DOI:
10.1039/C8OB01895B
(Review Article)
Org. Biomol. Chem., 2019,
17, 1595-1607
Recent advances in homogeneous borrowing hydrogen catalysis using earth-abundant first row transition metals
Received
3rd August 2018
, Accepted 4th September 2018
First published on 17th September 2018
Abstract
The review highlights the recent advances (2013-present) in the use of earth-abundant first row transition metals in homogeneous borrowing hydrogen catalysis. The utility of catalysts based on Mn, Fe, Co, Ni and Cu to promote a diverse array of important C–C and C–N bond forming reactions is described, including discussion on reaction mechanisms, scope and limitations, and future challenges in this burgeoning area of sustainable catalysis.
 From left to right: Benjamin G. Reed-Berendt, Kurt Polidano and Louis C. Morrill | Ben Reed-Berendt obtained a Masters in Chemistry at Cardiff University, where he is now pursuing a PhD. |
| Kurt Polidano obtained a Masters in Science at the University of Malta. He is now pursuing a PhD at Cardiff University as part of the EPSRC-funded Bath/Bristol/Cardiff Catalysis Centre for Doctoral Training. Ben and Kurt are both researching in the area of homogeneous borrowing hydrogen catalysis using earth-abundant first row metals. |
| Louis Morrill received his PhD from the University of St Andrews in 2014 under the direction of Prof. Andrew Smith and undertook postdoctoral research at UC Berkeley with Prof. Richmond Sarpong. In June 2015, he was appointed as a University Research Fellow at Cardiff University. Research in the group is focused on inventing new reactions in organic chemistry and developing sustainable catalytic methodologies for synthesis. |
1. Introduction
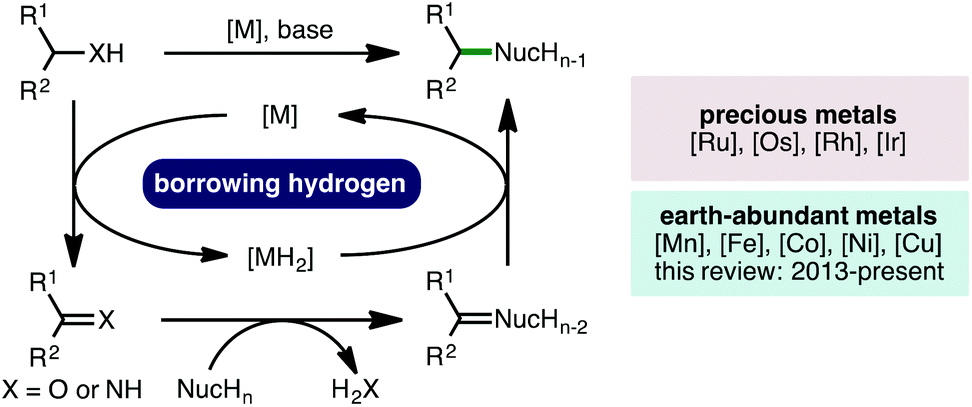
The borrowing hydrogen (BH) approach, also known as hydrogen autotransfer, combines a transfer hydrogenation process with a concurrent reaction on the in situ generated reactive intermediate.1 This one-pot oxidation–reaction–reduction sequence has received much attention due to its inherent high atom economy, permitting alcohols or amines to be employed as alkylating agents, generating water or ammonia as the sole by-products, respectively (Scheme 1).2 Traditionally, borrowing hydrogen processes have employed precious second and third row transition metal catalysts, specifically those based on Ru, Os, Rh and Ir, to perform hydrogen transfer.3 However, in line with the increasing global challenge to develop more sustainable catalytic methodologies for the production of chemicals for society, there has been a concerted effort within the scientific community to seek alternatives to catalysts based on precious metals by diversifying the utility of earth-abundant first row transition metals in catalysis.4 In this context, this review will describe the recent advances (2013-present) in homogeneous borrowing hydrogen catalysis using earth-abundant first row transition metal catalysts based on Mn, Fe, Co, Ni and Cu for various C–C and C–N bond forming processes.5 Heterogeneous borrowing hydrogen catalysis,6 in addition to net (de)hydrogenative processes,7 which have been previously reviewed,8 are outside the scope of this review.
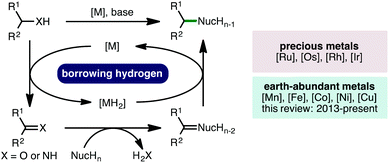 |
| | Scheme 1 Generalisation of the borrowing hydrogen approach. | |
2. Iron-catalysed borrowing hydrogen
2.1
N-Alkylation processes
The ability to access higher amines via the N-alkylation of primary and secondary amines is a fundamental transformation in synthetic chemistry. However, the reaction of primary amines with toxic alkyl halides or alkyl sulfonates generates waste and can result in over alkylation, whereas reductive amination requires the use of a stoichiometric reducing agent. Employing the borrowing hydrogen approach, amines can undergo selective mono-N-alkylation using bench stable and inexpensive alcohols as the alkylating agent, generating water as the sole by-product.9
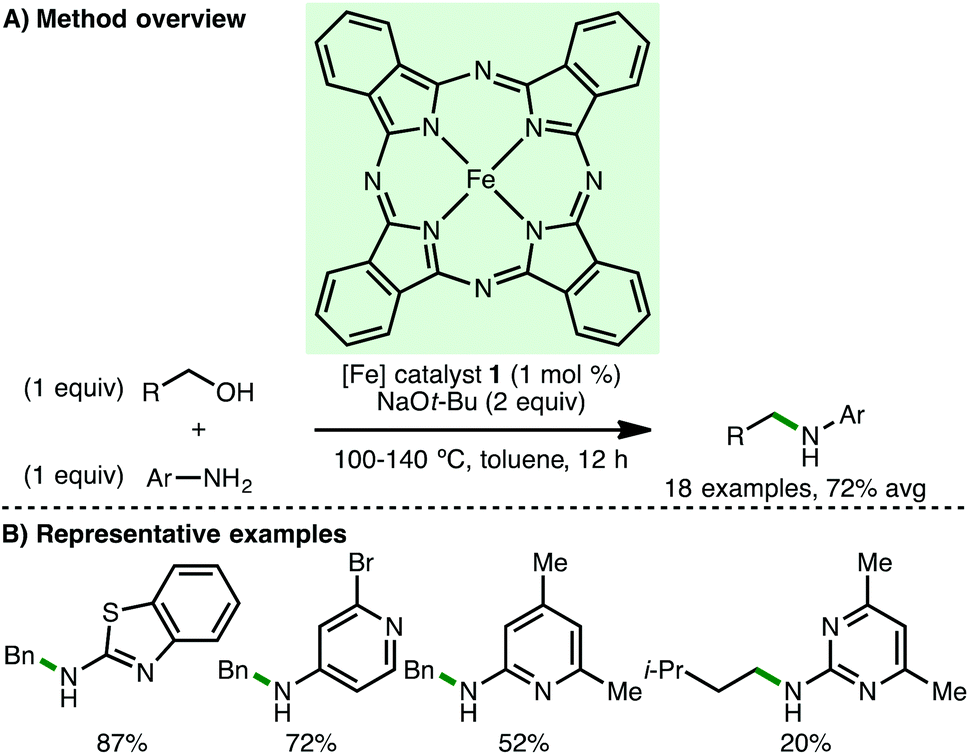
In 2013, Singh and co-workers reported Fe(II) phthalocyanine 1 as catalyst for the N-alkylation of various heteroaryl amines using primary benzylic alcohols, accessing the corresponding secondary amines in high isolated yields (Scheme 2).10 When isoamyl alcohol was used as the alkylating agent, the corresponding secondary amine was obtained in only 20% isolated yield. No background reaction was observed in the absence of Fe(II) phthalocyanine 1. Mechanistic experiments confirmed that both the oxidation of benzyl alcohol to benzylaldehyde and the transfer hydrogenation between benzyl alcohol and an aldimine occur under the specified reaction conditions, providing some evidence for the proposed borrowing hydrogen mechanism.
