
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigating the breakdown of the nerve agent simulant methyl paraoxon and chemical warfare agents GB and VX using nitrogen containing bases†
Craig
Wilson
a,
Nicholas J.
Cooper
b,
Michael E.
Briggs
 a,
Andrew I.
Cooper
*a and
Dave J.
Adams
*c
a,
Andrew I.
Cooper
*a and
Dave J.
Adams
*c
aMaterials Innovation Factory and Department of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK
bDstl, Porton Down, Salisbury, SP4 0JQ, Wiltshire, UK
cSchool of Chemistry, College of Science and Engineering, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: dave.adams@glasgow.ac.uk
First published on 21st November 2018
Abstract
A range of nitrogen containing bases was tested for the hydrolysis of a nerve agent simulant, methyl paraoxon (MP), and the chemical warfare agents, GB and VX. The product distribution was found to be highly dependant on the basicity of the base and the quantity of water used for the hydrolysis. This study is important in the design of decontamination technology, which often involve mimics of CWAs.
Introduction
Chemical warfare agents (CWAs) have a devastating effect on the body and will disable or kill on exposure. Blister agents, such as sulfur mustard (HD, bis(2-chloroethyl) sulfide), target the skin and respiratory system causing severe pain and damage to the body whereas nerve agents, such as sarin (GB, isopropyl methylphosphonofluoridate) and VX (O-ethyl S-[2-(diisopropylamino)ethyl] methylphosphonothioate), target the central nervous system resulting in death through asphyxiation.1–5 In the modern era, the first large-scale use of CWAs occurred during the 1st World War with the widespread use of chlorine and mustard gas.6 The continuing use of CWAs throughout the world today, highlights the importance and need to develop effective decontamination systems that are applicable to a wide range of CWAs as rapid identification of the CWA is not always possible.7Depending on the type of CWA, deactivation can proceed via a number of pathways (Scheme S1†).6 The most common deactivation pathway for the G-type nerve agents such as sarin (GB) and tabun (GA) (Fig. 1a & b), is via hydrolysis, resulting in cleavage of the labile P–F or P–CN bond and a substantial reduction in toxicity.8,9 Although hydrolysis can also occur with the V-type nerve agents, these often contain multiple hydrolytically labile bonds and care must be taken to ensure the desired bond is cleaved.10,11 In the case of VX (Fig. 1c), hydrolysis of the P–O bond instead of the P–S bond, leads to production of EA-2192 (S-(2-diisopropylaminoethyl) methylphosphonothioic acid), which exhibits roughly the same toxicity as VX itself.12 The blister agent HD (Fig. 1d) can also undergo deactivation via hydrolysis.13 However, its poor water solubility reduces the efficiency of this decontamination method.14 As a result, oxidation to the sulfoxide, or the addition of a co-solvent is most commonly used for HD deactivation.15–17
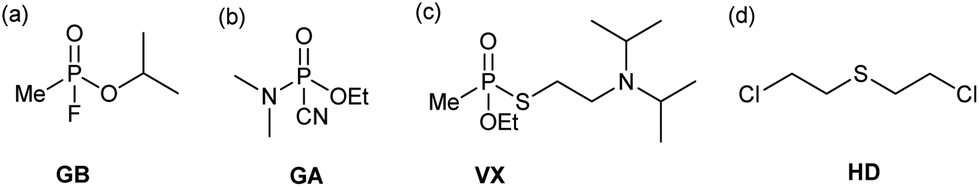 | ||
| Fig. 1 Chemical structures for chemical warfare agents (CWAs); (a) sarin (GB), (b) tabun (GA), (c) VX, and (d) sulfur mustard (HD). | ||
The high toxicity of CWAs means that research into their deactivation is often carried out using simulants. Paraoxon, the active metabolite of the insecticide parathion, is commonly used as a simulant for G-type nerve agents. It retains a similar mode of action to the CWAs and targets the central nervous system.18,19 Three main pathways have been proposed for the breakdown of paraoxon, and similar simulants such as fenitrothion, upon reaction with a range of oxygen and nitrogen containing nucleophiles or α-nucleophiles, and surfactants.20–23 A mechanistic study of the reaction of paraoxon with piperidine, found nucleophilic attack can occur at the phosphorus centre resulting in P–OAr cleavage (SN2(P)), the aliphatic carbon with C–O cleavage (SN2(C)), and/or the aromatic group with Ar–O cleavage (SNAr) (Scheme S2†).24 The product distribution, and hence the toxicity of the decomposition products, depends strongly on the conditions of the reaction, with the C–O bond cleavage pathway resulting in the formation of the toxic breakdown product, ethyl 4-nitrophenyl phosphate.25 The ethyl 4-nitrophenyl phosphate breakdown product is likely to display the same mode of action as paraoxon and CWAs upon the enzyme acetylcholinesterase,26 due to the continued presence of the reactive 4-nitrophenyl group, which can cause the organophosphate to bind irreversibly to the enzyme. This is in contrast to the diethyl phosphate breakdown product, formed from the P–OAr bond cleavage pathway.
