highest for bisacylgermanes. Germyl radicals produced upon triplet-state α-cleavage of the acylgermanes react remarkably fast with monomers. Addition rate constants to (meth)acrylates range from 0.4–4.5 × 108 M−1 s−1, depending on the substitution pattern. These values are clearly higher than those reported for related phosphorus-centered radicals derived from acylphosphane oxides. We have further established the nature of the products and side-products formed at initial stages of the polymerizations using chemically induced dynamic nuclear polarization (CIDNP) experiments.
Introduction
Shifting absorptions to specifically chosen wavelengths, particularly in the visible region, has been a major challenge in photoinitiator (PI) research. Additionally, a favorable quantum efficiency for the initiating process is highly desired. For medical applications such as dental fillings, low toxicity is another important issue. It has been shown that acylgermanes have the potential to meet these requirements.1–6 Recently, we have introduced a straightforward synthetic route for the preparation of tetraacylgermanes, having the advantage of a simplified synthesis and red-shifted n–π* absorptions compared to mono- and bisacylgermanes.7,8 In this contribution, we wish to systematically assess the photoinitiating efficiency of specifically modified tetraacylgermanes (5–8, Chart 1), and to provide a comparison with mono- to trisacylgermanes (1–4), as well as to the widely used mono- and bisacylphosphane oxide initiators 9 and 10.9–16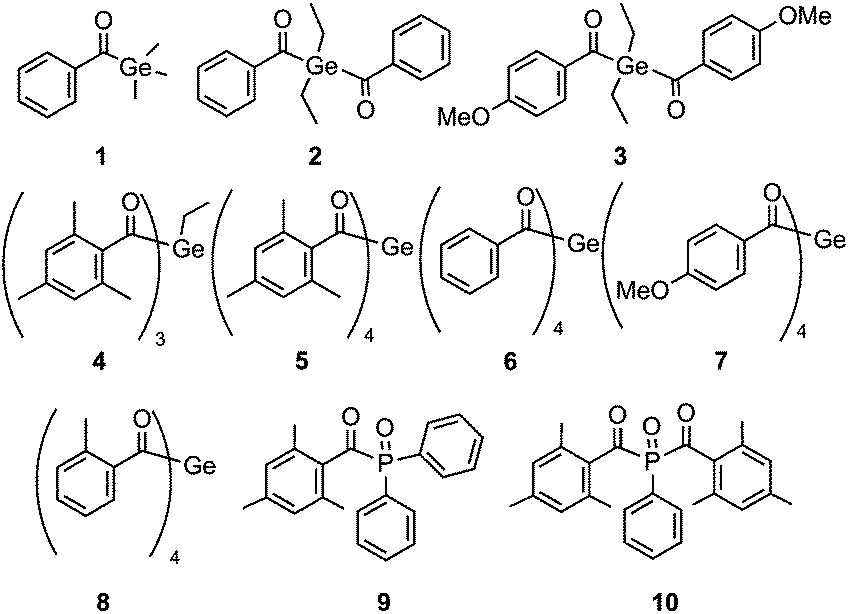 | ||
| Chart 1 Chemical structures of acylgermanes 1–8 and acylphosphane oxides 9 and 10. | ||
Trisacylgermanes are very rarely known in literature17 and have – to the best of our knowledge – not been investigated as photoinitiators yet. In order to complete the array of germanium-based photoinitiators displaying one to four acyl moieties, we have therefore developed a convenient synthetic protocol to obtain a trisacylgermane derivative (4).
Compounds 1, 2 and 6 have been selected for this study, since they feature the same aromatic moiety (unsubstituted benzoyl group) one, two and four times, respectively. Similarly, bisacylgermane 3 and tetraacylgermane 7 allow studying the influence of the p-methoxy substituent on the photoreactivity. Additionally, we have decided to investigate tetraacylgermane 8 – an asymmetrically substituted derivative, providing good solubility in organic solvents and a strongly red-shifted absorption band.8 Trisacylgermane 4 and tetraacylgermane 5 both feature mesitoyl groups.
The goal of our work is to provide insights into structure–reactivity relationships, to promote further strategies for the development of tailor-made germanium-based initiators.
Generally, photolysis of acylgermanes (Scheme 1) leads to triplet-state α-cleavage of a Ge–C(O) bond, yielding a benzoyl-type and a germyl radical. We have investigated the α-cleavage reaction shown in Scheme 1 using time-resolved (TR) EPR spectroscopy. The photobleaching efficiency of acylgermanes 1–8 was analysed both qualitatively upon irradiation at different wavelengths, and quantitatively in terms of decomposition quantum yields. Moreover, we have studied the kinetics and reaction mechanisms of polymerizations utilizing laser-flash photolysis (LFP), and chemically induced dynamic nuclear polarization (CIDNP-NMR) spectroscopy.
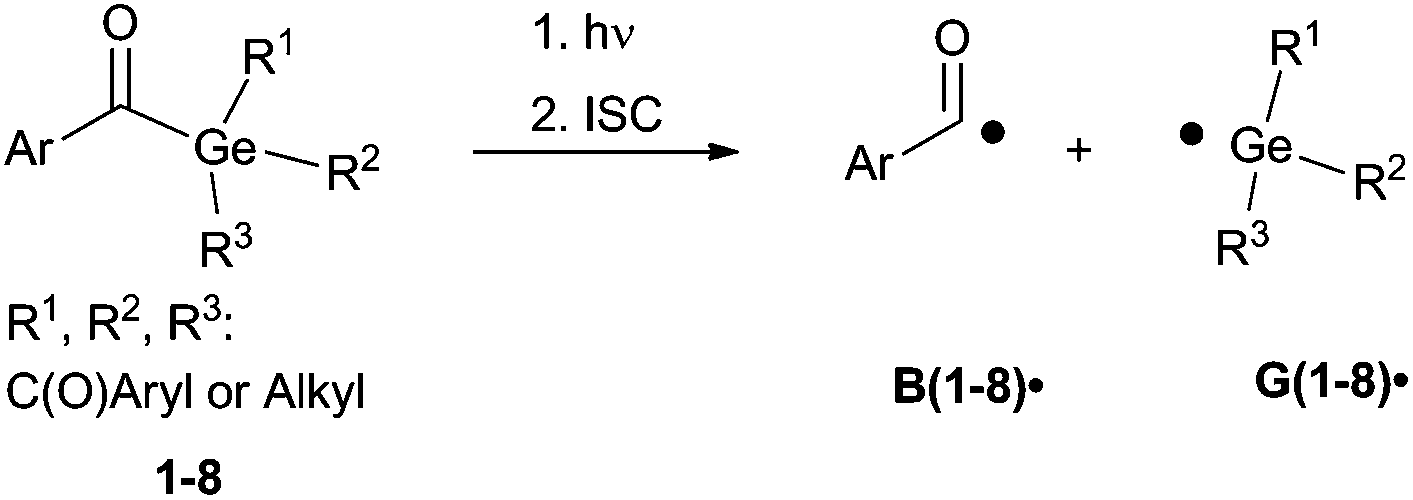 | ||
| Scheme 1 Photocleavage of acylgermanes, leading to the formation of a benzoyl-type radical B˙ and a germyl radical G˙. R1, R2 and R3 abbreviate the substituents displayed in the formulas in Chart 1. | ||
Experimental section
General considerations
All synthesis steps were performed under a nitrogen atmosphere using standard Schlenk techniques. Solvents were dried using a column solvent purification system.18 Commercial KOtBu (97%) was used without further purification. Tetrakis(trimethylsilyl)germane ((Me3Si)4Ge),19 ethyltris-(trimethylsilyl)germane ((Me3Si)3GeEt)20 and the acid fluorides21 were prepared according to published procedures. 1H (299.95 MHz) and 13C (75.43 MHz) NMR spectra were recorded on a Varian INOVA 300 spectrometer in CDCl3 solution and referenced versus TMS using the internal 2H-lock signal of the solvent. Tetraacylgermanes Ge[C(O)R]4 (R = mesityl (5),7 R = phenyl (6),7 R = (4-methoxy)phenyl (7),8 R = (2-methyl)phenyl (8)8 were synthesized as described previously).7,8Synthesis of ethyltrismesitoylgermane (4)
A flask was charged with 1.25 g (3.9 mmol) of (Me3Si)3GeEt and 0.48 g of KOtBu (4.3 mmol). 10 mL of DME were added. The reaction mixture was stirred for one hour. In a second flask, 3.2 g (19.0 mmol) of mesitoyl fluoride were dissolved in 5 mL DME and the potassium germanide solution was slowly added via a syringe. After complete addition, the reaction mixture was stirred for another 12 h. After aqueous work-up with saturated NH4Cl the phases were separated and the aqueous phase was extracted with dichloromethane. The combined organic layers were dried over Na2SO4, filtered and the volatile components were evaporated. The crude product was recrystallized from acetone giving 1.5 g (2.8 mmol, 71%) of pure 4. Melting point: mp 100–103 °C. UV-Vis: λmax (acetonitrile)/nm 382.5 (ε/L mol−1 cm−1 1303). Elemental analysis: found C, 70.73; H, 7.07. Calc. for C32H38GeO3: C, 70.75; H, 7.05%. 1H NMR (300 MHz, CDCl3, ppm): δ 6.66 (s, 2H, Aryl–H), 2.21 (s, 3H, p-CH3), 2.09 (s, 6H, o-CH3), 1.37 (q, 2H, Ge–CH2–CH3), 1.01 (t, 3H, Ge–CH2–CH3), 13C NMR (75.43 MHz, CDCl3, ppm): δ 237.24 (GeC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 142.13, 139.02, 132.30, 128.71 (Aryl–C), 21.04 (Mes-p-CH3), 18.92 (Mes-o-CH3), 10.31 (GeCH2–CH3), 8.62 (GeCH2–CH3). X-Ray crystallography: CCDC number 1561720.