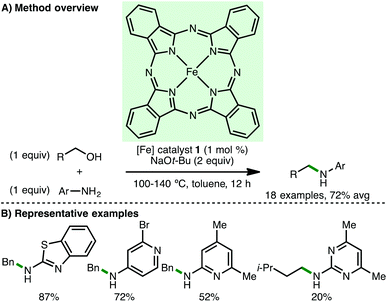 |
| | Scheme 2 Fe-Catalysed N-alkylation of heteroaryl amines (Singh). | |
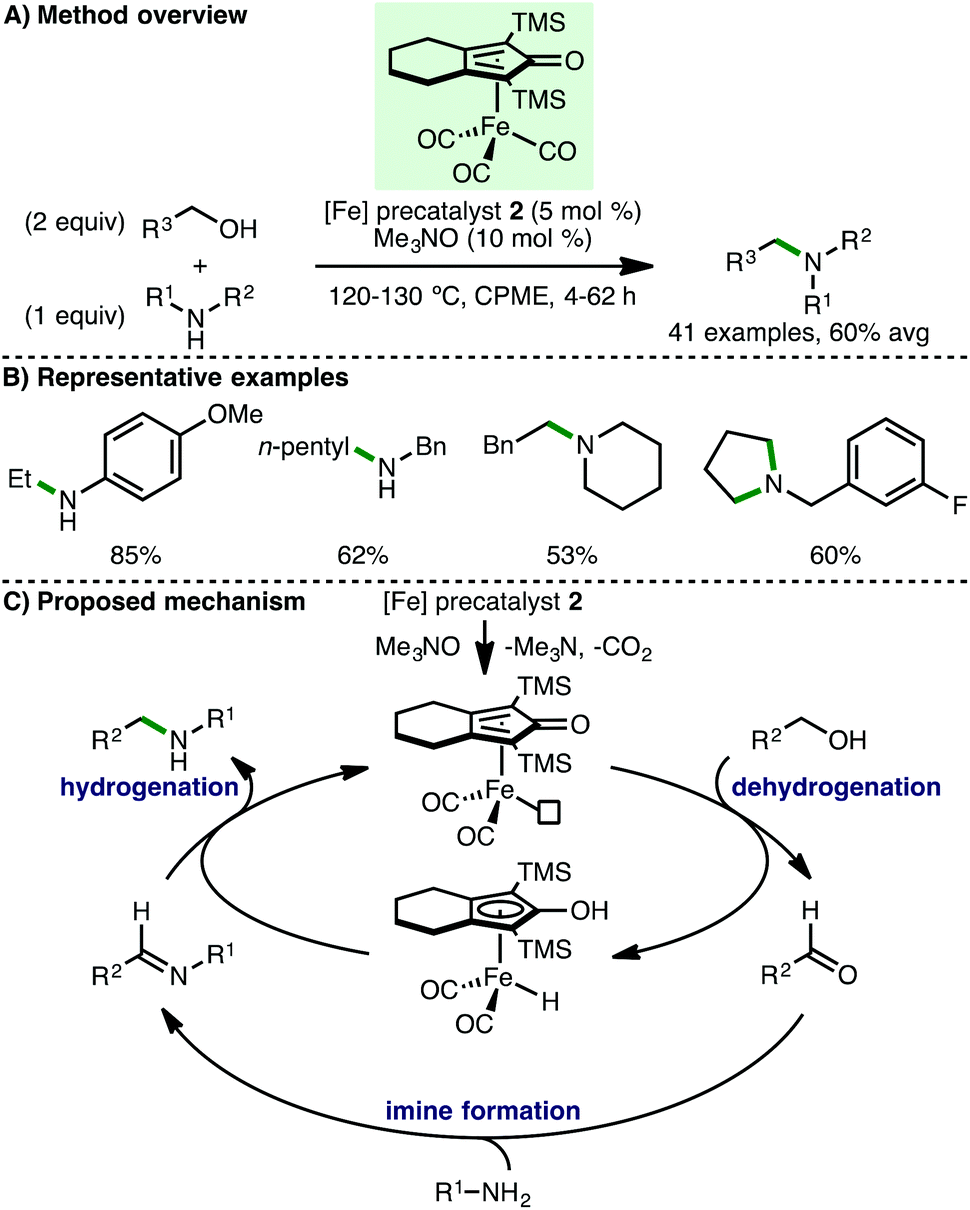
Feringa, Barta and co-workers subsequently employed (cyclopentadienone)iron carbonyl precatalyst 2 for the N-alkylation of amines using alcohols (Scheme 3).11 Employing CPME as a green solvent, it was found that electron-rich anilines undergo N-alkylation using primary aliphatic alcohols, with the exception of methanol. When secondary alcohols including isopropanol and cyclohexanol were used, only low conversions to the N-alkylated amines were observed. Various benzylamines were also shown to undergo N-alkylation with pentan-1-ol, and diols were employed to access 5-, 6- and 7-membered N-heterocycles. It was also noted that secondary amines undergo N-alkylation with benzylic alcohols and this was applied to the synthesis of N-arylpiperazine drug Piribedil, a dopamine antagonist used in the treatment of Parkinson's disease. Employing closely related catalytic systems, the same group subsequently described a generalised approach for the N-alkylation of various secondary and primary amines with benzylic alcohols,12 and the direct N-alkylation of unprotected α-amino acids (glycine, alanine and proline) with long-chain aliphatic fatty alcohols to form bio-derived surfactants.13 The proposed mechanism for borrowing hydrogen N-alkylation processes involving (cyclopentadienone)iron carbonyl complexes begins with CO decoordination, which can be achieved thermally, photochemically, and/or by using activators such as Me3NO and PPh3. The resulting active iron complex abstracts hydrogen from the alcohol to form a transient reactive aldehyde intermediate. A subsequent condensation with the amine forms an imine, which then undergoes hydrogenation by the iron-hydrogen complex to give the N-alkylated product with regeneration of the active iron complex. In order to support the proposed borrowing hydrogen mechanism, the authors performed a detailed in situ NMR study, which detected the simultaneous presence of the alcohol starting material, the aldehyde and imine intermediates, and the amine product.
