
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and application of a highly branched, mechanism-based 2-deoxy-2-fluoro-oligosaccharide inhibitor of endo-xyloglucanases†
Namrata
Jain
 ab,
Mohamed A.
Attia
ab,
Wendy A.
Offen
c,
Gideon J.
Davies
c and
Harry
Brumer
*abd
ab,
Mohamed A.
Attia
ab,
Wendy A.
Offen
c,
Gideon J.
Davies
c and
Harry
Brumer
*abd
aMichael Smith Laboratories, University of British Columbia, 2185 East Mall, Vancouver, British Columbia V6T 1Z4, Canada. E-mail: brumer@msl.ubc.ca; Fax: (+1) 6048222114; Tel: (+1) 6048273738
bDepartment of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, British Columbia V6T 1Z1, Canada
cDepartment of Chemistry, University of York, Heslington, York YO10 5DD, UK
dDepartment of Biochemistry and Molecular Biology, University of British Columbia, 2350 Health Sciences Mall, Vancouver, British Columbia V6T 1Z3, Canada
First published on 24th October 2018
Abstract
Xyloglucan (XyG) is a complex polysaccharide that is ubiquitous and often abundant in the cell walls of terrestrial plants. XyG metabolism is therefore a key component of the global carbon cycle, and hence XyG enzymology is of significant fundamental and applied importance in biomass conversion. To facilitate structure–function analyses of XyG-specific endo-glucanases, we have synthesized a 2′,4′-dinitrophenyl 2-deoxy-2-fluoro-β-glycoside mechanism-based inhibitor based on the highly branched XyG repeating motif XXXG (Xyl3Glc4: ([α-D-Xylp-(1→6)]-β-D-Glcp-(1→4)-[α-D-Xylp-(1→6)]-β-D-Glcp-(1→4)-[α-D-Xylp-(1→6)]-β-D-Glcp-(1→4)-D-Glcp. Key steps in the chemo-enzymatic synthesis included selective enzyme hydrolysis of XyG polysaccharide to produce the core heptasaccharide, per-O-acetylation, α-bromination, reductive glycal formation, electrophilic fluorination, SNAr glycosylation, and Zemplen deprotection. The resulting compound, XXXG(2F)-β-DNP, specifically labelled the active sites of several endo-(xylo)glucanases by accumulation of a covalent glycosyl-enzyme intermediate, as revealed by intact protein mass spectrometry. Crystallography of a complex with a Cellvibrio japonicus Glycoside Hydrolase Family 5 (GH5) endo-xyloglucanase corroborated the covalent nature of the intermediate, and further revealed the anticipated specificity for the catalytic nucleophile of this anomeric-configuration-retaining glycosidase. This specificity complements that of an analogous XXXG N-bromoacetylglycosylamine inhibitor, which labelled the catalytic acid–base sidechain in the same enzyme [Attia, et al., Biotechnol. Biofuels, 2018, 11, 45]. We anticipate that these inhibitors may find continued use in mechanistic analyses of endo-(xylo)glucanases from diverse GH families.
Introduction
Terrestrial and marine biomass represents a large and currently underutilized source of carbon in the transition from fossil petroleum to sustainable fuels, chemicals, and materials.1,2 However, the complexity and recalcitrance of terrestrial plant cell walls, in particular, significantly impede the extraction and (bio)chemical transformation of individual component polymers, including cellulose, diverse matrix hetero-polysaccharides (e.g. hemicelluloses and pectins), and lignin. To help overcome inherent limitations in strictly chemical processes, including a lack of precise molecular control, there has been a widespread and sustained interest in the discovery and characterization of efficient enzymes for biomass transformation in the food, feed, and bioproducts industries.3,4 In particular, Glycoside Hydrolases (GHs), which currently comprise over 150 structurally related families in the Carbohydrate-Active Enzymes (CAZymes) database,5 have a particularly rich history of fundamental and applied research. Detailed mechanistic and structural studies have been essential in delineating CAZyme specificity determinants, which in turn underpin the surgical application of individual enzymes and enzyme cocktails in bioprocesses.6–8The use of small molecule glycomimetic inhibitors has been central to GH structure–function analyses in glycobiology.9–11 In particular, irreversible, covalent inhibitors of GHs have been extensively used to identify key substrate-binding and catalytic residues through analytical biochemistry and enzyme crystallography, and to screen for new GHs. A wide array of generally reactive, photo-activatable, or mechanism-based inhibitors based on bespoke glycan specificity motifs has been deployed, including carbasugar-epoxides (e.g. conduritol-β-epoxide, cyclophellitol), cyclopropylcarbasugars, epoxyalkyl glycosides, N-bromoacetylglycosylamines, bromoketone C-glycosides, glucosylthio-hydroquinones, aziridines, cyclosulfates, glycosylmethyl triazenes, activated phenylmethyl glycosides, various photoaffinity labels, and glycosides fluorinated at the 2- or 5-position (see comprehensive reviews and references therein,12–14 and recent primary literature15–19).
