
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biomimetic synthesis of the bisindole framework present in sciodole, an alkaloid from Tricholoma sciodes†
Joshua A.
Homer
and
Jonathan
Sperry
 *
*
School of Chemical Sciences, 23 Symonds Street, University of Auckland, Auckland, New Zealand. E-mail: j.sperry@auckland.ac.nz
First published on 14th September 2018
Abstract
A synthesis of the unique bisindole framework present in the mushroom-derived alkaloid sciodole has been achieved, validating a biosynthesis proposal that the C–N bisindole bond present in the natural product is forged by amination of an azafulvenium.
The Tricholoma genus of mushrooms are known to produce several indole natural products harbouring a methyl group at the C2 position.1 Perhaps the most structurally intriguing member of this family is (+)-sciodole (1),2 a reduced N1′–C7 dimer of 5-methoxy-2,4-dimethylindole (2)3 which itself is also found in Tricholoma1c,f (Fig. 1).
 | ||
| Fig. 1 (+)-Sciodole (1) and 5-methoxy-2,4-dimethylindole (2). | ||
Unlike most indole alkaloids, sciodole (1) is not biosynthetically derived from tryptophan. It can be proposed that sciodole (1) is derived from lascivol (3), a predation deterrant present in Tricholoma lascivum4 but also likely to exist in the sciodole source, Tricholoma sciodes (Scheme 1). Cleavage of the glutamic acid residue4 from lascivol would afford the cyclohexenone 4 that upon cyclisation and double dehydration would give tetrahydroindole 5. Elimination of methanol generates azafulvenium 6 that upon attack by 4 would forge the key C–N bond to give 7 possessing the requisite anti-stereochemistry. Cyclisation–aromatisation of the ‘lower half’ would then afford sciodole (1). The key dimerisation event raises interesting questions from a biosynthesis perspective; dimerisation via an SN2 pathway (i.e., from 4 or 5) would not lead to the anti-stereochemistry present in sciodole (1). Moreover, it is likely that an amine (i.e., 4) serves as the nucleophile in the dimerisation, as the indole 2 (formed from 4 and/or 5) would attack the azafulvenium 6via C3 and not N1.
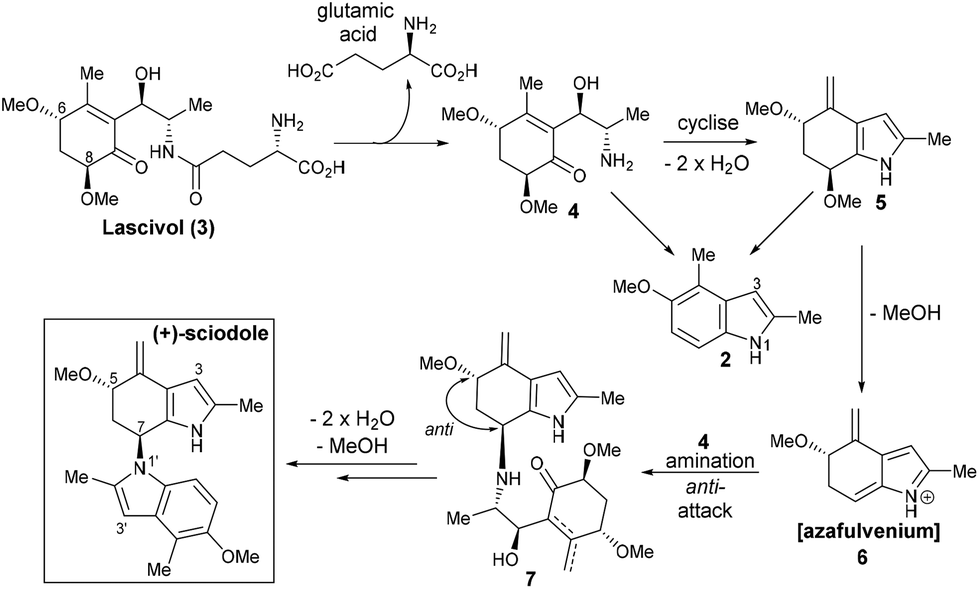 | ||
| Scheme 1 Proposed biosynthesis of sciodole (1) from lascivol (3). | ||
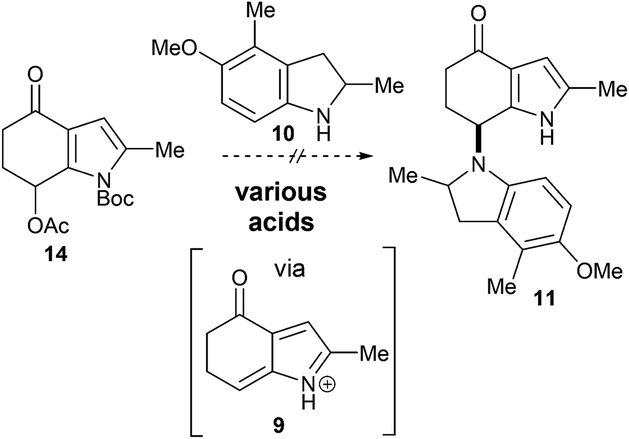
