
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly reactive bis-cyclooctyne-modified diarylethene for SPAAC-mediated cross-linking†
Alexander V.
Strizhak
ab,
Krishna
Sharma
 a,
Oleg
Babii
c,
Sergii
Afonin
c,
Anne S.
Ulrich
cd,
Igor V.
Komarov
*ef and
David R.
Spring
*a
a,
Oleg
Babii
c,
Sergii
Afonin
c,
Anne S.
Ulrich
cd,
Igor V.
Komarov
*ef and
David R.
Spring
*a
aUniversity Chemical Laboratory, University of Cambridge, Lensfield Road, CB2 1EW Cambridge, UK. E-mail: spring@ch.cam.ac.uk
bEnamine Ltd., Vul. Chervonotkatska 78, 02094 Kyiv, Ukraine
cInstitute of Biological Interfaces (IBG-2), Karlsruhe Institute of Technology (KIT), POB 3640, 76021 Karlsruhe, Germany
dInstitute of Organic Chemistry (IOC), KIT, Fritz-Haber-Weg 6, 76131 Karlsruhe, Germany
eTaras Shevchenko National University of Kyiv, Institute of High Technologies, Vul. Volodymyrska 64/13, 01601 Kyiv, Ukraine. E-mail: ik214@yahoo.com
fLumobiotics GmbH, Auer Str. 2, 76227, Karlsruhe, Germany
First published on 26th October 2018
Abstract
Photoisomerizable diarylethenes equipped with triple bonds are promising building blocks for constructing bistable photocontrollable systems. Here we report on the design, synthesis and application of a cross-linking reagent which is based on a diarylethene core and features two strained cyclooctynes. High reactivity of the cyclooctyne rings in catalyst-free 1,3-dipolar cycloaddition reactions was suggested to stem from the additional strain imposed by the fused thiophene rings. This hypothesis was confirmed by quantum chemical calculations.
Introduction
Reversibly photoisomerizable diarylethene (DAE) units have been inserted in countless molecules and systems, whose properties and functions can be controlled with light.1 DAEs undergo pericyclic transformations under irradiation with UV or visible light (Fig. 1). The “open” and “closed” photoforms are stable at ambient temperatures that makes them attractive for numerous applications, especially in biology and medicine.1i,j,m,o | ||
| Fig. 1 Diarylethene fragment used in photocontrollable molecules and systems, R1, R2 = alkyl; R3, R4 = H, alkyl, aryl; Y = S, O or N. The core unit undergoing photoinduced pericyclic transformations is highlighted in red. | ||
One of the popular synthetic approaches utilizes functionalized DAE-derived reagents (“building blocks”) for modular construction of photocontrollable molecules. For example, bis-diethynyl-substituted diarylethene 1 (Fig. 2) has been proposed as an entry to new photochromic DAE materials through palladium-catalyzed Sonogashira cross-coupling reaction2 or for Cu-catalysed cycloaddition to azides (a “click”-reaction) involving its triple bonds.3
 | ||
| Fig. 2 The cross-linking reagents (1–3), a pyrrole-containing cyclooctyne (4) described in the literature and the new reagent developed in this work (5). | ||
We were interested in 1 and similar compounds because they could be used for cross-linking the azide-substituted side chains in biologically active peptides in order to stabilize their conformation via so-called “stapling”. This could be done, for example, using a two-component strategy, employing the click-reaction.4 Stapling can increase the biostability, improve binding affinities and pharmacokinetic properties of peptides,5 and photoisomerizable cross-linkers can additionally make their bioactivities photocontrollable – a feature which is attracting much interest due to potential applications in biotechnology and medicine.1i,j,o
Cross-linking of peptides with azobenzene-derived, mainly thiol-reactive photoisomerizable cross-linkers was extensively studied since the beginning of 2000th. Efficient photocontrol of peptide conformation,6a–i folding,6j,k and affinity of binding to DNA6l,m was documented. It was demonstrated that biologically relevant processes like protein–protein interaction (PPI)6n–q and insulin secretion6r could be “switched on” and “switched off” reversibly with the use of the azobenzene-derived cross-linked peptides. Photoisomerizable spiropyrane6s and rhodopsin-like fragments6t were also utilized in peptide cross-linkers to enable the photocontrol of peptide conformation and properties. These studies have laid the ground for the development of practically useful photocontrollable biologically active compounds for biotechnology and in vivo applications.1i
DAE-derived peptide cross-linkers are much less studied. The only proof-of-principle experiment demonstrating successful photocontrol of peptides stapled by the DAE building block 2 have been reported for DNA-binding peptides (Fig. 2).6u The activated carboxylic groups in 2 reacted with the amino groups of ornithine side chains to form amide bonds in the stapled peptides. To the best of our knowledge, no DAE building blocks equipped for a click-reaction have been used so far for peptide stapling. In this paper, we report on the design and synthesis of such a building block, and its validation for peptide stapling applications.