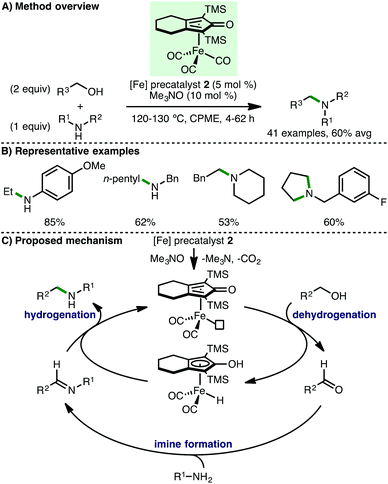 |
| | Scheme 3 Fe-Catalysed N-alkylation of aryl- and benzylamines (Feringa, Barta). | |
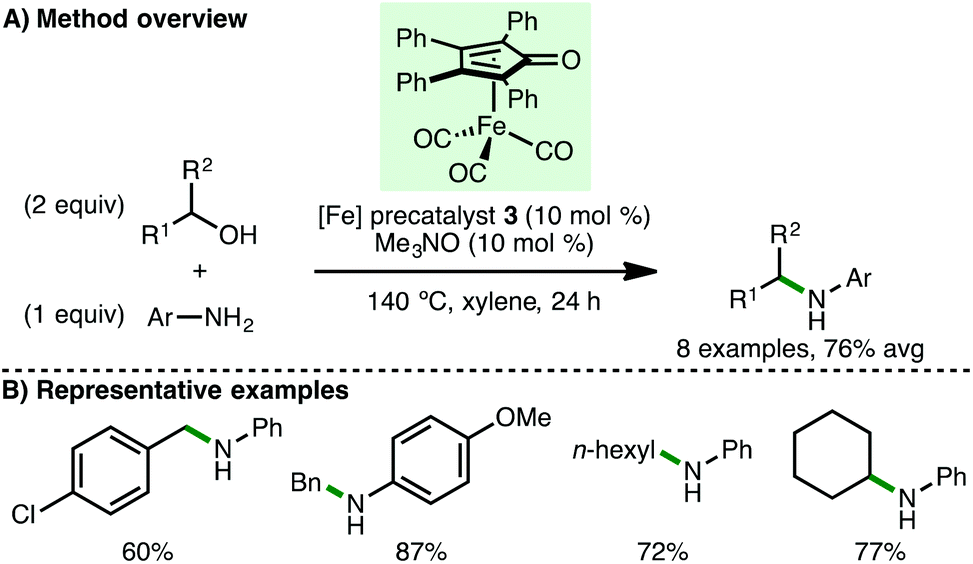
Soon after the initial report from Feringa and Barta,11 Wills and co-workers reported the use of (tetraphenylcyclopentadienone)iron carbonyl precatalyst 3 for the N-alkylation of amines via borrowing hydrogen catalysis (Scheme 4).14 The N-alkylation of aniline and 4-methoxyaniline with benzylic and aliphatic alcohols was demonstrated, accessing the corresponding secondary amines in high yields. Secondary alcohols were tolerated, as illustrated by the N-alkylation of aniline with cyclohexanol, giving product in 77% isolated yield. In a subsequent detailed study by the same group, a family of (cyclopentadienone)iron carbonyl precatalysts were synthesised containing various aromatic groups flanking the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the cyclopentadienone to probe the effect of electronic variation on the efficiency of N-alkylation reactions.15 In general, no clear reactivity trends could be identified across a selection of substrates, although the N-alkylation of aniline with a broader selection of secondary alcohols and diols was demonstrated. In many cases, it was found that the Knölker precatalyst 2 (cf.Scheme 3), containing trimethysilyl groups flanking the CO of the cyclopentadienone, proved optimal across a broader range of substrates.
O of the cyclopentadienone to probe the effect of electronic variation on the efficiency of N-alkylation reactions.15 In general, no clear reactivity trends could be identified across a selection of substrates, although the N-alkylation of aniline with a broader selection of secondary alcohols and diols was demonstrated. In many cases, it was found that the Knölker precatalyst 2 (cf.Scheme 3), containing trimethysilyl groups flanking the CO of the cyclopentadienone, proved optimal across a broader range of substrates.
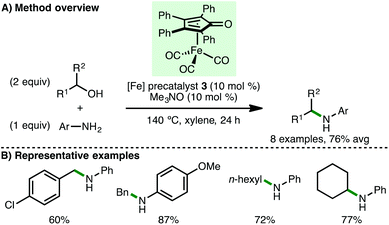 |
| | Scheme 4 Fe-Catalysed N-alkylation of arylamines (Wills). | |
In 2015, Zhao and co-workers developed a general approach for N-alkylation of amines with secondary alcohols by using AgF (40 mol%) as a Lewis acid additive, accessing the corresponding secondary amines in good yields (Scheme 5).16 The method was somewhat limited to the use of electron-rich arylamines, with benzylamine and piperidine giving lower isolated yields. N-Alkylation was also demonstrated using primary aliphatic alcohols, benzylic alcohols and diols. Importantly, no background reaction was observed in the absence of Knölker's complex 4, supporting a borrowing hydrogen mechanism. It was proposed that AgF facilitates both imine formation and imine hydrogenation via coordination to the ketone and imine respectively.