Here, we report our attempts to identify suitable bases to promote hydrolysis of the simulant methyl paraoxon (MP, dimethyl 4-nitrophenyl phosphate) and the CWAs GB and VX. We focused on optimising the basic hydrolysis of MP to produce exclusively the non-toxic product, dimethyl phosphate (Scheme 1). For the more successful bases, the conditions for breakdown were varied by using less water and base to establish whether the desired product distribution still holds. The best performing bases were also tested under various conditions against the CWAs, GB and VX, to establish how their hydrolysis pathway compares to that of the simulant, MP. Surprisingly, despite hydrolysis being an effective means for decontamination of nerve agents, there are no previous studies of simple bases for hydrolysis nor investigations into how breakdown of a nerve agent simulant compares to that of real agents for such bases.
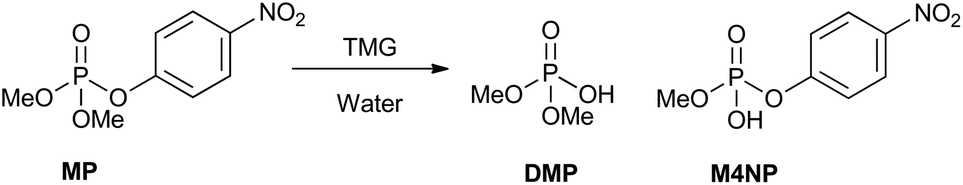 | ||
| Scheme 1 The possible breakdown products for the degradation of methyl paraoxon (MP), when using base in the presence of water. Breakdown products include; dimethyl phosphate (DMP) and methyl 4-nitrophenyl phosphate (M4NP). | ||
Results and discussion
Screening of bases
Initially, the reaction of fourteen bases (Fig. 2) with MP was examined in a water/acetonitrile solution. The product distribution and conversion were monitored by 31P[1H] NMR (Fig. S1.1–1.15†). The desired non-toxic product, dimethyl phosphate, and the toxic product, methyl 4-nitrophenyl phosphate, have peaks at 2.8 and −4.2 ppm, respectively, while MP has a peak at −4.5 ppm. The position of the peak corresponding to the dimethyl phosphate was in accordance with that previously reported for MP breakdown.27 The stronger bases TBD, MTBD, DBU, and TMG all resulted in very fast breakdown of MP to produce exclusively dimethyl phosphate (Table 1). The majority of these bases displayed complete conversion of MP in less than 1 hour. By contrast, quinuclidine resulted in very fast conversion to produce 98% of the toxic breakdown product, methyl 4-nitrophenyl phosphate. A similar result was also observed for its derivative, DABCO, which afforded 100% conversion to the toxic product. This suggested that the nucleophilicity of the base is an important factor in dictating the decomposition pathway taken by MP, with the two highly nucleophilic bases, quinuclidine and DABCO, favouring rapid formation of the toxic product via nucleophilic attack at the aliphatic carbon of the methoxy group. This mechanism is supported by the positive ionisation mass spectrometry of each reaction, which shows the presence of the methylated quinuclidine or DABCO bases (Fig. S2†). In addition, negative ionisation confirmed the presence of methyl 4-nitrophenyl phosphate in each reaction. The remaining bases tested in the screen are all nucleophilic, albeit to varying degrees. This is expressed in the product distribution observed at the end of the reaction, with all the bases predominately forming the toxic product (Table 1). Using inverse-gated 31P[1H] NMR, we were able to quantify the product distribution. Aside from TBD, MTBD, DBU, and TMG, which showed complete conversion