O), 142.13, 139.02, 132.30, 128.71 (Aryl–C), 21.04 (Mes-p-CH3), 18.92 (Mes-o-CH3), 10.31 (GeCH2–CH3), 8.62 (GeCH2–CH3). X-Ray crystallography: CCDC number 1561720.
Materials and solvents for spectroscopic investigations
Photoinitiators 1–3 were kindly provided by Ivoclar Vivadent AG. The phosphorus-based initiators Irgacure® TPO (9) and Irgacure® 819 (10) were obtained from TCI. The monomers butyl acrylate (BA, purity ≥ 99.0%), methyl methacrylate (MMA, purity ≥ 99.0%) and methyl acrylate (MA, purity ≥ 99.5%) were obtained from Fluka and freed from stabilizers by passing through a column of basic alumina before use. Propiophenone (99%) was obtained from Sigma Aldrich and used as received. The solvents acetonitrile (Sigma Aldrich, ≥99.9%), acetonitrile-d3 (Euriso-top) and toluene (Fluka, ≥99.8%) were employed as received. All experiments were performed at ambient temperature.UV-Vis spectroscopy
UV-Vis spectra were recorded on a TIDAS UV-Vis spectrometer (J&M, Germany). For photobleaching experiments (steady state photolysis, SSP), samples were irradiated perpendicular to the light beam of the UV-Vis spectrometer in a fluorescence quartz cuvette (1 cm × 1 cm) while UV-Vis spectra were recorded. Irradiation was performed using low-power LEDs (Roithner Lasertechnik, Austria): LED470 (maximum at 462.5 nm, 30 nm half width, 0.013 W at 20 mA) and LED385 (maximum at 385 nm, 15 nm half width, 0.013 W at 20 mA). A spectrophotometer (GL Spectis, GL Optics, Germany) was used to measure the output power of the LEDs and to ensure an equal photon flux for SSP experiments. All LEDs were operated at a photon flux of 0.05 μmol s−1. Samples were prepared to achieve an absorbance within the linear range of the spectrometer during the course of the experiment (0.8–2 mM in a solvent mixture of acetonitrile and MMA (1/1 v/v ratio)). All samples were purged with argon for 5 minutes before the measurement and were magnetically stirred during the experiment to ensure sufficient mixing.Quantum yields of decomposition
Quantum yields of decomposition have been determined using the same setup as described for the photobleaching studies (see above). The evaluation procedure for determining photochemical quantum yields via on-line UV-Vis spectroscopy has been described in detail by our group recently.22 We compute the quantum yield Φ by using the concentration change and the exact amount of absorbed light at the very beginning of the reaction. The quantum yield Φ is equal to the initial reaction rate divided by the product of the incident light intensity (photon flux I0) and the fraction of absorbed light f. The initial reaction rate (k) corresponds to the first derivative of the exponential fit of concentration vs. time curves, obtained from the absorbance traces using the extinction coefficients of the photoinitiators.22,23 For irradiation, the LED operated at 385 nm (LED385) was used. The photon flux I0 was determined using the spectrophotometer. The factor f is easily calculated from the initial absorbance A of the sample at the irradiation wavelength (f = 1–10−A).23 All samples were prepared in a solvent mixture of acetonitrile and MMA (1/1 v/v ratio) to achieve an initial absorbance of ∼0.6 at 385 nm. Samples were purged with argon for 5 minutes before the measurement and magnetically stirred. Knowledge of the exact volume of the sample is required for calculating the photon flux of the LED in the unit (mol L−1 s−1). Measuring single absorbance spectra before and after bubbling with argon allows to account for solvent evaporation during the bubbling procedure and to determine the correct sample volume.22Laser flash photolysis (LFP)
The experiments were performed on a LKS80 Laser Flash Photolysis Spectrometer (Applied Photophysics, UK). Samples were excited with the frequency tripled light from a Spitlight Compact 100 (InnoLas, Germany) solid state Nd:YAG laser at 355 nm (∼10 mJ per pulse, 8 ns). Rate constants for the addition of the germyl radicals to the monomer double bonds were determined in pseudo-first-order experiments; solutions of the compounds in acetonitrile containing monomer concentrations in the range of 0.025 M to 0.5 M and providing absorbance of ∼0.3 at 355 nm were prepared. Static solutions were saturated with argon before the measurements. The decay of the germyl radicals was recorded at the absorption maximum determined from the transient absorption spectra.TR-EPR spectroscopy
Continuous-wave time-resolved EPR (TR-EPR) experiments were performed on a Bruker ESP 300E X-band spectrometer (unmodulated static magnetic field) equipped with a 125 MHz dual channel digital oscilloscope (Le Croy 9400). As the light source, the frequency tripled light of a Nd:YAG laser was used (InnoLas Spitlight 400, 355 nm, operating at 20 Hz, ∼7 mJ per pulse, 8 ns). The setup is controlled by the fsc2 software developed by Dr J. T. Toerring (Berlin). Spectra were recorded by acquiring the accumulated (50 accumulations) time responses to the incident laser pulses at each magnetic field value of the chosen field range (field steps: 0.2 G). Argon-saturated solutions in toluene (∼15 mM in photoinitiator concentration) were pumped through a cylindrical quartz cell positioned in the cavity of the EPR spectrometer using a flow system (flow rate: 2–3 mL min−1).1H CIDNP-NMR spectroscopy
