
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transition metal-free coupling of terminal alkynes and hypervalent iodine-based alkyne-transfer reagents to access unsymmetrical 1,3-diynes†
J.
Schörgenhumer
 and
M.
Waser
*
and
M.
Waser
*
Institute of Organic Chemistry, Johannes Kepler University Linz, Altenbergerstrasse 69, 4040 Linz, Austria. E-mail: mario.waser@jku.at
First published on 2nd October 2018
Abstract
A variety of unsymmetrical 1,3-diynes can easily be accessed in good yields under catalyst- and transition metal-free conditions by reacting terminal alkynes with hypervalent iodine-based electrophilic alkyne-transfer reagents.
The 1,3-diyne motif is a frequently found structural element that either serves as a valuable synthon for further manipulations, or can be found as such in biologically active compounds as well as functional materials.1 Thus, it comes as no surprise that the development of efficient synthesis strategies has attracted considerable interest.2 The most classical strategy to access symmetric 1,3-diynes is the Cu-mediated oxidative homo-coupling of terminal alkynes, also known as Glaser coupling, which was first reported almost 150 years ago.3 A lot of efforts have been made since that to introduce different and complementary strategies to carry out the transition metal-catalysed homo-coupling of alkynes.4 In addition, the syntheses of unsymmetrically substituted 1,3-diynes became a very important topic recently.2,5–7 The groups of Liu7a and Patil7b independently described very efficient protocols for the gold-catalysed coupling of terminal alkynes with hypervalent iodine-based alkyne transfer reagents in 2017. The use of these electrophilic alkyne transfer reagents for organic syntheses in general is well-established,8 however their use to access 1,3-diynes has so far received less attention.7,9 Besides the aforementioned very recent hetero-coupling relying on gold-catalysis,7 the groups of Lee and Kitamura reported early examples for the coupling of alkynylcuprates with alkynyliodonium salts in 1997 already.9 We have recently reported the transition metal-free homocoupling of terminal alkynes in the presence of hypervalent iodine reagents.10 Control experiments suggested the presence of in situ formed alkynyliodonium species, which then undergo the C(sp)–C(sp) coupling upon reaction with an in situ formed Li-acetylide. Based on the emerging interest in unsymmetrical 1,3-diynes we now investigated the development of an operationally simple and generally applicable strategy to access a variety of diynes 3 by reacting simple terminal alkynes 1 with preformed hypervalent iodine reagents 2 under transition metal- and catalyst-free conditions (Scheme 1).