to the non-toxic reaction product, the remaining bases afforded between 83 and 100% conversion to the toxic product (Table 1). Aniline and pyridine gave exclusively the toxic product displaying the same behaviour as DABCO, albeit on a much longer timescale. MP did not show any conversion, in water alone after 4 days, as confirmed from the 31P[1H] NMR spectra showing the retention of the MP peak (Fig. S1.16†). This indicates the hydrolysis of MP does not occur in water alone, at least on the timescale in which the majority of the bases showed complete conversion. Even after approximately 7 months, the MP showed only a 24% conversion to the toxic product (Fig. S1.16C†).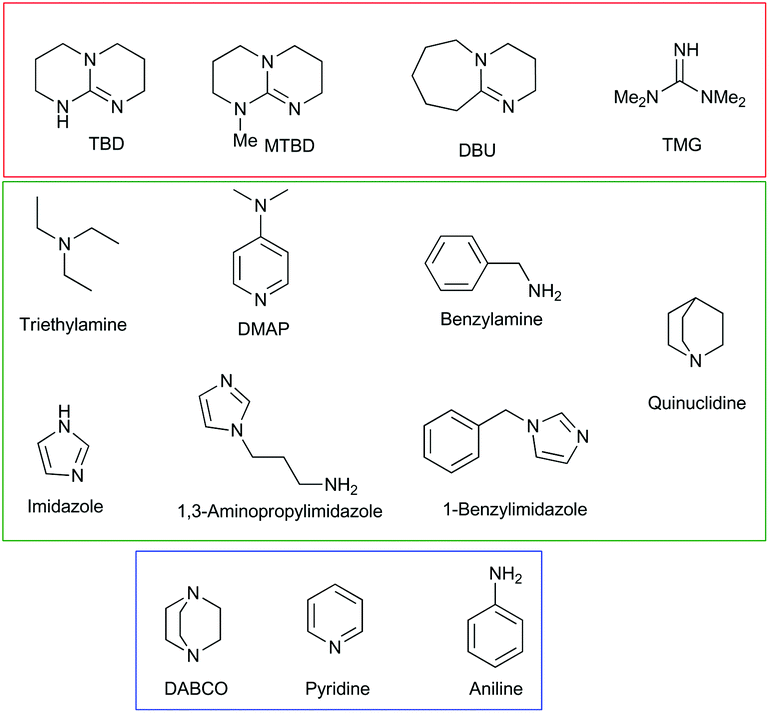 | ||
| Fig. 2 Structures of all the bases used in the study. Bases highlighted in red gave exclusively the non-toxic product, bases highlighted in blue gave exclusively the toxic product, and the bases highlighted in green gave a mixture of toxic and non-toxic products. The reaction was carried out using 8.8 equivalents of base and 827 equivalents of water, with respect to MP. An equal volume of acetonitrile was employed to ensure the homogeneity of the reaction mixture. Breakdown occurred between 1 hour and 68 days, depending upon the base (Table 1). | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v), with product distribution (%) observed via31P[1H] NMR. Base strength represented by the pKa of the bases conjugate acid, measured in water28–32
:1 v/v), with product distribution (%) observed via31P[1H] NMR. Base strength represented by the pKa of the bases conjugate acid, measured in water28–32
| pKa (conjugate acid) | Nitrogen base | Complete conversion observed within | Percentage of dimethyl phosphate (non-toxic) | Percentage of methyl 4-nitrophenyl phosphate (toxic) |
|---|---|---|---|---|
| 15.2 | 1,5,7-Triazabicyclo[4.4.0]dec-5-ene (TBD) | 1 hour | 100 | 0 |
| 15.0 | 7-Methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene (MTBD) | 1 hour | 100 | 0 |
| 13.5 | 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) | 1 hour | 100 | 0 |
| 13.0 | 1,1,3,3-Tetramethylguanidine (TMG) | 2.5 hours | 100 | 0 |
| 11.3 | Quinuclidine | 1 hour | 2 | 98 |
| 10.7 | Triethylamine | 4 days | 9 | 91 |
| 9.6 | 4-Dimethylaminopyridine (DMAP) | 4 days | 10 | 90 |
| 9.3 | Benzylamine | 4 days | 3 | 97 |
| 8.8 | 1,4-Diazabicyclo[2.2.2]octane (DABCO) | 1 hour | 0 | 100 |
| 7.0 | Imidazole | 25–28 days | 17 | 83 |
| 6.9 | 1,3-Aminopropylimidazole | 4 days | 6 | 94 |
| 6.7 | 1-Benzylimidazole | 50–68 days | 10 | 90 |
| 5.3 | Pyridine | 25–34 days | 0 | 100 |
| 4.6 | Aniline | 35–68 days | 0 | 100 |
In general, good correlation was observed between the strength of the base, as determined from the pKa of the conjugate acid,28–32 and the reaction rate and product distribution. The stronger bases (TBD, MTBD, DBU, and TMG), show a preference for the alkaline hydrolysis pathway. For weaker bases, the nucleophilicity of the base is more significant in determining the decomposition pathway; stronger nucleophiles promote rapid formation of the toxic product, while weaker nucleophiles predominately promote formation of the toxic product, albeit at a slower rate.