1H NMR and CIDNP experiments were carried out on a 200 MHz Bruker AVANCE DPX spectrometer equipped by a custom-made CIDNP probe head. A Quantel Nd-YAG Brilliant B laser (355 nm, ∼70 mJ per pulse, pulse length 8–10 ns) operating at 20 Hz was employed as the light source. The timing sequence of the experiment consists of a series of 180° radiofrequency (RF) pulses (applied to suppress the NMR signals of the parent compounds), the laser flash, the 90° RF detection pulse and the acquisition of the FID. “Dummy” CIDNP spectra employing the same pulse sequence but without the laser pulse were always measured. Samples were prepared in deuterated acetonitrile and deoxygenated by bubbling with argon before the experiment. Chemical shifts (δ) are reported in ppm relative to tetramethylsilane (TMS) using the residual deuterated solvent signal as an internal reference (CD3CN, δH = 1.94 ppm).Results and discussion
Ge-Based initiators
The systematic assessment of acylgermanes from mono- to tetraacyl-derivatives requires the preparation of trisacylgermanes such as 4. The synthetic pathways leading to trisacylgermane 4 and to tetraacylgermanes 5–8 are summarized in Scheme 2. To obtain the desired germanide, the corresponding precursors, (Me3Si)3GeEt or (Me3Si)4Ge, were treated with KOtBu. Upon addition of the respective acid fluoride, the desired product was formed (4, yield: 71%). Concerning trisacylgermanes, a stable product could only be isolated in the case of compound 4, featuring mesitoyl groups. We are currently working on the preparation of further functionalized derivatives. X-ray crystallography revealed the structure of 4 as shown in Fig. 1. Structural features such as bond lengths and angles (see Fig. 1) are compatible with previously reported values for tetraacylgermanes. For a more detailed discussion the reader is referred to the ESI† and ref. 7 and 8.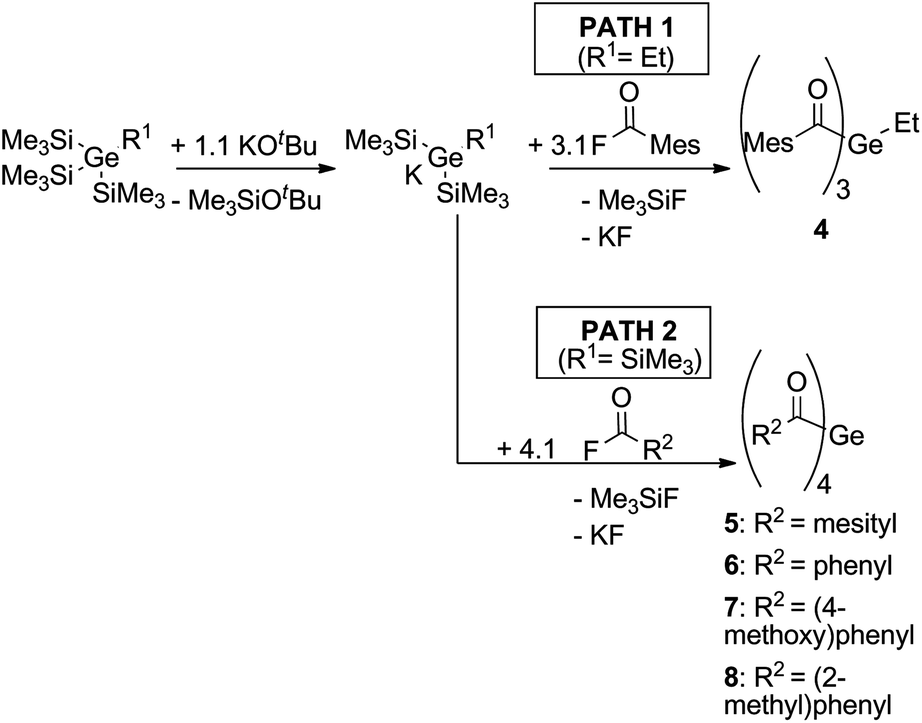 | ||
| Scheme 2 Synthesis of trisacylgermane 4 and tetraacylgermanes 5–8.7,8 | ||
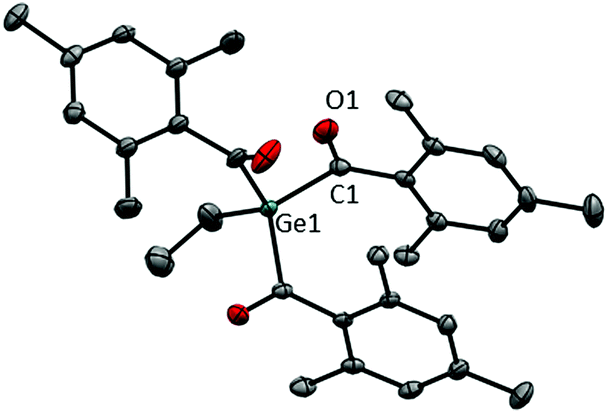 | ||
| Fig. 1 ORTEP representation of 4. Thermal ellipsoids are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: Ge–C(mean) 2.031, C–O(mean) 1.214; C–Ge–C (mean) 109.45, O–C–Caryl–Caryl (mean) 90.1. | ||
UV-Vis absorption spectra
Fig. 2 depicts the UV-Vis absorption spectra of compounds 1–8 with intense π–π* absorption bands below 350 nm and weaker bands above 360 nm, attributable to n–π* transitions. Excitation of the latter ones is responsible for the photoinduced α-cleavage of the Ge–C(O) bond, leading to germyl and benzoyl-type radicals (Scheme 1). The extinction coefficients ε increase from mono- to tetraacylgermanes, in line with the extension of the π-system. The absorption bands of 1–8 are clearly influenced by the substituents at the aromatic rings.8 Compounds featuring a p-methoxy substituent (bisacylgermane 3 and tetraacylgermane 7) show blue-shifted absorption spectra and higher extinction coefficients when compared to the corresponding unsubstituted acylgermanes 2 and 6 (benzoyl derivatives). For the mesitoylderivatives 4 (ethyltrismesitoylgermane) and 5 (tetramesitoylgermane), the strongest hypsochromic shifts are observed. The o-tolyl substituent of tetraacylgermane 8 induces a bathochromic shift, leading to the strongest absorbance among the studied initiators in the visible range above 450 nm. The UV-Vis spectra of phosphorus-based photoinitiators 9 and 10 are provided in the ESI,† showing blue-shifted absorption spectra when compared to acylgermanes 1–8. No absorbance is observed above 420 nm and 440 nm for compounds 9 and 10, respectively, making these initiators unsuitable for long-wavelength curing applications.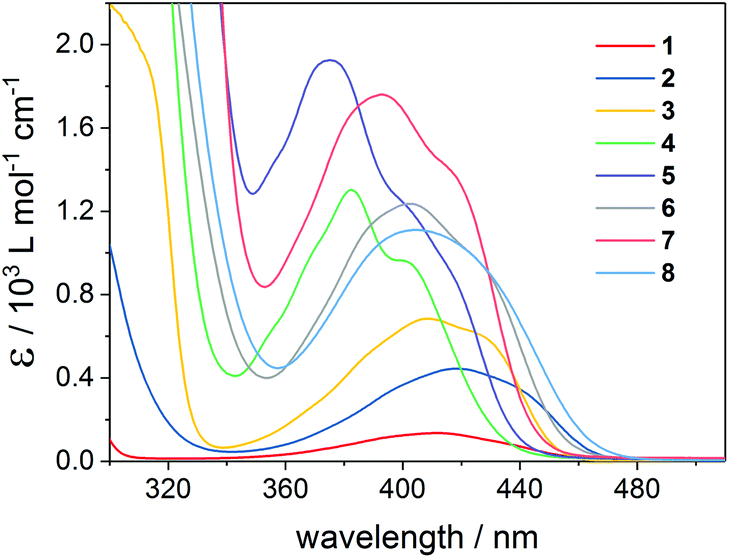 | ||
| Fig. 2 UV-Vis absorption spectra of mono-, bis- and tris- and tetraacylgermanes 1–8 recorded in acetonitrile. | ||
Photobleaching behavior
Efficient bleaching of the photoinitiator is an essential requirement to achieve high curing depth and to avoid colored polymers. Fast photobleaching at irradiation wavelengths in the visible range is particularly desirable for applications such as dental fillings. We have tested the photobleaching of 1–8via steady-state-photolysis (SSP) at 385 nm and 470 nm. The LED operated at 385 nm was chosen as an example of shorter-wavelength irradiation, whereas LED light at 470 nm corresponds to the longest-wavelength emission band of typical dental lamps.24Photobleaching experiments were performed in the presence of methyl methacrylate (MMA) monomer. The monomer acts as a radical trap, leading to the formation of photoproducts which do not absorb in the visible wavelength range. Further, samples were saturated with argon before the measurements to avoid side reactions of the excited photoinitiators and generated radicals with oxygen.
Fig. 3 depicts time-decays of the absorbance of mono- to tetraacylgermanes followed at the n–π* absorption maxima upon irradiation with LEDs at 385 nm and 470 nm. For a relative (qualitative) comparison of the photobleaching efficiency, these plots were normalized. The corresponding UV-Vis spectra are provided in the ESI.† Photobleaching is more efficient upon irradiation with LED light at 385 nm than at 470 nm for all investigated compounds. Monoacylgermane 1 shows the least efficient bleaching at both wavelengths. Additionally, trisacylgermane 4 and tetraacylgermane 7 display rather inefficient photobleaching at 470 nm, despite their good bleaching performance at 385 nm. This might be due to low absorbance of these compounds at 470 nm (see the UV-Vis spectra, Fig. 2). In contrast, tetraacylgermane 8 exhibits remarkably fast photobleaching upon irradiation at 470 nm, indicating efficient initiation under high-wavelength visible light (Fig. 3b).