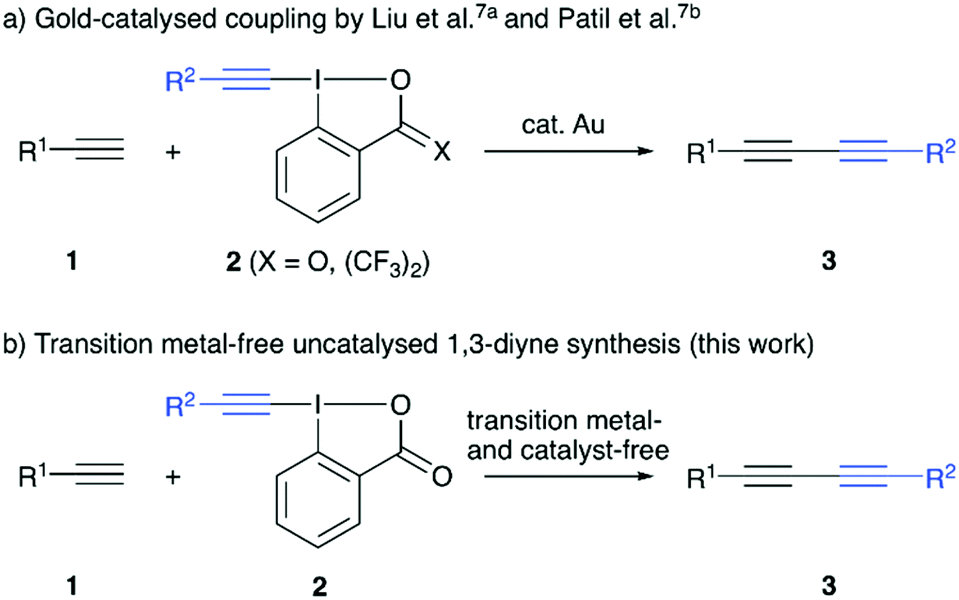 | ||
| Scheme 1 Previous gold-catalysed reports for the synthesis of dissymmetric 1,3-diynes 3 (a) and targeted heterocoupling of alkynes 1 and alkyne-transfer reagents 2 under transition metal-free non-catalysed conditions. | ||
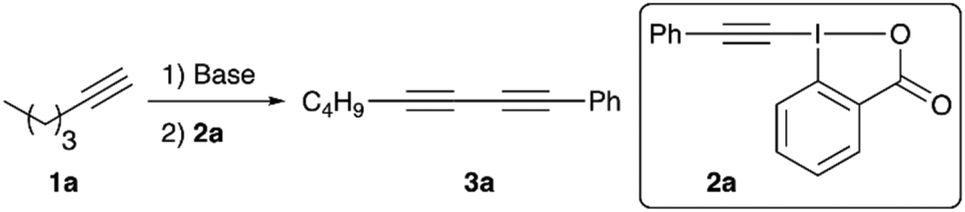
We started our investigations by optimizing the reaction of n-hexyne (1a) with the phenylacetylene-based benziodoxolone 2a (Table 1 gives an overview of the most relevant screening results). All reactions were carried out by deprotonating the terminal alkyne 1a first, followed by subsequent addition of the iodine reagent 2a. Dry THF was used as a solvent for all screening reactions (toluene or CH2Cl2 were very low yielding only), and n-BuLi (1.6 M in hexanes) was the base of choice for the initial experiments. We immediately realized that both steps, deprotonation and electrophile addition, are best carried out at −78 °C (entries 1–3). Furthermore, n-BuLi should be used only in a very subtle excess, as addition of 1.5 equiv. of n-BuLi lead to lower yields accompanied by the formation of notable quantities of by-products already (compare entries 3 and 4). It should be noted that we generally observed two side-reactions that may potentially occur, depending on the reaction conditions: (a) homocoupling of n-hexyne 1a and (b) homocoupling of phenylacetylene (which may be rationalized by cleavage off phenylacetylene from 2a followed by coupling with a second molecule of 2a). However, these side-reactions could be suppressed rather efficiently by using only 1.1 equivalent of n-BuLi combined with a slight excess of alkyne-transfer reagent 2a, which gave the dissymmetric diyne 3a in a satisfying and easily reproducible isolated yield of 87% (entry 5). Other bases were tested as well (see entries 6 and 7 for two examples), but neither of them was found to be satisfying and we thus used the conditions shown in entry 5 (Table 1) to investigate the application scope for this reaction by using different terminal alkynes 1 with a small variety of different hypervalent iodine reagents 2 (Scheme 2).
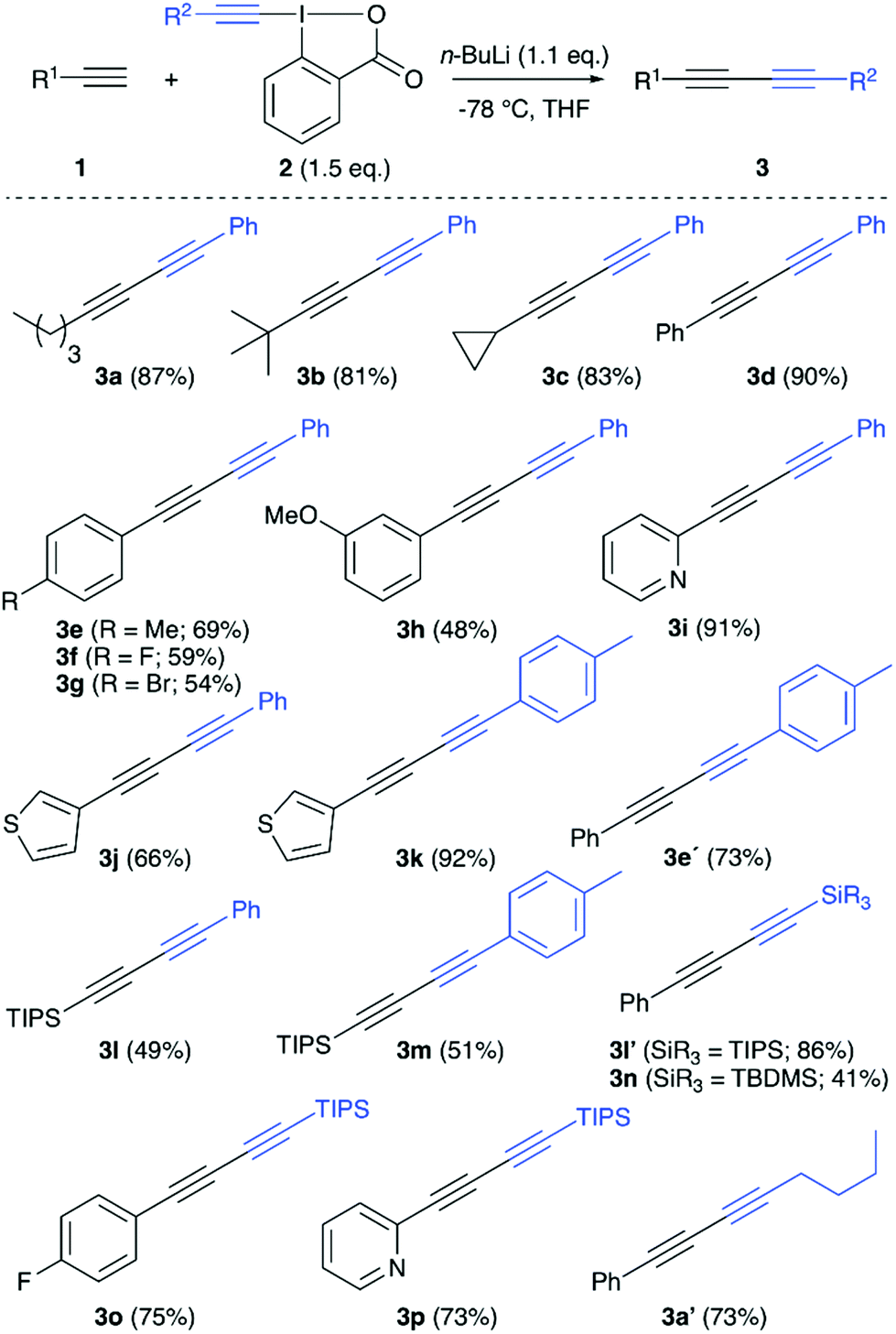 | ||
| Scheme 2 Application scope (all reactions were carried out by adding 1.1 eq. n-BuLi to 0.2 mmol 1 in dry THF at −78 °C, stirring for 2 hours, followed by adding 1.5 eq. of 2 at −78 °C and slowly warming the reaction mixture to r.t. over 3 h; TIPS = triisopropylsilyl; TBDMS = t-butyldimethylsilyl). | ||
|
|
|||||
|---|---|---|---|---|---|
| Entrya | Base (equiv.) | Deprotonation (T) | 2a (equiv.) | Cond. (T, t) | Yieldb (%) |
| a All reactions were carried out using 0.2 mmol 1a in dry THF (1 mL) by first adding the base at the indicated temperature and stirring for 1–2 hours, followed by adding 2a at −78 °C and slowly warming the reaction mixture to r.t. over the indicated period. b Isolated yields. | |||||
| 1 | n-BuLi (1.1) | −40 °C | 1.1 | −78 °C–r.t., 2 h | 46 |
| 2 | n-BuLi (1.1) | −40 °C | 1.1 | −78 °C–r.t., 5 h | 47 |
| 3 | n-BuLi (1.1) | −78 °C | 1.1 | −78 °C–r.t., 2 h | 65 |
| 4 | n-BuLi (1.5) | −78 °C | 1.1 | −78 °C–r.t., 3 h | 53 |
| 5 | n-BuLi (1.1) | −78 °C | 1.5 | −78 °C–r.t., 3 h | 87 |
| 6 | LDA (1.1) | −78 °C | 1.5 | −78 °C–r.t., 3 h | 10 |
| 7 | NaHMDS (1.1) | −78 °C | 1.5 | −78 °C–r.t., 3 h | 21 |
Addition of different aliphatic terminal alkynes to the phenylacetylene-based electrophilic reagent 2a proceeded rather high yielding (see products 3a–3c), and also aryl-substituted alkynes allowed for the synthesis of the diaryl-containing 1,3-diynes 3d–3j in reasonable yields, although some influence of the aryl substituents was observed (i.e. for products 3g and 3h, in those cases notable amounts of homocoupling sideproducts were observed). By using a tolylacetylene-based iodine reagent instead, a similar, maybe even slightly higher, reactivity as for the phenyl-based one could be observed (see products 3k and 3e′). The use of simple TIPS-acetylene as the nucleophilic reaction partner was possible as well (giving products 3l and 3m) albeit it was found that the inverse approach by adding an arylacetylene nucleophile to the TIPS-acetylene-containing iodine reagent allows for a clearly higher yield (compare the results for 3l and 3l′). Surprisingly, the TBDMS-acetylene-based iodine reagent gave the diyne 3n in significantly lower yield, and we hereby observed quite a large amount of the homocoupling of TBDMS-acetylene. Finally, also the hexyne-based iodine reagent could be successfully employed as demonstrated in the synthesis of 3a′.
Summing up the observations made during the investigations of the scope, it becomes obvious that the nature of the employed terminal alkyne plays an important role (which supports the crucial role of the intermediate Li-acetylide). Thus this reaction works best for more electron-neutral terminal alkynes, while the presence of more polar groups or silyl groups unfortunately reduces the reactivity and increasing amounts of homocoupling products are observed in those cases.
In conclusion, we have found that dissymmetric 1,3-diynes 3 can be synthesized without the need of any (transition metal) catalysts by reacting terminal alkynes 1 (which are in situ deprotonated with n-BuLi) with hypervalent iodine-based electrophilic alkyne-transfer reagents 2. This procedure works in reasonable yields for different terminal alkynes 1 as well as iodine reagents 2 and thus may provide a complementary protocol to the recently developed powerful gold-catalysed approaches.7
General reaction procedure: 140 μL (0.22 mmol, 1.1 eq.) of a solution of n-BuLi (1.6 M in hexane) were added to a solution of the corresponding terminal alkyne 1 (0.20 mmol, 1.0 eq.) in dry THF (1 mL) at −78 °C. After stirring for 2 h, the corresponding ethynyl-benziodoxolone 2 (0.30 mmol, 1.5 eq.) was added in one portion. The mixture was allowed to reach room temperature over 3 h while stirring rapidly. The resulting suspension was quenched with 2 mL of a saturated solution of NaHCO3 and extracted three times with 5 mL dichloromethane. After evaporation of the solvent, the crude product was purified by column chromatography (silica gel) to afford the targeted diyne 3 in the reported yield.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The NMR spectrometers used were acquired in collaboration with the University of South Bohemia (CZ) with financial support from the European Union through the EFRE INTERREG IV ETC-AT-CZ program (project M00146, “RERI-uasb”).Notes and references
- (a) F. Bohlmann, T. Burkhardt and C. Zdero, Naturally Occurring Acetylenes, Academic Press, London, 1973 Search PubMed; (b) L. Hansen and P. M. Boll, Phytochemistry, 1986, 25, 285–293 CrossRef CAS; (c) H. Matsunaga, M. Katano, H. Yamamoto, H. Fujito, M. Mori and K. Takata, Chem. Pharm. Bull., 1990, 38, 3480–3482 CrossRef CAS PubMed; (d) J. M. Tour, Chem. Rev., 1996, 96, 537–553 CrossRef CAS PubMed; (e) R. E. Martin and F. Diederich, Angew. Chem., Int. Ed., 1999, 38, 1350–1377 CrossRef; (f) A. L. K. S. Shun and R. R. Tykwinski, Angew. Chem., Int. Ed., 2006, 45, 1034–1057 CrossRef CAS PubMed.
