
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePurine-substituted imidazolium mesomeric betaines and their tautomeric N-heterocyclic carbenes. Formation of a cyclic borane adduct†
Jiaxi
Zhang
a,
Eike G.
Hübner
 a,
Jan C.
Namyslo
a,
Martin
Nieger
b and
Andreas
Schmidt
*a
a,
Jan C.
Namyslo
a,
Martin
Nieger
b and
Andreas
Schmidt
*a
aClausthal University of Technology, Institute of Organic Chemistry, Leibnizstrasse 6, D-38678 Clausthal-Zellerfeld, Germany. E-mail: schmidt@ioc.tu-clausthal.de
bUniversity of Helsinki, Department of Chemistry, Laboratory of Inorganic Chemistry, P.O. Box 55 and FIN-00014 University of Helsinki, Finland
First published on 5th September 2018
Abstract
6-Chloropurine and 2,6-dichloropurine were reacted with N1-substituted imidazoles to give purin-6-yl substituted imidazolium salts, respectively. Deprotonation of the 1-methylimidazolium derivative resulted in the formation of the corresponding stable conjugated mesomeric betaine, whereas the 1-phenyl,- 1-vinyl- and 1-(2-hydroxyethyl) derivatives proved to be unstable. In situ generation of the mesomeric betaines by caesium carbonate in the presence of sulfur and selenium, however, gave the thiones and the selenones of the tautomeric purine-substituted imidazol-2-ylidene, respectively. Its anionic N-heterocyclic carbene was formally trapped by reaction with triethylborane at high temperatures as a cyclic boron adduct which is the first representative of a new heterocyclic ring system. DFT calculations gained insight into the electronic properties of the N-heterocyclic carbenes substituted by π-electron donators. Results of a single crystal X-ray analysis of the boron adduct are presented.
Introduction
Anionic N-heterocyclic carbenes are interesting target molecules, because their σ-donicities as well as their π-electron characteristics can be adjusted by their molecular architecture in combination with their type of conjugation. Structures, syntheses and applications of first anionic N-heterocyclic carbenes have been summarized in a review article1 and some publications appeared since then.2 A closer look at the hitherto described examples reveals that the term “anionic N-heterocyclic carbene” encompasses fundamentally different types of structures. Thus, the first anionic N-heterocyclic dicarbene 1 (Scheme 1), containing both normal (C2) and abnormal carbene (C4) partial structures, was prepared by lithiation of the corresponding imidazole-2-ylidene and was obtained as the polymer [:C{[N(2,6-iPr2C6H3)]2CHCLi(THF)}]n.3 Reviews summarizing the chemical properties of carbenes of this type4 and results of studies on coordination of metals in general5 as well as main group elements have been published.6 Neither the negative charge of the anionic N-heterocyclic carbene 1 nor that of anion 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 are delocalized in terms of resonance. In contrast to the abnormal carbene structure of 1, the anionic NHC 2 possesses a deprotonated nitrogen atom of a N3-unsubstituted imidazole. The anionic N-heterocyclic carbene 3 is substituted by a siliconate group at C48 and the corresponding boron derivatives belong to a similar molecular architecture.9 The anions 410 and 511 were prepared by deprotonation of conjugated mesomeric betaines (CMB), i.e. imidazolium-olates and sydnones, respectively. As a consequence of their origin, the negative charge is delocalized within the π-conjugated system which sets them apart from the anionic N-heterocyclic carbenes 1, 2, and 3. The canonical formulae of 4 and 5 identify the carbene carbon atoms as sites of the negative charges. Consequently the carbene carbon atoms display considerable atomic orbital coefficients of the calculated highest occupied molecular orbitals (HOMO or HOMO−1) which are π-orbitals, respectively. Thus, the π-electron donor/acceptor properties of the NHCs can be influenced by the architecture of the molecules. With respect to this, the pyrimidine derivative 6 clearly belongs to another type of anionic N-heterocyclic carbene, because it was generated from a cross-conjugated mesomeric betaine (CCMB). Consequently the negative charge is exclusively delocalized in the 3-oxobut-1-en-1-olate partial structure according to the resonance forms without mesomeric charge distribution into the diaminocarbene moiety.12 No conjugation at all between the anionic substituent and the N-heterocyclic carbene moiety of 7 exists, as this anionic N-heterocyclic carbene was prepared by deprotonation of a zwitterionic starting material with isolated anionic and positive charges.13 Thus, carbene 7 is an example of a type of anionic N-heterocyclic carbene which differs from all aforementioned molecules. The chemistry of N-heterocyclic carbenes in the light of history14 as well as the intersection between the substance classes of mesomeric betaines and N-heterocyclic carbenes have been reviewed.15
