
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereoselective synthesis of 1,3-disubstituted dihydroisoquinolines viaL-phenylalanine-derived dihydroisoquinoline N-oxides†
Jesús
Flores-Ferrándiz
,
Nicholas
Carter
,
Maria José
González-Soria
,
Malgorzata
Wasinska
,
Daniel
Gill
,
Beatriz
Maciá
 * and
Vittorio
Caprio
*
* and
Vittorio
Caprio
*
School of Science and the Environment, Manchester Metropolitan University, All Saints Campus, Oxford Road, Manchester, M15 6BH, UK. E-mail: b.macia-ruiz@mmu.ac.uk; v.caprio@mmu.ac.uk
First published on 12th September 2018
Abstract
The preparation of chiral pool-derived nitrone 3 and its use in the protecting-group free, stereoselective synthesis of a range of 1,3-disubstituted tetrahydroisoquinolines is described. Grignard reagent additions to nitrone 3 yielded trans-1,3-disubstituted N-hydroxytetrahydroisoquinolines 6 with good levels of selectivity, while 1,3-dipolar cycloadditions to this nitrone provided access to 3-(2-hydroxyalkyl)isoquinolines 12 as single diastereomers.
Introduction
Tetrahydroisoquinoline (THIQ) systems occur in a wide range of natural products and therapeutic drugs1 and have been highlighted both as privileged scaffolds2 and as useful moieties in fragment-based drug discovery.3 Moreover, there is interest in the use of tetrahydroisoquinolines as ligands in asymmetric catalysis4 and as chiral bases.5 The methods for accessing 1,3-disubstituted THIQ's, can be broadly grouped into two strategies, based on either cyclisations of suitably substituted aromatic precursors6 or functionalisation of isoquinoline-based scaffolds.7 In an effort to develop a strategy towards enantiodefined tetrahydroisoquinolines, of general use, we became interested in an approach of the latter type utilizing a chiral pool-derived isoquinoline unit with a potentially broad reactivity profile. The only previous approaches with these criteria utilize either 1-lithiated derivative 18 or imine 25 – with reactivity restricted to additions to electrophiles or nucleophiles respectively (Fig. 1).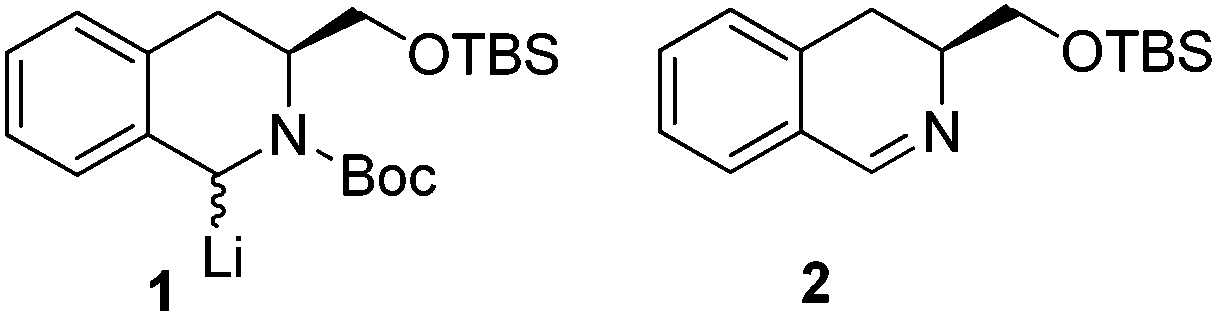 | ||
| Fig. 1 Previous chiral pool-derived THIQ precursors. | ||
We envisaged that nitrone 3 would exhibit much utility in this regard. The core scaffold is readily accessible, in enantioenriched form from L-phenylalanine, and the reactivity of the nitrone group, functionalised via 1,3-dipolar cycloaddition,9 nucleophilic addition10 and reductive coupling with electrophiles,11 confers wide scope to this system (Scheme 1). Furthermore, the hydroxymethyl group functions as a readily removable stereocontrol element.12
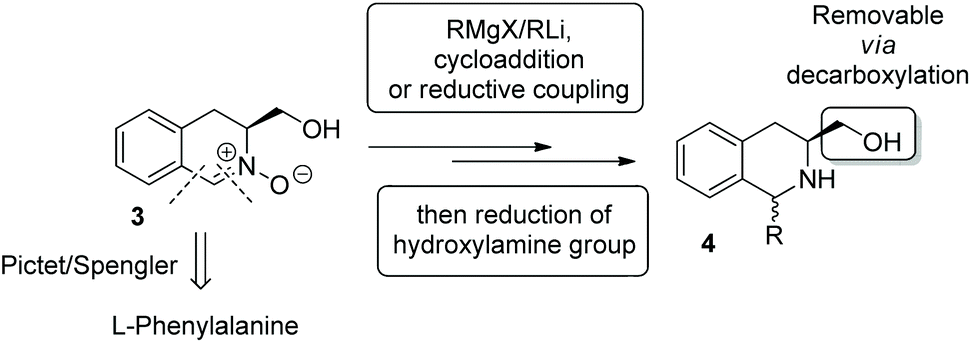 | ||
| Scheme 1 Synthetic approaches to 1,3-disubstituted tetrahydroisoquinolines utilising nitrone 3. | ||
Results and discussion
Preparation of nitrone 3