Our ongoing interest in biomimetic synthesis5 led us to instigate a synthetic programme to test the hypothesis outlined in Scheme 1, focusing in particular on the amination event.6 Generating an azafulvenium in the presence of the exo-methylene group (i.e., 5 → 6) was predicted to be troublesome under acidic conditions commonly used to access this electrophile.7 In view of this, the 7-acetoxy-4-indolone 88 was chosen as the precursor from which the azafulvenium 9 would be generated (Scheme 2). With regard to the N-nucleophile, indoline 10 was chosen as the surrogate for the amine 4. Thus, successful amination of the azafulvenium 9 at C7 with indoline 10 would lead to the bisindole derivative 11 bearing the unique bisindole core structure of sciodole (1). This transformation poses strategic and methodological challenges; the indoline 10 could feasibly react at any of the C2, C3 and C4 sites in the 1-azafulvenium 9 and the C–N bond in the desired product 11 is somewhat hindered.
 | ||
| Scheme 2 Initial synthetic plan. | ||
It was decided that the azafulvenium precursor 8 would be accessible by a C–H acetoxylation strategy. When attempting this transformation on the known 4-indolone 129 using lead tetracetate,10–12 a plethora of products were formed, likely resulting from competing α-acetoxylation13 and oxidation of the pyrrole ring.14 We reasoned that de-activating the pyrrole ring would help reduce side-reactions at this site and as a result, the 4-indolone 12 was converted into its N-Boc derivative 13. Subjecting 13 to lead tetracetate pleasingly led to regioselective C–H acetoxylation at the desired C7 site to give 14, as confirmed by NOE analysis (Scheme 3).15
 | ||
| Scheme 3 Synthesis of the azafulvenium precursor 14. | ||
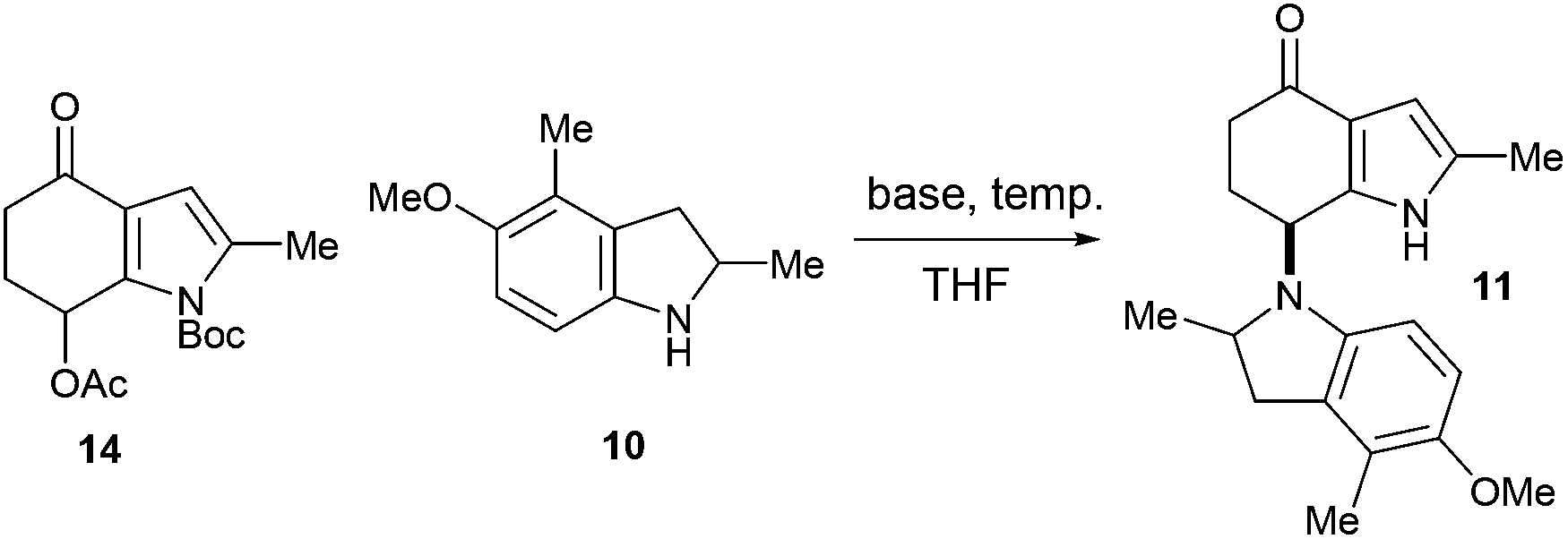
With the azafulvenium precursor 14 in hand, attention turned to the biomimetic amination (Scheme 4). Subjecting 14 to a variety of Brønsted and Lewis acidic conditions, both prior to the addition of, and in the presence of indoline 10,15 was met with failure. Extensive degradation was observed, with 1H NMR analysis of the crude reaction mixtures indicating trace quantities of indoles were present (likely formed by aromatisation of both 10 and 14); no sign of 11 was detected.
 | ||
| Scheme 4 Failed biomimetic dimerisation under acidic conditions. | ||