Of particular interest for applications are bio-orthogonal cross-linking reagents utilizing the strain-promoted azide–alkyne cycloaddition (SPAAC) as the click-reaction, which avoids toxic Cu-catalysts (Fig. 3).7 SPAAC has already been successfully used for peptide stapling: a cyclooctadiyne derivative 3 (Fig. 2) was employed as the stapling reagent and demonstrated excellent performance.8 Here, we aimed at developing a DAE-based cross-linking building block suitable for the Cu-free SPAAC.
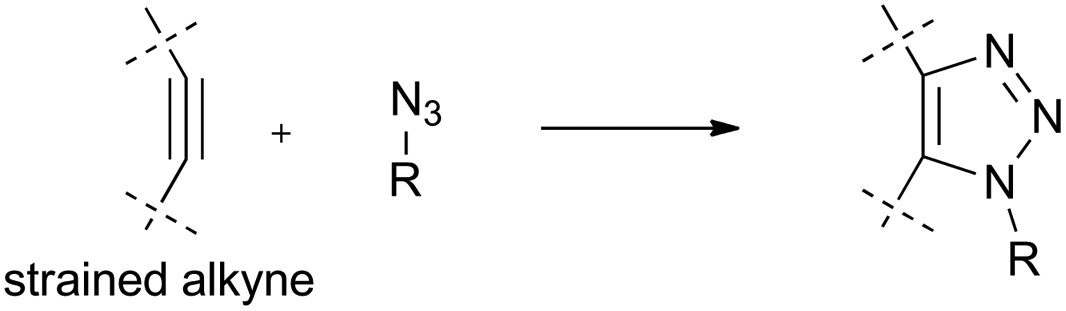 | ||
| Fig. 3 Strain-promoted azide–alkyne cycloaddition (SPAAC) reaction. | ||
Results and discussion
Design of the target compound
The utility of SPAAC in biological systems critically depends on the reactivity of the strained alkynes involved in the reaction: more reactive reagents can address faster biological processes and can react with the azide-modified biomolecules even if they are present at low concentrations in living systems. After the pioneering work describing relatively slow-reacting cyclooctynes,7a much effort was put into the development of stable, but more reactive fluoro- and difluoro-substituted analogues,7b heteroatom-containing cyclooctynes,7e dibenzo-annulated cyclooctadiynes9 and twisted systems10 (see a recent review11).When designing our DAE-based building block, we were inspired by a recent report demonstrating that the addition of one mole of azide to dibenzo[a,e]cyclooctadiyne 3 made the second triple bond 500-fold more reactive.12a This can be attributed to additional strain imposed on the medium-sized carbocycle by the annulation of the five-membered triazole ring, resulting in the enhanced reactivity of the remaining triple bond. An exceptionally high reactivity was also reported for compound 4, a cyclooctyne mono-annulated to a five-membered pyrrole ring.12b Taking into account these findings and aiming at enhanced reactivity of our reagent, we targeted structure 5 (Fig. 2), in which two cyclooctyne residues are symmetrically annulated to the five-membered thiophene rings of the DAE fragment. Comparing to the known DAE-derived building block 2, compound 5 will form less conformationally flexible cross-linker (due to the presence of two additional cycles) which might help to better convey its structural changes to the cross-linked molecular unit upon photoisomerization.
Synthesis
The key intermediate in our synthesis of 5 was the cycloheptanone derivative 6 (Fig. 4). This compound is easily available through the technically simple 1,1,1,3,3,3-hexafluoro-2-propanol-promoted intramolecular Friedel–Crafts acylation reaction of 7, as described for its non-methyl-substituted analogue.13 Compound 7 was obtained in good yield starting from 2-methyl-4-bromothiophene 8, following the procedures first described more than half a century ago.14 All the synthetic sequence can be scaled up to multigram quantities of the compound 6, which can be prepared in a reasonable time (1–2 weeks). | ||
| Fig. 4 Synthesis of the model cyclooctyne 12 and its reaction with benzyl azide. | ||
Easy synthetic availability of 6 prompted us to explore the cyclooctyne ring construction first using this compound as a model, and then applying the elaborated procedure to the more complex and expensive DAE-derived bis-cyclooctyne precursor of 5, which could also be synthesized from 6.