Our approach to nitrone 3 centred on the oxidation of the known amine 5 (Scheme 2), accessed by Pictet–Spengler reaction of L-phenylalanine,13 followed by borane-mediated reduction of the resulting acid.5 Trialing a range of methods for the direct oxidative conversion of amines to nitrones, revealed that the corresponding isoquinoline N-oxide was a significant byproduct. Best results were achieved using Oxone as oxidant, providing dihydroisoquinoline derived nitrone 3 cleanly, in moderate yield (58%), with no evidence of overoxidation to the corresponding isoquinoline N-oxide.14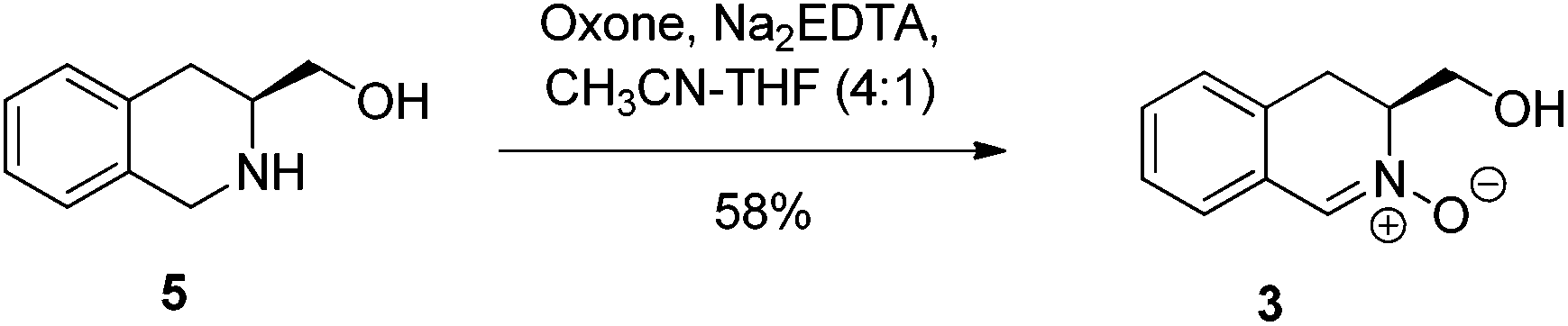 | ||
| Scheme 2 Synthesis of nitrone 3. | ||
Addition of organometallic reagents to nitrone 3
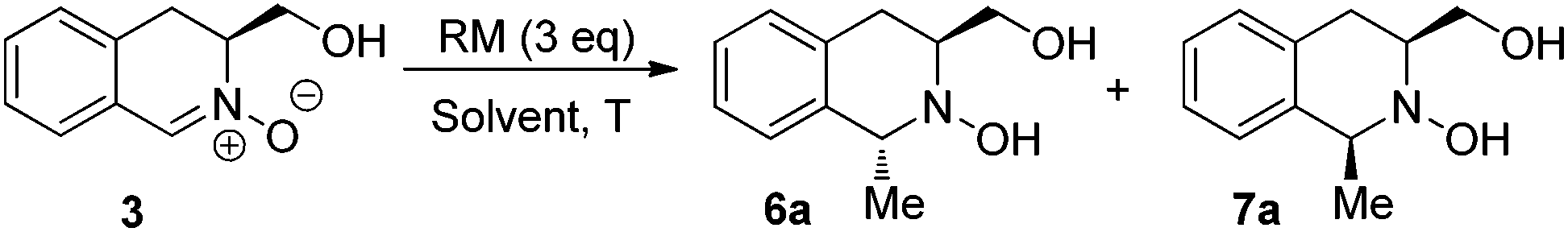
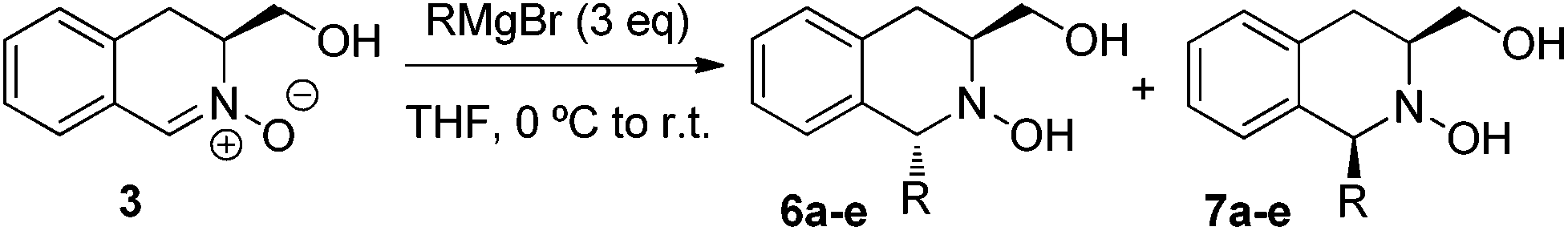
With nitrone 3 in hand, the nucleophilic addition of organometallic reagents was initially investigated by screening of a range of solvents and temperatures, using methylmagnesium bromide and methyllithium as test reagents (Table 1). In all cases, full conversion was achieved using 3 equivalents of nucleophile, with the 1,3-trans isomer 6a as the major product. THF provided the best diastereomeric ratios and no advantage was conferred by performing the reaction under cryogenic conditions see (entries 7–9). It is noteworthy that, while the addition of the more reactive methyllithium proceeded with full conversion, stereoselectivity was lower than that obtained with the corresponding Grignard reagent (entry 10 vs. entry 7). Consequently, subsequent studies were performed using Grignard species under the conditions outlined in entry 7.|
|
|||||
|---|---|---|---|---|---|
| Entry | RM | Solvent | T (°C) | 6a/7ab | Conv.c (%) |
| a Reaction conditions: 3 (0.1 mmol), RM (3 eq.), T, solvent. b Ratio determined by analysis of 1H-NMR spectra of crude products. Relative stereochemistry determined by 2D-NOESY studies; see ESI for further details. c Determined by 1H-NMR analysis of crude products. | |||||
| 1 | MeMgBr | CH2Cl2 | 0 | 4.5/1 | >99 |
| 2 | MeMgBr | PhMe | 0 | 7.1/1 | >99 |
| 3 | MeMgBr | Dioxane | 0 | 3.0/1 | >99 |
| 4 | MeMgBr | t BuOMe | 0 | 3.5/1 | >99 |
| 5 | MeMgBr | Et2O | 0 | 5.5/1 | >99 |
| 6 | MeMgBr | THF | 20 | 6.0/1 | >99 |
| 7 | MeMgBr | THF | 0 | 7.5/1 | >99 |
| 8 | MeMgBr | THF | −20 | 7.5/1 | >99 |
| 9 | MeMgBr | THF | −40 | 7.5/1 | >99 |
| 10 | MeLi | THF | 0 | 3.8/1 | >99 |
The addition of different Grignard reagents to nitrone 3 provided, in all cases, the trans-isomer 6 as the major product (Table 2). No discernable trends are evident in Table 2, although the best levels of selectivity were obtained with the larger phenylmagnesium bromide (entry 5). Cis/trans stereochemistry of products 6 and 7 was assigned by 2D NOESY analysis (see Fig. 2 for an example). Strong correlations between the protons of the C1-substituent and H3 were observed in the trans-adduct, while the trans-diaxial correlations between H1 and H3 revealed the cis-diequatorial substitution pattern of the minor adduct, as illustrated on the ethyl-adduct 6b/7b.