Since their introduction by Withers and coworkers 30 years ago,20 2-deoxy-2-fluoroglycosides bearing activated aglycones (and the related 2-deoxy-2,2-difluoroglycosides21–23) have been widely used for mechanistic and structural studies of diverse GHs.14 By virtue of their mechanism-based inhibition and conservative steric substitution, this class of compounds has been exceptionally useful for identifying the catalytic nucleophile by protein mass spectrometry and/or crystallography in GH families that utilize a double-displacement, anomeric-configuration-retaining mechanism (Scheme 1a).24 During catalysis, the presence of the fluorine group at C-2 destabilizes the transition state of both chemical steps, while the incorporation of a good nucleofuge, such as fluorine or 2,4-dinitrophenol (2,4-DNP), increases the rate of leaving group departure sufficiently to enable the accumulation of the 2-deoxyfluoroglycosyl-enzyme intermediate. For some GHs, inhibition is essentially complete, while in others, these inhibitors act as “slow substrates” due to demonstrable turnover to release the free enzyme,25 especially in the presence of sugars as alternate glycosyl acceptor substrates.20,21,26–31
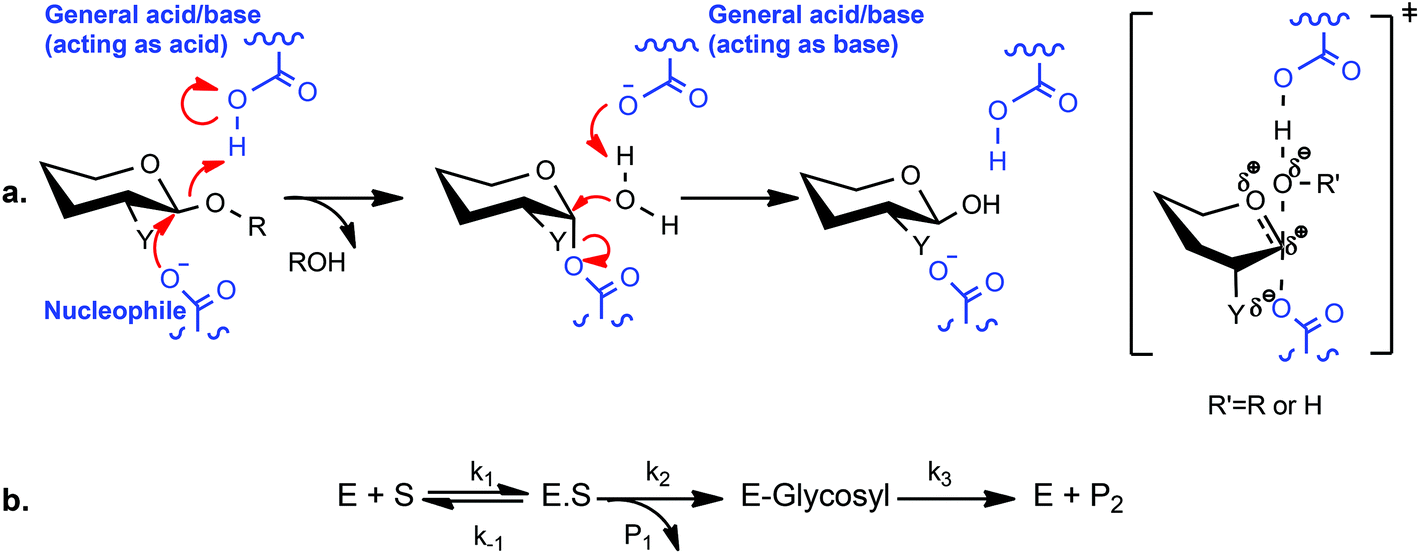 | ||
| Scheme 1 Canonical double-displacement mechanism and kinetic scheme of anomeric-configuration-retaining glycoside hydrolases. In the case of 2-deoxy-2-fluoroglycoside mechanism-based inhibitors (a, Y = F, R = 2,4-dinitrophenyl, e.g. XXXG(2F)-β-DNP), the rate of k3 is reduced relative to k2 (b), resulting in the accumulation of the covalent glycosyl-enzyme intermediate. | ||
The xyloglucans (XyGs) comprise a family of complex heteropolysaccharides, whose members are ubiquitous in land plants,32 where they can constitute up to one-quarter of the dry weight of the primary cell wall.33 A central structural feature of XyGs is a linear β(1,4)-glucan backbone that is regularly substituted with α(1,6)-xylosyl residues, which can be further extended by various other saccharide residues depending on the source tissue.34 In dicot XyGs, the core repeating unit comprises a heptasaccharide motif (Xyl3Glc4), in which three of four contiguous backbone glucosyl units are branched (Fig. 1). Consequently, such XyGs are referred to as “XXXG-type”35 in the standard shorthand, in which “G” represents an unbranched β(1,4)-linked backbone glucosyl residue, and “X” represents the disaccharide motif comprising a β(1,4)-linked backbone glucosyl residue bearing an α(1,6)-linked xylosyl sidechain.36
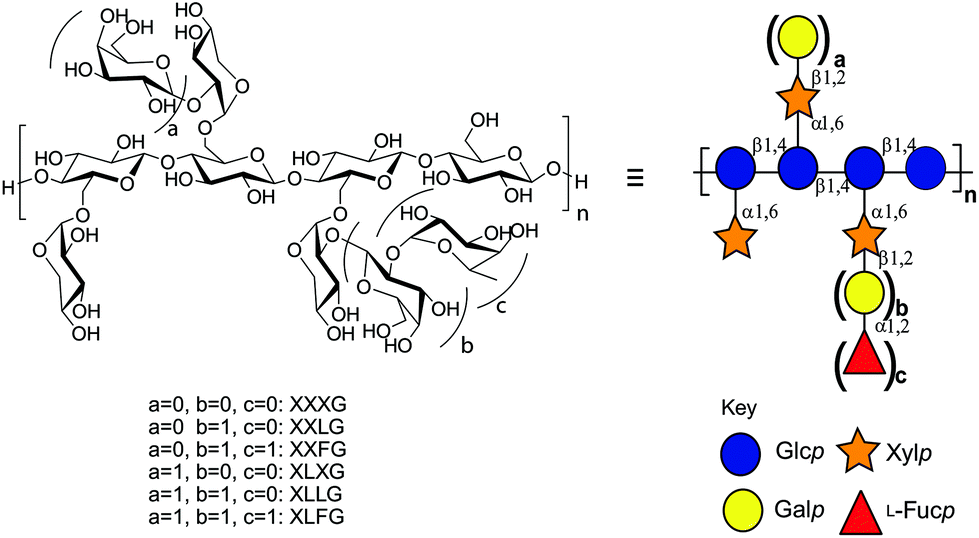 | ||
| Fig. 1 Structure of dicot xyloglucan. Substructure nomenclature is according to Tuomivaara et al.36 and symbols are according to the Consortium for Functional Glycomics.65 | ||
Inspired by the success of 2-deoxy-2-fluoroglycosides as mechanism-base inhibitors and motivated by a long-standing interest in the enzymology of xyloglucan metabolism by plants and microorganisms,37–40 we present here the chemo-enzymatic synthesis of XXXG(2F)-β-DNP as a specific mechanism-based inhibitor of endo-(xylo)glucanases (EC 3.2.1.151). In particular, we demonstrate the application of this compound for the covalent labelling of exemplar configuration-retaining endo-(xylo)glucanases by protein mass spectrometry and crystallography.
Experimental
General synthetic and analytic techniques
All reagents and solvents were analytical or HPLC grade and were purchased from Sigma-Aldrich, Alfa-Aesar or ACROS Organics. For anhydrous reactions, glassware was dried overnight in a 100–150 °C oven and purged with argon prior to use. Solvents were dried by stirring with activated 4 Å molecular sieves overnight under argon.TLC was performed using aluminum sheet TLC plates (0.25 mm) pre-coated with Merck silica gel 60 F254, using ethyl acetate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexanes or water:isopropanol:ethyl acetate as solvent systems (particular solvent ratios specified below), and visualized by a UV lamp and/or 10% sulfuric acid in water with charring using a heat gun. Flash chromatography was performed using Merck silica gel 60 with ethyl acetate:hexanes or water:isopropanol:ethyl acetate as mobile phases. Fractions were analyzed by TLC, and those with the desired compounds were pooled together and evaporated under reduced pressure.
:hexanes or water:isopropanol:ethyl acetate as solvent systems (particular solvent ratios specified below), and visualized by a UV lamp and/or 10% sulfuric acid in water with charring using a heat gun. Flash chromatography was performed using Merck silica gel 60 with ethyl acetate:hexanes or water:isopropanol:ethyl acetate as mobile phases. Fractions were analyzed by TLC, and those with the desired compounds were pooled together and evaporated under reduced pressure.
All 19F-, 13C- and 1H-NMR data were collected on a Bruker Avance 400 MHz spectrometer at room temperature (100.6 MHz and 376.5 MHz for 13C- and 19F-, respectively). The NMR spectra were referenced to solvent as follows: HOD = 4.79 ppm, CHCl3 = 7.27 ppm, 13CHCl3 = 71 ppm, C19FCl3 = 0 ppm. MALDI-MS data were collected on a Bruker Autoflex instrument in reflectron mode over m/z 700–3500 using 6-Aza-2-thiothymine (ATT) as the matrix. HRMS data were obtained using either a Waters Xevo G2-S Q-ToF or Waters/Micromass LCT ToF mass spectrometer in positive-ion mode, via direct infusion through an electrospray ion source.
Synthesis of XXXG(2F)-β-DNP