Considering the incompatibility of the biomimetic amination with acid, an alternative strategy was considered. The generation 1-azafulveniums from hydroxy-, alkoxy- and acetoxymethylpyrroles under acidic conditions is a well-established method to generate this electrophile,7,8 but we pictured a base-mediated process following a mechanistic scenario akin that outlined in Scheme 5. Base-mediated cleavage16 of the Boc group in 14 would drive elimination of acetate, generating the 1-azafulvene 1517 that upon reaction with indoline 10 (likely the anion) would give the desired product 11.
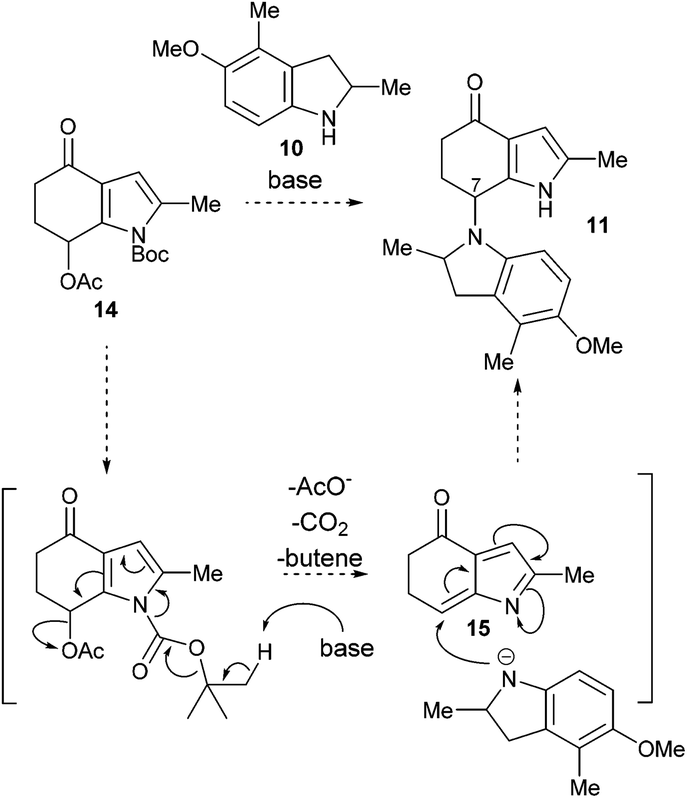 | ||
| Scheme 5 Proposed biomimetic amination by base-mediated azafulvene generation. | ||
When considering how best to approach the proposal in Scheme 5, several factors were considered. Azafulvene formation and indoline deprotonation would each require one equivalent of base and in view of this, three equivalents of base were used, representing an excess (1.5 equiv.) for each of these two operations. Moreover, in order to avoid side-reactions with the carbonyl, acetoxy group and the azafulvene, it was prudent to employ non-nucleophilic bases in the first instance. The condensed details of a study examining the base-mediated dimerisation are outlined in Table 1. The non-nucleophilic bases sodium hexamethyldisilazide (NaHMDS), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and 2,6-lutidine all led to no reaction, with degradation observed upon increasing the reaction temperature (entries 1–3). Trialling more nucleophilic bases was initially disappointing, with potassium carbonate, KF-alumina18 and tripotassium phosphate all leading to a similar outcome (entries 4–6). The first positive result was obtained when sodium methoxide was used, affording the desired product 11 in 5% yield, alongside significant amounts of 16 resulting from reaction of the azafulvene with the base (entry 7). In an effort to reduce the amount of 16 being produced, the amount of indoline 10 was increased and the base was added portionwise, which pleasingly led to a significantly improved yield of 11 and only trace amounts of 16 being formed (entry 8). Employing an excess of the indoline 10 with respect to the base had a negligible effect on the yield of 11 (entry 9). Using equimolar quantities of indoline 10 and base led to the best yield of 11 (entry 10). The acetate eliminated during azafulvene formation is unlikely to be acting as a base in this process, as no reaction was observed when using sodium acetate (entry 11).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Indoline (10) (equiv.) | Base (equiv.) | Temp (°C) | Time (h) | Yield (11)a (%) |
a
11 was isolated as a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereomers (entries 7–10).
b :1 mixture of diastereomers (entries 7–10).
b
 .
c Base added in 0.5 eq. portions and 16 was not detected. .
c Base added in 0.5 eq. portions and 16 was not detected.
|
|||||
| 1 | 1 | NaHMDS (3) | −78–rt | 4 | — |
| 2 | 1 | DBU (3) | rt–80 | 6.5 | — |
| 3 | 1 | 2,6-Lutidine (3) | rt–80 | 6 | — |
| 4 | 1 | K2CO3 (3) | rt–80 | 4 | — |
| 5 | 1 | KF-Alumina (3) | rt–80 | 4 | — |
| 6 | 1 | K3PO4 (3) | rt–80 | 6 | Trace |
| 7 | 1 | NaOMe (3) | rt | 0.5 | 11 (5%) 16b (35%) |
| 8c | 3 | NaOMe (5) | rt | 5 | 26 |
| 9c | 5 | NaOMe (3) | rt | 6 | 23 |
| 10 | 3 | NaOMe (3) | rt | 6 | 40 |
| 11 | 3 | NaOAc (3) | rt–80 | 8 | — |
We are confident 10 and 14 are not reacting via an SN2 pathway. It has been reported7c that 2-hydroxymethylpyrroles will only divert from an azafulvenium (SN1) to an SN2 pathway upon deactivation of the pyrrole by strong electron withdrawing groups (i.e., triflate), while the corresponding N-Boc derivative still reacts via the SN1 pathway. Moreover, the reaction between 10 and 14 turns from colourless to orange upon addition of sodium methoxide, indicative of azafulvene formation.6d
With the bisindole 11 in hand, dehydrogenation of the ‘lower half’ was considered (Scheme 6). Upon subjecting 11 to one equivalent of DDQ, selective dehydrogenation successfully occurred, affording 17 comprising the sciodole framework. This selective aromatisation is another result that supports the biosynthesis proposal outlined in Scheme 2. Although the conversion of 11 and/or 17 into sciodole (1) appears relatively straightforward, we were unable to complete the total synthesis. Unfortunately upon subjecting 11 and 17 to methylenation and α-oxidation procedures, no reaction or degradation was observed under a variety of different reaction conditions.
 | ||
| Scheme 6 Selective dehydrogenation and attempted completion of the synthesis. | ||
In conclusion, we have validated a proposal that the biosynthesis of sciodole (1) involves the amination of an azafulvene followed by selective aromatisation. Further studies will focus on employing alternative nucleophiles in the biomimetic dimerization and hence completing the total synthesis of sciodole. Moreover, the base-mediated azafulvene generation reported herein may form the basis for a conceptually distinct method for generating this electrophile.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are indebted to the University of Auckland for a doctoral scholarship (J. A. H) and the Royal Society of New Zealand for a Rutherford Discovery Fellowship (J. S). We thank Professor Martin Banwell (Australian National University) for helpful discussions.Notes and references
- (a) L. Garlaschelli, Z. Pang, O. Sterner and G. Vidari, Tetrahedron, 1994, 50, 3571 CrossRef; (b) Z. Pang and O. Sterner, J. Nat. Prod., 1994, 57, 852 CrossRef; (c) Z. Pang and O. Sterner, Acta Chem. Scand., 1996, 50, 303 CrossRef; (d) F. Fons, S. Rapior, A. Fruchier, P. Saviuc and J.-M. Bessiere, Cryptogam.: Mycol., 2006, 27, 45 Search PubMed; (e) W. Qiu, H. Kobori, T. Suzuki, J.-H. Choi, V. K. Deo, H. Hirai and H. Kawagishi, Biosci., Biotechnol., Biochem., 2014, 78, 755 CrossRef PubMed; (f) W. Qiu, H. Kobori, J. Wu, J.-H. Choi, H. Hirai and H. Kawagishi, Biosci., Biotechnol., Biochem., 2017, 81, 441 CrossRef PubMed.
- O. Sterner, Nat. Prod. Lett., 1994, 4, 9 CrossRef.
- For a synthesis of 2, see: J. A. Homer and J. Sperry, ARKIVOC, 2018, iv, 6 Search PubMed.
- T. Eizenhöfer, B. Fugmann, W. S. Sheldrick, B. Steffan and W. Steglich, Liebigs Ann. Chem., 1990, 1115 CrossRef . This paper reports that lascivol (3) is converted into indole 2 and dimethyl glutamate upon acid methanolysis.
- (a) E. M. Boyd and J. Sperry, Org. Lett., 2015, 17, 1344 CrossRef PubMed; (b) M. B. Calvert and J. Sperry, Chem. Commun., 2015, 51, 6202 RSC; (c) E. K. Davison, P. A. Hume and J. Sperry, Org. Lett., 2018, 20, 3545 CrossRef PubMed.
- For natural product biosynthesis invoking an azafulvenium, see: (a) A. Al Mourabit and P. Potier, Eur. J. Org. Chem., 2001, 237 CrossRef; (b) C. Pöverlein, G. Breckle and T. Lindel, Org. Lett., 2006, 8, 819 CrossRef PubMed; (c) A. E. Ondrus and M. Movassaghi, Chem. Commun., 2009, 4151 RSC; (d) A. E. Ondrus and M. Movassaghi, Tetrahedron, 2006, 62, 5287 CrossRef PubMed.
- For acid-mediated generation of azafulveniums from hydroxyl- and alkoxymethylpyrroles, see: (a) D. M. Wallace, S. H. Leung, M. O. Senge and K. M. Smith, J. Org. Chem., 1993, 58, 7245 CrossRef; (b) L. F. Tietze, G. Kettschau and K. Heitmann, Synthesis, 1996, 4151 Search PubMed; (c) A. D. Abell, B. K. Nabbs and A. R. Battersby, J. Am. Chem. Soc., 1998, 120, 1741 CrossRef; (d) A. D. Abell and J. C. Litten, Chem. Commun., 1998, 919 RSC; (e) A. D. Abell and B. K. Nabbs, Org. Lett., 1999, 1, 1403 CrossRef; (f) A. Dinsmore, K. Mandy and J. P. Michael, Org. Biomol. Chem., 2006, 4, 1032 RSC.
- For the generation of azafulveniums from 2-acetoxymethypyrroles, see: (a) T. D. Lash, K. Miyake, L. Xu and G. M. Ferrence, J. Org. Chem., 2011, 76, 6295 CrossRef PubMed; (b) M. Nakamura, H. Tahara, K. Takahashi, T. Nagata, H. Uoyama, D. Kuzuhara, S. Mori, T. Okujima, H. Yamada and H. Uno, Org. Biomol. Chem., 2012, 10, 6840 RSC; (c) P. M. G. Sabido and D. A. Lightner, Monatsh. Chem., 2014, 145, 775 CrossRef.
- Y. Peng, J. Luo, Q. Feng and Q. Tang, Eur. J. Org. Chem., 2016, 5169 CrossRef.
- C7–H acetoxylation of a 4-indolone using KMnO4, see: Z. Z. Caliskan and M. S. Ersez, J. Mol. Catal. B: Enzym., 2015, 111, 64 CrossRef.
- C7–H acetoxylation of a tetrahydroindole using Pb(OAc)4, see: T. D. Lash, K. A. Bladel and M. C. Johnson, Tetrahedron Lett., 1987, 28, 1135 CrossRef.
- C7–H autoxidation of 2-diphenyldiazenyl-N-vinyltetrahydroindole: E. Y. Schmidt, E. Y. Senotrusova, I. A. Ushakov, O. N. Kazheva, O. A. Dyachenko, G. G. Alexandrov, A. V. Ivanov, A. I. Mikhaleva and B. A. Trofimov, ARKIVOC, 2010, ii, 352 Search PubMed.
- (a) G. W. K. Cavill and D. H. Solomon, J. Chem. Soc., 1955, 4426 RSC; (b) M. Lj. Mihailović, J. Foršek and Lj. Lorenc, Tetrahedron, 1977, 33, 235 CrossRef; (c) C. Fäh, L. A. Hardegger, L. Baitsch, W. B. Schweizer, S. Meyer, D. Bur and F. Diederich, Org. Biomol. Chem., 2009, 7, 3947 RSC.
- J. K. Howard, K. J. Rihak, A. C. Bissember and J. A. Smith, Chem. – Asian J., 2016, 11, 155 CrossRef PubMed.
- See ESI† for all NOESY spectra and the procedure for the preparation of 10.
- (a) I. Hasan, E. R. Marinelli, L.-C. C. Lin, F. W. Fowler and A. B. Levy, J. Org. Chem., 1981, 46, 157 CrossRef; (b) K. Ravinder, A. V. Reddy, K. C. Mahesh, M. Narasimhulu and Y. Venkateswarlu, Synth. Commun., 2007, 37, 281 CrossRef.
- Base-mediated azafulvene generation from pyrrole Mannich bases, see. (a) F. Schnierle, H. Reinhard, N. Dieter, E. Lippacher and H. von Dobeneck, Liebigs Ann. Chem., 1968, 715, 90 CrossRef; (b) H. von Dobeneck and T. Messerschmitt, Liebigs Ann. Chem., 1971, 751, 32 CrossRef; (c) H. von Dobeneck, T. Messerschmitt, E. Brunner and U. Wunderer, Liebigs Ann. Chem., 1971, 751, 40 CrossRef; (d) P. E. Sonnet, J. L. Flippen and R. D. Gilardi, J. Heterocycl. Chem., 1974, 11, 811 CrossRef; (e) S. L. Berthiaume, B. L. Bray, P. Hess, Y. Liu, M. L. Maddox, J. M. Muchowski and M. E. Scheller, Can. J. Chem., 1995, 73, 675 CrossRef.
- B. E. Blass, Tetrahedron, 2002, 58, 9301 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ob02142b |
| This journal is © The Royal Society of Chemistry 2018 |