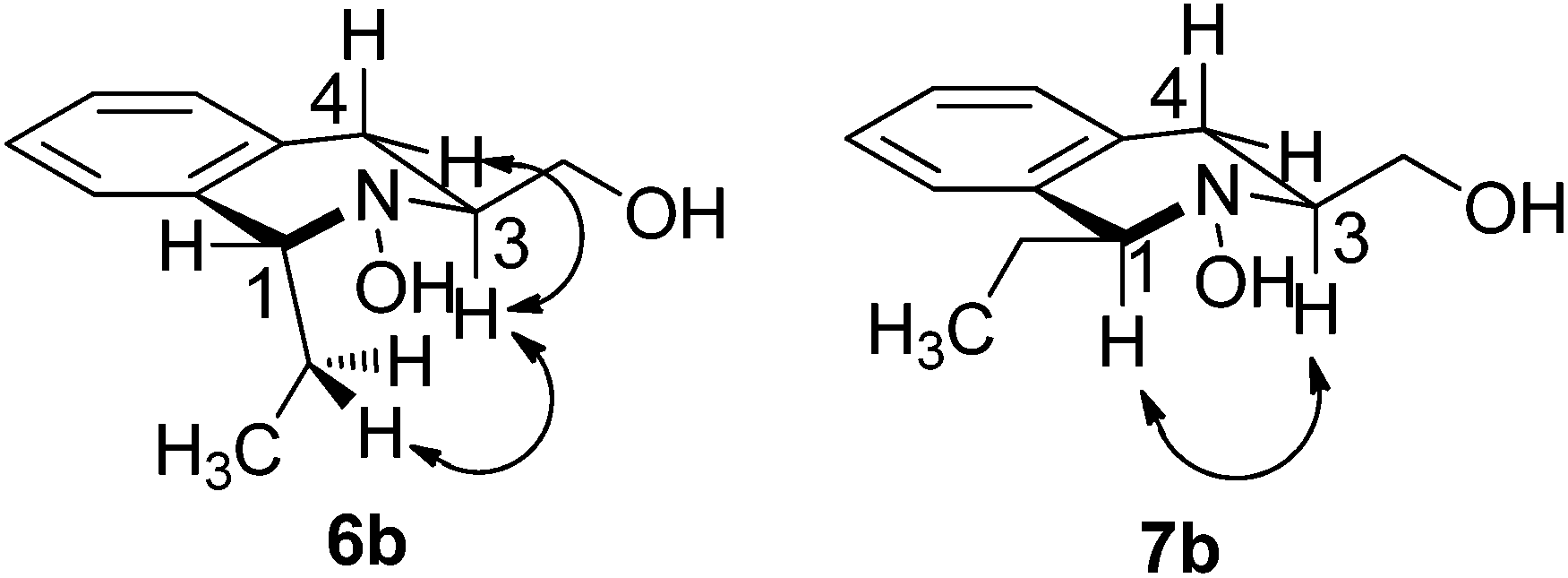 | ||
| Fig. 2 Selected correlations observed in the 2D-NOESY spectra of tetrahydroisoquinolines 6b and 7b. | ||
|
|
||||
|---|---|---|---|---|
| Entry | RM | Products |
6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7a,b :7a,b |
Yieldc (%) |
| a Reaction conditions: 3 (0.3 mmol), RMgBr (3 eq.), THF, 0 °C to r.t. b Ratio determined by analysis of crude 1H-NMR spectra. Relative stereochemistry determined by 2D-NOESY studies, see Fig. 2 and ESI for further details. c Isolated combined yield after column chromatography. | ||||
| 1 | MeMgBr | 6a/7a | 7.5/1 | 75 |
| 2 | EtMgBr | 6b/7b | 6.5/1 | 87 |
| 3 | i PrMgBr | 6c/7c | 5.9/1 | 85 |
| 4 | AllylMgBr | 6d/7d | 2.8/1 | 90 |
| 5 | PhMgBr | 6e/7e | 10/1 | 96 |
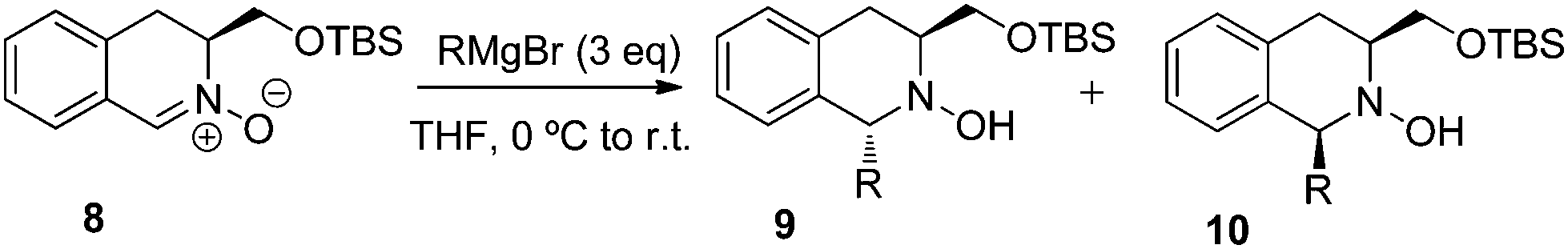
The effect of protection of the 3-hydroxymethyl group, in combination with increasing size of the 3-substituent, was investigated by studying the addition of different Grignard reagents to the TBS-protected nitrone 8 (Table 3), prepared by sodium tungstate-catalysed-oxidation of the known TBS-derivative of hydroxymethyl-THIQ 5.5,15 The addition of both ethyl- and phenylmagnesium bromide to 8 proceeded in favour of the trans-adducts 9, but with much lower selectivities than those obtained with the unprotected nitrone 3 (compare Table 3, entries 1 and 2 with Table 2, entries 2 and 5, respectively).
|
|
||||
|---|---|---|---|---|
| Entry | RM | Product | 9/10b | Yieldc (%) |
| a Reaction conditions: 8 (0.3 mmol), RMgBr (3 eq.), THF, 0 °C to r.t. b Ratio determined by 1H-NMR on the reaction crude. Relative stereochemistry determined by 2D-NOESY studies. c Isolated combined yield after column chromatography. | ||||
| 1 | EtMgBr | 9a/10a | 2.5/1 | 56 |
| 2 | PhMgBr | 9b/10b | 3/1 | 53 |
These results indicate that, in addition to the metal, (see Table 1, entries 7 and 10, for the addition of MeMgBr and MeLi, respectively), the nitrone oxygen and the free hydroxyl group play an important role in determining the mode and level of diastereoselectivity in the addition to 3. This hypothesis is supported by the results obtained with imine 2, where the selectivities of the addition of organolithium species are generally lower than those reported in Table 2 for all but the largest nucleophiles.5 We propose the model depicted in Fig. 3 to account for this selectivity, which involves a cyclic magnesium chelate similar to that hypothesized during additions to N-glycosyl nitrones.16 In our model, the syn addition is hindered by the pseudo-axial proton at C4.
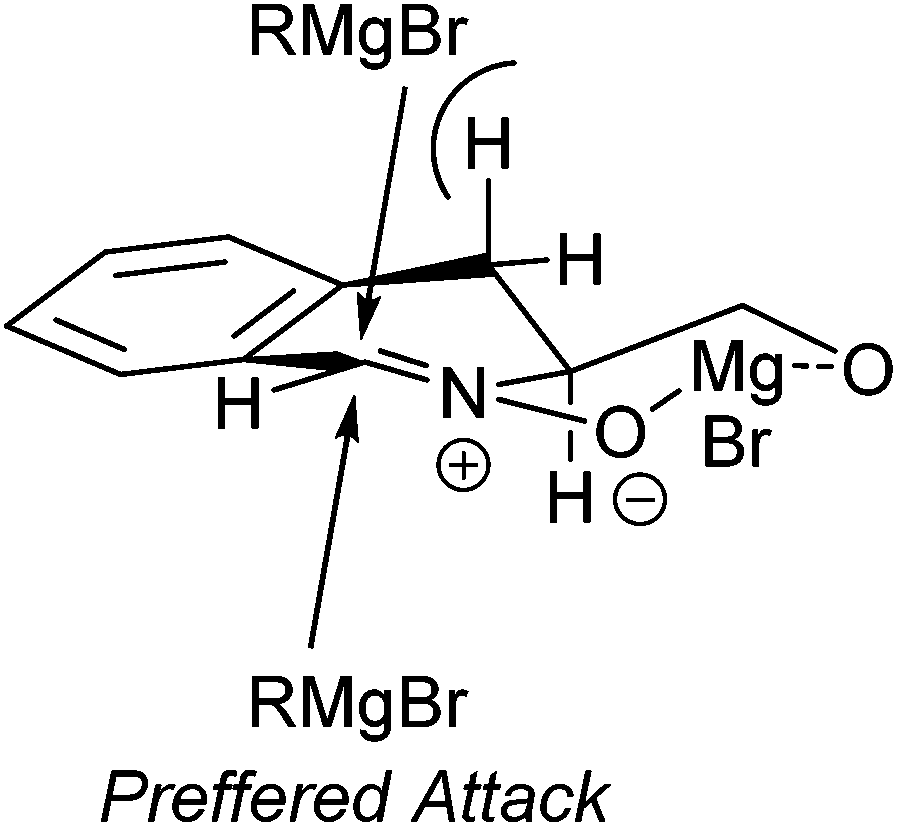 | ||
| Fig. 3 Proposed mode of addition of Grignard reagents to nitrone 3. | ||
Cycloaddition reactions of nitrone 3
Next, the preparation of more functionalized, stereodefined 1,3-THIQ systems was investigated via 1,3-dipolar cycloaddition of 3 with a range of alkenes, followed by reduction of the resulting isoxazolidines 11 (Table 4). Styrene and hex-1-ene were chosen as the prototypical model dipolarophiles (entries 1 and 2), alongside functionalized butenes, selected to access products of subsequent synthetic utility (entries 3 and 4). Optimum yields/selectivity were obtained in toluene under reflux conditions. The stereochemistry of the cycloaddition was readily deduced by analysis of 2D-NOESY spectra of cycloadducts 11 (see an example in Fig. 4), which revealed strong correlations between H2/H6 and H10b/CH2(OH). In all cases, the exo-1,3-trans-isoxazolidine was exclusively obtained in moderate to good yield and no traces of any other isomers were detected in the 1H NMR spectra of crude products. The cycloaddition reactions of nitrone 3 with a range of electron deficient systems, including acrylates, were trialed but, unfortunately, failed to provide the corresponding cycloadducts.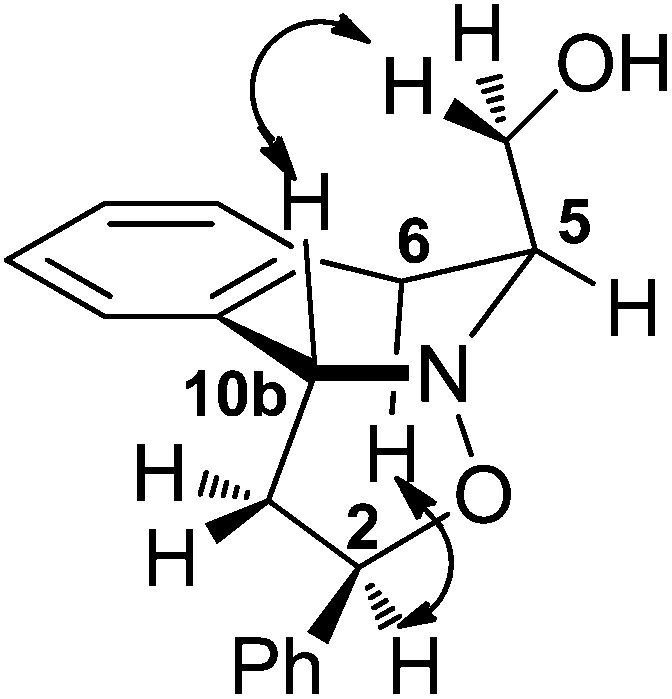 | ||
| Fig. 4 Selected correlations observed in the 2D-NOESY spectrum of 11a. | ||
|
|
||||
|---|---|---|---|---|
| Entry | R | Product | 11 (% yield) | 12 (% yield) |
a Reaction conditions: (i) CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2R (5 eq.), toluene, 110 °C; (ii) AcOH, Zn, reflux.
b Isolated yield after flash chromatography. Relative stereochemistry determined by 2D-NOESY studies, see Fig. 4 and ESI for further details. CH2R (5 eq.), toluene, 110 °C; (ii) AcOH, Zn, reflux.
b Isolated yield after flash chromatography. Relative stereochemistry determined by 2D-NOESY studies, see Fig. 4 and ESI for further details.
|
||||
| 1 | Ph | 11a/12a | 86 | 33 |
| 2 | (CH2)3CH3 | 11b/12b | 74 | 90 |
| 3 | (CH2)2OBn | 11c/12c | 61 | 51 |
| 4 | (CH2)2SPh | 11d/12d | 64 | 47 |
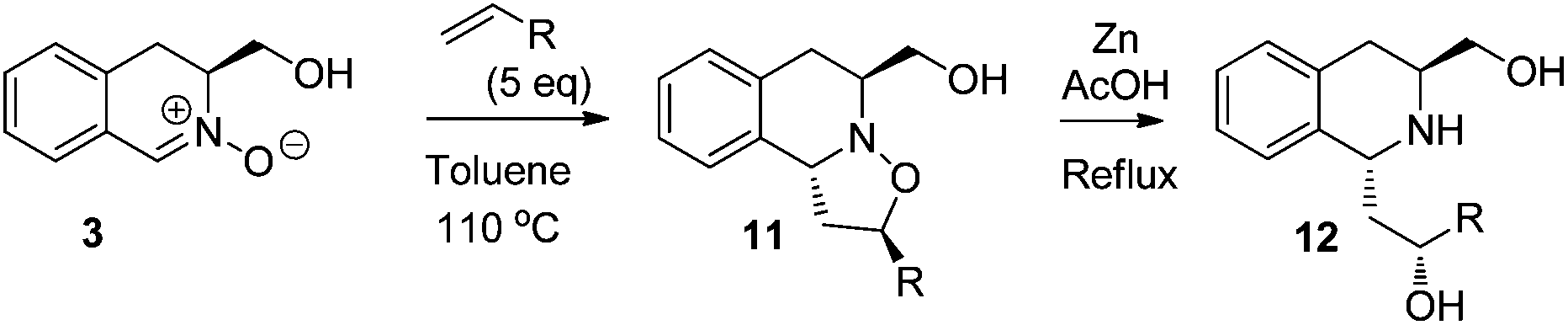
The subsequent reductive N–O bond cleavage of isoxazolidine 11, with zinc powder in AcOH, proceeded with no stereochemical degradation,17 providing the corresponding 1,3-THIQ 12a–d in good to moderate yields (Table 2).
Conclusions
In conclusion, we have developed an approach to the synthesis of a range of optically active 1,3-disubstituted tetrahydroisoquinolines from (S)-3-(hydroxymethyl)-3,4-dihydroisoquinoline 2-oxide 3. The addition of Grignard reagents to 3 proceeded with good yields and stereoselectivities, and unprecedented levels of stereoselection were found for the dipolar cycloaddition reaction of alkenes to nitrone 3. This strategy allows the direct assembly of highly functionalized tetrahydroisoquinoline units with up to three stereogenic centres in a simple and effective manner, starting from a readily available chiral substrate.Experimental
General
1H NMR and 13C NMR have been recorded on a JEOL® ECS-400 (400 and 100.6 MHz, respectively) using CDCl3 as solvent. Chemical shift values are reported in ppm with TMS as internal standard (CDCl3: δ 7.26 for 1H-NMR, δ 77.0 for 13C-NMR). Data are reported as follows: chemical shifts, multiplicity (s = singlet, quint = quintuplet, m = multiplet), coupling constants (Hz), and integration. IR spectra were recorded on Nicolet® 380 FT/IR – Fourier Transform Infrared Spectrometer. Only the most significant frequencies have been considered during the characterization, and have been reported in cm−1. High resolution mass spectra were measured on an Agilent Technologies® 6540 Ultra-High-Definition (UHD) Accurate-Mass equipped with a time of flight (Q-TOF) analyzer and the samples were ionized by ESI techniques and introduced through a high pressure liquid chromatography (HPLC) model Agilent Technologies® 1260 Infinity Quaternary LC system. Optical rotations were measured on a Bellingham + Stanley® ADP 440 + Polarimeter with a 0.5 cm cell (c given in g per 100 mL).All reactions were monitored by thin-layer chromatography using precoated sheets of silica gel 60, 0.25 mm thick (F254 Merck KGaA®). The components were visualized by UV light (254 nm) and phosphomolybdic acid. Flash column chromatography was done using Geduran® Silica gel 60, 40–63 microns RE. The eluent used is mentioned in each particular case. All glassware employed during inert atmosphere experiments was flame-dried under a stream of dry argon. Alkenes reagents were purchased from Sigma-Aldrich or Fisher Scientific and used without further purification. Grignard and organolithium reagents were purchased from Sigma-Aldrich. Anhydrous THF was obtained from a Pure Solv™ Solvent Purification Systems.
:4 (28 mL) and 0.01 M aqueous EDTA solution (22.4 mL) at 5 °C. Oxone (10.3 g, 16.8 mmol) was then added portionwise over 2 h, and the mixture stirred for 20 min, at 5 °C. Ethyl acetate (80 mL) was added and the aqueous layer further extracted with ethyl acetate (2 × 80 mL). The combined organic extracts were dried over anhydrous MgSO4 and concentrated under reduced pressure to give nitrone 3 (1.66 g, 59%) as a pale yellow solid; m.p. decomposes T > 145 °C; [α]25D 174 (c = 3.3, CHCl3); IR (ATR): ν = 3242, 3054, 2945, 2897 cm−1; 1H NMR (400 MHz, CDCl3): δH = 7.80 (s, 1H), 7.35–7.25 (m, 2H), 7.25–7.20 (m, 1H), 7.18–7.14 (m, 1H), 4.51 (br s, 1H), 4.26–4.16 (m, 1H), 4.04 (dd, J = 12.0, 6.9 Hz, 1H), 3.94 (dd, J = 12.0, 3.6 Hz, 1H), 3.22–3.07 (m, 2H) ppm; 13C NMR (101 MHz, CDCl3): δC = 135.4, 130.0, 129.8, 127.6, 127.5, 127.2, 125.6, 67.2, 62.7, 29.8 ppm; MS (EI, 70 eV): m/z (%) = 177 (M+, 8), 130 (100), 115 (34), 103 (22), 77 (26); HRMS (ESI-MeOH): m/z calcd for C10H12NO2 [M + H]+: 178.0863 found 178.0862.
General procedure for the diastereoselective addition of Grignard reagents
A solution of RMgBr (0.9 mmol, 3 eq.) was added to a solution of nitrone 3 (53.1 mg, 0.3 mmol) in THF (1.5 mL). The mixture was stirred at 0 °C for 1 h and then warmed to room temperature and quenched with water. The mixture was extracted with EtOAc (3 × 10 mL) and the combined organic phases dried over MgSO4, and concentrated under reduced pressure. The crude product, was purified by flash column chromatography eluting with n-hexane/EtOAc (gradient from 10 to 60%) to give the corresponding 1,3-disubstituted tetrahydroisoquinoline-2-ol as a yellow oil.:1) as eluent, to give nitrone 10 (0.46 g, 57%) as a viscous oil. [α]25D −36 (c = 1, CHCl3); IR (ATR): ν = 2952, 2927, 2883, 2855 cm−1; 1H NMR (400 MHz, CDCl3): δH = 7.73 (s, 1H), 7.28–7.18 (m, 3H), 7.08 (d, J = 6.9 Hz, 1H), 4.16–4.08 (m, 1H), 3.96 (dd, J = 9.9, 3.8 Hz, 1H), 3.84 (dd, J = 9.9, 7.7 Hz, 1H), 3.42 (dd, J = 16.7, 7.3 Hz, 1H), 3.25 (dd, J = 16.8, 3.2 Hz, 1H), 0.79 (s, 9H), −0.01 (s, 3H), −0.03 (s, 3H) ppm; 13C NMR (101 MHz, CDCl3): δC = 134.8, 129.6, 129.2, 127.7, 127.6, 127.2, 125.1, 68.8, 61.8, 29.2, 25.5, 18.0, −5.7, −5.78 ppm; HRMS (ESI-MeOH): m/z calcd for C16H26NO2Si [M + H]+: 292.1727 found 292.1734.
General procedure for the cycloaddition reactions of nitrone 3
A solution of nitrone 3 (0.1 g, 0.56 mmol) in dry toluene (5 mL) was heated to 80 °C. A solution of the corresponding alkene (5 eq.) in dry toluene (5 mL) was added dropwise and the reaction was heated at 110 °C overnight. Removal of the solvent under reduced pressure yielded the crude product as a dark brown oil. Purification by flash chromatography eluting with EtOAc/cyclohexane (1:1) gave the corresponding products 11.
General procedure for the reduction of cycloadducts 1116
Zinc dust (5.0 eq.) was added to a solution of cycloadduct 11 (1 eq.) in 50% acetic acid:water at room temperature. The mixture was stirred under reflux for 3 h. After cooling, the mixture was neutralized with saturated aqueous NaHCO3 and the aqueous phase was extracted with CH2Cl2 (3 × 10 mL). The combined organic phases were dried over MgSO4, concentrated under reduced and the residue purified by column chromatography.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. F.-F. thanks the University of Alicante for a placement studentship. M. W. thanks the Erasmus+programme for funding her internship. B. M. thanks the European Commission for a Marie Curie Career Integration Grant, the EPSRC for a First Grant and the RS for a travel grant.Notes and references
- (a) A. H. Jackson, in Chemistry and Biology of Isoquinoline Alkaloids, ed. J. D. Phillipson, M. F. Margaret and M. H. Zenk, Springer, Berlin, Germany, 1985, pp. 62–78 Search PubMed; (b) J. D. Scott and R. M. Williams, Chem. Rev., 2002, 102, 1669–1730 CrossRef PubMed; (c) K. W. Bentley, Nat. Prod. Rep., 2006, 23, 444–463 RSC; (d) S. Kotha, D. Deodhar and P. Khedkar, Org. Biomol. Chem., 2014, 12, 9054–9091 RSC; (e) V. H. Le, R. M. Williams and T. Kan, Nat. Prod. Rep., 2015, 32, 328–347 RSC.
- (a) M. J. Fisher, R. Backer, S. Husain, H. M. Hsiung, J. T. Mullaney, T. P. O'Brian, P. L. Ornstein, R. R. Rothhaar, J. M. Zgombick and K. Briner, Bioorg. Med. Chem. Lett., 2005, 15, 4459–4462 CrossRef PubMed; (b) M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347–361 CrossRef PubMed; (c) B. A. Granger, K. Kaneda and S. F. Martin, Org. Lett., 2011, 13, 4542–4545 CrossRef PubMed.
- C. W. Murray and D. C. Rees, Angew. Chem., Int. Ed., 2016, 55, 488–492 CrossRef PubMed.
- (a) J. Feng, S. Dastgir and C.-J. Li, Tetrahedron Lett., 2008, 49, 668–671 CrossRef; (b) B. K. Peters, S. K. Chakka, T. Naicker, G. E. M. Maguire, H. G. Kruger, P. G. Andersson and T. Govender, Tetrahedron: Asymmetry, 2010, 21, 679–687 CrossRef; (c) T. Naicker, K. Petzold, T. Singh, P. I. Arvidsson, H. G. Kruger, G. E. M. Maguire and T. Govender, Tetrahedron: Asymmetry, 2010, 21, 2859–2867 CrossRef; (d) S. K. Chakka, Z. E. D. Cele, S. C. Sosibo, V. Francis, P. I. Arvidsson, H. G. Kruger, G. E. M. Maguire and T. Govender, Tetrahedron: Asymmetry, 2010, 21, 846–852 CrossRef; (e) P. D. McCloud, A. M. Reckling and C.-J. Li, Heterocycles, 2010, 80, 1319–1337 CrossRef; (f) S. K. Chakka, P. G. Andersson, G. E. M. Maguire, H. G. Kruger and T. Govender, Eur. J. Org. Chem., 2010, 972–980 CrossRef; (g) T. Naicker, P. I. Arvidsson, H. G. Kruger, G. E. M. Maguire and T. Govender, Eur. J. Org. Chem., 2011, 6923–6932 CrossRef; (h) Z. E. D. Cele, S. C. Sosibo, P. G. Andersson, H. G. Kruger, G. E. M. Maguire and T. Govender, Tetrahedron: Asymmetry, 2013, 24, 191–195 CrossRef; (i) D. T. Kong and Z. M. A. Judeh, Synthesis, 2016, 48, 2271–2279 CrossRef.
- V. K. Aggarwal, P. S. Humphries and A. Fenwick, J. Chem. Soc., Perkin Trans. 1, 1999, 2883–2889 RSC.
- For selected recent examples see: (a) D. Enders, J. X. Liebich and G. Raabe, Chem. – Eur. J., 2010, 16, 9763–9766 CrossRef PubMed; (b) D. L. Priebbenow, S. G. Stewart and F. M. Pfeffer, Tetrahedron Lett., 2012, 53, 1468–1471 CrossRef; (c) S. Fustero, I. Ibanez, P. Barrio, M. A. Maestro and S. Catalan, Org. Lett., 2013, 15, 832–835 CrossRef PubMed; (d) B. Dulla, N. D. Tangellamudi, S. Balasubramanian, S. Yellanki, R. Medishetti, R. K. Banote, G. H. Chaudhari, P. Kulkarni, J. Iqbal, O. Reiser and M. Pal, Org. Biomol. Chem., 2014, 12, 2552–2558 RSC; (e) L. R. Peacock, R. S. L. Chapman, A. C. Sedgwick, M. F. Mahon, D. Amans and S. D. Bull, Org. Lett., 2015, 17, 994–997 CrossRef PubMed; (f) K. Mori, N. Umehara and T. Akiyama, Adv. Synth. Catal., 2015, 357, 901–906 CrossRef; (g) S. Raghavan and P. Senapati, J. Org. Chem., 2016, 81, 6201–6210 CrossRef PubMed.
- For selected recent examples see: (a) N. Chatani, T. Asaumi, S. Yorimitsu, T. Ikeda, F. Kakiuchi and S. Murai, J. Am. Chem. Soc., 2001, 123, 10935–10941 CrossRef PubMed; (b) N. Y. Kuznetsov, V. N. Khrustalev, I. A. Godovikov and Y. N. Bubnov, Eur. J. Org. Chem., 2006, 113–120 CrossRef; (c) A. Peschiulli, V. Smout, T. E. Storr, E. A. Mitchell, Z. Elis, W. Herrebout, D. Berthelot, L. Meerpoel and B. U. Maes, Chem. – Eur. J., 2013, 19, 10378–10387 CrossRef PubMed; (d) G. Lahm and T. Opatz, Org. Lett., 2014, 16, 4201–4203 CrossRef PubMed; (e) Y. Kita, K. Yamaji, K. Higashida, K. Sathaiah, A. Iimuro and K. Mashima, Chem. – Eur. J., 2015, 21, 1915–1927 CrossRef PubMed.
- A. Monsees, S. Laschat and I. Dix, J. Org. Chem., 1998, 63, 10018–10021 CrossRef.
- For reviews covering cycloadditions to nitrones see: (a) M. Fredrickson, Tetrahedron, 1997, 53, 403–425 CrossRef; (b) P. de March, M. Figueredo and J. Font, Heterocycles, 1999, 50, 1213–1226 CrossRef; (c) H. M. I. Osborn, N. Gemmell and L. M. Harwood, J. Chem. Soc., Perkin Trans. 1, 2002, 2419–2438 RSC; (d) A. E. Koumbis and J. K. Gallos, Curr. Org. Chem., 2003, 7, 585–628 CrossRef; (e) L. M. Stanley and M. P. Sibi, Chem. Rev., 2008, 108, 2887–2902 CrossRef PubMed; (f) S. Kanemasa, Heterocycles, 2010, 82, 87–200 CrossRef; (g) T. M. V. D. Pinho e Melo, Eur. J. Org. Chem., 2010, 3363–3376 CrossRef; (h) G. Molteni, Heterocycles, 2016, 92, 2115–2140 CrossRef; Also, see: (i) W. S. Jen, J. J. M. Weiner and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 9874–9875 CrossRef; (j) P. H. Poulsen, S. Vergura, A. Monleon, D. K. B. Jørgensen and K. A. Jørgensen, J. Am. Chem. Soc., 2016, 138, 6412–6415 CrossRef PubMed; (k) J. Vesely, R. Rios, I. Ibrahem, G.-L. Zhao, L. Eriksson and A. Cordova, Chem. – Eur. J., 2008, 14, 2693–2698 CrossRef PubMed.
- For reviews covering nucleophilic additions to nitrones see: (a) R. Matute, S. Garcia-Viñuales, H. Hayes, M. Ghirardello, A. Darù, T. Tejero, I. Delso and P. Merino, Curr. Org. Synth., 2016, 13, 669–686 CrossRef; (b) M. Lombardo and C. Trombini, Curr. Org. Chem., 2002, 6, 695–713 CrossRef; (c) M. Lombardo and C. Trombini, Synthesis, 1999, 905–917 Search PubMed.
- (a) G. Masson, S. Py and Y. Vallee, Angew. Chem., Int. Ed., 2002, 41, 1772–1775 CrossRef; (b) D. Riber and T. Skrydstrup, Org. Lett., 2003, 5, 229–231 CrossRef PubMed; (c) M. Szostak, N. J. Fazakerley, D. Parmar and D. Procter, Chem. Rev., 2014, 114, 5959–6039 CrossRef PubMed.
- (a) I. M. P. Huber and H. Seebach, Helv. Chim. Acta, 1987, 70, 1944–1954 CrossRef; (b) M. Gurram, B. Gyimothy, R. Wang, S. Q. Lam, F. Ahmed and R. J. Herr, J. Org. Chem., 2011, 76, 1605–1613 CrossRef PubMed.
- M. P. Chelopo, S. A. Pawar, M. K. Sokhela, T. Govender, H. G. Kruger and G. E. M. Maguire, Eur. J. Med. Chem., 2013, 66, 407–414 CrossRef PubMed.
- C. Gella, E. Ferrer, R. Alibes, F. Busque, P. de March, M. Figueredo and J. A. Font, J. Org. Chem., 2009, 74, 6365–6367 CrossRef PubMed.
- D. Page, A. Naismith, R. Schmidt, R. Coupal, E. Labarre, M. Gosselin, D. Bellemare, K. Payza and W. Brown, J. Med. Chem., 2001, 44, 2387–2390 CrossRef PubMed.
- M. Bonanni, M. Marradi, S. Cicchi, C. Faggi and A. Goti, Org. Lett., 2005, 7, 319–322 CrossRef PubMed.
- P. Aschwanden, L. Kværnø, R. W. Geisser, F. Kleinbeck and E. M. Carreira, Org. Lett., 2005, 7, 5741–5742 CrossRef PubMed.
- S. Bag, R. Jayarajan, R. Mondal and D. Maiti, Angew. Chem., Int. Ed., 2017, 56, 3182–3186 CrossRef PubMed.
- H. Leicht, I. Göttker-Schnetmann and S. Mecking, J. Am. Chem. Soc., 2017, 139, 6823–6826 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ob02007h |
| This journal is © The Royal Society of Chemistry 2018 |