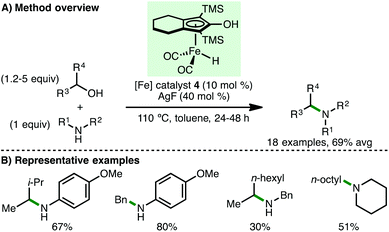 |
| | Scheme 5 Fe-Catalysed N-alkylation using silver fluoride as an additive (Zhao). | |
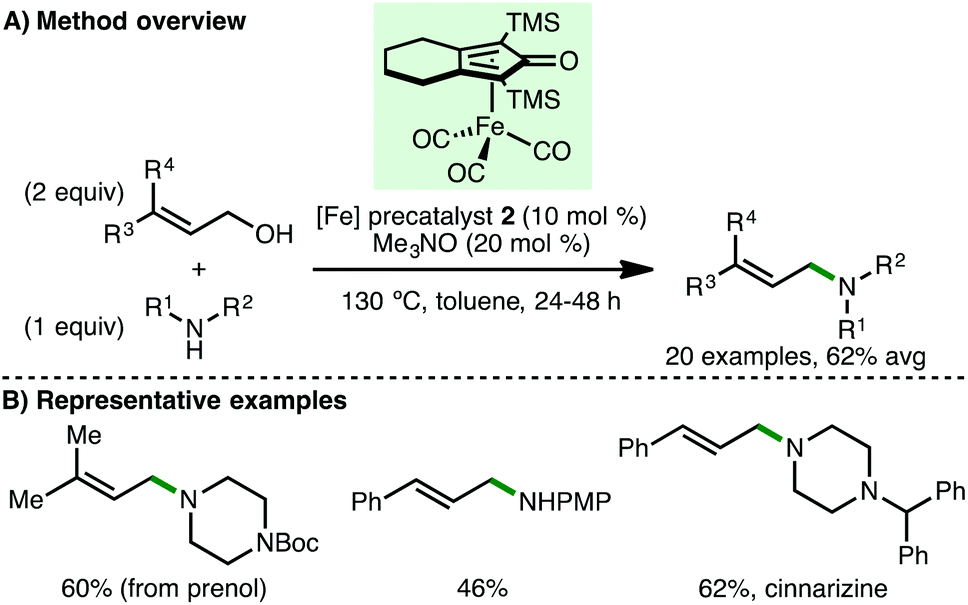
Building on these reports, Sundararaju and co-workers employed (cyclopentadienone)iron carbonyl precatalyst 2 for the N-allylation of a diverse array of primary and secondary amines using allylic alcohols via the borrowing hydrogen approach (Scheme 6).17 This process is noteworthy due to the possible side reactions that could occur, but do not, including isomerisation of the allylic alcohol followed by reductive amination, or 1,4-conjugate addition of the metal hydride to the α,β-unsaturated aldehyde intermediate followed by reductive amination. Impressively, terpenoids prenol, geraniol and farnesol were employed as allylating agents, giving the corresponding tertiary amines in synthetically useful yields. Enal intermediates were commonly observed as by-products, providing evidence for a borrowing hydrogen mechanism over a possible π-allyl species. The authors directed their methodology towards the synthesis of antihistaminic drug, cinnarizine, and antifungal drug, nafetifine, in good yields. The same group later extended this approach towards the synthesis of pyrroles, which involves an initial borrowing hydrogen N-allylation of amines (aryl, benzylic and aliphatic) with substituted but-2-ene-1,4-diols followed by dehydrogenative cyclisation and subsequent aromatisation.18
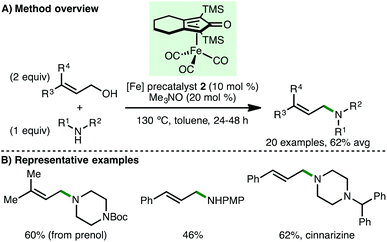 |
| | Scheme 6 Fe-Catalysed N-allylation using allylic alcohols (Sundararaju). | |
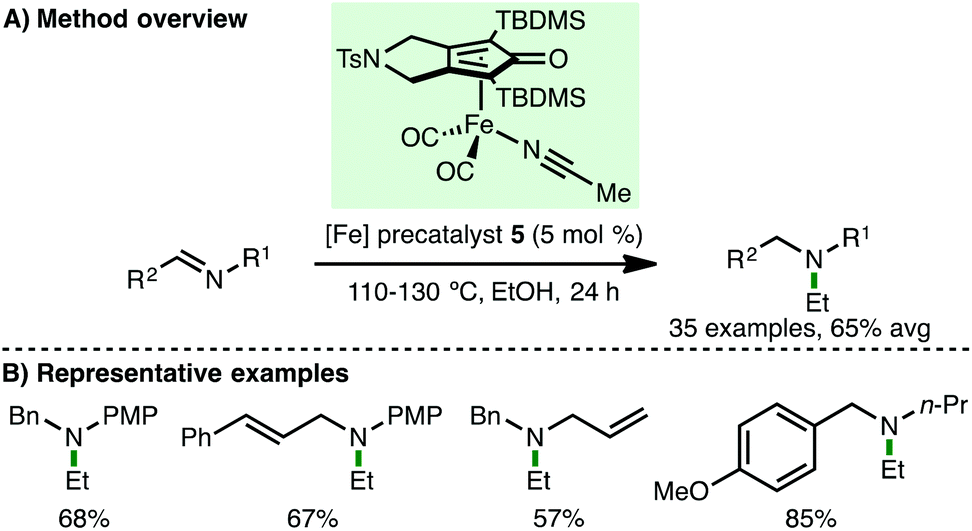
Very recently, Gandon, Bour and co-workers reported the reductive ethylation of imines with ethanol, catalyzed by (cyclopentadienone)iron carbonyl precatalyst 5 (Scheme 7).19 A variety of electron-rich aryl and alkyl N-substituted aldimines participate in the reductive ethylation protocol, giving the corresponding tertiary amines in good isolated yields. It is proposed that an initial transfer hydrogenation between ethanol and an aldimine generates acetaldehyde and the corresponding secondary amine, which react together to form an enamine that undergoes hydrogenation to afford the N-ethyl tertiary amine products. The authors suggest that the interesting exclusive selectivity for ethanol may be attributed to the need for rapid trapping of the formed aldehyde (which would be slower for larger alcohols) and enamine hydrogenation (which would not be possible for methanol). Density functional theory (DFT) calculations confimed that the required free energy of activation for the proposed enamine hydrogenation was only 8.0 kcal mol−1, indicating its feasibility.