7 are delocalized in terms of resonance. In contrast to the abnormal carbene structure of 1, the anionic NHC 2 possesses a deprotonated nitrogen atom of a N3-unsubstituted imidazole. The anionic N-heterocyclic carbene 3 is substituted by a siliconate group at C48 and the corresponding boron derivatives belong to a similar molecular architecture.9 The anions 410 and 511 were prepared by deprotonation of conjugated mesomeric betaines (CMB), i.e. imidazolium-olates and sydnones, respectively. As a consequence of their origin, the negative charge is delocalized within the π-conjugated system which sets them apart from the anionic N-heterocyclic carbenes 1, 2, and 3. The canonical formulae of 4 and 5 identify the carbene carbon atoms as sites of the negative charges. Consequently the carbene carbon atoms display considerable atomic orbital coefficients of the calculated highest occupied molecular orbitals (HOMO or HOMO−1) which are π-orbitals, respectively. Thus, the π-electron donor/acceptor properties of the NHCs can be influenced by the architecture of the molecules. With respect to this, the pyrimidine derivative 6 clearly belongs to another type of anionic N-heterocyclic carbene, because it was generated from a cross-conjugated mesomeric betaine (CCMB). Consequently the negative charge is exclusively delocalized in the 3-oxobut-1-en-1-olate partial structure according to the resonance forms without mesomeric charge distribution into the diaminocarbene moiety.12 No conjugation at all between the anionic substituent and the N-heterocyclic carbene moiety of 7 exists, as this anionic N-heterocyclic carbene was prepared by deprotonation of a zwitterionic starting material with isolated anionic and positive charges.13 Thus, carbene 7 is an example of a type of anionic N-heterocyclic carbene which differs from all aforementioned molecules. The chemistry of N-heterocyclic carbenes in the light of history14 as well as the intersection between the substance classes of mesomeric betaines and N-heterocyclic carbenes have been reviewed.15
 | ||
| Scheme 1 | ||
In continuation of our interest in mesomeric betaines16 and their conversion into N-heterocyclic carbenes,14,15 and as part of our studies directed toward the chemical and physical consequences of different types of conjugation, we describe here imidazol-2-ylidenes which are in π-conjugation with an anionic purinate which serves as π-electron donator via N1 of the imidazole.
Results and discussion
A series of purin-6-yl-imidazolium chlorides was prepared starting from substituted imidazoles, 6-chloropurine 8a and 2,6-dichloropurine 8b, respectively (Scheme 2). The reaction of 8a required chlorobenzene as solvent, whereas reactions of 8b proceeded best in ethyl acetate. The salts 10a–h, except for 10d, were obtained in medium to excellent yields as colourless solids under these conditions, respectively. The 1H NMR signals of the H/D exchangeable 2-Himidazolium appear between 10.39 ppm and 10.99 ppm. We tried a series of bases to deprotonate the salts 10. The anion exchange resins Amberlite IRA-400 and IRA-96 in their hydroxide forms, respectively, proved to be unsuited reagents to deprotonate the salts, because the resulting mesomeric betaines or their tautomeric N-heterocyclic carbenes were bound on the resins and could not be eluted. The system consisting of silica gel, ethyl acetate, and petroleum ether with small amounts of triethylamine proved to be a suited medium to deprotonate the salts to the corresponding mesomeric betaines under mild conditions. However, the mesomeric betaines proved to be unstable on trying to remove the solvents or on storage in solution. An exception is the mesomeric betaine 11a which is remarkably stable. It was best obtained in 95% yield on deprotonation with potassium hydroxide in methanol under cooling with ice (Scheme 3). | ||
| Scheme 2 | ||
 | ||
| Scheme 3 | ||
On betaine formation, considerable chemical shift changes of the purinyl substituent can be observed. Thus, Δδ2-H and Δδ8-H of the purine moiety are shifted by 0.44 ppm and 0.69 ppm to higher field, respectively. The signal of 2-H of the imidazolium ring appears at δ = 10.37 ppm in DMSO-d6 so that exclusively tautomer 11aA seems to be present under these conditions. No traces of the N-heterocyclic carbene tautomer 11aB was detected and this observation is in analogy to imidazolium-phenolates,17 imidazolium-isocytosinates,18 imidazolium-indolates,19 imidazolium-azaindolates,19 triazolium-indolates,19 and nitrone.20 Deprotonation of the mesomeric betaine 11a resulted in the formation of an anionic N-heterocyclic carbene 12 which was clearly detected as the base peak at m/z = 201.0885 (calcd 201.0883) in high resolution electrospray ionization mass spectrometry. In accordance with the rules of resonance two negative charges can be formulated at C2 of the imidazole moiety, one of which is caused by the pair of electrons in the sp2 hybrid orbital in the plane of the heteroaromatic ring and the other one is due to the delocalization within the common π-electron system.
The sites of negative charges and positive charges in the canonical formulae of the mesomeric betaine 11a are shown in Fig. 1. As C2 of the imidazolium ring is a site of either charge, this mesomeric betaine can unambiguously be classified as a conjugated system (CMB).21 The carbene carbon atom C2 of the anionic N-heterocyclic carbene 12 remains a site of negative charge in the resonance forms.
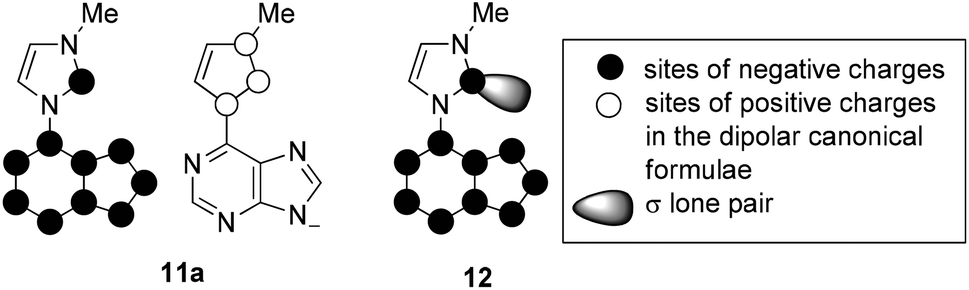 | ||
| Fig. 1 Charge distribution according to the resonance forms. | ||
According to DFT calculations the highest occupied molecular orbital (HOMO) of the mesomeric betaine tautomer 11aA is essentially located in the purine ring, and the same is true for the HOMO−1 (Fig. 2). By contrast, the LUMO is distributed over the entire π-electron system. A large coefficient of the LUMO is located on the bond which connects the purine ring with the imidazole ring which has a calculated bond length of 1.43 Å. The betaine was calculated to be almost perfectly planar (τ = 0.031°). The HOMO of the corresponding N-heterocyclic carbene tautomer 11aB is a σ lone pair located at the carbene carbon atom, whereas the HOMO−1's atomic orbital coefficients are essentially located in the π-electronic system of the imidazole ring with smaller contributions in the purine's π-electron system. The LUMO, by contrast, possesses its major coefficients in the purine ring and on C2 of the imidazole ring. The bond length between the two ring systems was calculated to be slightly shortened (1.39 Å) in comparison to the mesomeric betaine tautomer, but the molecule remains planar on tautomerisation (τ = 0.59°). According to the calculation, the mesomeric betaine tautomer 11aA is by approximately dE = 53 kJ mol−1 (electronic energy) more stable in vacuo than the N-heterocyclic carbene tautomer 11aB. As DMSO stabilizes more dipolar structures, the mesomeric betaine tautomer is even more stable in DMSO solution than in the vacuum (approximately dE = 91 kJ mol−1). The anionic N-heterocyclic carbene 12 is not planar in vacuo according to the calculation. In contrast to the N-heterocyclic carbene tautomer 11aB mentioned above, its HOMO is a π orbital which is located in the purine and the imidazole ring including contributions at C2imidazol-2-ylidene. By contrast, the HOMO−1 is a σ lone pair at the carbene carbon atom plus considerable atomic orbital coefficients especially in the five-membered fragment of the purine ring. The coefficient at N7purine obviously causes the dihedral angle (19,9°) between the two ring systems by electronic repulsion to the σ lone pair at C2imidazole. The torsion, however, is small enough to allow conjugation between the two rings. The LUMO of the N-heterocyclic carbene is distributed all over the π-electron system. The bond length between the two ring systems was calculated to be 1.42 Å.
 | ||
| Fig. 2 Frontier orbitals of 11aA, 11aB, and 12. | ||
When the purine salts are treated with elemental sulphur and selenium in the presence of bases, respectively, they behave as N-heterocyclic carbenes and yield imidazole-thiones and imidazole-selenones in acceptable yields (Scheme 4).
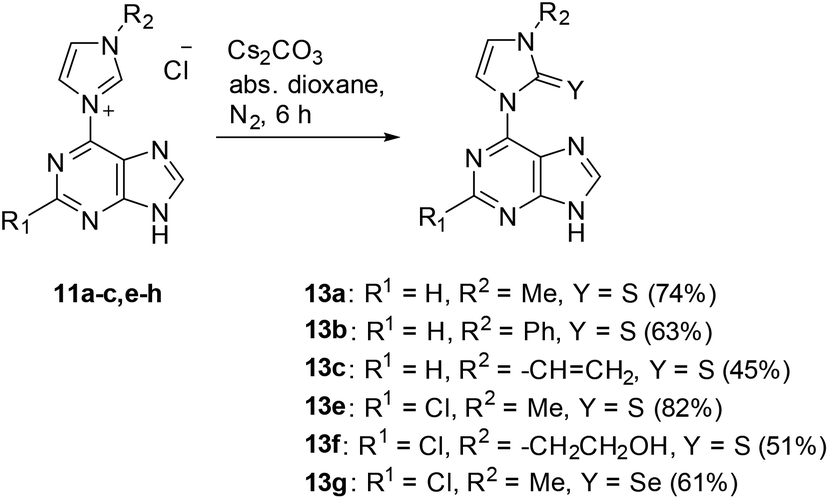 | ||
| Scheme 4 | ||
The anionic N-heterocyclic carbene can formally be trapped as boron adduct. Thus, reaction of the mesomeric betaine 11a with triethylborane at 140 °C in anhydrous dioxane in an autoclave resulted in the formation of the first representative of a new heterocyclic ring system, imidazo[2′,1′:3,4][1,4,2] diazaborinino[1,6,5-gh]purinium-7-ide 14 (Scheme 5).
 | ||
| Scheme 5 | ||
To exclude the formation of its regioisomer 15, a combination of H,H-COSY-, H,C-HSQC-, H,C-HMBC-, and NOESY-experiments was carried out. NOESY correlations between C8purine and the ethyl groups attached to the boron atom proved the formation of the regioisomer 14 possessing the six-membered ring with a bond between N7purine and the boron atom (Fig. 3).
 | ||
| Fig. 3 Diagnostic NOESY correlations. Values of 1H NMR shifts (in black) and 13C NMR shifts (in red) [ppm], respectively. | ||
Single crystals of the boron adduct 14 were obtained by slow evaporation of a concentrated solution in ethyl acetate. The boron adduct crystallized triclinic (Fig. 4). The torsion angle between imidazole and the purine ring [C7A–C7–N8–C12, crystallographic numbering] was determined to be −1.76(15)°. The dihedral angles C12–B1–N1–C7A and N1–B1–C12–N8 were found to be −17.39(12)° and 19.10(14)°, respectively. The bond length between the boron atom and the former carbene carbon atom is 1.6643(16) Å, whereas the bond between the boron atom and the methylene carbon atoms of the ethyl groups was found to have a bond length of 1.6226(16) Å and 1.6315(17) Å. The bond distance between the boron atom and the nitrogen atom is 1.6003(14) Å.
 | ||
| Fig. 4 Molecular drawing of the boron adduct 14 of the anionic N-heterocyclic carbene 12 (displacement parameters are drawn at 50% probability level). | ||
According to a DFT calculation, the HOMO of the boron adduct consists of π orbital contributions of the purine ring plus the B–C σ bonds between the boron atom and the methylene moiety of the ethyl groups (Fig. 5). The LUMO is distributed all over the π electron system. The angles C7–N8–C12–B1 and B1–N1–C7A–C7 were determined to be −10.66(16) and 9.97(18), respectively.
 | ||
| Fig. 5 Frontier orbitals of 14. | ||
Experimental
The reactions were carried out under an atmosphere of nitrogen in oven-dried glassware. Nuclear magnetic resonance (NMR) spectra were obtained with a Bruker Avance 400 and Bruker Avance III 600 MHz. 1H NMR spectra were measured at 400 MHz or 600 MHz and 13C NMR spectra were measured at 100 MHz or 150 MHz, with the solvent peak or tetramethylsilane used as the internal reference. Multiplicities are described by using the following abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, and m = multiplet, and the signal orientations in DEPT experiments were described as follows: o = no signal; + = up (CH, CH3); − = down (CH2). ATR-IR spectra were obtained on a Bruker Alpha in the range of 400 to 4000 cm−1. The mass spectra were measured with a Varian 320 MS Triple Quad GC/MS/MS with a Varian 450-GC. The electrospray ionization mass spectra (ESIMS) were measured with an Agilent LCMSD series HP 1100 with APIES. The compound samples were sprayed from MeCN at 4000 V capillary voltage and fragmentor voltages of 30 V, unless otherwise noted. The HR-MS spectra were obtained with a Bruker Impact II or a Bruker Daltonik Tesla-Fourier transform-ion cyclotron resonance mass spectrometer with electrospray ionization or a Waters Micromass LCT with direct inlet. Melting points are uncorrected and were determined in an apparatus according to Dr Tottoli (Büchi). Yields are not optimized.Calculations
All density-functional theory (DFT)-calculations were carried out by using the Firefly 8.2.0 QC package,22 which is partially based on the GAMESS (US)23 source code, running on Linux 2.6.18-238.el5 SMP (x86_64) on five AMD Phenom II X6 1090T processor workstations (Beowulf-cluster) with Infiniband interconnect and parallelized with MPICH 1.2.7p1. MM2 optimized structures were used as starting geometries. Complete geometry optimizations were carried out on the implemented N31G6* basis set and with the PBE0 density functional. All calculated structures were proven to be true minima by the absence of imaginary frequencies. Solvent effects were estimated by help of the polarizable continuum model (PCM) implemented in Firefly. Partial charges were obtained with NBO 5.924 from the results of the DFT calculations. Orbital plots were obtained using Jmol 14.27.2.
Crystal structure determination of 14
The single-crystal X-ray diffraction study was carried out on a Bruker D8 Venture diffractometer with Photon100 detector at 123(2) K using Cu-Kα radiation (l = 1.54178 Å). Direct Methods (SHELXS-97)25 were used for structure solution and refinement was carried out using SHELXL-2014 (full-matrix least-squares on F2).26 Hydrogen atoms were localized by difference electron density determination and refined using a riding model. A semi-empirical absorption correction and an extinction correction were applied.
14: colourless crystals, C13H17BN6, Mr = 268.13, crystal size 0.36 × 0.36 × 0.12 mm, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 7.2834(4) Å, b = 7.5733(4) Å, c = 12.2004(7) Å, α = 92.486(2)°, β = 94.926(2)°, γ = 96.678(2)°, V = 664.96(6) Å3, Z = 2, ρ = 1.339 Mg m−3, μ(Cu-Kα) = 0.680 mm−1, F(000) = 284, 2θmax = 144.4°, 10331 reflections, of which 2589 were independent (Rint = 0.022), 183 parameters, R1 = 0.036 (for 2467 I > 2σ(I)), wR2 = 0.095 (all data), S = 1.04, largest diff. peak/hole = 0.300/−0.178 e Å−3.
(no. 2), a = 7.2834(4) Å, b = 7.5733(4) Å, c = 12.2004(7) Å, α = 92.486(2)°, β = 94.926(2)°, γ = 96.678(2)°, V = 664.96(6) Å3, Z = 2, ρ = 1.339 Mg m−3, μ(Cu-Kα) = 0.680 mm−1, F(000) = 284, 2θmax = 144.4°, 10331 reflections, of which 2589 were independent (Rint = 0.022), 183 parameters, R1 = 0.036 (for 2467 I > 2σ(I)), wR2 = 0.095 (all data), S = 1.04, largest diff. peak/hole = 0.300/−0.178 e Å−3.
CCDC 1845098 (14)† contains the supplementary crystallographic data for this paper.
1-Methyl-3-(9H-purin-6-yl)-1H-imidazolium chloride 10a
Samples of 618 mg (4.0 mmol) of 6-chloropurine and 984 mg (12.0 mmol) of 1-methylimidazole were dissolved in 20 mL of chlorobenzene. Then, the solution was heated under reflux over a period of 7 h. After cooling to rt, the resulting precipitate was filtered off, washed with diethylether and recrystallized from ethyl acetate. Yield: 584 mg (62%) of a colourless solid, dec. 358 °C (from EtOAc). νmax/cm−1 2241, 3235, 3122, 3086, 1615, 1573, 1533, 1441, 1346, 1293, 1184, 1129, 1106, 916, 841, 767, 646, 573, 534, 406. δH (400 MHz; DMSO-d6) 10.39 (s, 1H), 8.99 (s, 1H), 8.92 (s, 1H), 8.84 (m, 1H), 8.06 (m, 1H), 4.09 (s, 3H) ppm. δC (100 MHz; DMSO-d6) 155.6, 151.4, 147.4, 141.7, 137.2, 125.0, 122.0, 119.8, 36.6 ppm. m/z(ESIMS) 201.1 (M)+. HR-ESI-MS [C9H9N6]: calcd 201.0883 [M]+, found 201.0886.1-Phenyl-3-(9H-purin-6-yl)-1H-imidazolium chloride 10b
1-Vinyl-3-(9H-purin-6-yl)-1H-imidazolium chloride 10c
(2-Chloro-9H-purin-6-yl)-1-methyl-1H-imidazolium chloride 10e
A sample of 189 mg (1.0 mmol) of 2,6-dichloropurine and 164 mg (2.0 mmol) of 1-methylimidazol were dissolved in 10 mL of ethyl acetate and heated under reflux for 7 h. After cooling to rt the resulting precipitate was filtered off, washed with diethylether and recrystallized from ethyl acetate. Yield: 168 mg (62%) of a colourless solid, dec. 367 °C (from EtOAc). νmax/cm−1 3057, 2636, 1694, 1586, 1331, 1178, 1015, 859, 839, 791, 634, 617, 598, 547. δH (400 MHz; D2O) 8.63 (s, 1H), 8.62 (d, J = 2.2 Hz, 1H), 7.81 (d, J = 2.2 Hz, 1H), 4.15 (s, 3H) ppm. δC (100 MHz; D2O) 160.1, 155.4, 151.2, 145.3, 140.1, 127.8, 124.7, 123.1, 39.8 ppm. m/z (ESIMS) 235.0 (M)+. HR-ESI-MS [C9H8N6]: calcd 235.0499 [M]+, found 235.0498.1-Phenyl-3-(2-chloro-9H-purin-6-yl)-1H-imidazolium chloride 10f
3-(2-Chloro-9H-purin-6-yl)-1-vinyl-1H-imidazolium chloride 10g
3-(2-Chloro-9H-purin-6-yl)-1-(2-hydroxyethyl)-1H-imidazolium chloride 10h
6-(1-Methyl-1H-imidazolium-3-yl)purin-7-ide 11a
A sample of 237 mg (1.0 mmol) of the salt 10a was suspended in 50 mL of anhyd methanol and cooled to 0 °C. Then, 50 mL of KOH in water (1.1 mol L−1) was added dropwise. After 30 minutes of stirring at that temperature, the suspension was evaporated to dryness. The crude product was washed with ice-cold water and dried in vacuo. Yield: 190 mg (95%) of a colourless solid, dec. 308 °C (from H2O). νmax/cm−1 3306, 3160, 2109, 3086, 1657, 1612, 1575, 1533, 1441, 1345, 1293, 1240, 1184, 1105, 1089, 915, 841, 766, 645, 572, 534, 406. δH (400 MHz; D2O) 10.37 (s, 1H), 8.87 (m, 1H), 8.55 (s, 1H), 8.23 (s, 1H), 7.94 (m, 1H), 4.04 (s, 3H) ppm. δC (100 MHz; D2O) 167.6, 159.8, 146.4, 137.7, 136.3, 124.4, 124.2, 119.4, 36.3 ppm. m/z (ESIMS) 201.1 (M + H)+. HR-ESI-MS [C9H8N6]: calcd 201.0883 [M + H]+, found 201.0885.1-Methyl-3-(9H-purin-6-yl)-1,3-dihydro-2H-imidazole-2-thione 13a
A suspension of 326 mg (1.0 mmol) of caesium carbonate, 109 mg (0.5 mmol) of salt 10a, and 22 mg (0.7 mmol) of sulfur in 15 mL of anhyd acetonitrile was heated under nitrogen over a period of 7 h under reflux. After the solvent was distilled off in vacuo, the crude reaction product was chromatographed (silica gel, EtOAc). Yield: 86 mg (74%) of a yellow solid, dec. 258 °C (from EtOAc). νmax/cm−1 3108, 2922, 1623, 1554, 1460, 1446, 1383, 1368, 1351, 1249, 1179, 1100, 914, 844, 801, 732, 701, 666, 651, 612, 598, 547, 534. δH (400 MHz; DMSO-d6) 8.93 (s, 1H), 8.69 (s, 1H), 7.47 (d, J = 2.5 Hz, 1H), 7.42 (d, J = 2.5 Hz, 1H), 3.58 (s, 3H) ppm. δC (100 MHz; DMSO-d6) 162.7, 159.4, 151.3, 147.5, 144.4, 120.3, 117.4, 34.5 ppm. HR-ESI-MS for [C9H8N6S]: calcd 255.0423 [M + Na]+. Found 255.0414.1-Phenyl-3-(9H-purin-6-yl)-1,3-dihydro-2H-imidazole-2-thione 13b
1-(9H-Purin-6-yl)-3-vinyl-1,3-dihydro-2H-imidazole-2-thione 13c
1-(2-Chloro-9H-purin-6-yl)-3-methyl-1,3-dihydro-2H-imidazole-2-thione 13e
1-(2-Chloro-9H-purin-6-yl)-3-(2-hydroxyethyl)-1,3-dihydro-2H-imidazole-2-thione 13f
1-(2-Chloro-9H-purin-6-yl)-3-methyl-1,3-dihydro-2H-imidazole-2-selenone 13g
7,7-Diethyl-8-methyl-7,8-dihydroimidazo[2′,1′:3,4][1,4,2] diazaborinino[1,6,5-gh]purinium-7-ide 14
A solution of 242 mg (1.0 mmol) of triethylborane in 2 mL of anhyd dichloromethane was added dropwise into a suspension of 96 mg (0.5 mmol) of 11a in 5 mL of the same solvent. The mixture was then heated to 200 °C under a nitrogen atmosphere for 24 h in a Schlenk tube. After cooling to rt, the solvent was distilled off in vacuo and the crude reaction product was chromatographed (silica gel, EtOH:EtOAc = 2:1). Yield: 91 mg (39%) of a colorless solid, dec. 314 °C (from EtOH/EtOAc). νmax/cm−1 2940, 2905, 2862, 2818, 1669, 1581, 1473, 1461, 1427, 1404, 1313, 1304, 1192, 1082, 1048, 847, 802, 641, 623, 571. δH (400 MHz; DMSO-d6) 8.77 (s, 1H), 8.47 (s, 1H), 8.38 (d, J = 2.1 Hz, 1H), 7.90 (d, J = 2.1 Hz, 1H), 3.92 (s, 1H), 0.79–0.74 (m, 2H), 0.72–0.68 (m, 2H), 0.32 (t, J = 7.8 Hz, 6H) ppm. δC (100 MHz; DMSO-d6) 161.7, 151.3, 150.3, 141.4, 126.7, 118.1, 114.8, 36.4, 16.7, 10.8 ppm. m/z (ESIMS) 269.1 (M + H)+, 559.3 (2M + Na)+. HR-ESI-MS for [C13H17BN6] calcd 269.1681 (M + H)+. Found 269.1685.