The exocyclic alkene 9 was obtained from 6 in excellent yield using the Wittig reaction. Rearrangement of 9 to the corresponding cyclooctanone 10 proceeded smoothly under action of hydroxy(tosyloxy)iodobenzene followed by aqueous-methanol work-up, a procedure reported recently for β-benzocycloalkenones.15 Formation of the enol triflate 11 followed by elimination completed the synthesis of the model cyclooctyne 12, in overall 18% yield.
We were pleased to find that compound 12 was highly reactive in SPAAC, yet stable enough to allow performing the reaction with azides in situ at ambient temperature. Although attempts at isolating of pure 12 failed in our hands due to compound decomposition, storage of this compound at −20 °C in the solution after preparation preserved the compound almost not degraded for several days. Reaction of 12 with benzyl azide proceeded completely in less than 30 minutes at 1 mM concentration and 0 °C giving isomeric 13a and 13b (syn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :anti 24:76). Both triazole isomers were separated by preparative HPLC and their structure was assigned through series of HMBC and NOESY 2D-NMR experiments (see ESI†).
:anti 24:76). Both triazole isomers were separated by preparative HPLC and their structure was assigned through series of HMBC and NOESY 2D-NMR experiments (see ESI†).
DFT calculations of the structure of 12 and its reaction with methyl azide confirmed that the high reactivity of this compound stems from the additional strain imposed by the five-membered heterocycle fused to the cyclooctyne ring (Fig. 5).
 | ||
| Fig. 5 (a) Energy level diagram depicting SPAAC between 12 and methyl azide in methanol (energy in kcal mol−1). (b) Calculated strain energies and alkyne bond angles of 12, cyclooctyne 14, and benzo-fused cyclooctyne 15. | ||
Transition state activation barriers for SPAAC between 12 and methyl azide were calculated using Gaussian 0916 with the B3LYP density functional and the 6-31 G(d) basis set within the CPCM model for methanol as solvent at standard conditions (see ESI† for full computational details). We found that the activation barrier for the formation of the anti-regioisomer (21.8 kcal mol−1) was lower than for the syn-regioisomer (22.6 kcal mol−1), which is consistent with the experimental observation of the anti-isomer being the predominant product (Fig. 5a). Furthermore, the activation barrier for SPAAC with 12 (21.8 kcal mol−1) was found to be lower than the activation barrier reported for both cyclooctyne 14 and benzocyclooctyne 15 (24.9 kcal mol−1),17 suggesting an increased reactivity. We also calculated the strain energy for 12 (18.4 kcal mol−1) which was found to be higher than the calculated strain energy of cyclooctyne 14 (13.6 kcal mol−1) and benzocyclooctyne 15 (14.4 kcal mol−1), implying an increased reactivity due to the fusion of the five-membered thiophene ring (Fig. 5b). This increase in the ring strain due to the fused five-membered thiophene ring was also reflected by the decreased alkyne bond angles in 12 (151.1°, 156.7°) compared to 14 (157.4°, 157.5°) and 15 (155.1°, 157.2°).
With these encouraging results at hands, we performed the synthesis of the target DAE-derived bis-cyclooctyne 5, also starting from 6. The corresponding synthetic route is shown in Fig. 6.
 | ||
| Fig. 6 Synthesis of the target compound 5 and its SPAAC with benzyl azide. | ||
The cycloheptanone 6 was brominated, and the obtained bromo-derivative 16 was converted (through alkene 17) into the cyclooctanone 18, similarly to the transformation of 6 to 10 described above. The carbonyl group in 18 was protected to allow synthesizing the DAE derivative 20 using the procedure reported previously by Irie et al.18 Then the carbonyl groups were liberated (forming compound 21). Formation of the enol triflate (compound 22) followed by the HOTf elimination completed the synthesis of the DAE building block 5 in about 10% overall yield, calculating on 6.
As expected, the bis-cyclooctyne 5 was as reactive as 12 towards benzyl azide, a colourless 23a was formed as a major product in its SPAAC with benzyl azide (the storage of 12 after the preparation should be done at −20 °C due to its relative instability). Compound 23a was isolated in the open form and fully characterized. Blue-coloured 23a(closed) was prepared from the colourless 23a(open) by irradiation with UV light (256 nm) and characterized; 23a(closed) could, in turn, be transformed back to 23a(open) by irradiation with visible light (590 nm). Both transformations proceeded in quantitative yield, demonstrating a high efficiency of the photoconversion. Notably, the conversion of 23a(closed) to 23a(open) can be achieved by red light: the low-energy absorption band of the closed isomer is intense enough at 630–650 nm (see the UV-VIS absorption spectrum in Fig. 7a). This feature is important for the application of the DAE-derived compounds in vivo, because red light penetrates deeply into live tissues. Hence, photocontrol of biologically active derivatives of 23 should be feasible non-invasively as deep as 1–2 cm beneath the tissue surface.19
 | ||
| Fig. 7 (a) Photoisomerization of 23 and UV-VIS absorption spectra of both photoforms; (b) kinetics of the photoconversion of 23a(closed) to 23a(open) (MeOH solution, C = 1 × 10−5 M, 25 °C) by irradiation with UV light (256 nm) at ∼10 mW cm−2. | ||
The photoisomerization of 23a(closed) to 23a(open) proceeded within minutes (Fig. 7b), which is typical for DAEs.1a–d Observation of a perfect isosbestic point (298 nm) confirmed that no other chemical processes other than the photo-isomerization took place during irradiation.
Finally, to check the utility of the new building block 5 for peptide stapling, we prepared a stapled version of a peptide that originally stemmed from the PDI sequence (LTFEHYWAQLTS), which had been identified by phage display as an efficient inhibitor of p53/MDM2 and p53/MDMX protein–protein interactions.20 These interactions are known as important targets for anti-cancer drug candidates.21 Peptide inhibitors of p53/MDM2 and p53/MDMX are among the most promising compounds currently under investigation and development.22 Recently, PDI analogues stapled by SPAAC employing the cyclooctadiyne 3 as a linker have been prepared.8 For one of the most potent MDM2 binders identified, the linear precursor 24 bearing two azide-substituted side-chains in positions (i,i + 7) was used to prepare a stapled peptide 25 (Fig. 8). In this work, we also used precursor 24 to prepare the DAE-modified stapled peptides.
 | ||
| Fig. 8 Linear peptide used for stapling with 3 (ref. 8) and 5 (this work) and its SPAAC products 25 and 26. | ||
SPAAC between 24 and the cross-linker 5 was performed at 1 mM concentration of both reactants and was found to be complete within less than one hour (methanol, 25 °C, LCMS monitoring of the reaction mixture). Three different stapled peptide isomers (of the general formula 26, Fig. 8) were easily separated by preparative HPLC using standard chromatographic approaches (see the ESI† for the details).
Conclusions
A novel DAE-derived bis-cyclooctyne 5 was synthesized for use as a cross-linking reagent by SPAAC. The stable compound was shown to be highly reactive towards azides due to additional strain imposed on the cyclooctyne rings by the fused thiophene rings. The high azide reactivity of 5 makes it a useful building block for azide cross-linking, e.g. for obtaining stapled peptides, as demonstrated on a peptide inhibitor of the p53/MDM2 and p53/MDMX protein–protein interactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge EU funding by the EU H2020-MSCA-RISE-2015 through the PELICO project (grant 690973). DRS acknowledges support from the Engineering and Physical Sciences Research Council (EP/P020291/1) and Royal Society (Wolfson Research Merit Award). K. S. would like to thank Trinity College, Cambridge Trust, Cambridge Nehru Trust and the Cambridge Philosophical Society for providing fellowships. Part of this work was performed using the Darwin Supercomputer of the University of Cambridge High Performance Computing Service (http://www.hpc.cam.ac.uk/), provided by Dell Inc. using Strategic Research Infrastructure Funding from the Higher Education Funding Council for England and funding from the Science and Technology Facilities Council. ASU and SA thank the DFG for supporting GRK 2039, ASU and OB acknowledge the BMBF for VIP-PLUS funding. IVK acknowledges the Alexander von Humboldt Foundation (Germany) for financial support as a recipient of the Georg Forster Research Prize.Notes and references
-
(a)
Photochromism: molecules and systems, ed. H. Dürr and H. Bouas-Laurent, Elsevier, Amsterdam, 1990 Search PubMed
; (b) M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS PubMed
- A. Osuka, D. Fujikane, H. Shinmori, S. Kobatake and M. Irie, J. Org. Chem., 2001, 66, 3913–3923 CrossRef CAS PubMed
-
(a) O. Tosic and J. Mattay, Eur. J. Org. Chem., 2011, 371–376 CrossRef CAS
-
(a) Y. H. Lau, P. de Andrade, S.-T. Quah, M. Rossmann, L. Laraia, N. Sköld, T. J. Sum, P. J. E. Rowling, T. L. Joseph, C. Verma, M. Hyvönen, L. S. Itzhaki, A. R. Venkitaraman, C. J. Brown, D. P. Lane and D. R. Spring, Chem. Sci., 2014, 5, 1804–1809 RSC
- P. M. Cromm, J. Spiegel and T. N. Grossmann, ACS Chem. Biol., 2015, 10, 1362–1375 CrossRef CAS PubMed
-
(a) J. R. Kumita, O. S. Smart and G. A. Woolley, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3803–3808 CrossRef CAS PubMed
-
(a) N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed
- Y. H. Lau, Y. Wu, M. Rossmann, B. X. Tan, P. de Andrade, Y. S. Tan, C. Verma, G. J. McKenzie, A. R. Venkitaraman, M. Hyvönen and D. R. Spring, Angew. Chem., Int. Ed., 2015, 54, 15410–15413 CrossRef CAS PubMed
- L. Kii, A. Shiraishi, T. Hiramatsu, T. Matsushita, H. Uekusa, S. Yoshida, M. Yamamoto, A. Kudo, M. Hagiwara and T. Hosoya, Org. Biomol. Chem., 2010, 8, 4051–4055 RSC
- T. Harris, G. dos Passos Gomes, S. Ayad, R. J. Clark, V. V. Lobodin, M. Tuscan, K. Hanson and I. V. Alabugin, Chem., 2017, 3, 1–12 Search PubMed
- J. Dommerholt, F. P. J. T. Rutjes and F. L. van Delft, Top. Curr. Chem. (Z), 2016, 374, 1–20 CrossRef CAS PubMed
-
(a) D. A. Sutton and V. V. Popik, J. Org. Chem., 2016, 81, 8850–8857 CrossRef CAS PubMed
- H. F. Motiwala, R. H. Vekariya and J. Aubé, Org. Lett., 2015, 17, 5484–5487 CrossRef CAS PubMed
- Ya. L. Gol'dfarb, Yu. B. Vol'kenshtein and B. V. Lopatin, J. Gen. Chem. USSR, 1964, 34, 961–961 Search PubMed
- M. W. Justik and G. F. Koser, Molecules, 2005, 10, 217–225 CrossRef CAS PubMed
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09 Rev. D.01, Wallingford, CT, 2013 Search PubMed
- K. Chenoweth, D. Chenoweth and W. A. Goddard III, Org. Biomol. Chem., 2009, 7, 5255–5258 RSC
- K. Higashiguchi, K. Matsuda, Y. Asano, A. Murakami, S. Nakamura and M. Irie, Eur. J. Org. Chem., 2005, 91–97 CrossRef CAS
- O. Babii, S. Afonin, L. V. Garmanchuk, V. V. Nikulina, T. V. Nikolaienko, O. V. Storozhuk, D. V. Shelest, O. I. Dasyukevich, L. I. Ostapchenko, V. Iurchenko, S. Zozulya, A. S. Ulrich and I. V. Komarov, Angew. Chem., Int. Ed., 2016, 55, 5493–5496 CrossRef CAS PubMed
- B. Hu, D. M. Gilkes and J. Chen, Cancer Res., 2007, 67, 8810–8817 CrossRef CAS PubMed
- C. J. Brown, S. Lain, C. S. Verma, A. R. Fersht and D. P. Lane, Nat. Rev. Cancer, 2009, 9, 862–873 CrossRef CAS PubMed
-
(a) C. Zhan and W. Lu, Curr. Pharm. Des., 2011, 17, 603–609 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, analytical data for novel compounds & details of the calculations. See DOI: 10.1039/c8ob02428f |
| This journal is © The Royal Society of Chemistry 2018 |