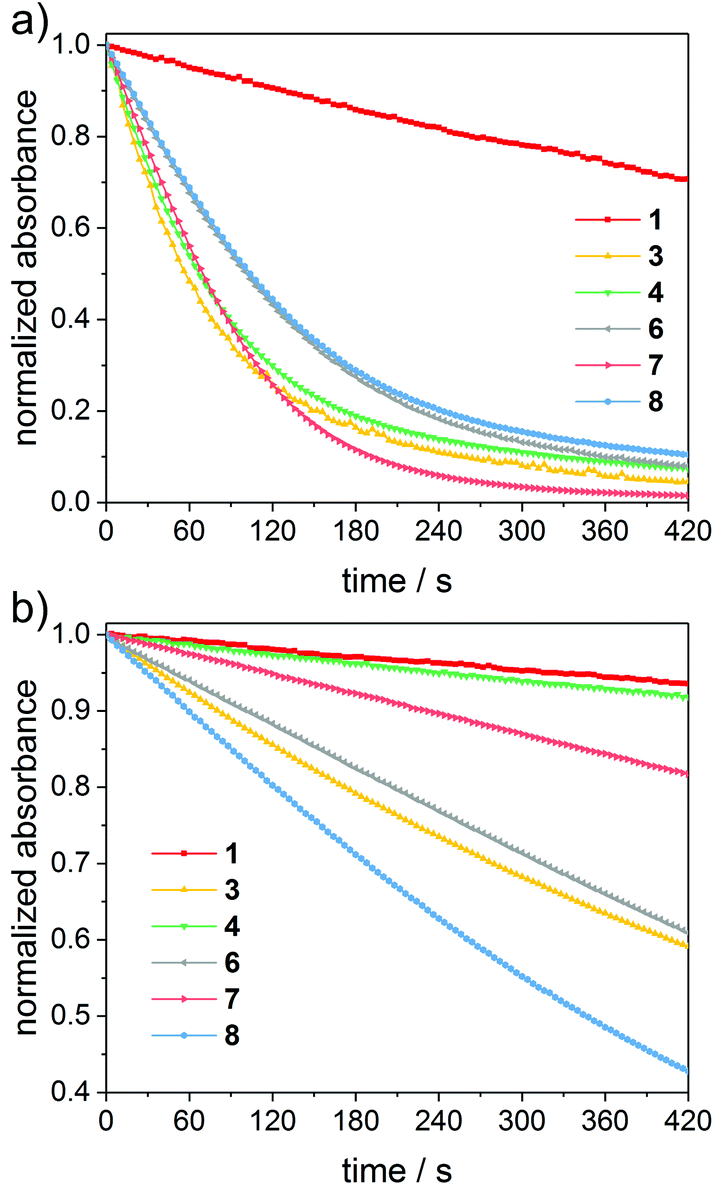 | ||
| Fig. 3 Steady-state photolysis of mono-, bis-, tris- and tetraacylgermanes in acetonitrile/MMA (1/1 v/v). Plots of normalized absorbance versus time for irradiation at (a) 385 nm and (b) 470 nm, monitored at the n–π* absorption maxima (1: 412.5 nm, 3: 409 nm, 4: 383 nm, 6: 404 nm, 7: 393 nm, 8: 405.5 nm). | ||
To further rationalize the wavelength-dependent photobleaching efficiency, we have utilized the Beer–Lambert law to calculate the amount of absorbed light and the penetration depth of the incident light at conditions similar to the SSP experiments. Plots of the absorbed light at an optical path length l of 1 cm as well as the path length at which 90% of the incident light is absorbed (penetration depth, l90%) are presented in Fig. 4 for compounds 1 and 8 (1 mM solutions). These calculations have been performed at wavelengths of 385 nm, 410 nm, 430 nm and 470 nm (see the ESI† for the data for compounds 3–7). Fig. 4 reflects the vast differences in the absorption properties between compounds 1 and 8. The significantly lower molar extinction coefficients ε of 1 compared to 8 result in much lower amounts of absorbed light for 1. The penetration depth of the incident light is inversely related to the amount of absorbed light, leading to a penetration depth approximately ten times higher for compound 1 than for 8 at 470 nm. Accordingly, the amount of absorbed light for irradiation at 470 nm is substantially higher for compound 8 than for 1, in line with the significantly more efficient photobleaching of 8.
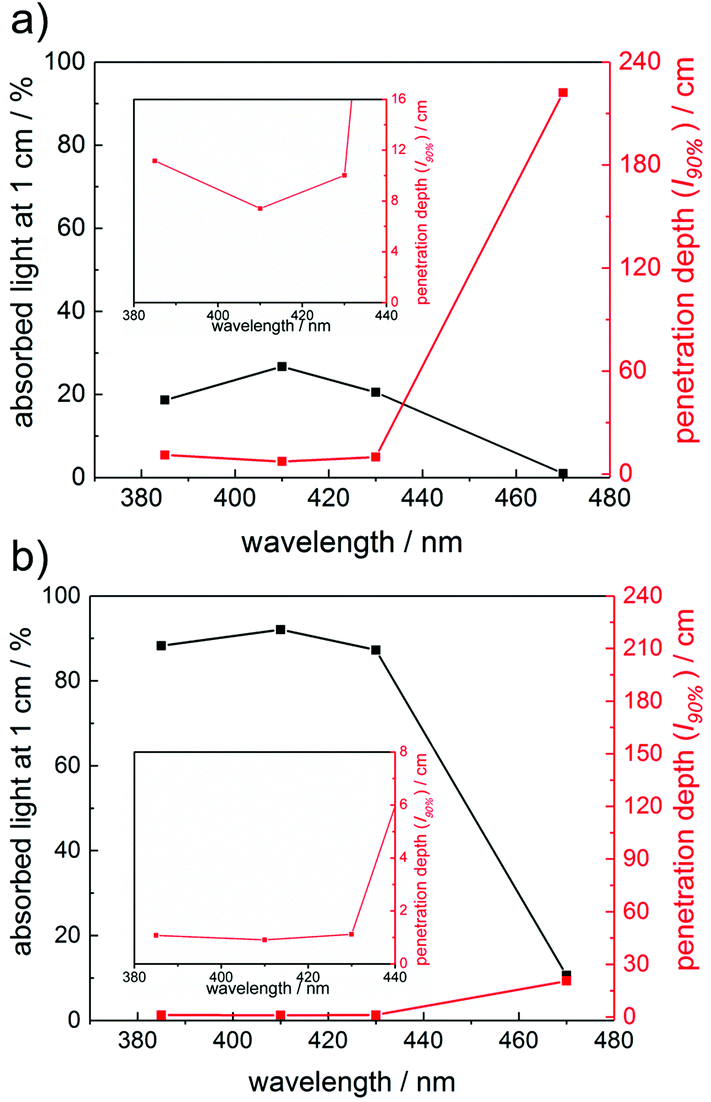 | ||
| Fig. 4 Plots of absorbed light at a path length l of 1 cm and calculated penetration depth l90% for 1 mM solutions of (a) compound 1 and (b) compound 8. The insets show a zoom into the lower penetration depth area. Lines are to guide the eye. | ||
For practical applications, a good match between the absorption maxima and the applied irradiation wavelengths (corresponding to a high amount of absorbed light), as well as a high penetration depth of the incident light are desired, especially when curing thick layers.25–27 The photobleaching effect leads to increasing penetration depths in the course of the polymerization. Therefore, photoinitiators with optimal bleaching efficiency at the irradiation wavelengths are beneficial, especially in cases where high curing depths are required. Further, high initial absorbance is advantageous, since this increases the probability of generating a sufficient amount of primary radicals, which can effectively start the polymerization chain reactions, preventing that oxygen inhibition becomes dominant.28 High initial absorbance can be achieved either by increasing the PI concentration and/or by choosing a PI with high extinction coefficients. Since the concentration of the photoinitiator in a formulation should be kept low (for reasons of reduced costs etc.), PIs with high extinction coefficients are particularly desired. These considerations suggest that tetraacylgermanes (highest extinction coefficients, good bleaching behavior) might be most suitable among the studied compounds for several curing applications.
Quantum yields of decomposition
Fig. 3 shows a relative comparison of the photobleaching behavior (the initial absorbance was normalized to 1 for all compounds). This represents a qualitative description of the photobleaching efficiency, but does not consider the differences in the extinction coefficients at the irradiation wavelengths. A quantitative evaluation has been achieved by measuring the photodecomposition quantum yields via on-line UV-Vis spectroscopy.22As described for the photobleaching experiments above, we have determined these quantum yields in a solvent mixture of acetonitrile and MMA (1/1 v/v ratio), in order to evaluate the performance of the photoinitiators at conditions similar to the application in polymerization formulations. Furthermore, by adding the monomer as a radical trap, absorbing photoproducts in the n–π* region of the parent initiators, which may interfere with the evaluation, are avoided. As a light source, we have chosen an LED operated at 385 nm, since all compounds show sufficient absorbance at this wavelength, allowing for a precise evaluation. For good comparability, all samples were prepared to achieve equal initial light absorbance at the irradiation wavelength (A385 ∼ 0.6, LED385 operated at 20 mA forward current). To account for solvent effects, the extinction coefficients of the investigated photoinitiators have been re-determined in the solvent mixture acetonitrile/MMA in order to allow exact determination of concentration changes.
Fig. 5 presents the kinetic trace (concentration versus time) obtained upon photolysis of bis(acyl)germane 3 at 385 nm, monitored at the n–π* absorption maximum (409.5 nm, ε = 763 L mol−1 cm−1, see the inset of Fig. 5 for the corresponding UV-Vis spectra). As described in the Experimental section, the initial reaction rate k is obtained from the first derivative of the exponential fit of this trace.23 The quantum yield Φ is calculated via dividing −k by the photon flux I0 (1.89 × 10−5 mol L−1 s−1) and the factor f (determined from the initial absorbance at 385 nm, f = 0.73).
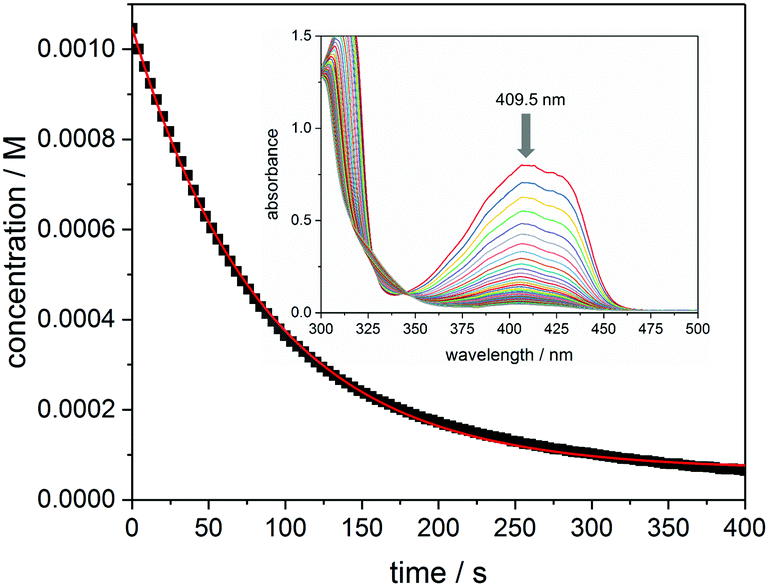 | ||
| Fig. 5 Decay of concentration versus irradiation time for compound 3, monitored at 409.5 nm, together with the exponential fit of the data (red line). The inset shows the corresponding UV-Vis spectra recorded upon irradiation at 385 nm (time steps between each spectrum: 12 s, sample: 1.1 mM 3 in acetonitrile/MMA (1/1 v/v)). | ||
The experimental data for compounds 1–2, 6–8 and 10 are presented in the ESI.†Table 1 summarizes the monitoring wavelengths, extinction coefficients, reaction rates k and the obtained decomposition quantum yields Φ for all investigated compounds and provides a comparison with previously published values, if available.
| Compound | λ/nm (ε/M−1 cm−1) | k/10−6 (M s−1) | Φ (measured) | Φ (published) |
|---|---|---|---|---|
| a Our work, see ref. 22. | ||||
| 1 | 413 (136) | −2.91 | 0.21 ± 0.02 | 0.403 |
| 2 | 420 (493) | −11.59 | 0.83 ± 0.01a | 0.853 |
| 3 | 409.5 (763) | −11.84 | 0.83 ± 0.01 | — |
| 6 | 403.5 (1224) | −5.50 | 0.38 ± 0.01 | — |
| 7 | 394 (1902) | −6.57 | 0.44 ± 0.02 | — |
| 8 | 405.5 (1370) | −4.72 | 0.34 ± 0.01 | — |
| 10 | 370 (996) | −8.95 | 0.64 ± 0.04a | 0.69 |
The decomposition quantum yield obtained for bisacylphosphane oxide 10 (Φ = 0.64 ± 0.04) is in good agreement with the published value of 0.6.9,10 Moreover, for bisacylgermane 2 excellent agreement is obtained with the value determined previously via HPLC analysis (Φmeasured = 0.83, Φpublished = 0.85).3 In contrast, our value for compound 1 (Φmeasured = 0.21) is lower than the published quantum yield (Φpublished = 0.40).3
In general, bisacylgermanes 2 and 3 exhibit higher quantum yields of decomposition than the tetraacylgermanes 6–8. This might, however, be compensated by the capability of tetraacylgermanes to undergo four subsequent photocleavage steps to produce in total four benzoyl radicals in a stepwise procedure.
Additionally, apart from the decomposition quantum yields, other factors contribute to the photoinitiating efficiency as well, including the match of the UV-Vis absorption spectra with the available light sources, the photobleaching efficiency (see also the discussion in the previous section), the reactivity of the generated radicals to monomers, and the probability of undergoing side-reactions, such as quenching by oxygen.
Time-resolved EPR spectra of primary radicals
Time-resolved EPR spectroscopy allows the simultaneous observation of both radicals produced upon photolysis of acylgermanes 1–8 within short time scales (50 ns time regime).6,7,13,29 As the light source, a laser operated at 355 nm was used. This wavelength lies within the n–π* bands of the photoinitiators, as well as the wavelengths chosen for the photobleaching experiments (LED light up to 470 nm). Therefore, the photochemical reaction pathways are expected to be analogous for all light sources applied in this study (triplet-state α-cleavage). Performing photolysis experiments using tunable laser sources could however provide additional insights into bond cleavage efficiencies. This will be subject of future work.Laser-flash photolysis of bisacylgermane 3 affords the TR-EPR spectrum shown in Fig. 6a. The spectrum is dominated by a quintet attributed to the germyl radical G(3)˙ (g = 2.006, hyperfine coupling constant (hfc) = 0.5 mT). The quintet (intensity ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:6:4:1) stems from the four equivalent methylene protons of the ethyl substituents at the Ge-center (β hydrogen atoms).6 This signal partly overlaps with the unresolved signal of the benzoyl-type radical B(3)˙ (g = 2.000). Fig. 6b depicts the TR-EPR spectrum of trisacylgermane 4. Due to the two β hydrogen atoms, radical G(4)˙ exhibits a triplet (intensity ratio 1:2:1, g = 2.002, hfc = 0.44 mT), which partly overlaps with the signal of radical B(4)˙ (g = 2.000).
:4:6:4:1) stems from the four equivalent methylene protons of the ethyl substituents at the Ge-center (β hydrogen atoms).6 This signal partly overlaps with the unresolved signal of the benzoyl-type radical B(3)˙ (g = 2.000). Fig. 6b depicts the TR-EPR spectrum of trisacylgermane 4. Due to the two β hydrogen atoms, radical G(4)˙ exhibits a triplet (intensity ratio 1:2:1, g = 2.002, hfc = 0.44 mT), which partly overlaps with the signal of radical B(4)˙ (g = 2.000).
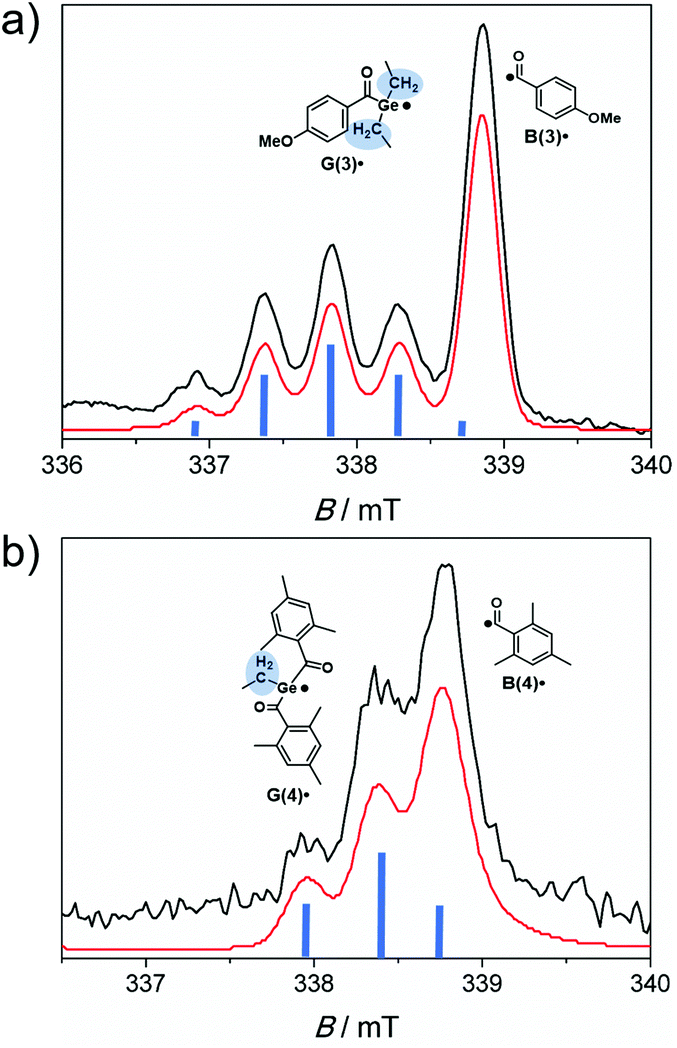 | ||
| Fig. 6 TR-EPR spectra recorded 300–400 ns after laser-flash photolysis (355 nm) of argon-saturated toluene solutions (10 mM) of (a) 3 and (b) 4, together with their simulations (red lines). Blue bars indicate the splitting of the germyl radical signals due to β hydrogen atoms. | ||
Similarly, the TR-EPR spectra of tetraacylgermane derivatives show two signals in absorption, being assigned to the germyl radicals G˙ and the benzoyl-type radicals B˙ (for the experimental spectra the reader is referred to ref. 7 and 8). The TR-EPR spectrum of monoacylgermane 1 could not be obtained. This might be due to unfavorable triplet-polarization effects (magnetic field effects) and/or to inefficient α-cleavage, resulting in a radical concentration below the detection limit.29
Germanium-centered radicals and their reactivity
Laser-flash photolysis (LFP) is ideally suited for studying short lived species and their kinetics. A detailed overview is given by J. C. Scaiano.30 Transient optical absorption spectra obtained upon LFP (355 nm excitation) of acylgermanes 2–8 reveal distinct absorption bands between 470 and 530 nm (see Fig. 7a and the ESI†). These bands are attributed to the germyl radicals G(2–8)˙ (see Scheme 1).6,31 Benzoyl-derived radicals do not reveal distinct absorptions in the wavelength range of our LFP experiments, and can only be observed by time-resolved IR and EPR spectroscopy.12,32 In the case of monoacylgermane 1, the Ge-centered radical was not distinguishable, which might be due to unfavorable absorption properties of the Me3Ge˙ radical (G(1)˙). Both Ge-centered and benzoyl-type radicals have been shown to add to the double bonds of monomers, the Ge-centered radicals being ca. two to three orders of magnitude faster.6,31,32 The distinct transient absorptions of the germyl radicals G(2–8)˙ allow to determine the rate constants for their addition to various monomers via pseudo-first-order analysis. Exponential fitting of the decay traces obtained at various quencher concentrations yielded the pseudo-first-order rate constants kexp. The second order addition rate constants (kmonomer) are obtained from the slopes of their linear dependence on the monomer concentration cmonomer (see eqn (1)).6,9| kexp = k0 + kmonomer·cmonomer | (1) |
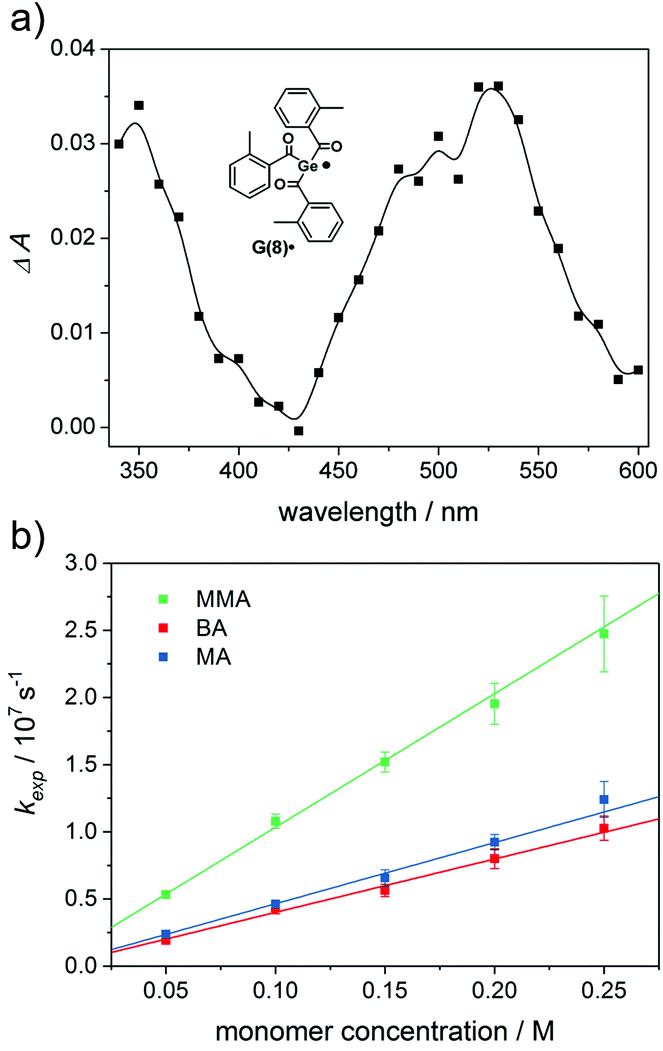 | ||
| Fig. 7 (a) Transient optical absorption spectra (absorbance change ΔA versus wavelength) of radical G(8)˙ recorded 200–300 ns after laser excitation (355 nm, 8 ns) of 8 in argon-saturated acetonitrile solutions (A355 ∼ 0.3). (b) LFP of 8 in the presence of monomers: pseudo-first-order decay rate constant (kexp) of radical G(8)˙versus monomer concentration (excitation wavelength: 355 nm, monitoring wavelength: 530 nm). | ||
In eqn (1), k0 represents the estimated rate constant for the decay of the radicals in the absence of a quencher.
We have investigated the reactivity of G(2–8)˙ toward the monomers butyl acrylate (BA), methyl acrylate (MA) and methyl methacrylate (MMA) (Chart 2). The corresponding curves for radical G(8)˙ are shown in Fig. 7b (for G(2–7)˙ see the ESI†).
 | ||
| Chart 2 Chemical structures of the investigated monomers. | ||
The addition rate constants for G(2–8)˙ are summarized in Fig. 8 and Table 2. Generally, all Ge-centered radicals show higher reactivity toward methyl methacrylate than toward butyl acrylate and methyl acrylate. The higher reactivity toward methacrylates than to acrylates suggests that Ge-centered radicals prefer to react with electron rich double bonds. Analogous results have been obtained with phosphorus-centered radicals derived from bisacylphosphane oxide initiators.33
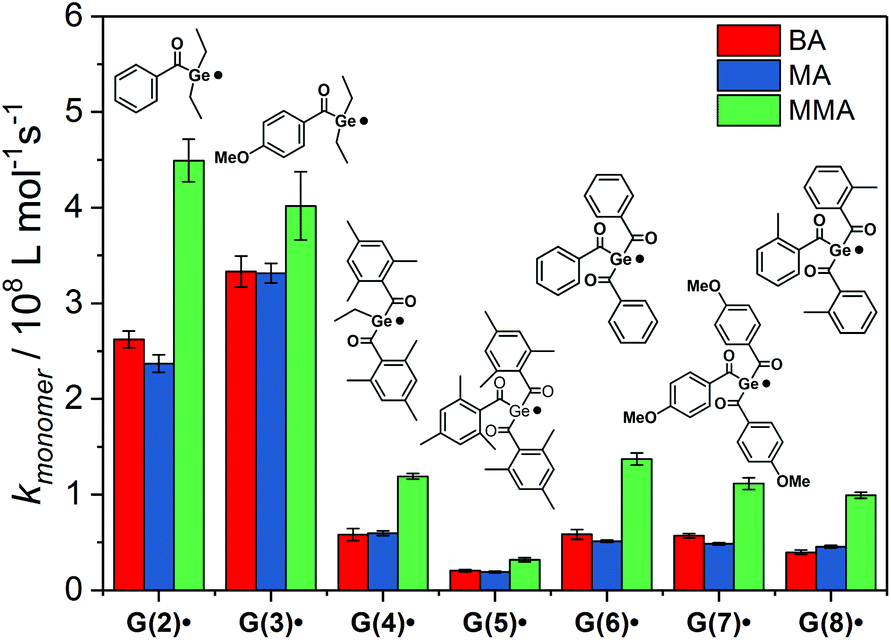 | ||
| Fig. 8 Second-order rate constants kmonomer for the addition of G(2–8)˙ to butyl acrylate (BA), methyl acrylate (MA) and methyl methacrylate (MMA). | ||
| k monomer/108 M−1 s−1 | |||
|---|---|---|---|
| Radical | BA | MA | MMA |
| a Value taken from ref. 9. | |||
| G(1)˙ | — | — | — |
| G(2)˙ | 2.6 ± 0.1 | 2.4 ± 0.1 | 4.5 ± 0.3 |
| G(3)˙ | 3.2 ± 0.2 | 3.3 ± 0.1 | 4.0 ± 0.4 |
| G(4)˙ | 0.58 ± 0.06 | 0.59 ± 0.02 | 1.2 ± 0.05 |
| G(5)˙ | 0.20 ± 0.01 | 0.19 ± 0.01 | 0.32 ± 0.02 |
| G(6)˙ | 0.59 ± 0.05 | 0.51 ± 0.01 | 1.4 ± 0.07 |
| G(7)˙ | 0.57 ± 0.03 | 0.48 ± 0.02 | 1.1 ± 0.07 |
| G(8)˙ | 0.40 ± 0.01 | 0.45 ± 0.02 | 0.99 ± 0.03 |
| P(9)˙ | 0.32 ± 0.03 | — | — |
| 0.28a | |||
| P(10)˙ | 0.12 ± 0.04 | — | — |
| 0.11a | |||
Addition rate constants toward butyl acrylate are higher for G(2–8)˙ than for phosphorus-centered radicals P(9)˙ and P(10)˙ (see Table 2).9,33Table 2 shows excellent agreement of our measured rate constant with the published value for radical P(10)˙. For radical P(9)˙, our rate constant is slightly higher than the value reported by Turro and coworkers.9
Germyl radicals derived from bisacylgermanes 2 and 3 display three to four times higher reactivity toward monomer double bonds than corresponding radicals derived from tris- and tetraacylgermanes 4–8. Steric congestion by the bulky aromatic substituents around the radical center might be the reason for the lower reactivity of G(4–8)˙ when compared to G(2–3)˙. In derivative G(5)˙, where the radical center is substantially shielded by three mesitoyl groups, the addition rate constants are significantly lower than for the less sterically congested radicals G(6–8)˙. The influence of steric factors is further evident by comparing the addition rate constants of G(4)˙ and G(5)˙. Radical G(4)˙, stemming from ethyltrismesitoylgermane 4 adds ca. three times faster to monomers than G(5)˙, stemming from the more bulky tetramesitoylgermane 5. Apart from the less reactive mesitoyl derivative (5), all derivatives within the group of tetraacylgermanes show similar reactivity toward monomers. Thus, substituents introduced on the aromatic rings do not interfere with the photoinitiating reactivity, while allowing to specifically tune solubility or absorption properties.
Radical reaction pathways
Photo-induced α-cleavage of 1–8 and the corresponding follow-up reactions can be conveniently followed by using 1H CIDNP spectroscopy.6,16,29,34 This NMR-based method reveals products formed exclusively via radical pairs (B˙/G˙). Fig. 9 compares the 1H NMR and CIDNP spectra of 2 recorded in presence of butyl acrylate (BA). The signals of the methylene protons adjacent to the germanium center (marked with d) appear in strong enhanced absorption in the CIDNP spectrum due to a geminate (or cage) re-formation of 2 from G˙ and B˙. The emissive quartet at δ = 3.03 ppm (J = 7.2 Hz) and the absorptive triplet at δ = 1.14 ppm (J = 7.2 Hz) are assigned to propiophenone P. This assignment is substantiated by an external reference NMR measurement of propiophenone, yielding the above-mentioned chemical shifts and J-couplings. We suggest that P is formed via recombination of B˙ and an ethyl radical Et˙, which is produced in a second cleavage step at the stage of G˙ (Scheme 3). The singlet at δ = 10.01 ppm appearing in enhanced absorption corresponds to the aldehyde proton of the benzaldehyde derivative BH. Analogous signals have been observed in the CIDNP spectra of several photoinitiating systems containing a benzoyl moiety.35 The formation of BH is usually attributed to a disproportionation reaction (β-hydrogen transfer) of a benzoyl-type radical and radicals, which are able to donate hydrogen atoms.6,35 In the absence of monomer, hydrogen atoms can be transferred from the ethyl group of G˙, resulting in a weak aldehyde signal as described previously.6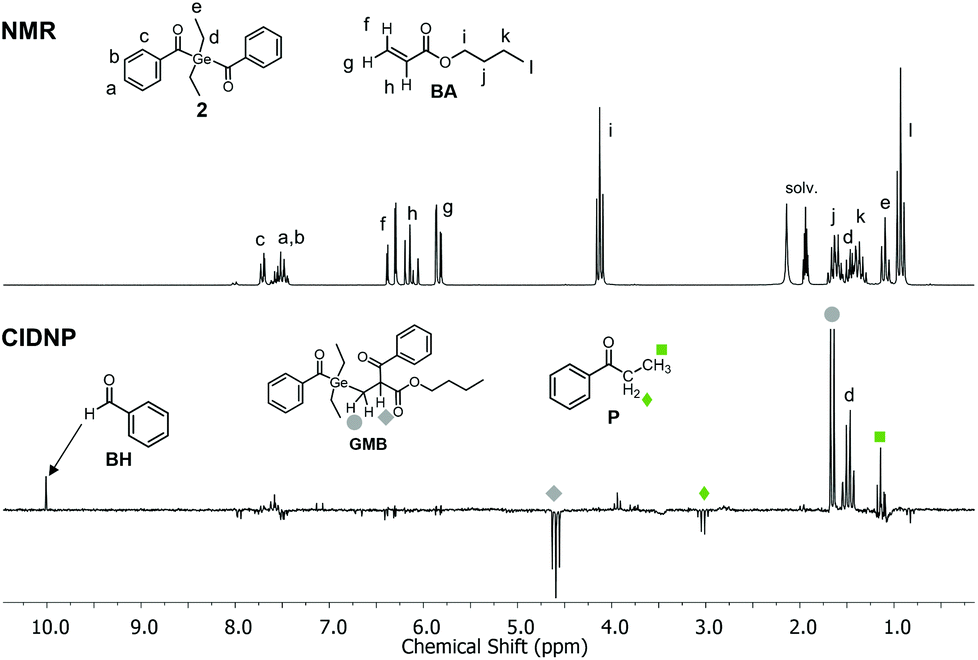 | ||
| Fig. 9 1H NMR and CIDNP spectra (excitation at λ = 355 nm, ca. 70 mJ per pulse) of 2 in the presence of butyl acrylate BA (10 mM 2, 50 mM BA in acetonitrile-d3). | ||
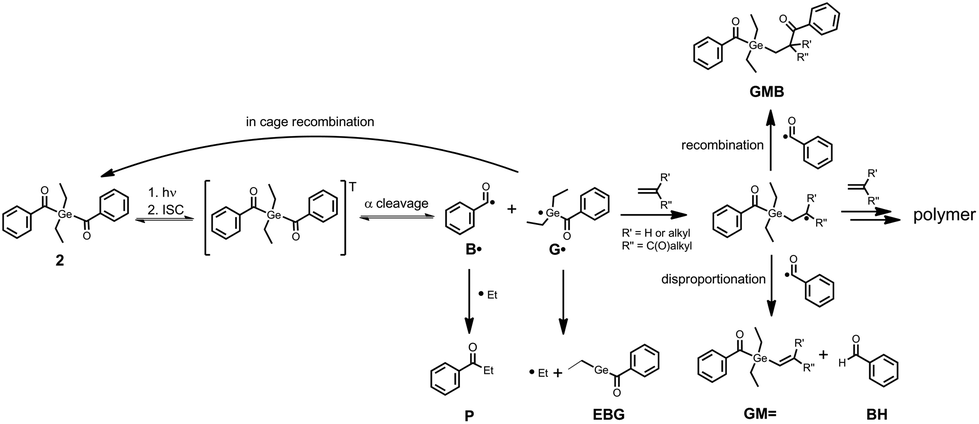 | ||
| Scheme 3 Proposed reaction scheme of initiator 2 in the presence of monomers. | ||
In the presence of a monomer, the aldehyde signal becomes significantly more pronounced since radicals from the growing polymer chain can serve as additional hydrogen donors, resulting in the formation of BH and chain termination products (i.e. species GM=, Scheme 3). Fig. 9 additionally shows two strongly polarized signals consisting of an emissive triplet at δ = 4.59 ppm (J = 7.7 Hz) and an absorptive doublet at δ = 1.65 ppm (J = 7.7 Hz). These signals can potentially be assigned to species GMB, which is formed upon addition of benzoyl radical B˙ to a chain radical initiated by G˙ (Scheme 3). Photoinitiators 3–8 show analogous reactivity toward monomers, whereas aldehyde formation and radical-to-monomer addition products were not observed with monoacylgermane 1, suggesting inefficient initiation (see the ESI†).
Conclusions
In this contribution, we have presented a detailed investigation of the photochemistry and photoinitiating properties of a series of mono-, to tetraacylgermanes. We have additionally reported on the synthesis and characterization of a trisacylgermane photoinitiator.TR-EPR spectroscopy confirms the formation of germyl and benzoyl-type radicals (G˙ and B˙) upon triplet-state α-cleavage of acylgermanes 1–8. The reaction patterns of the primary radicals G˙ and B˙ in the presence of monomers have been established using CIDNP spectroscopy.