Variation of water content
The reaction of the bases with MP was carried out using an excess of base and water, 8.8 and 827 equivalents, respectively, with respect to MP. These amounts were chosen based on initial screens using bases for MP hydrolysis and kept constant throughout the screen of the bases. The presence of water was necessary for the base catalysed hydrolysis of MP to produce dimethyl phosphate.24 For real world CWA decontamination systems, it would be desirable to reduce the amount of base and water used for the hydrolysis. We therefore looked to optimise the water content required to hydrolyse MP using TMG, the simplest base to afford 100% selectivity for dimethyl phosphate.While maintaining the amount of base at 5 equivalents with respect to MP, the quantity of water was varied between 1 and 800 equivalents, and the outcome monitored by 31P[1H] NMR (Fig. 3 and Table S1†). A stepwise reduction in the amount of water from 800 to 50 equivalents afforded 97–100% conversion to the non-toxic product, DMP. Within this range of water, the alkaline hydrolysis pathway predominates leading to attack of the hard nucleophilic oxygen from hydroxide on the oxophilic phosphorus centre.23 This leads to DMP formation and loss of the 4-nitrophenol (pKa = 7.1), in preference over methanol (pKa = 15.4), due to the lower pKa of the 4-nitrophenol group.25 Nucleophilic attack of the phosphotriester is most likely to occur via a weakly associative, pentacoordinate transition state, with the more electronegative and more acidic 4-nitrophenyl group adopting the apical position, as hydroxide attacks in a SN2 manner.33,34 A peak shift in the 31P[1H] NMR, from 2.8 to 1.7 ppm, was observed during the reduction in water, however the splitting pattern in the 31P NMR, from the phosphorus-proton coupling, remained unchanged (Fig. S3†). The peak positions in the 1H NMR spectrum remained unchanged on reducing the water content from 800 to 50 equivalents (Fig. S4†).
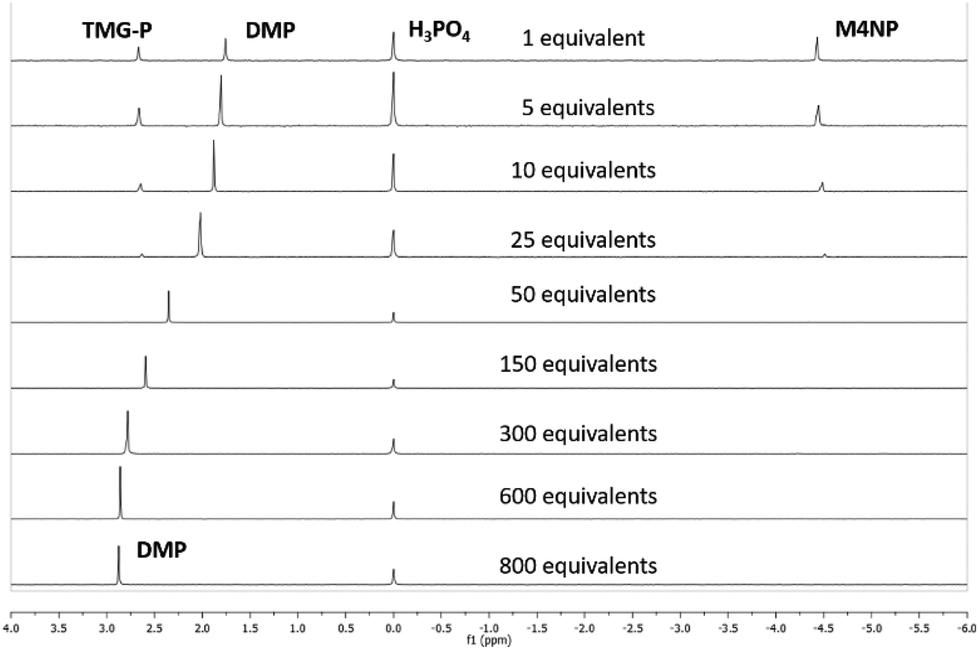 | ||
| Fig. 3 31P[1H] NMR spectra obtained for the breakdown of methyl paraoxon (MP) with varying amounts of water (1–800 equivalents) and 5 equivalents of the TMG base, after 24 hours at room temperature. The peak at 0.0 ppm is from the phosphoric acid standard. Tabulated data presented in Table S1.† | ||
On reducing the water content below 50 equivalents we observed the appearance of a peak for the toxic product at −4.5 ppm in the 31P NMR; this peak occurs as a quartet in the NMR spectra and increases in intensity as the water content decreases (Fig. S3†). In addition, a third breakdown product at 2.7 ppm was also observed when using 1, 5, 10, and 25 equivalents of water. This new peak has the same splitting pattern in the 31P NMR as the peak assigned to dimethyl phosphate (Fig. S3†). Mass spectrometry for the reactions performed using 1 and 5 equivalents of water revealed the presence of an additional phosphorus containing product, with a m/z peak at 224 (Fig. S5a & b†), which corresponds to a product formed by the SN2 attack of TMG at the phosphorus centre (denoted as TMG–P), with displacement of 4-nitrophenol. This mass ion was absent from the mass spectrum obtained for the reactions conducted using 50 and 800 equivalents of water (Fig. S5c & d†). Upon reducing the water content below 50 equivalents, the TMG begins to act as a competitive nucleophile, presumably due to a decrease in the amount of free hydroxide present in the reaction mixture. Attack of the TMG preferentially occurs at the aliphatic carbon from the methoxy group, due to the softer nucleophilic nitrogen favouring attack at the softer electrophilic carbon,23 and results in the formation of M4NP. A small amount of nucleophilic attack by the TMG was also observed at the hard phosphorus centre, giving rise to TMG-P.
4-Nitroanisole was also detected in the 1H NMR with 1, 5, and 10 equivalents of water, with peaks observed at 3.9, 7.1, and 8.2 ppm (Fig. S4†). Its presence was confirmed by the addition of 4-nitroansiole to a 1H NMR sample from the reaction performed using 1 equivalent of water (Fig. S6†). This product is thought to originate from methylation of 4-nitrophenolate, which is formed as the by-product of MP hydrolysis alongside dimethyl phosphate; methylation of the 4-nitrophenolate is thought to occur by reaction with MP to generate the toxic product as a result. Addition of dimethyl phosphate and methyl 4-nitrophenyl phosphate to NMR samples, synthesis detailed in ESI, further confirmed the presence of these two products (Fig. S6†).
Variation of base content
With excess TMG base (5 equivalents), the water content can be reduced from 800 to 50 equivalents while still maintaining a decomposition pathway that produces >97% dimethyl phosphate. The effect of reducing the number of equivalents of TMG on the reaction pathway was also studied. We found that on reducing the base to 2 equivalents, 200 equivalents water was required to maintain complete conversion to the non-toxic product after 24 hours at room temperature (Fig. S7†).We also varied the amount of TMG from 1.0 to 9.0 equivalents, with respect to MP, while keeping the water content fixed at 800 equivalents (Fig. S8†). With the exception of 1 equivalent of TMG, all reactions gave exclusive conversion to the non-toxic product within 24 hours. Using 1 equivalent of TMG we observed 53% conversion after 24 hours and 59% conversion after 25 days, by 31P NMR. The slow conversion is likely due to the acidic breakdown products, such as 4-nitrophenol and dimethyl phosphate, protonating the base and preventing further hydrolysis from taking place. This explains why excess base is usually required for hydrolysis of G-type nerve agents.6,9 A similar trend was also observed when using MTBD as the base (Fig. S9†).
Hydrolysis of GB and VX
Next, we examined how the real nerve agents, GB and VX, were hydrolysed with varying amounts of water (Fig. 4). GB was hydrolysed in water alone, producing 96–97% of the desired breakdown product, isopropyl methylphosphonic acid, using 1, 50, and 800 equivalents water (Fig. 4a). The hydrolysis of VX in water alone was less effective, with 75 and 97% VX remaining after a 24 hours period at room temperature, when using 800 and 50 equivalents of water, respectively. The 25% conversion observed for VX using 800 equivalents water, produced only 9% of the desired non-toxic product, ethyl methyl phosphonate (EMPA). The remaining two breakdown products consisted of 13% the toxic breakdown product, EA-2192, along with 3% of the non-toxic thioic acid (Fig. S12a†), formed via C–S bond cleavage.12 Hydrolysis of VX using 1 equivalent of water was ineffective with no conversion. Similarly, the MP simulant proved ineffective for hydrolysis in water alone across all three equivalents of water (Fig. 4a). The more labile P–F bond in GB, compared to the P–S bond in VX and P–O bond in MP, makes GB more susceptible for hydrolysis in water alone.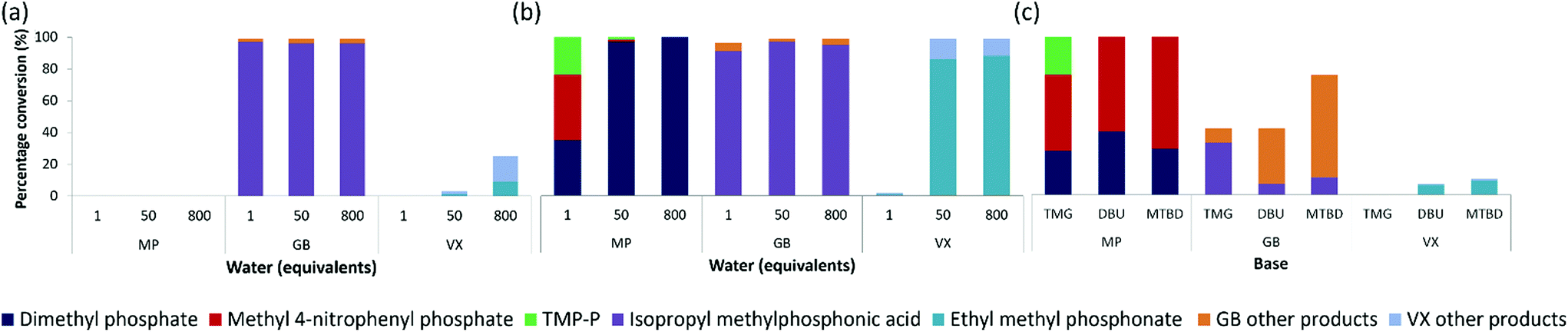 | ||
| Fig. 4 Testing of 1, 50, and 800 equivalents of water, with respect to the simulant MP and CWAs GB and VX, in (a) the absence and (b) the presence (5 equivalents) of TMG base. (c) Testing bases; TMG, DBU, and MTBD, 2 equivalents with respect to MP, GB, and VX, in the absence of water, after 24 hours at room temperature. Products highlighted on spectra for (a) Fig. S10–12, (b) Fig. S13–15, and (c) Fig. S16–18.† Tabulated data presented in Tables S2 & 3.† | ||
On addition of 5 equivalents of TMG, complete hydrolysis of GB was observed after 24 hours with 50 and 800 equivalents of water, while 1 equivalent of water afforded 97% conversion to predominately the non-toxic isopropyl methylphosphonic acid (Fig. 4b). This is in contrast to MP hydrolysis using 1 equivalent of water which afforded only 35% of the non-toxic product, dimethyl phosphate. Hydrolysis of VX in the presence of 5 equivalents of TMG showed complete conversion resulting in 86 and 88% EMPA, when using 50 and 800 equivalents of water, respectively. The other product formed was the toxic product, EA-2192, (Fig. S15†). The presence of a much greater amount of hydroxide when using 50 and 800 equivalents of water favours the alkaline hydrolysis of VX. The lifetime of the pentacoordinate phosphorus transition state, from hydroxide attack, allows for either the P–S bond or P–O bond to occupy the apical position, via pseudorotation.33,35 The less electronegative but bulkier thiolate group will have a higher preference for being in the apical position, as far away as possible from the incoming hydroxide nucleophile. This results in preferential cleavage of the P–S bond, and formation of EMPA as the major breakdown product, over P–O bond cleavage and EA-2192 formation.36,37 Using 1 equivalent of water with TMG showed only 1% conversion to exclusively EMPA (Fig. 4b). Such a low conversion when 1 equivalent of water was used is likely due to insufficient amounts of hydroxide being generated to promote alkaline hydrolysis.
Breakdown in the absence of water
The bases MTBD, DBU, and TMG were also tested, against MP, in the absence of water (Fig. 4c). For TMG, all three breakdown products were observed, with at least 48% conversion to the toxic product. This further supports our hypothesis that in the absence of water the formation of the toxic product is strongly favoured, even with the stronger, less nucleophilic TMG base. Nucleophilic attack of the base on the phosphorous was more prevalent with TMG than MTBD or DBU, as indicated by the 31P NMR and 1H NMR (Fig. S16†). The greater steric hindrance of the DBU and MTBD units compared to TMG likely hinders their nucleophilic attack at the phosphorus centre.The bases TMG, DBU, and MTBD were also tested against GB and VX in the absence of water (Fig. 4c). By comparison with MP, lower conversion was observed for GB and VX, with between 23 and 58% GB and between 89 and 92% VX remaining after 24 hours at room temperature. The breakdown of VX was less effective when treated with base alone, compared to GB and MP. Breakdown of VX using TMG without water proved ineffective, while only 7 and 10% conversion, to predominately the non-toxic EMPA, was observed using DBU and MTBD, respectively. GB breakdown with TMG, DBU, and MTBD (Fig. 4c) showed conversion to products other than the desired isopropyl methylphosphonic acid. This is similar to the breakdown of the simulant MP with the bases alone.
Most of the other GB breakdown products are likely to be non-toxic; that is, containing no fluorine. For example, methylphosphonic acid, formed by further hydrolysis of isopropyl methylphosphonic acid. The absence of fluorine in the breakdown products is confirmed by the lack of phosphorus-fluorine splitting in the product peaks (Fig. S11, 14, and 17†) compared to the splitting observed for the peaks corresponding to GB (Fig. S17†). The high electronegativity of the fluorine in GB makes this group most likely to occupy the apical position in the pentacoordinate transition state, resulting in the preferential cleavage of the P–F bond, when hydroxide attacks at phosphorus.38
Conclusions
In summary, on screening a range of bases for the hydrolysis of the CWA simulant MP, a correlation was observed between the breakdown product distribution and the properties of the base. The stronger, less nucleophilic bases resulted in alkaline hydrolysis of MP to produce dimethyl phosphate as the desired non-toxic breakdown product, whereas weaker, more nucleophilic bases afforded a toxic by-product. By varying the conditions for the hydrolysis, the amount of water was shown to have the greatest impact on the breakdown product distribution.MP hydrolysis in water alone was not effective, showing no breakdown of the simulant after 4 days. However, the addition of 5 equivalents of the TMG base using 50 equivalents of water resulted in conversion to 97% dimethyl phosphate, the desired breakdown product, after 24 hours at room temperature. In contrast, GB hydrolysis was effective both in the absence and the presence of base. GB hydrolysis could occur in water alone, producing 97% isopropyl methylphosphonic acid, as the desired product when using just 1 equivalent of water. Whereas, similar to MP, VX was not effective for hydrolysis in water alone. Addition of the TMG base resulted in complete conversion to produce 86% of the desired breakdown product, ethyl methyl phosphonate, using 50 equivalents of water and 5 equivalents of TMG as base.
We have demonstrated that treatment of the simulant, MP, and the CWAs GB and VX with 5 equivalents of a strong non-nucleophilic base in the presence of 50 equivalents of water should result in full conversion to predominately non-toxic products. Such a simple, low-cost approach for CWA destruction, which tests for breakdown of CWAs under practically relevant conditions, is likely to improve the practical application of such a decontamination method in the stockpile destruction of CWAs. This study also highlights the differences in both reactivity and product distribution that can exist between simulants and CWAs. Clearly, the simulant is less toxic due to its enhanced chemical stability. This can make studying breakdown more challenging, for instance by forming unwanted breakdown products compared to the real agents. However, benefits should be found when testing developed systems against real agents, in particular G agents such as GB, which are inherently more susceptible towards hydrolysis.
Experimental
Materials and methods
All breakdown reactions were performed at ambient temperature. 31P NMR for breakdown experiments, using MP, was performed on a Bruker 500 MHz spectrometer. 31P NMR for breakdown experiments, using GB and VX, was performed on a Bruker 400 MHz spectrometer. Deuterated acetonitrile was used as NMR solvent for all experiments. An internal reference, 1% H3PO4 in deuterium oxide (0.0 ppm, singlet), sealed in a glass capillary, was used in all 31P NMR experiments unless stated otherwise.The synthesis of methyl paraoxon and lithium methyl 4-nitrophenyl phosphate is reported in the ESI.†
Breakdown studies
The reactions involving the CWAs, GB, and VX, were carried out under the same conditions as with MP. All reactions with GB and VX were carried out at Dstl by trained personnel using appropriate care and equipment due to the lethal toxicity of the agents.:1 v/v). To this solution, MP (3 μL, 0.0168 mmol, 1 equivalent) was added. The solution was transferred to an NMR tube, fitted with a glass capillary (containing 1% H3PO4 as internal standard). 31P [1H] NMR was recorded within the first hour for each sample. Further monitoring of the reaction solution was carried out until complete conversion was achieved, established by the disappearance of the MP peak at −4.5 ppm.
:1 v/v) containing either TMG (1.0, 2.5, 5.0, or 9.0 equivalents) or MTBD (1.0, 2.5, 5.0, or 9.0 equivalents). The resulting solution was stirred in a vial at room temperature for 24 hours. After such time, the solution was transferred to an NMR tube, fitted with a glass capillary (containing 1% H3PO4 as internal standard). The 31P[1H] NMR was recorded within 1 hour of transfer of the solution from the vial.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Defence Science and Technology Laboratory (Dstl) for funding. Glasgow University mass spectrometry service. DJA thanks the EPSRC for a Fellowship (EP/L021978/1).Notes and references
- A. N. Bigley and F. M. Raushel, Biochim. Biophys. Acta, Proteins Proteomics, 2013, 1834, 443–453 CrossRef CAS PubMed.
- K. G. Davis and G. Aspera, Ann. Emerg. Med., 2001, 37, 653–656 CrossRef CAS PubMed.
- K. Kehe and L. Szinicz, Toxicology, 2005, 214, 198–209 CrossRef CAS PubMed.
- F. R. Sidell and J. Borak, Ann. Emerg. Med., 1992, 21, 865–871 CrossRef CAS PubMed.
- A. Watson, D. Opresko, R. Young, V. Hauschild, J. King and K. Bakshi, Organophosphate Nerve Agents, Elsevier Academic Press Inc, San Diego, 2009 Search PubMed.
- Y. C. Yang, J. A. Baker and J. R. Ward, Chem. Rev., 1992, 92, 1729–1743 CrossRef CAS.
- M. Enserink and J. Kaiser, Science, 2013, 341, 1050–1051 CrossRef CAS PubMed.
- S. Farquharson, F. E. Inscore and S. Christesen, Top. Appl. Phys., 2006, 103, 447 CrossRef CAS.
- K. Kim, O. G. Tsay, D. A. Atwood and D. G. Churchill, Chem. Rev., 2011, 111, 5345–5403 CrossRef CAS PubMed.
- S.-Y. Moon, G. W. Wagner, J. E. Mondloch, G. W. Peterson, J. B. DeCoste, J. T. Hupp and O. K. Farha, Inorg. Chem., 2015, 54, 10829–10833 CrossRef CAS PubMed.
- A. N. Bigley, C. Xu, T. J. Henderson, S. P. Harvey and F. M. Raushel, J. Am. Chem. Soc., 2013, 135, 10426–10432 CrossRef CAS PubMed.
- Y.-C. Yang, Acc. Chem. Res., 1999, 32, 109–115 CrossRef CAS.
- Q.-Q. Wang, R. A. Begum, V. W. Day and K. Bowman-James, Org. Biomol. Chem., 2012, 10, 8786–8793 RSC.
- R. M. Herriott, J. Gen. Physiol., 1947, 30, 449–456 CrossRef CAS PubMed.
- Y. Y. Liu, A. J. Howarth, J. T. Hupp and O. K. Farha, Angew. Chem., Int. Ed., 2015, 54, 9001–9005 CrossRef CAS PubMed.
- A. J. Howarth, C. T. Buru, Y. Liu, A. M. Ploskonka, K. J. Hartlieb, M. McEntee, J. J. Mahle, J. H. Buchanan, E. M. Durke, S. S. Al-Juaid, J. F. Stoddart, J. B. DeCoste, J. T. Hupp and O. K. Farha, Chem. – Eur. J.-, 2017, 23, 214–218 CrossRef CAS PubMed.
- Y. Liu, S. Y. Moon, J. T. Hupp and O. K. Farha, ACS Nano, 2015, 9, 12358–12364 CrossRef CAS PubMed.
- J. E. Casida and G. B. Quistad, Chem. Res. Toxicol., 2004, 17, 983–998 Search PubMed.
- J. Misik, R. Pavlikova, J. Cabal and K. Kuca, Drug Chem. Toxicol., 2015, 38, 32–36 CrossRef CAS PubMed.
- E. A. Castro, D. Ugarte, M. F. Rojas, P. Pavez and J. G. Santos, Int. J. Chem. Kinet., 2011, 43, 708–714 CrossRef CAS.
- X. Han, V. K. Balakrishnan and E. Buncel, Langmuir, 2007, 23, 6519–6525 CrossRef CAS PubMed.
- X. Han, V. K. Balakrishnan, G. W. vanLoon and E. Buncel, Langmuir, 2006, 22, 9009–9017 CrossRef CAS PubMed.
- N. M. Rougier, R. V. Vico, R. H. de Rossi and E. I. Buján, J. Org. Chem., 2010, 75, 3427–3436 CrossRef CAS PubMed.
- P. Pavez, D. Millán, J. I. Morales, E. A. Castro, A. C. López and J. G. Santos, J. Org. Chem., 2013, 78, 9670–9676 CrossRef CAS PubMed.
- D. J. Kennedy, B. P. Mayer, S. E. Baker and C. A. Valdez, Inorg. Chim. Acta, 2015, 436, 123–131 CrossRef CAS.
- R. T. Delfino, T. S. Ribeiro and J. D. Figueroa-Villar, J. Braz. Chem. Soc., 2009, 20, 407–428 CrossRef CAS.
- S. Y. Moon, Y. Y. Liu, J. T. Hupp and O. K. Farha, Angew. Chem., Int. Ed., 2015, 54, 6795–6799 CrossRef CAS PubMed.
- V. K. Aggarwal, I. Emme and S. Y. Fulford, J. Org. Chem., 2003, 68, 692–700 CrossRef CAS PubMed.
- I. Kaljurand, A. Kutt, L. Soovali, T. Rodima, V. Maemets, I. Leito and I. A. Koppel, J. Org. Chem., 2005, 70, 1019–1028 CrossRef CAS PubMed.
- E. Vedejs and S. Denmark, Lewis Base Catalysis in Organic Synthesis, Wiley, 2016 Search PubMed.
- H. Walba and R. W. Isensee, J. Org. Chem., 1961, 26, 2789–2791 CrossRef CAS.
- P. Wiczling, P. Kawczak, A. Nasal and R. Kaliszan, Anal. Chem., 2006, 78, 239–249 CrossRef CAS PubMed.
- K. C. Kumara Swamy and N. Satish Kumar, Acc. Chem. Res., 2006, 39, 324–333 CrossRef CAS PubMed.
- S. H. Gellman, R. Petter and R. Breslow, J. Am. Chem. Soc., 1986, 108, 2388–2394 CrossRef CAS PubMed.
- P. Vayron, P. Y. Renard, F. Taran, C. Creminon, Y. Frobert, J. Grassi and C. Mioskowski, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 7058–7063 CrossRef CAS.
- J. Šečkutė, J. L. Menke, R. J. Emnett, E. V. Patterson and C. J. Cramer, J. Org. Chem., 2005, 70, 8649–8660 CrossRef PubMed.
- K. A. Daniel, L. A. Kopff and E. V. Patterson, J. Phys. Org. Chem., 2008, 21, 321–328 CrossRef CAS.
- F. Zheng, C.-G. Zhan and R. L. Ornstein, J. Chem. Soc., Perkin Trans. 2, 2001, 2355–2363 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ob02475h |
| This journal is © The Royal Society of Chemistry 2018 |