XXXG heptasaccharide ([α-D-Xylp-(1→6)]-β-D-Glcp-(1→4)-[α-D-Xylp-(1→6)]-β-D-Glcp-(1→4)-[α-D-Xylp-(1→6)]-β-D-Glcp-(1→4)-D-Glcp; abbreviated nomenclature according to Tuomivaara et al.36) was prepared enzymatically from tamarind kernel powder, per-O-acetylated, and converted to the corresponding α-glycosyl bromide (Scheme 2) as per our previously established procedure.41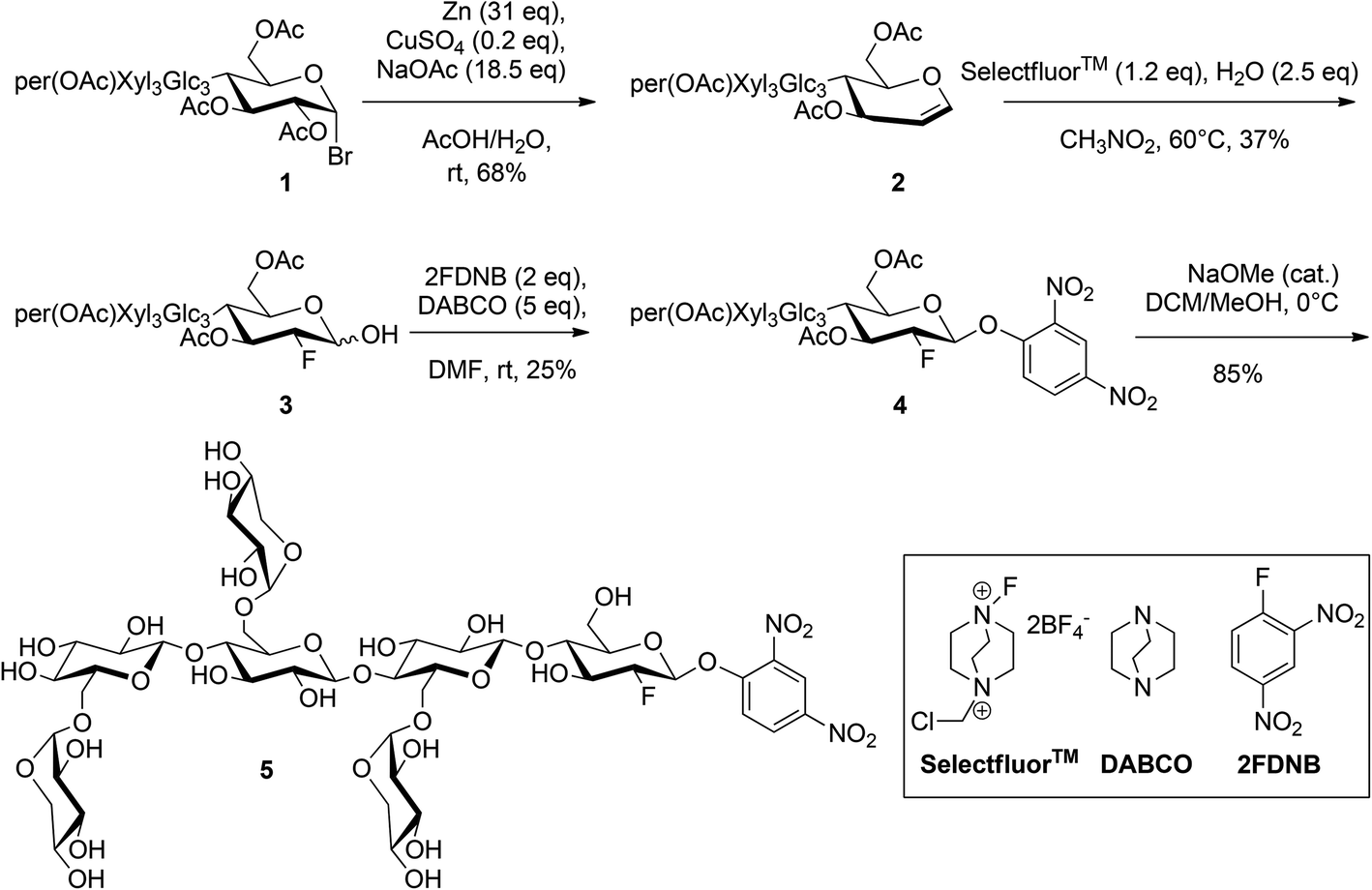 | ||
| Scheme 2 Synthesis of XXXG(2F)-β-DNP (5). | ||
The synthesis of per-O-acetylated XXXG glycal (2) was performed by adapting the method of Xu et al.42 (per-OAc)XXXG α-bromide (1, 1 g) was dissolved in 30 ml in acetic acid. A solution of Zn (31 eq.), NaOAc (18.5 eq.), and CuSO4·5H2O (0.2 eq.) was suspended in 20 mL of water and stirred for 5 min. The solution of 1 in acetic acid was slowly added to the suspension. The reaction was stirred for 3 h at room temperature, and then filtered through a Celite pad. The solvent was concentrated under reduced pressure, re-dissolved in CH2Cl2 and washed with NaHCO3 (3×) and brine (1×). The organic layer was concentrated, and flash chromatography (mobile phase ethyl acetate/hexanes 2:1, Rf = 0.3 in the same solvent mixture) followed by evaporation of the solvent under reduced pressure was used to isolate a white solid (0.63 g, 68% yield). 1H-NMR (Fig. S1,† 400 MHz, CDCl3): δ 6.42 (d, J = 6.14 Hz, 1H, H1), 5.42–5.29 (m, 7H), 5.20–3.63 (m), 1.98–2.03 (m, 54H, COCH3). 13C-NMR (Fig. S2,† 100.6 MHz, CDCl3): δ 170.76, 170.60, 170.41, 170.36, 170.34, 170.24, 170.17, 170.12, 170.09, 170.01, 169.97, 169.88, 169.85, 169.71, 169.63, 169.45, 168.97, 168.68, 168.64 (18 × CO-Ac), 145.58 (C1), 101.07, 100.72, 100.57, 98.24, 97.55, 97.17, 95.94 (C2, 6 × C1), 75.66, 75.36, 74.85, 74.78, 74.45, 74.09, 73.42, 7.38, 72.94, 72.29, 72.06, 71.90, 71.80, 71.70, 70.92, 70.77, 70.49, 70.44, 69.54, 69.49, 69.36, 69.33, 69.23, 69.18, 69.10, 67.45, 67.17 (6 × C2, 7 × C3, 7 × C4, 7 × C5), 65.87, 61.77, 59.22, 58.85 (4 × C6), 20.58–21.01 (18 × CH3). Monoisotopic m/z calculated for C75H100O49K+: 1823.4975; MALDI-ToF MS found: 1823.3. m/z calculated for C75H100O49Na+: 1807.5236; ESI-HRMS found: 1807.5282.
The synthesis of 1-hydroxy-2-deoxy-2-fluoro-(per-OAc)XXXG (3) was adapted from previously reported methods.43,44 Compound 2 (0.63 g) was dissolved in dry nitromethane (20 ml) and 1.2 eq. of Selectfluor™ was added to the solution to create a suspension, which was stirred at room temperature for 6 h, at which time water (2.4 eq.) was added. The solution was then heated to 60 °C and stirred at that temperature for 16 h. The reaction was filtered over Celite to remove the Selectfluor™ by-product and the filtrate was concentrated by rotary evaporation. Elution of the crude product through a silica column (ethyl acetate/hexanes, 3.5:1) to remove polar impurities and remaining by-products yielded a mixture of anomers (yield 0.24 g, 37%), which was used directly in the next step without further separation. 19F-NMR (Fig. S3,† 376.5 MHz, CDCl3) gluco β-anomer: −197.91 (ddd, JH2–F2 = 51.1 Hz, JH3–F2 = 13.7 Hz, JH1–F2 = 1.6 Hz, F2), gluco α-anomer: −199.30 (dd, JH2–F2 = 49.5 Hz, JH3–F2 = 12.0 Hz, JH1–F2 = 0 Hz, F2), consistent with values for the corresponding cellobioside.21,45 Monoisotopic m/z calculated for C75H101FO50Na+: 1843.5248; MALDI-ToF MS found: 1843.3; ESI-HRMS found: 1843.5494.
The synthesis of the per-O-acetylated 2′4′-dinitrophenyl β-glycoside of 2-deoxy-2-fluoro-XXXG (4) was adapted from a previously reported glycosylation method.21 A solution of 2-fluoro-dinitrobenzene (2FDNB, 2 eq.) was dissolved in dry dimethylformamide (DMF) and stirred over activated 4 Å molecular sieves overnight. Crude 3 (0.24 g) was dissolved in dry DMF (10 mL) and under dry conditions 5 eq. of 1,4-diazabicyclo[2.2.2]octane (DABCO) were added to the solution as a solid powder. The reaction was stirred for 15 min, after which it was added to the solution of 2FDNB through a syringe with an oven-dried steel needle. The reaction was stirred for 3.5 h, after which time the molecular sieves were filtered away and the DMF in the filtrate was evaporated under reduced pressure. The solution was re-dissolved in CH2Cl2 and washed with NaHCO3 (3×) and brine (1×). Flash chromatography (mobile phase ethyl acetate/hexanes 2.5:1) was used to isolated the pure compound (TLC Rf = 0.35 in the same solvent mixture) with a yield of 66 mg, 25%. 1H-NMR (Fig. S4,† 400 MHz, CDCl3): δ 8.73 (m, 1H, H′3), 8.45 (m, 1H, H′4), 7.42 (m, 1H, H′5), 5.56 (d, 1H, H1), 5.49–5.24 (m), 4.10–3.42 (m), 1.97–2.16 (m, 54H, COCH3). 13C-NMR (Fig. S5,† 100.6 MHz, CDCl3): δ 170.76, 170.60, 170.41, 170.36, 170.34, 170.24, 170.17, 170.12, 170.09, 170.01, 169.97, 169.88, 169.85, 169.71, 169.63, 169.45, 168.97, 168.68, 168.64 (18 × CO-Ac), 153.32 (C′3), 142.03 (C′4), 140.08 (C′5), 128.63 (C′1), 121.43 (C′2), 116.49 (C′6), 145.58 (C1), 101.07, 100.72, 100.57, 98.24, 97.55, 97.17, 95.94 (C2, 6 × C1), 75.66, 75.36, 74.85, 74.78, 74.45, 74.09, 73.42, 7.38, 72.94, 72.29, 72.06, 71.90, 71.80, 71.70, 70.92, 70.77, 70.49, 70.44, 69.54, 69.49, 69.36, 69.33, 69.23, 69.18, 69.10, 67.45, 67.17 (6 × C2, 7 × C3, 7 × C4, 7 × C5), 65.87, 61.77, 59.22, 58.85 (4 × C6), 20.58–21.01 (18 × CH3). 19F-NMR (Fig. S6,† 376.5 MHz, CDCl3) −194.75 (ddd, JH2–F2 = 47.7 Hz, JH3–F2 = 15.7 Hz, JH1–F2 = 2.5 Hz, F2). Monoisotopic m/z calculated for C81H103FN2O54Na+: 2009.5257; MALDI-ToF MS found: 2009.4; ESI-HRMS found: 2009.5463.