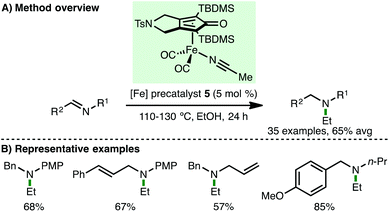 |
| | Scheme 7 Fe-Catalysed reductive N-ethylation of imines (Gandon, Bour). | |
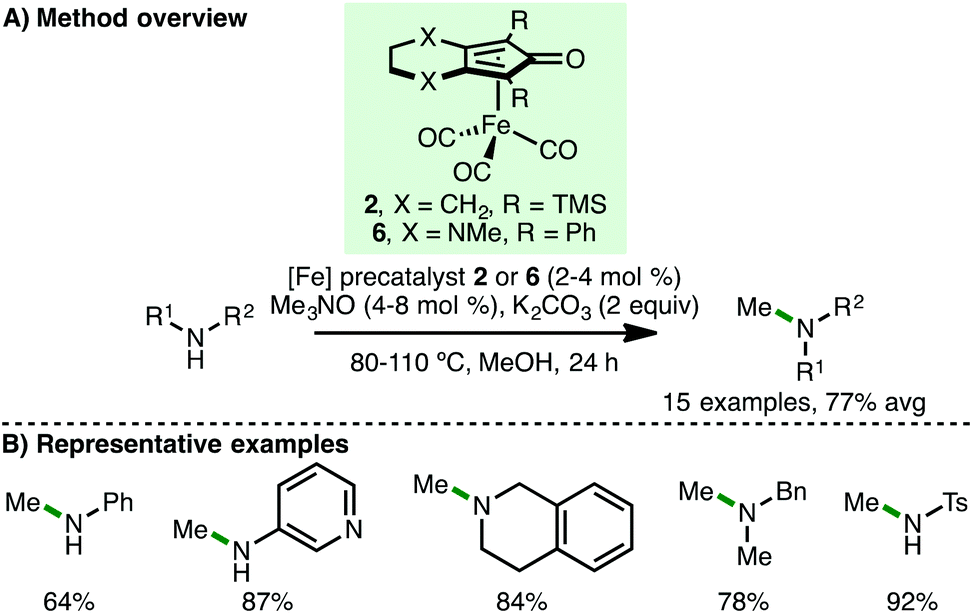
Inspired by these reports, our group subsequently disclosed the iron-catalysed N-methylation of amines using methanol via the borrowing hydrogen approach (Scheme 8).20 Despite the increased energy of methanol dehydrogenation (ΔH = +84 kJ mol−1) in addition to Feringa, Barta and co-workers highlighting that methylation was not successful in their study using (cyclopentadienone)iron carbonyl precatalyst 2,11 we found that simply employing methanol as solvent and using K2CO3 (2 equiv.) as base permitted the efficient N-methylation of various primary arylamines in high yields. Furthermore, a selection of cyclic and acyclic secondary amines also undergo N-methylation. Interestingly, these reactions do no proceed in the absence of base. Employing Renaud's (cyclopentadienone)iron carbonyl precatalyst 6, which contains a more electron-rich cyclopentadienone framework, the N-monomethylation of sulfonamides was also demonstrated.
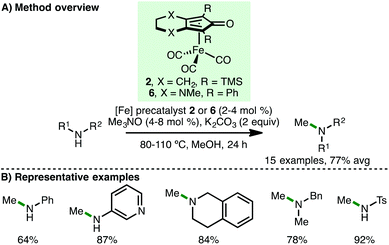 |
| | Scheme 8 Fe-Catalysed N-methylation using methanol (Morrill). | |
In 2016, Kirchner and co-workers disclosed the use of a Fe(II) PNP pincer precatalyst 7 for the N-alkylation of amines using alcohols (Scheme 9).21 A variety of primary amines (aryl, benzylic and aliphatic) and primary alcohols (benzylic and aliphatic) were employed in the substrate scope, accessing a selection of secondary amines in excellent yields. It was noted that the addition of 3 Å molecular sieves, which promote imine formation by the removal of water and may also serve as a Lewis acid, was important for obtaining the products in high yield. The proposed mechanism involves initial treatment of precatalyst 7 with base to generate a coordinately unsaturated iron complex, which contains a deprotonated PNP ligand, one hydride ligand and one inert CO ligand. This active complex