Tetraacylgermanes exhibit higher molar extinction coefficients than mono-, bis- and trisacylgermanes, especially in the long-wavelength region. This is particularly relevant for the application as visible light photoinitiators. Substituents on the aromatic rings lead to a shift of the n–π* absorption bands, with the o-tolyl derivative 8 showing the most red-shifted UV-Vis spectrum of all studied compounds. While shifting the absorption bands, substituents do not markedly influence the reactivity of the germanium-centered radicals toward monomers, as shown using laser-flash photolysis experiments. The reactivity of these radicals toward monomer double bonds is mostly influenced by the type of acylgermane. Germyl radicals derived from bisacylgermanes add faster to monomer double bonds than germyl radicals derived from tetraacylgermanes, both compound classes however being more reactive than related phosphorus-centered radicals.
Photobleaching experiments demonstrate the wavelength-dependent photobleaching behavior of 1–8, with compound 8 displaying the most efficient bleaching upon irradiation with high-wavelength visible light (470 nm LED). Concerning quantum yields of decomposition, bisacylgermanes have been found to be most efficient. We have further discussed the interplay of several factors contributing to the efficiency of photoinitiators, including decomposition quantum yields, absorption spectra and their match with the desired irradiation wavelengths, photobleaching efficiency, amount and reactivity of generated radicals, side reactions, solubility and toxicity.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from Ivoclar Vivadent AG and NAWI Graz. A. E. thanks Dr Dmytro Neshchadin (TU Graz, Institute of Physical and Theoretical Chemistry) for valuable discussions.References
- N. Moszner, F. Zeuner, I. Lamparth and U. K. Fischer, Macromol. Mater. Eng., 2009, 294, 877–886 CrossRef CAS.
- N. Moszner, U. K. Fischer, B. Ganster, R. Liska and V. Rheinberger, Dent. Mater., 2008, 24, 901–907 CrossRef CAS PubMed.
- B. Ganster, U. K. Fischer, N. Moszner and R. Liska, Macromolecules, 2008, 41, 2394–2400 CrossRef CAS.
- Y. Catel, U. Fischer, P. Fässler and N. Moszner, Macromol. Chem. Phys., 2016, 1–6 Search PubMed.
- N. Moszner, I. Lamparth, J. Angermann, U. K. Fischer, F. Zeuner, T. Bock, R. Liska and V. Rheinberger, Beilstein J. Org. Chem., 2010, 6, 1–9 Search PubMed.
- D. Neshchadin, A. Rosspeintner, M. Griesser, B. Lang, S. Mosquera-Vazquez, E. Vauthey, V. Gorelik, R. Liska, C. Hametner, B. Ganster, R. Saf, N. Moszner and G. Gescheidt, J. Am. Chem. Soc., 2013, 135, 17314–17321 CrossRef CAS PubMed.
- J. Radebner, A. Eibel, M. Leypold, C. Gorsche, L. Schuh, R. Fischer, A. Torvisco, D. Neshchadin, R. Geier, N. Moszner, R. Liska, G. Gescheidt, M. Haas and H. Stueger, Angew. Chem., Int. Ed., 2017, 56, 3103–3107 CrossRef CAS PubMed.
- J. Radebner, M. Leypold, A. Eibel, J. Maier, L. Schuh, A. Torvisco, R. Fischer, N. Moszner, G. Gescheidt, H. Stueger and M. Haas, Organometallics, 2017, 36, 3624–3632 CrossRef CAS.
- S. Jockusch and N. J. Turro, J. Am. Chem. Soc., 1998, 120, 11773–11777 CrossRef CAS.
- S. Jockusch, I. V. Koptyug, P. F. McGarry, G. W. Sluggett, N. J. Turro and D. M. Watkins, J. Am. Chem. Soc., 1997, 119, 11495–11501 CrossRef CAS.
- G. W. Sluggett, P. F. McGarry, I. V. Koptyug and N. J. Turro, J. Am. Chem. Soc., 1996, 118, 7367–7372 CrossRef CAS.
- G. W. Sluggett, C. Turro, M. W. George, I. V. Koptyug and N. J. Turro, J. Am. Chem. Soc., 1995, 117, 5148–5153 CrossRef CAS.
- I. Gatlik, P. Rzadek, G. Gescheidt, G. Rist, B. Hellrung, J. Wirz, K. Dietliker, G. Hug, M. Kunz and J.-P. Wolf, J. Am. Chem. Soc., 1999, 121, 8332–8336 CrossRef CAS.
- G. Müller, M. Zalibera, G. Gescheidt, A. Rosenthal, G. Santiso-Quinones, K. Dietliker and H. Grützmacher, Macromol. Rapid Commun., 2015, 36, 553–557 CrossRef PubMed.
- D. E. Fast, M. Zalibera, A. Lauer, A. Eibel, C. Schweigert, A.-M. Kelterer, M. Spichty, D. Neshchadin, D. Voll, H. Ernst, Y. Liang, K. Dietliker, A.-N. Unterreiner, C. Barner-Kowollik, H. Grützmacher and G. Gescheidt, Chem. Commun., 2016, 9917–9920 RSC.
- M. Griesser, D. Neshchadin, K. Dietliker, N. Moszner, R. Liska and G. Gescheidt, Angew. Chem., Int. Ed., 2009, 48, 9359–9361 CrossRef CAS PubMed.
- A. Castel, P. Rivière, J. Satgé and D. Desor, J. Organomet. Chem., 1992, 433, 49–61 CrossRef CAS.
- A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518–1520 CrossRef CAS.
- A. G. Brook, F. Abdesaken and H. Söllradl, J. Organomet. Chem., 1986, 299, 9–13 CrossRef CAS.
- J. Fischer, J. Baumgartner and C. Marschner, Organometallics, 2005, 24, 1263–1268 CrossRef CAS.
- L. R. Subramanian and G. Siegemund, in Houben-Weyl Methods of Organic Chemistry Supplement: Organo-Fluorine Compounds Fluorinating Agents and Their Application in Organic Synthesis, Thieme, Stuttgart, 1998 Search PubMed.
- E. Stadler, A. Eibel, D. E. Fast, H. Freißmuth, C. Holly, M. Wiech, N. Moszner and G. Gescheidt, Photochem. Photobiol. Sci. Search PubMed , under revision.
- K. L. Willett and R. A. Hites, J. Chem. Educ., 2000, 77, 900–902 CrossRef CAS.
- Scientific documentation bluephase® family – LED for every use, Ivoclar Vivadent®.
- K. Dietliker, T. Jung, J. Benkhoff, H. Kura, A. Matsumoto, H. Oka, D. Hristova, G. Gescheidt and G. Rist, Macromol. Symp., 2004, 217, 77–98 CrossRef CAS.
- D. E. Fast, A. Lauer, J. P. Menzel, A. Kelterer, G. Gescheidt and C. Barner-Kowollik, Macromolecules, 2017, 50, 1815–1823 CrossRef CAS.
- A. Lauer, D. E. Fast, J. Steinkoenig, A.-M. Kelterer, G. Gescheidt and C. Barner-Kowollik, ACS Macro Lett., 2017, 952–958 CrossRef CAS.
- S. Ligon, B. Husár, H. Wutzel, R. Holman and R. Liska, Chem. Rev., 2014, 557–589 CrossRef CAS PubMed.
- A. Yurkovskaya, O. Morozova and G. Gescheidt, in Encyclopedia of Radicals in Chemistry, Biology and Materials, John Wiley & Sons, Ltd, Chichester, UK, 2012 Search PubMed.
- J. C. Scaiano, in Reactive Intermediate Chemistry, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2005, pp. 847–871 Search PubMed.
- J. Lalevée, X. Allonas and J. P. Fouassier, Chem. Phys. Lett., 2009, 469, 298–303 CrossRef.
- C. S. Colley, D. C. Grills, N. A. Besley, S. Jockusch, P. Matousek, A. W. Parker, M. Towrie, N. J. Turro, P. M. W. Gill and M. W. George, J. Am. Chem. Soc., 2002, 124, 14952–14958 CrossRef CAS PubMed.
- A. Eibel, M. Schmallegger, M. Zalibera, A. Huber, Y. Bürkl, H. Grützmacher and G. Gescheidt, Eur. J. Inorg. Chem., 2017, 2017, 2469–2478 CrossRef CAS.
- U. Kolczak, G. Rist, K. Dietliker and J. Wirz, J. Am. Chem. Soc., 1996, 118, 6477–6489 CrossRef CAS.
- A. Borer, R. Kirchmayr and G. Rist, Helv. Chim. Acta, 1978, 61, 305–324 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1561720. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7py01590a |
| This journal is © The Royal Society of Chemistry 2018 |