The 2′4′-dinitrophenyl β-glycoside of 2-deoxy-2-fluoro-XXXG (XXXG(2F)-β-DNP (5)) was produced by Zemplen deprotection of 4 (66 mg) in 10 ml methanol/CH2Cl2 9:1, to which 0.5 equivalents of NaOMe (25% in MeOH) were added. The reaction was stirred at 4 °C and monitored by TLC overnight. The product was purified by flash chromatography using water/isopropanol/ethyl acetate (1:3:4) as the mobile phase. The purified product was re-dissolved in water and freeze-dried to give a pale, fluffy powder in 85% yield (34 mg). 1H-NMR (Fig. S7,† 400 MHz, CDCl3): δ 8.73 (m, 1H, H′3), 8.45 (m, 1H, H′4), 7.42 (m, 1H, H′5), 5.56 (d, 1H, H1), 5.49–5.24 (m), 4.10–3.42 (m), 1.97–2.16 (m, 54H, COCH3). 13C-NMR (Fig. S8,† 100.6 MHz, CDCl3): δ 170.76, 170.60, 170.41, 170.36, 170.34, 170.24, 170.17, 170.12, 170.09, 170.01, 169.97, 169.88, 169.85, 169.71, 169.63, 169.45, 168.97, 168.68, 168.64 (18 × CO-Ac), 153.32 (C′3), 142.03(C′4), 140.08 (C′5), 128.63 (C′1), 121.43 (C′2), 116.49(C′6), 145.58 (C1), 101.07, 100.72, 100.57, 98.24, 97.55, 97.17, 95.94 (C2, 6 × C1), 75.66, 75.36, 74.85, 74.78, 74.45, 74.09, 73.42, 7.38, 72.94, 72.29, 72.06, 71.90, 71.80, 71.70, 70.92, 70.77, 70.49, 70.44, 69.54, 69.49, 69.36, 69.33, 69.23, 69.18, 69.10, 67.45, 67.17 (6 × C2, 7 × C3, 7 × C4, 7 × C5), 65.87, 61.77, 59.22, 58.85 (4 × C6), 20.58–21.01 (18 × CH3). 19F-NMR (Fig. S9,† 376.5 MHz, CDCl3) −201.29 (ddd, JH2–F2 = 51.4 Hz, JH3–F2 = 15.2 Hz, JH1–F2 = 2.5 Hz, F2). Monoisotopic m/z calculated for C45H67FN2O36Na+: 1253.3360; MALDI-ToF MS found: 1253.3; ESI-HRMS found: 1253.3354.
endo-Xyloglucanase production
The endo-xyloglucanases Cellvibrio japonicus GH5D (CjGH5D),46Cellvibrio japonicus GH74 (CjGH74),47Bacteroides ovatus GH5 (BoGH5),38 and Prevotella bryantii GH5 (PbGH5)48 were produced recombinantly and purified as previously described.The catalytic acid/base mutant CjGH5D(E255A) was generated using the PCR-based QuickChange II Site-Directed Mutagenesis Kit (Agilent, USA) following the manufacturer's protocol. PCR amplification was conducted using the forward primer 5′-TTTGCCGGCACTAACGCCCCCAATGCGGAAAAT-3′ and the reverse primer 5′-ATTTTCCGCATTGGGGGCGTTAGTGCCGGCAAA-3′, utilizing pET28a::CjGH5D46 as the template. The resulting plasmid pET28a:: CjGH5D(E255A) was sequenced to confirm the desired mutation.
Chemically competent E. coli Rosetta DE3 cells were transformed with the plasmid pET28a:: CjGH5D(E255A). The resulting colonies were grown on LB solid medium containing kanamycin (50 μg mL−1) and chloramphenicol (30 μg mL−1). Gene overexpression and recombinant protein purification were then performed as described for the wild-type enzyme.46 The concentration of the purified recombinant CjGH5D(E255A) was determined using an Epoch Micro-Volume Spectrophotometer System (BioTek®, USA) at 280 nm. The presence of the desired mutation was confirmed by intact protein mass spectrometry.49 The purified protein was aliquoted and stored at −80 °C until needed.
Screening for active-site labelling
Individual 20 μL solutions of CjGH5D, PbGH5, BoGH5, CjGH74, and CjGH5D(E255A) (2.3 μM) in 0.5 mM sodium phosphate buffer, pH 7.0, containing 2.5 mM XXXG(2F)-β-DNP were incubated for 12 h. Intact protein masses were determined on a Waters Xevo LC-ESI-MS Q-ToF with a NanoAcuity UPLC system essentially as previously described,49 and analyzed using the software Masslynx 4.0.Inhibition kinetics measurements
All kinetic data were obtained using an Agilent Cary 60 UV-Vis Spectrophotometer equipped with a Peltier temperature-controlled cell holder. The data were fit by linear least-squares analysis using the bundled Cary Kinetics software or nonlinear regression using Origin 8 (OriginLab, Northampton, MA, USA).To measure time-dependent enzyme inactivation, 2.5 mM of XXXG(2F)-β-DNP (5) was incubated with 20 μM CjGH5D in 50 mM sodium phosphate buffer, pH 7.5, in a total volume of 100 μL (also including 0.1 mg mL−1 bovine serum albumin (BSA) to prevent non-specific loss of activity) for a period of 420 minutes at 40 °C. A control experiment was run in parallel, in which inhibitor was omitted from the buffered enzyme/BSA solution. Periodically, 10 μL of each solution was withdrawn and diluted 1:100 in 50 mM sodium phosphate buffer, pH 7.5, and 100 μL of the diluted solution was added to 100 μL of 0.4 mM XXXG-β-CNP,41 which was previously dissolved in water and preincubated at 40 °C. Linear initial-rate kinetics of 2-chloro-4-nitrophenolate release were measured at 405 nm (ε = 17.74 mM−1 cm−1) over 1 min in a quartz cuvette (l = 1 cm) maintained at 40 °C, essentially as previously described.46
Prior to measuring burst kinetics, the background hydrolysis rate of 235 μL of 5 mM XXXG(2F)-β DNP (5) in 50 mM phosphate buffer, pH 7.5, was measured at 40 °C for 75 min (ε405 = 11.17 mM−1 cm−1, determined using a standard curve of absorbance as a function of dinitrophenolate concentration at the specified buffer concentration and pH conditions. To make the standard solution, 2,4-dinitrophenol was desiccated overnight in presence of phosphorus pentoxide, and thereafter dissolved in the specified buffer). Thereafter, CjGH5D (15 μL, 0.45 mM), which had been preincubated at 40 °C in the same buffer, was added to the cuvette and mixed rapidly (final volume 250 μL, final enzyme concentration 27 μM). The rate of dinitrophenolate (DNP−) release was monitored for an additional 700 min and eqn (1)25 was fit to the data using Origin 8 software, according to the inhibition mechanism shown in Scheme 1b. The cuvette was covered with parafilm to limit evaporation during the entire course of the experiment.
| [DNP−] = [DNP−]0(1−e−k2t) + k3t | (1) |
X-ray crystallography and structure solution
CjGH5D (E255A) was crystallized with protein at 20 mg mL−1 in 20 mM sodium phosphate buffer, pH 7.0, containing 10% (v/v) glycerol, by employing similar conditions to those used to crystallize the wild-type protein.46 A crystal grown from a protein stock solution supplemented with 5 mM inhibitor was harvested directly into liquid nitrogen using a CryoLoop™ (Hampton Research, Aliso Viejo, CA, USA). In addition, an unliganded crystal was soaked with 2 mM inhibitor for 45 min, and harvested similarly.Data were collected at Diamond beamline IO3 and processed using XDS50 for the co-crystallised complex and collected at IO4-1 and processed using DIALS51 for the soaked complex. Both datasets were put through the data reduction pipeline in ccp4i252 which uses POINTLESS, AIMLESS and CTRUNCATE,53 and cut-off at resolutions of 1.7 and 2.0 Å (judged by Rpim < 0.60 and CC1/2 > 0.50 in outer bin), respectively (ESI Table S1†). Both structures were solved using the unliganded structure of the wild-type enzyme (PDB ID 5OYC) as the model for refinement with REFMAC.54 After refinement with water molecules added, the ligands were built into difference electron density in the weighted 2Fo − Fc maps using COOT,55 and validated using PRIVATEER56 prior to deposition at the Protein Data Bank with accession codes 6HAA and 6HA9.