can abstract hydrogen from the alcohol to form the corresponding aldehyde (dehydrogenation cycle) and reduce the in situ formed imine to access the N-alkylated product (hydrogenation cycle) via an insertion mechanism. The same group subsequently employed a related Fe(II) complex containing a triazine-based PNP pincer ligand for N-alkylation of amines with alcohols.22
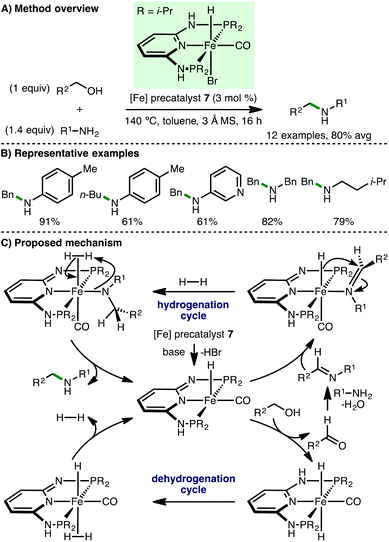 |
| | Scheme 9 Fe-Catalysed N-alkylation of amines (Kirchner). | |
2.2
C-Alkylation processes
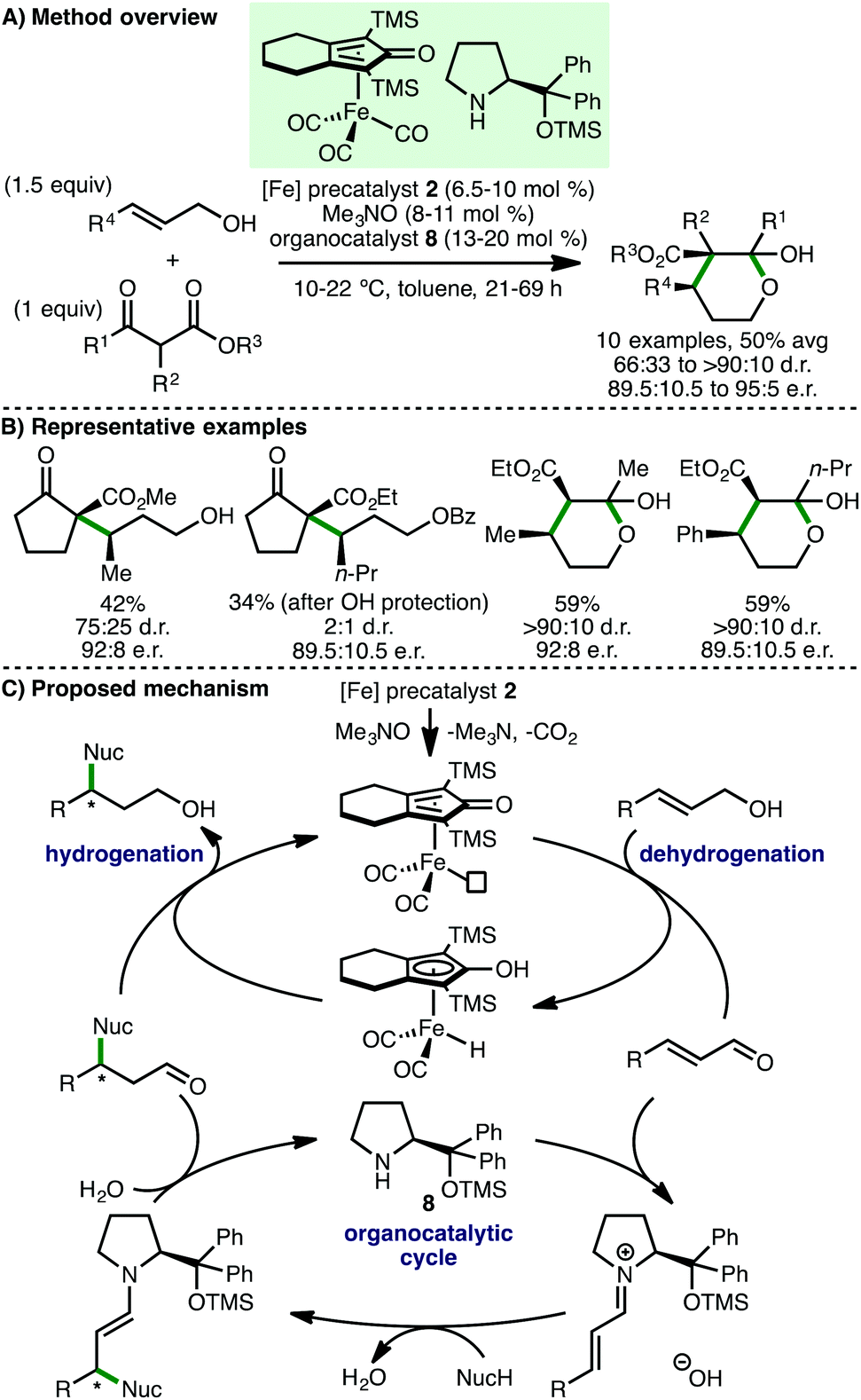
In 2013, Quintard and Rodriguez developed an innovative iron/organo dual catalysis approach for the enantioselective γ-functionalisation of allylic alcohols.23 Employing (cyclopentadienone)iron carbonyl precatalyst 2 in combination with a Jørgensen-Hayashi type secondary amine organocatalyst 8, a selection of γ-functionalised alcohols were formed in modest yields and enantioselectivities (Scheme 10). It is remarkable that this process occurs at only 10–22 °C, which is likely crucial for obtaining high enantioselectivies, whilst being responsible for the long reaction times (21–69 h). The alcohol products, which exist in equilibrium with the corresponding lactol, could be converted to enantioenriched spiro-δ-lactones upon treatment with DBU.24 The authors proposed a dual cooperative catalytic cycle that begins with CO decoordination of precatalyst 2 with Me3NO to form an active iron complex that abstracts hydrogen from the allylic alcohol to form an α,β-unsaturated aldehyde intermediate. Condensation with secondary amine catalyst 8 forms an α,β-unsaturated iminium ion, which undergoes an enantioselective Michael addition with the β-ketoester nucleophile. Subsequent enamine protonation and iminium ion hydrolysis completes the organocatalytic cycle. The β-substituted chiral aldehyde undergoes a final chemoselective reduction to afford the γ-functionalised chiral alcohol, with regeneration of the active iron complex. The same group extended