Conclusions
(Purin-6-yl)-1-methyl-imidazolium salts can be deprotonated to give a stable mesomeric betaine, the N-heterocyclic tautomer of which can be trapped by sulphur and selenium. The corresponding anionic N-heterocyclic carbene formally gave a cyclic borane adduct which is the first representative of a new heterocyclic ring system, imidazo[2′,1′:3,4] [1,4,2]diazaborinino[1,6,5-gh]purine.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Gerald Dräger, university of Hannover (Germany) is gratefully acknowledged for measuring a part of the HRMS spectra.References
- A. Nasr, A. Winkler and M. Tamm, Coord. Chem. Rev., 2016, 316, 68 CrossRef.
- (a) H. Niu, R. J. Mangan, A. V. Protchenko, N. Phillips, W. Unkrig, C. Friedmann, E. L. Kolychev, R. Tirfoin, J. Hicks and S. Aldridge, Dalton Trans., 2018, 47, 7445 RSC; (b) J. B. Waters, L. S. Tucker and J. M. Goicoechea, Organometallics, 2018, 37, 655 CrossRef; (c) A.-L. Lücke, S. Wiechmann, T. Freese, M. Nieger, T. Földes, I. Pápai, M. Gjikaj, A. Adam and A. Schmidt, Tetrahedron, 2018, 74, 2092 CrossRef; (d) G. Kleinhans, G. Guisado-Barrios, E. Peris and D. I. Bezuidenhout, Polyhedron, 2018, 143, 43 CrossRef; (e) G. Nagai, T. Mitsudome, K. Tsutsumi, S. Sueki, T. Ina, M. Tamm and K. Nomura, J. Jpn. Pet. Inst., 2017, 60, 256 CrossRef; (f) M. Ruamps, N. Lugan and V. César, Eur. J. Inorg. Chem., 2017, 36, 4167 CrossRef.
- Y. Wang, Y. Xie, M. Y. Abraham, P. Wei, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2010, 132, 14370 CrossRef PubMed.
- J. B. Waters and J. M. Goicoechea, Coord. Chem. Rev., 2015, 293–294, 80 CrossRef; R. S. Ghadwal, Dalton Trans., 2016, 45, 16081 RSC.
- (a) D. Specklin, C. Fliedel, C. Gourlaouen, J.-C. Bruyere, T. Avilés, C. Boudon, L. Ruhlmann and S. Dagorne, Chem. – Eur. J., 2017, 23, 5509 CrossRef PubMed; (b) A. Winkler, K. Brandhorst, M. Freytag, P. G. Jones and M. Tamm, Organometallics, 2016, 35, 1160 CrossRef; (c) A. J. Martínez-Martínez, M. A. Fuentes, A. Hernán-Gómez, E. Hevia, A. R. Kennedy, R. E. Mulvey and C. T. O'Hara, Angew. Chem., Int. Ed., 2015, 54, 14075 CrossRef PubMed; (d) L. C. H. Maddock, T. Cadenbach, A. R. Kennedy, I. Borilovic, G. Aromí and E. Hevia, Inorg. Chem., 2015, 54, 9201 CrossRef PubMed; (e) J. B. Waters and J. M. Goicoechea, Dalton Trans., 2014, 43, 14239 RSC; (f) C. Pranckevicius and D. W. Stephan, Chem. – Eur. J., 2014, 20, 6597 CrossRef PubMed; (g) A. El-Hellani and V. Lavallo, Angew. Chem., Int. Ed., 2014, 53, 4489 CrossRef PubMed; (h) D. R. Armstrong, S. E. Baillie, V. L. Blair, N. G. Chabloz, J. Diez, J. Garcia-Alvarez, A. R. Kennedy, S. D. Robertson and E. Hevia, Chem. Sci., 2013, 4, 4259 RSC.
- (a) P. K. Majhi, K. C. F. Chow, T. H. H. Hsieh, E. G. Bowes, G. Schnakenburg, P. Kennepohl, R. Streubel and D. P. Gates, Chem. Commun., 2016, 52, 998 RSC; (b) A. Winkler, M. Freytag, P. G. Jones and M. Tamm, Z. Anorg. Allg. Chem., 2016, 642, 1295 CrossRef; (c) M. Uzelac, A. R. Kennedy, A. Hernán-Gómez, M. A. Fuentes and E. Hevia, Z. Anorg. Allg. Chem., 2016, 642, 1241 CrossRef; (d) R. S. Ghadwal, S. O. Reichmann, E. Carl and R. Herbst-Irmer, Dalton Trans., 2014, 43, 13704 RSC; (e) M. Chen, Y. Wang, R. J. Gilliard Jr., P. Wei, N. A. Schwartz and G. H. Robinson, Dalton Trans., 2014, 43, 14211 Search PubMed.
- P. Ai, A. A. Danopoulos and P. Braunstein, Inorg. Chem., 2015, 54, 3722 CrossRef PubMed.
- F. Medici, G. Gontard, E. Derat, G. Lemière and L. Fensterbank, Organometallics, 2018, 37, 517 CrossRef.
- A. Igarashi, E. L. Kolychev, M. Tamm and K. Nomura, Organometallics, 2016, 35, 1778 CrossRef.
- V. César, V. Mallardo, A. Nano, G. Dahm, N. Lugan, G. Lavigne and S. Bellemin-Laponnaz, Chem. Commun., 2015, 51, 5271 RSC.
- (a) T. Freese, A.-L. Lücke, J. C. Namyslo, M. Nieger and A. Schmidt, Eur. J. Org. Chem., 2018, 1646 CrossRef; (b) T. Freese, A.-L. Lücke, C. A. S. Schmidt, M. Polamo, M. Nieger, J. C. Namyslo and A. Schmidt, Tetrahedron, 2017, 73, 5350 CrossRef; (c) A.-L. Lücke, S. Wiechmann, T. Freese, Z. Guan and A. Schmidt, Z. Naturforsch., 2016, 71b, 643 Search PubMed; (d) S. Wiechmann, T. Freese, M. H. H. Drafz, E. G. Hübner, J. C. Namyslo, M. Nieger and A. Schmidt, Chem. Commun., 2014, 50, 11822 RSC.
- (a) G. Lavigne, V. César and N. Lugan, Chem. – Eur. J., 2010, 16, 11432 CrossRef PubMed; (b) V. César, N. Lugan and G. Lavigne, J. Am. Chem. Soc., 2008, 130, 11286 CrossRef PubMed.
- A. Ferry, K. Schaepe, P. Tegeder, C. Richter, K. M. Chepiga, B. J. Ravoo and F. Glorius, ACS Catal., 2015, 5, 5414 CrossRef.
- A. Schmidt, S. Wiechmann and C. F. Otto, Adv. Heterocycl. Chem., 2016, 119, 143 CrossRef.
- A. Schmidt, S. Wiechmann and T. Freese, ARKIVOC, 2013, i, 424 Search PubMed.
- A. Schmidt and M. Nieger, Heterocycles, 1999, 51, 2119 CrossRef.
- (a) M. Liu, J. C. Namyslo, M. Nieger, M. Polamo and A. Schmidt, Beilstein J. Org. Chem., 2016, 12, 2673 CrossRef PubMed; (b) M. Liu, M. Nieger, E. Hübner and A. Schmidt, Chem. – Eur. J., 2016, 22, 5416 CrossRef PubMed.
- J. Zhang, M. Franz, E. Hübner and A. Schmidt, Tetrahedron, 2016, 72, 525 CrossRef.
- N. Pidlypnyi, S. Wolf, M. Liu, K. Rissanen, M. Nieger and A. Schmidt, Tetrahedron, 2014, 70, 8672 CrossRef.
- C. Färber, M. Leibold, C. Bruhn, M. Maurer and U. Siemeling, Chem. Commun., 2012, 227 RSC.
- (a) C. A. Ramsden, Tetrahedron, 2013, 69, 4146 CrossRef; (b) C. A. Ramsden and W. P. Oziminski, Tetrahedron, 2014, 70, 7158 CrossRef; (c) W. P. Oziminski and C. A. Ramsden, Tetrahedron, 2015, 71, 7191 CrossRef; (d) W. D. Ollis, S. P. Stanforth and C. A. Ramsden, Tetrahedron, 1985, 41, 2239 CrossRef.
- A. A. Granovsky, Firefly version 8, http://www.classic.chem.msu.su/gran/firefly/index.html.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347 CrossRef.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales and F. Weinhold, NBO 5.9, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2012 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of calculations. CCDC 1845098 (14). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ob01916a |
| This journal is © The Royal Society of Chemistry 2018 |