Results and discussion
Inhibitor synthesis
The synthesis of the target compound XXXG(2F)-β DNP (5) was accomplished using a linear synthetic scheme starting from the heptasaccharide XXXG (Scheme 2). XXXG was produced via sequential endo-xyloglucanase and β-galactosidase hydrolysis of tamarind kernel XyG, per-O-acetylated, and converted to the corresponding α-bromide as per our established procedure.41 Transformation of per-O-acetyl XXXG α-bromide to the corresponding glycal via a reductive elimination was straightforward using Zn metal and CuSO4 in the presence of NaOAc/HOAc in water.42The crucial step of installing fluorine at the 2-position of the protected XXXG glycal was achieved using the electrophilic fluorinating agent Selectfluor™, followed by hydrolytic work-up to obtain the corresponding protected 1-hydroxy-2-deoxy-2-fluoro-oligosaccharide. Selectfluor™ has been widely used for the regiospecific 2-fluorination of a number of glycals.57 In the case of D-glucal, an equimolar mixture of gluco and manno 2F-epimers was obtained, while in the case of the corresponding disaccharides, e.g. cellobiose, the amount of the 2F-gluco epimer reached 80% in polar solvents.44,45 In our case, we were pleased to discover that only the desired gluco configured product was obtained in dry nitromethane,43 albeit as a mixture of α/β anomers, as indicated by 19F NMR (trace amounts of manno epimers were perhaps also present,45 Fig. S3†). As this represents the only example, to our knowledge, of the application of Selectfluor™ to such a large oligosaccharide glucal, the origins of this high stereoselectivity are not fully clear, but may be based in increased local steric congestion or altered access to boat-conformer addition products.45
The reaction of the anomeric mixture with 2,4-dinitrofluorobenzene in the presence of DABCO yielded exclusively the desired β-configured, kinetic product 4,58,59 as ascertained by the large H1–H2 coupling of 7.6 Hz and F2–H1 coupling of 3.0 Hz. With the key stereochemistry set, careful Zemplen deprotection followed by column chromatography yielded pure 5.
Active-site labelling of endo-xyloglucanases
To examine the efficacy of XXXG(2F)-β-DNP (5) as an inhibitor, we incubated a high concentration (2.5 mM) of the compound with three configuration-retaining GH5 endo-(xylo)glucanases with differing specificity profiles: BoGH5 from the human gut bacterium Bacteroides ovatus,38PbGH5 from the ruminant bacterium Prevotella bryantii,48 and CjGH5D from the soil saprophyte Cellvibrio japonicus.46 The configuration-inverting GH74 endo-xyloglucanase from Cellvibrio japonicus, CjGH74,47 which is not expected to be covalently inhibited because it uses a single step, direct water-attack mechanism,60 was also examined as a negative control. As anticipated, we observed time-dependent, 1:1 stoichiometric labelling of all three configuration-retaining GH5 enzymes by intact protein mass spectrometry, while CjGH74 remained entirely unmodified during a 12 h incubation (Fig. S1†). On the basis of this semi-quantitative analysis, the strict endo-xyloglucanase BoGH5 was completely labelled by 1 h (Fig. S10a†) and the promiscuous mixed-linkage β(1–3)/β(1–4)-endo-glucanase/endo-xyloglucanase PbGH5 showed near-complete labelling by 9 h (Fig. S10b†). In contrast, the predominant endo-xyloglucanase CjGH5D was only partially labelled after a 12 h incubation (Fig. S10c†). Further analysis, detailed below, revealed that this was due to turnover of the fluoroglycosyl-enzyme intermediate.
Kinetics of inhibition of CjGH5D with XXXG(2F)-β-DNP
To understand better the mechanism of inhibition of CjGH5D with XXXG(2F)-β-DNP, we performed kinetics measurements at the same high concentration of compound used in initial intact protein MS analyses. As shown in Fig. 2a, a time-dependent loss of activity toward the chromogenic substrate XXXG-β-2′-chloro-4′-nitrophenol is observed following incubation with 2.5 mM XXXG(2F)-β-DNP. However, it was consistently observed that the activity of the enzyme was not completely lost, even after extended incubation (e.g. 420 min, Fig. 2a). Similar results were observed in preliminary screening experiments using lower and higher inhibitor concentrations (up to 10 mM) with lengthy incubation times (e.g. 18 h, data not shown). These observations suggested the kinetically relevant turnover of the fluoroglycosyl-enzyme intermediate, as has been reported previously for other combinations of 2-deoxy-2-fluoroglycoside inhibitors and enzymes.20,21,26–31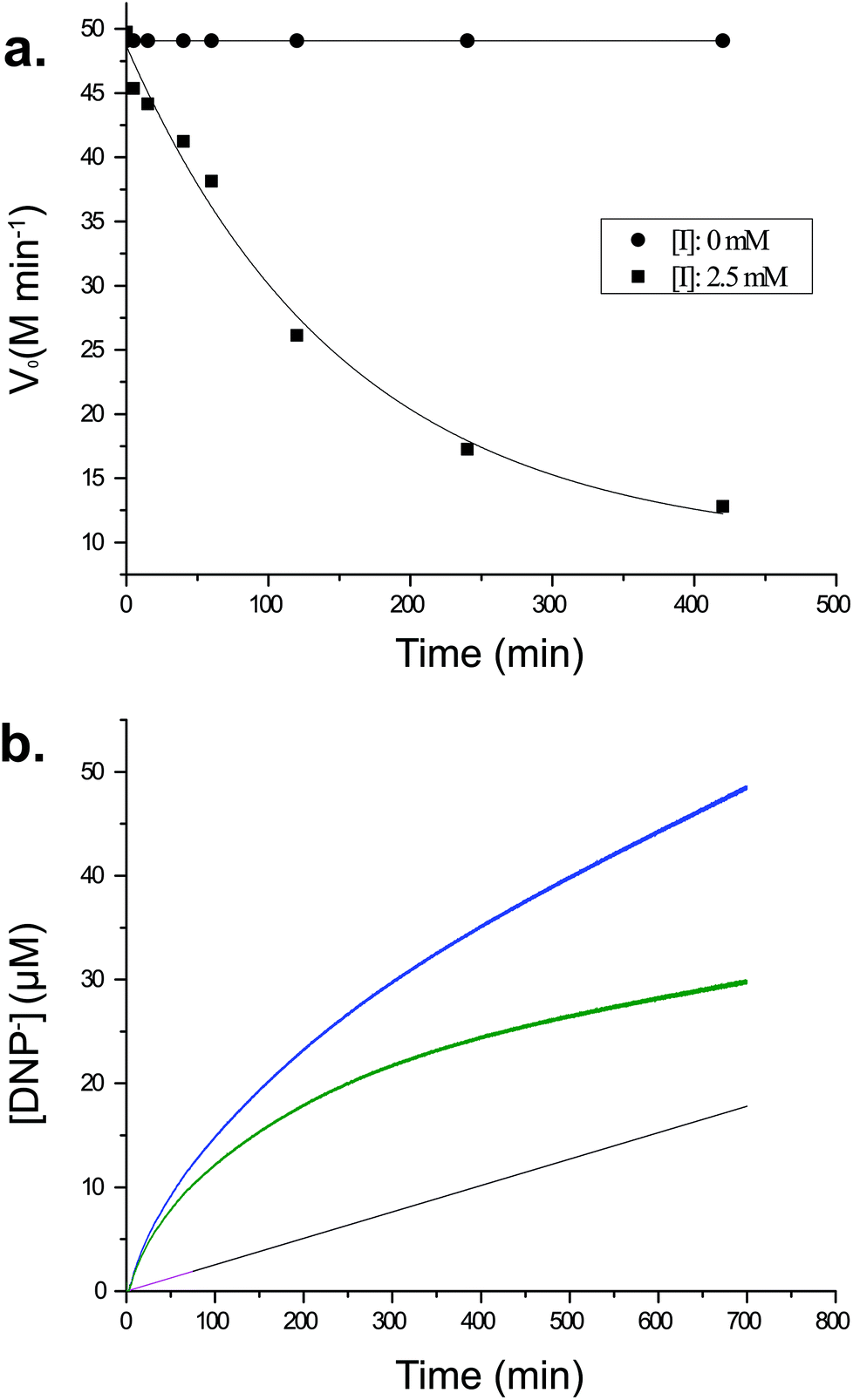 | ||
| Fig. 2 Inhibition of CjGH5D with XXXG(2F)-β-DNP. a. Time-dependent loss of activity upon incubation with 5. b. Real-time burst kinetics. Pink line: spontaneous hydrolysis of 5 over 75 minutes, with linear extrapolation shown in black. Blue line: 2,4-dinitrophenylate release after addition of CjGH5D. Green line: enzymic release of 2,4-dinitrophenylate obtained after subtraction of spontaneous hydrolysis. | ||
Further evidence to suggest turnover of the glycosyl enzyme was obtained by the observation of apparent pre-steady-state burst kinetics, by monitoring the release of 2,4-dinitrophenolate over time (Fig. 2b). The biphasic data were fit by eqn (1), which describes an initial pseudo-first-order exponential accumulation of the fluoroglycosyl-enzyme, followed by a linear phase dominated by steady-state turnover of the covalent intermediate (Fig. 2b).25 After accounting for an effectively linear background hydrolysis rate of 5 in buffer (0.28 × 10−3 min−1), values for k2 and k3 of 7.83 × 10−3 min−1 and 0.16 × 10−3 min−1, respectively, were obtained. The concentration of enzyme active sites, [Eo] = 17.60 μM, obtained from the magnitude of the burst (A405 = 0.20, DNP ε405 = 11.17 mM−1 cm−1) suggested that the CjGH5D preparation was not fully active ([P] = 27 μM in the assay).