this approach to 1,3-diketone nucleophiles, accessing enantioenriched ester products after an in situ retro-Claisen reaction of the initially formed γ-functionalised chiral alcohol.25 A subsequent detailed mechanistic study of this reaction revealed that using Cu(acac)2 (5 mol%) as additive improved enantioselectivity.26
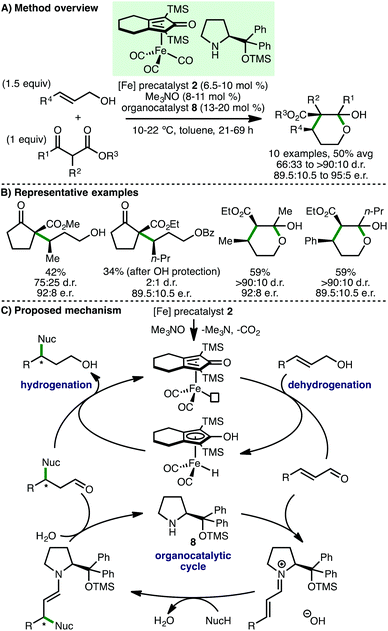 |
| | Scheme 10 γ-Functionalisation of allylic alcohols (Quintard, Rodriguez). | |
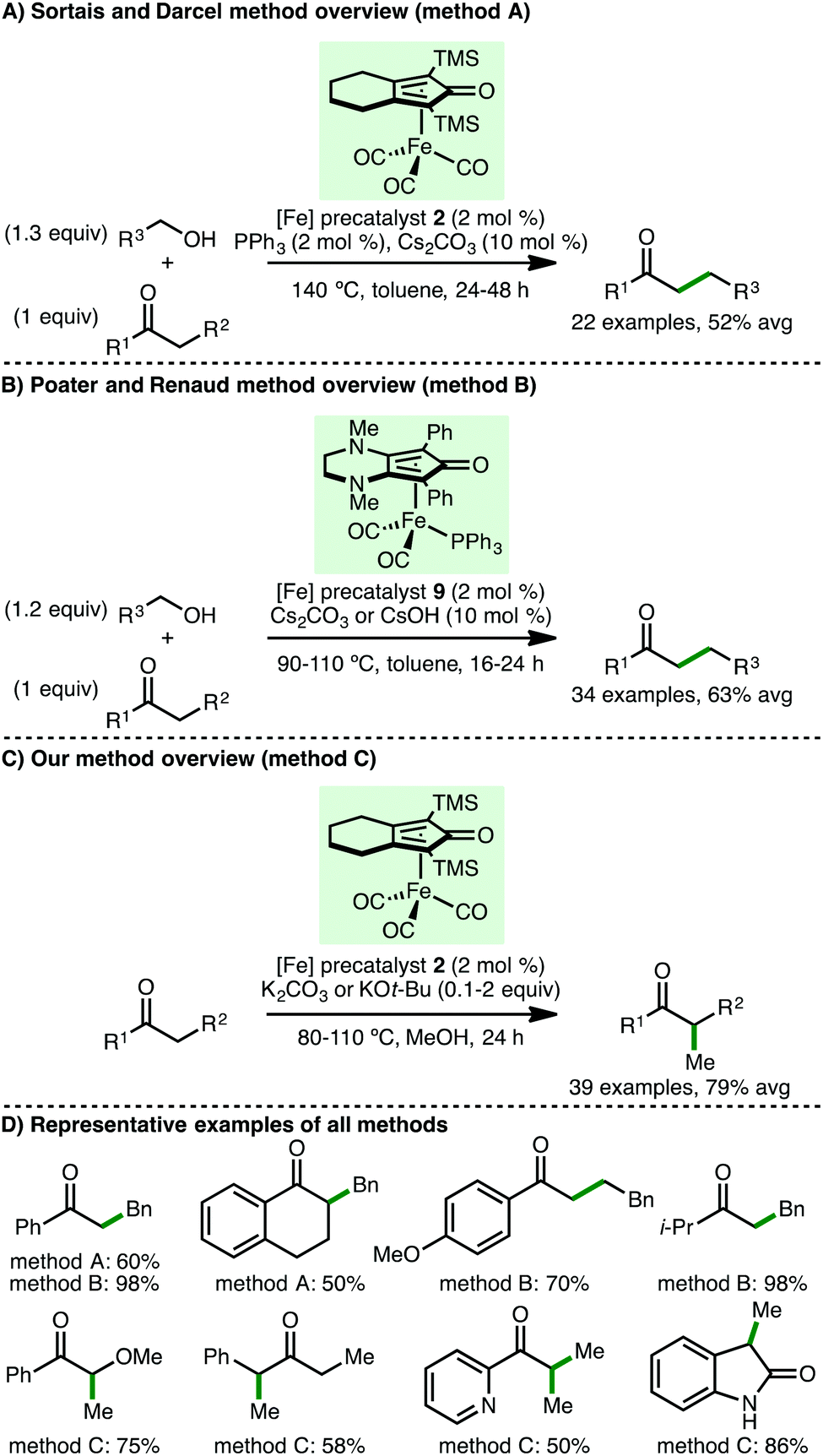
Subsequently, Sortais, Darcel and co-workers described the iron-catalysed α-alkylation of ketones via a borrowing hydrogen approach (Scheme 11).27 Employing (cyclopentadienone)iron carbonyl as precatalyst 2 (2 mol%), in the presence of PPh3 (2 mol%) as activator and Cs2CO3 (10 mol%) as base, at 140 °C in toluene for 24–48 h, a variety of acetophenone derivatives react with benzylic and aliphatic primary alcohols, accessing the α-alkylated products in modest yields. Poater, Renaud and co-workers subsequently showed that using iron complex 9, bearing an electron-rich cyclopentadienone ligand, permitted the α-alkylation of both aromatic and aliphatic ketones to be performed at only 90 °C for 16 h, forming the products with improved yields.28 DFT calculations revealed that the base plays an important role in alcohol dehydrogenation and alkene hydrogenation steps. Despite Sortais, Darcel and co-workers highlighting that methylation using methanol was not successful in their study using (cyclopentadienone)iron carbonyl precatalyst 2,27 by employing methanol as solvent, our group developed the efficient mono and di-methylation of various aromatic and aliphatic ketones, forming α-branched products in excellent yields.20 Furthermore, at 110 °C, C(3)-monomethylation of various oxindoles was possible. Mechanistic studies served to validate proposed reaction intermediates, with deuterium labelling studies supporting methanol as the methylating agent and the involvement of an iron-hydride species.