- W. Shi and A. Lei, Tetrahedron Lett., 2014, 55, 2763–2772 CrossRef CAS.
- C. Glaser, Ber. Dtsch. Chem. Ges., 1869, 2, 422–424 CrossRef.
- For some recent transition metal-catalysed approaches: (a) J.-H. Li, Y. Liang and X.-D. Zhang, Tetrahedron, 2005, 61, 1903–1907 CrossRef CAS; (b) J.-H. Li, Y. Liang and Y.-X. Xie, J. Org. Chem., 2005, 70, 4393–4396 CrossRef CAS PubMed; (c) J. Yan, J. Wu and H. Jin, J. Organomet. Chem., 2007, 692, 3636–3639 CrossRef CAS; (d) T. Kurita, M. Abe, M. Tomohiro, Y. Monguchi and H. Sajiki, Synlett, 2007, 2521–2524 CAS; (e) K. Kamata, S. Yamaguchi, M. Kotani, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2008, 47, 2407–2410 CrossRef CAS PubMed; (f) S. Chen, W. Wu and F. Tsai, Green Chem., 2009, 11, 269–274 RSC; (g) S. Zhang, X. Liu and T. Wang, Adv. Synth. Catal., 2011, 353, 1463–1466 CrossRef CAS; (h) A. Leyva-Perez, A. Domenech, S. I. Al-Resayes and A. Corma, ACS Catal., 2012, 2, 121–126 CrossRef CAS; (i) M. G. Leeming, G. N. Khairallah, S. Osburn, K. Vikse and R. A. J. O'Hair, Aust. J. Org. Chem., 2014, 67, 701–710 CrossRef CAS; (j) Z.-J. Wang, P.-H. Wang, J.-J. Lv, J.-J. Feng, X. Xu, A.-J. Wang, C.-T. Au and R. Qiu, RSC Adv., 2015, 5, 96372–96376 RSC; (k) X.-L. Shi, Q.-Q. Hu, F. Wang, W.-Q. Zhang and P.-G. Duan, J. Catal., 2016, 337, 233–239 CrossRef CAS.
- A. L. K. Shi Shun, E. T. Chernick, S. Eisler and R. R. Tykwinski, J. Org. Chem., 2003, 68, 1339–1347 CrossRef CAS PubMed.
- (a) H. Peng, Y. Xi, N. Ronaghi, B. Dong, N. G. Akhmedov and X. Shi, J. Am. Chem. Soc., 2014, 163, 13174–13177 CrossRef PubMed; (b) J. D. Myrtle, A. M. Beekman and R. A. Barrow, Org. Biomol. Chem., 2016, 14, 8253–8260 RSC; (c) S. Ghorai and D. Lee, Tetrahedron, 2017, 73, 4062–4069 CrossRef CAS; (d) E. Godin, A.-C. Bedard, M. Raymond and S. K. Collins, J. Org. Chem., 2017, 82, 7576–7582 CrossRef CAS PubMed; (e) B. S. Chinta and B. Baire, Org. Biomol. Chem., 2017, 15, 5908–5911 RSC.
- (a) X. Li, X. Xie, N. Sun and Y. Liu, Angew. Chem., Int. Ed., 2017, 56, 6994–6998 CrossRef CAS PubMed; (b) S. Banerjee and N. T. Patil, Chem. Commun., 2017, 53, 7937–7940 RSC.
- For a detailed review see: J. P. Brand and J. Waser, Chem. Soc. Rev., 2012, 41, 4165–4179 RSC.
- C.-H. Lee, N.-H. Jeong, S.-W. Lee and T. Kitamura, J. Ind. Eng. Chem., 1997, 3, 155–159 CAS.
- J. Schörgenhumer and M. Waser, Tetrahedron Lett., 2016, 57, 1678–1680 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ob02375a |
| This journal is © The Royal Society of Chemistry 2018 |