Crystallography of the XXXG(2F)-CjGH5D(E255A) covalent complex
The turnover of the fluoroglycosyl-enzyme (Fig. 3b) precluded the observation of a covalent complex by protein crystallography, despite MS evidence of its accumulation (Fig. S10†). To overcome this problem,30 we produced a site-directed mutation of the general acid/base residue Glu255 to alanine in order to further reduce the rate of hydrolysis of the fluoroglycosyl-enzyme intermediate (Fig. S10d†). Our first attempt to capture this intermediate was by soaking of E255A crystals with the inhibitor, which surprisingly led to an unreacted complex; with a −3 to +1 binding mode of the glycone with the aryl glycosidic bond still intact (Fig. S11†). For this reason, co-crystallisation was instead used to access the reacted tertiary structure of XXXG(2F)-CjGH5D(E255A) at 1.7 Å resolution (Fig. 3). The overall structures corresponded well to that of the wild-type enzyme in free, or “apo”, form (PBD ID 5OYC), which has a classic (β/α)8-barrel fold.46 Crystallographic data collection and refinement statistics are given in Table S1.† Privateer results showing validation for Glc (BGC) and Xyl (XYS) residues in XXXG(2F)-CjGH5D(E255A) covalent and XXXG(2F)-β-DNP are given in Table S2.†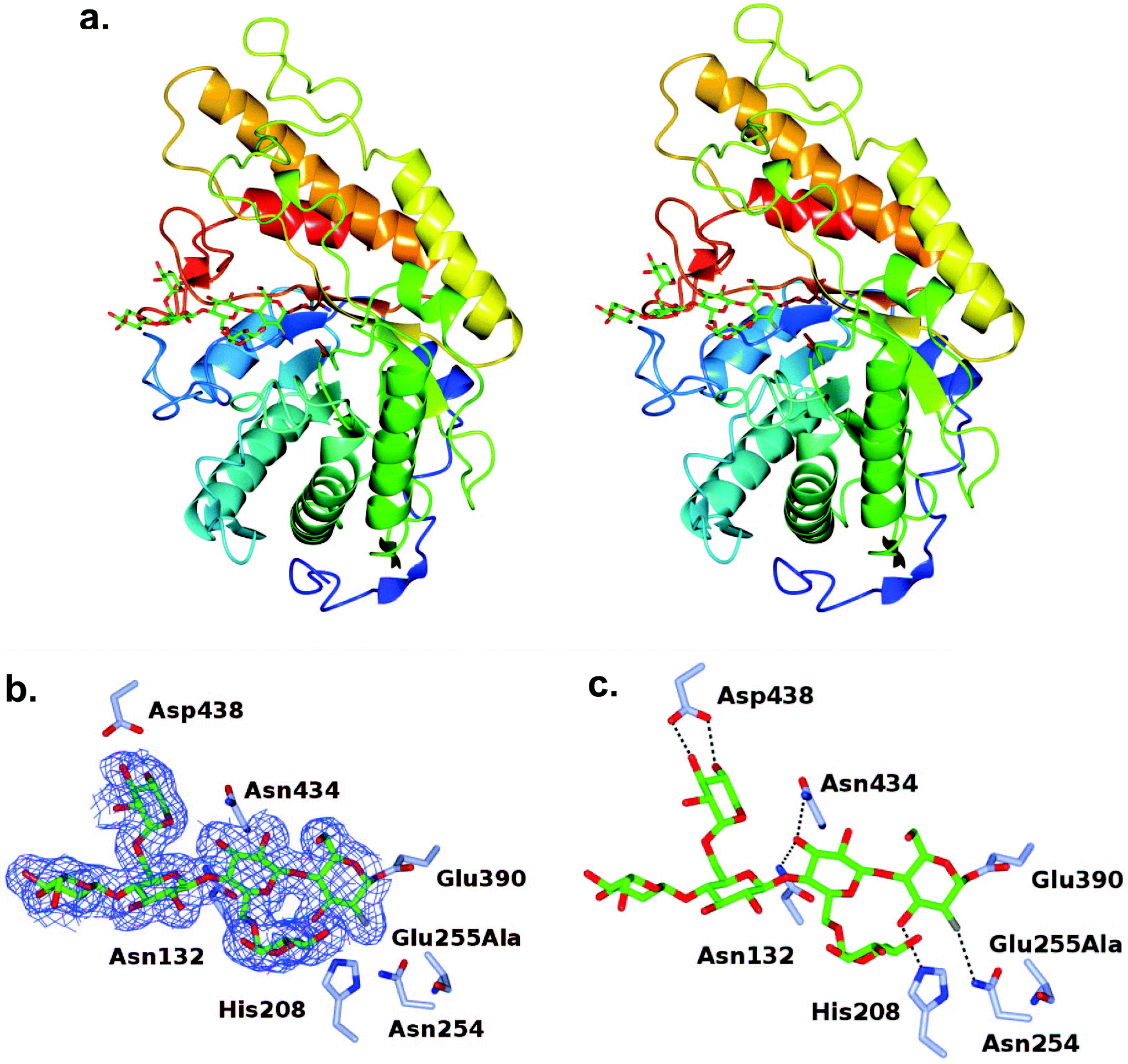 | ||
| Fig. 3 Tertiary structure of the covalent fluoroglycosyl-enzyme intermediate formed by cocrystallization of CjGH5D(E255A) with XXXG(2F)-β-DNP. a. Divergent (wall-eyed) stereo cartoon representation of the secondary structure, colour ramped from the N-terminus (blue) to the C-terminus (red), with the inhibitor represented as sticks with C atoms in green, and the catalytic residues with C atoms in tan. b: Maximum-likelihood/σA weighted 2Fobs − Fcalc electron density map contoured at an r.m.s.d. level of 1 σ for the ligand XXXG-2F. c: Hydrogen bonding interactions with the ligand. In panels b and c, side chains of interacting residues are shown in ice blue and hydrogen bonds are shown as dashed lines. | ||
In the co-crystallized complex, electron density corresponding to the near-complete glycone was observed spanning the −4 to −1 subsites61 within the active-site cleft, with the exception of the non-reducing-terminal xylosyl residue that was not modelled. In keeping with the mechanism-based design of the inhibitor, the electron density clearly revealed the covalent attachment of C1 of the 2-fluoroglucosyl residue in subsite −1 with Oε1 of the catalytic nucleophile Glu390 (distance ∼1.4 Å). The 2-fluoroglucosyl residue was in a relaxed 4C1 conformation, while the sidechain of Glu390 was rotated from its position in the apo enzyme structure, including an average 1.1 Å translation of Oε1 toward the sugar ring (Fig. 4).
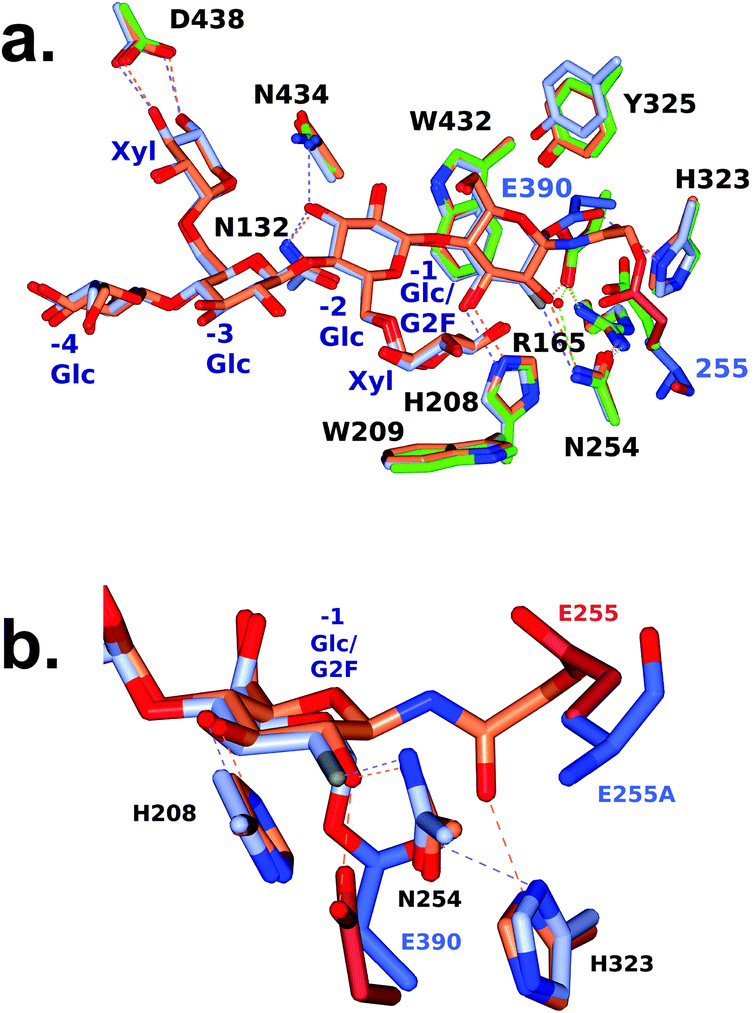 | ||
| Fig. 4 Active-site superposition of CjGH5D and corresponding covalent inhibitor complexes. a. Full view b. Close-up of the −1 subsite. Side chains are shown with carbon atoms coloured as follows: XXXG-NHAc-CjGH5D covalent complex (PDB ID: 5OYD) in coral, and XXXG(2F)-CjGH5D(E255A) covalent complex (PDB ID: 6HAA) in ice blue, and with catalytic residue carbon atoms shown in pale crimson and light blue respectively. Main chain atoms are shown for E255A for clarity. | ||
We previously determined the structure of a covalent complex of wild-type CjGH5D with the N-bromoacetylglycosylamine inhibitor XXXG-NHAcBr, in which the catalytic acid–base residue was labelled by substitution of the bromide (PDB ID 5OYD).46 Superposition of XXXG-NHAc-CjGH5D with the XXXG(2F)-CjGH5D(E255A) glycosyl-enzyme complex revealed the basis for the distinct specificity of the two inhibitor types (Fig. 4). Whereas the anomeric carbon of Glc-1 in the XXXG(2F)-β-DNP mechanism-based inhibitor was situated directly above the catalytic nucleophile (Glu390), the three-atom extension of the N-bromoacetyl group placed the α-carbon of the “warhead” in a position suitable for side-on attack by the catalytic acid/base (Glu255), and effectively out of reach of Glu390. Moreover, this latter reaction required the displacement of the sidechain of Glu255 (Cδ) by an additional 2 Å away from the ring atoms of Glc-1 to accommodate the reactive moiety. These steric and sidechain plasticity requirements are likely to underlie the current and previous observations that N-bromoacetylglycosylamine reagents tend to label catalytic acid/base residues in glycosidases (except in the case of off-active-site labelling62).
Conclusion
The chemo-enzymatic synthesis of the heptasaccharide XXXG(2F)-β-DNP has enabled the production of one of the most structurally complex 2-deoxy-2-fluorosugar mechanism-based inactivators to date. Possessing active-site-residue specificity advantages over analogous xyloglucan oligosaccharide bromoketone C-glycoside and N-bromoacetylglycosylamine inhibitors,63 we anticipate that XXXG(2F)-β-DNP may find continued use in structure–function analyses of endo-(xylo)glucanases from diverse GH families. In particular, N-bromoacetylglycosylamines generally appear to react with catalytic acid–base residues in configuration-retaining GHs,46,48,64 contrasting the exclusive catalytic nucleophile specificity of the Withers-type fluorosugar inhibitors.14Author contributions
NJ performed the chemo-enzymatic synthesis of inhibitor 5, as well as the active site labelling experiments. MAA performed recombinant protein production and site-directed mutagenesis of CjGH5D. WAO solved the crystal structures, which were analyzed under the supervision of GJD. HB devised the overall study and supervised research. All authors contributed to writing the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Nicholas McGregor and Dr. Gregory Arnal (Brumer Lab, UBC) for providing PbGH5 and BoGH5, respectively. NJ thanks Dr. Hongming Chen (Withers Lab, UBC) for assistance with 19F-NMR interpretation. Work in Vancouver was supported by NSERC via a Discovery Grant and a Strategic Partnership Grant for Networks (Industrial Biocatalysis Network), the Canada Foundation for Innovation, and the British Columbia Knowledge Development Fund. Work in York was supported by the BBSRC (grant BB/R001162/1). GJD holds the Royal Society Ken Murray Research Professorship. We thank Diamond Light Source for access to beamlines I03 and I04-1 (proposal number mx-13587) that contributed to the results presented here. We are grateful to Johan Turkenburg and Sam Hart for their assistance with synchrotron X-ray data collection. Waters Corporation is gratefully acknowledged for the provision of the LC-MS system used in this study.References
- J. K. Saini, R. Saini and L. Tewari, 3 Biotech, 2015, 5, 337–353 CrossRef PubMed.
- V. I. Popa, in Biomass as Renewable Raw Material to Obtain Bioproducts of High-Tech Value, ed. V. Popa and I. Volf, Elsevier, Cambridge, 2018, pp. 1–37 Search PubMed.
- E. M. Rubin, M. E. Himmel, S. Ding, D. K. Johnson and W. S. Adney, Nature, 2007, 454, 804–807 Search PubMed.
- J. Sun, S. Ding and J. Doran-Peterson, in Biological Conversion of Biomass for Fuels and Chemicals: Explorations from Natural Utilization Systems, ed. J. Sun, S. Ding and J. Doran-Peterson, The Royal Society of Chemistry, Cambridge, 2014, pp. 1–13 Search PubMed.
- V. Lombard, H. Golaconda Ramulu, E. Drula, P. M. Coutinho and B. Henrissat, Nucleic Acids Res., 2014, 42, D490–D495 CrossRef CAS PubMed.
- G. J. Davies and B. Henrissat, Structure, 1995, 3, 853–859 CrossRef CAS PubMed.
- J. D. Willis, M. Mazarei and C. N. Stewart, Front. Plant Sci., 2016, 7, 1–18 Search PubMed.
- S. J. Horn, G. Vaaje-Kolstad, B. Westereng and V. G. Eijsink, Biotechnol. Biofuels, 2012, 5, 45 CrossRef CAS PubMed.
- N. Asano, R. J. Nash, R. J. Molyneux and G. W. J. Fleet, Tetrahedron: Asymmetry, 2000, 11, 1645–1680 CrossRef CAS.
- G. Legler, in Advances in Carbohydrate Chemistry and Biochemistry, ed. S. Tipson, T. Derek and B. Horton, Academic Press, Cambridge, 1990, vol. 48, pp. 319–384 Search PubMed.
- T. M. Gloster and D. J. Vocadlo, Nat. Chem. Biol., 2012, 8, 683–694 CrossRef CAS PubMed.
- Y. Xu, N. Uddin and G. K. Wagner, in Methods in Enzymology, ed. A. M. Pyle and D. W. Christianson, Elsevier Inc., Cambridge, 1st edn, 2018, vol. 598, pp. 237–265 Search PubMed.
- L. I. Willems, J. Jiang, K. Y. Li, M. D. Witte, W. W. Kallemeijn, T. J. N. Beenakker, S. P. Schröder, J. M. F. G. Aerts, G. A. van der Marel, J. D. C. Codée and H. S. Overkleeft, Chemistry, 2014, 20, 10864–10872 CrossRef CAS PubMed.
- B. P. Rempel and S. G. Withers, Glycobiology, 2008, 18, 570–586 CrossRef CAS PubMed.
- J. Jiang, T. J. M. Beenakker, W. W. Kallemeijn, G. A. van der Marel, H. van den Elst, J. D. C. Codée, J. M. F. G. Aerts and H. S. Overkleeft, Chem. – Eur. J., 2015, 21, 10861–10869 CrossRef CAS PubMed.
- S. Chakladar, Y. Wang, T. Clark, L. Cheng, S. Ko, D. J. Vocadlo and A. J. Bennet, Nat. Commun., 2014, 5, 5590 CrossRef CAS PubMed.
- M. Artola, L. Wu, M. J. Ferraz, C. L. Kuo, L. Raich, I. Z. Breen, W. A. Offen, J. D. C. Codée, G. A. Van Der Marel, C. Rovira, J. M. F. G. Aerts, G. J. Davies and H. S. Overkleeft, ACS Cent. Sci., 2017, 3, 784–793 CrossRef CAS PubMed.
- C. Adamson, R. J. Pengelly, S. Shamsi Kazem Abadi, S. Chakladar, J. Draper, R. Britton, T. M. Gloster and A. J. Bennet, Angew. Chem., Int. Ed., 2016, 55, 14978–14982 CrossRef CAS PubMed.
- W. Ren, R. Pengelly, M. Farren-Dai, S. Shamsi Kazem Abadi, V. Oehler, O. Akintola, J. Draper, M. Meanwell, S. Chakladar, K. Świderek, V. Moliner, R. Britton, T. M. Gloster and A. J. Bennet, Nat. Commun., 2018, 9, 1–12 CrossRef PubMed.
- S. G. Withers, K. Rupitz and I. P. Street, J. Biol. Chem., 1988, 263, 17–20 Search PubMed.
- J. D. McCarter, M. J. Adam, C. Braun, M. Namchuk, D. Tull and S. G. Withers, Carbohydr. Res., 1993, 249, 77–90 CrossRef CAS PubMed.
- C. Braun, G. D. Brayer and S. G. Withers, J. Biol. Chem., 1995, 270, 26778–26781 CrossRef CAS PubMed.
- D. O. Hart, S. He, C. J. Chany, S. G. Withers, P. F. G. Sims, M. L. Sinnott and H. Brumer, Biochemistry, 2000, 39, 9826–9836 CrossRef CAS PubMed.
- S. Withers and R. Aebersold, Protein Sci., 1995, 4, 361–372 CrossRef CAS PubMed.
- I. P. Street, J. B. Kempton and S. G. Withers, Biochemistry, 1992, 31, 9970–9978 CrossRef CAS PubMed.
- S. Miao, L. Ziser, R. Aebersold and S. G. Withers, Biochemistry, 1994, 33, 7027–7032 CrossRef CAS PubMed.
- J. E. Blanchard and S. G. Withers, Chem. Biol., 2001, 8, 627–633 CrossRef CAS PubMed.
- S. G. Withers, A. J. Warren, I. P. Street, K. Rupitz, J. B. Kempton and R. Aebersold, J. Am. Chem. Soc., 1990, 112, 5887–5889 CrossRef CAS.
- J. C. Gebler, R. Aebersold and S. G. Withers, J. Biol. Chem., 1992, 267, 11126–11130 CAS.
- D. J. Vocadlo, G. J. Davies, R. Laine and S. G. Withers, Nature, 2001, 412, 835–838 CrossRef CAS PubMed.
- A. G. Watts, I. Damager, M. L. Amaya, A. Buschiazzo, P. Alzari, A. C. Frasch and S. G. Withers, J. Am. Chem. Soc., 2003, 125, 7532–7533 CrossRef CAS PubMed.
- Z. A. Popper, Curr. Opin. Plant Biol., 2008, 11, 286–292 CrossRef CAS PubMed.
- H. V. Scheller and P. Ulvskov, Annu. Rev. Plant Biol., 2010, 61, 263–289 CrossRef CAS PubMed.
- M. Pauly and K. Keegstra, Annu. Rev. Plant Biol., 2016, 67, 235–259 CrossRef CAS PubMed.
- J.-P. Vincken, W. S. York, G. Beldman and A. G. J. Voragen, Plant Physiol., 1997, 114, 9–13 CrossRef CAS PubMed.
- S. T. Tuomivaara, K. Yaoi, M. A. O'Neill and W. S. York, Carbohydr. Res., 2015, 402, 56–66 CrossRef CAS PubMed.
- M. A. Attia and H. Brumer, Curr. Opin. Struct. Biol., 2016, 40, 43–53 CrossRef CAS PubMed.
- J. Larsbrink, T. E. Rogers, G. R. Hemsworth, L. S. McKee, A. S. Tauzin, O. Spadiut, S. Klinter, N. A. Pudlo, K. Urs, N. M. Koropatkin, A. L. Creagh, C. A. Haynes, A. G. Kelly, S. N. Cederholm, G. J. Davies, E. C. Martens and H. Brumer, Nature, 2014, 506, 498–502 CrossRef CAS PubMed.
- J. M. Eklöf and H. Brumer, Plant Physiol., 2010, 153, 456–466 CrossRef PubMed.
- J. M. Eklöf, M. C. Ruda and H. Brumer, Methods Enzymol., 2012, 510, 97–120 Search PubMed.
- F. M. Ibatullin, M. J. Baumann, L. Greffe and H. Brumer, Biochemistry, 2008, 47, 7762–7769 CrossRef CAS PubMed.
- X. Xu, Q. Tan and M. Hayashi, Synthesis, 2008, 770–776 CAS.
- S. P. Vincent, M. D. Burkart, C. Y. Tsai, Z. Zhang and C. H. Wong, J. Org. Chem., 1999, 64, 5264–5279 CrossRef CAS.
- M. Albert, D. Karl and J. Ortner, Tetrahedron, 1998, 54, 4839–4848 CrossRef CAS.
- J. Ortner, M. Albert, H. Weber and K. Dax, J. Carbohydr. Chem., 1999, 18, 297–316 CrossRef CAS.
- M. A. Attia, C. E. Nelson, W. A. Offen, N. Jain, G. J. Davies, J. G. Gardner and H. Brumer, Biotechnol. Biofuels, 2018, 11, 45–61 CrossRef PubMed.
- M. Attia, J. Stepper, G. J. Davies and H. Brumer, FEBS J., 2016, 283, 1701–1719 CrossRef CAS PubMed.
- N. McGregor, M. Morar, T. H. Fenger, P. Stogios, N. Lenfant, V. Yin, X. Xu, E. Evdokimova, H. Cui, B. Henrissat, A. Savchenko and H. Brumer, J. Biol. Chem., 2015, 291, 1175–1197 CrossRef PubMed.
- G. Sundqvist, M. Stenvall, H. Berglund, J. Ottosson and H. Brumer, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 852, 188–194 CrossRef CAS PubMed.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed.
- D. G. Waterman, G. Winter, R. J. Gildea, J. M. Parkhurst, A. S. Brewster, N. K. Sauter and G. Evans, Acta Crystallogr., Sect. D: Struct. Biol., 2016, 72, 558–575 CrossRef CAS PubMed.
- L. Potterton, J. Agirre, C. Ballard, K. Cowtan, E. Dodson, P. R. Evans, H. T. Jenkins, R. Keegan, E. Krissinel, K. Stevenson, A. Lebedev, S. J. McNicholas, R. A. Nicholls, M. Noble, N. S. Pannu, C. Roth, G. Sheldrick, P. Skubak, J. Turkenburg, V. Uski, F. Von Delft, D. Waterman, K. Wilson, M. Winn and M. Wojdyr, Acta Crystallogr., Sect. D: Struct. Biol., 2018, 74, 68–84 CrossRef CAS PubMed.
- P. R. Evans and G. N. Murshudov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1204–1214 CrossRef CAS PubMed.
- G. N. Murshudov, P. Skubák, A. A. Lebedev, N. S. Pannu, R. A. Steiner, R. A. Nicholls, M. D. Winn, F. Long and A. A. Vagin, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 355–367 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS PubMed.
- J. Agirre, J. Iglesias-Fernández, C. Rovira, G. J. Davies, K. S. Wilson and K. D. Cowtan, Nat. Struct. Mol. Biol., 2015, 22, 833–834 CrossRef CAS PubMed.
- K. Dax, M. Albert, J. Ortner and B. J. Paul, Carbohydr. Res., 2000, 327, 47–86 CrossRef CAS PubMed.
- H. M. Chen and S. G. Withers, ChemBioChem, 2007, 8, 719–722 CrossRef CAS PubMed.
- H. J. Koeners, A. J. de Kok, C. Romers and J. H. van Boom, Recl. Trav. Chim. Pays-Bas, 1980, 99, 355–362 CrossRef CAS.
- M. L. Sinnott, Chem. Rev., 1990, 90, 1171–1202 CrossRef CAS.
- G. J. Davies, K. S. Wilson and B. Henrissat, Biochem. J., 1997, 321, 557–559 CrossRef CAS PubMed.
- T. S. Black, L. Kiss, D. Tull and S. G. Withers, Carbohydr. Res., 1993, 250, 195–202 CrossRef CAS PubMed.
- T. H. Fenger and H. Brumer, ChemBioChem, 2015, 16, 575–583 CrossRef CAS PubMed.
- D. Tull, D. L. Burgoyne, D. T. Chow, S. G. Withers and R. Aebersold, Anal. Biochem., 1996, 234, 119–125 CrossRef CAS PubMed.
- A. Varki, R. D. Cummings, M. Aebi, N. H. Packer, P. H. Seeberger, J. D. Esko, P. Stanley, G. Hart, A. Darvill, T. Kinoshita, J. J. Prestegard, R. L. Schnaar, H. H. Freeze, J. D. Marth, C. R. Bertozzi, M. E. Etzler, M. Frank, J. F. G. Vliegenthart, T. Lütteke, S. Perez, E. Bolton, P. Rudd, J. Paulson, M. Kanehisa, P. Toukach, K. F. Aoki-Kinoshita, A. Dell, H. Narimatsu, W. York, N. Taniguchi and S. Kornfeld, Glycobiology, 2015, 25, 1323–1324 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ob02250j |
| This journal is © The Royal Society of Chemistry 2018 |