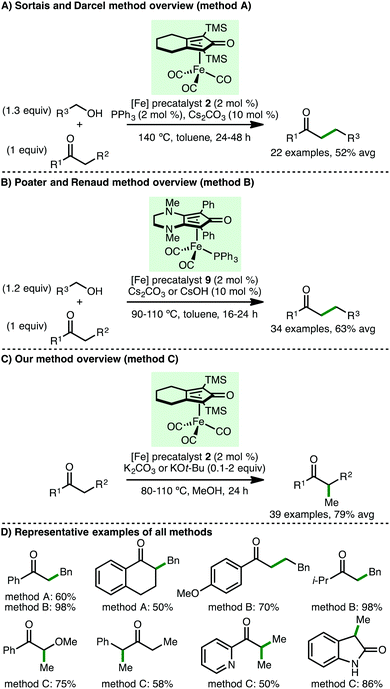 |
| | Scheme 11 Fe-Catalysed α-alkylation of ketones using alcohols (several authors). | |
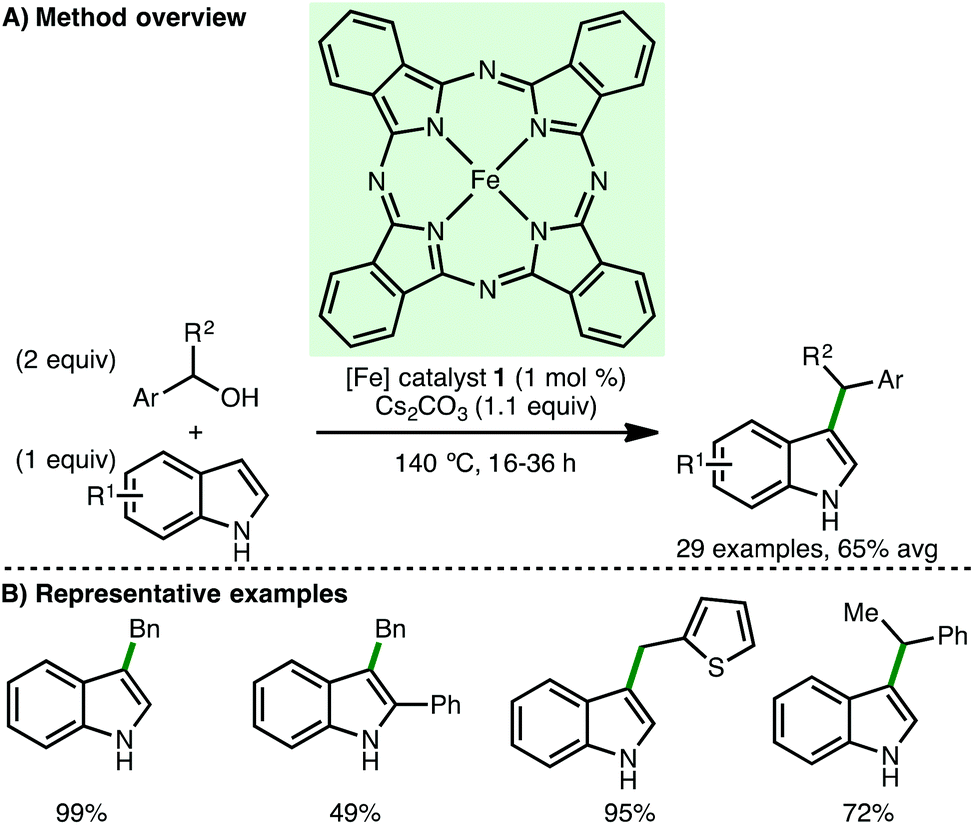
In 2017, Piersanti and co-workers described the Fe-catalysed C(3)-alkylation of indoles.29 Using Fe(II) phthalocyanine 1 (1 mol%) as catalyst and Cs2CO3 (1.1 equiv.) as base without solvent, a variety of N-unprotected indoles react with both primary and secondary benzylic alcohols, accessing the corresponding C(3)-alkylated products in good isolated yields (Scheme 12). N-Methylindole was inert under the described reaction conditions, implicating the indole anion in the alkylation. Mechanistic experiments provided some evidence to support a borrowing hydrogen mechanism over direct nucleophilic substitution or a radical pathway. Our group subsequently described the C(3)-methylation of several indoles using (cyclopentadienone)iron carbonyl precatalyst 2.20 As part of a larger study on Mn-catalysed aminomethylation, Kirchner and co-workers reported a single example of C(1)-methylation of 2-naphthol using precatalyst 7 (cf.Scheme 9).30
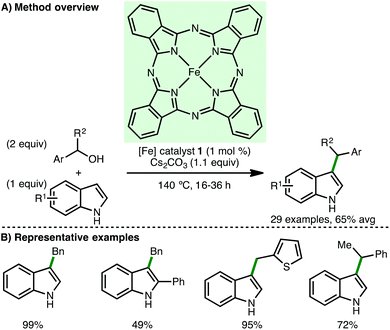 |
| | Scheme 12 Fe-Catalysed C(3)-alkylation of indoles (Piersanti). | |
3. Cobalt-catalysed borrowing hydrogen
3.1
N-Alkylation processes
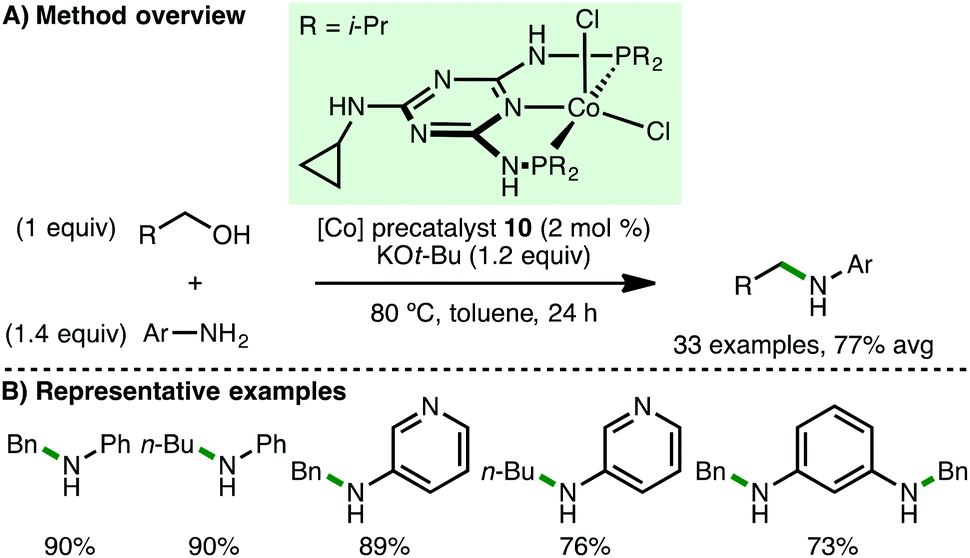
In 2015, Kempe and co-workers reported the first homogeneous cobalt-catalysed N-alkylation of aromatic amines using alcohols via the borrowing hydrogen approach (Scheme 13).31 Employing Co(II) PNP pincer precatalyst 10 (2 mol%) and KOt-Bu (1.2 equiv.) as base, a variety of electron-rich arylamines undergo N-alkylation at only 80 °C with benzylic and aliphatic alcohols, giving the corresponding secondary amine products in high yields. The authors also reported the selective sequential N-alkylation of benzene-1,3-diamine with two different alcohols, forming unsymmetrically substituted diamines.
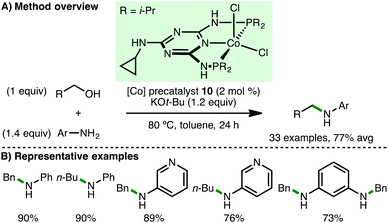 |
| | