
Vinylogous acyl triflates as an entry point to α,β-disubstituted cyclic enones via Suzuki–Miyaura cross-coupling†

Abstract
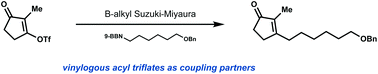
An alternative protocol for the B-alkyl Suzuki–Miyaura reaction to produce cyclic α,β-disubstituted enones is reported. The use of β-triflyl enones as coupling partners in lieu of their halogenated analogs provides enhanced substrate stability to light and chromatography without adversely affecting reactivity. This protocol allows efficient access to the synthetically challenging α,β-disubstituted enone motif under mild conditions.
- This article is part of the themed collections: Synthetic methodology in OBC and New Talent
 Please wait while we load your content...
Please wait while we load your content...

