
Rh-Catalyzed diastereoselective desymmetrization of enone tethered-cyclohexadienones via tandem arylative cyclization†‡
Rambabu
Chegondi  *ab
*ab
*ab
Abstract
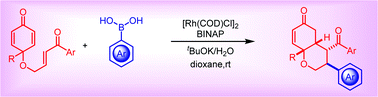
The rhodium-catalyzed arylative cyclization of enone tethered-cyclohexadienones has been developed with high efficiency, thus providing cis-fused bicyclic enones in good yields and with excellent diastereoselectivities. Furthermore, this mild transformation has a broad range of substrate scope and excellent functional group tolerance. In addition, bicyclic products have an enone functionality, which can be a synthetically valuable handle for further transformations.
- This article is part of the themed collections: Synthetic methodology in OBC and New Talent
 Please wait while we load your content...
Please wait while we load your content...

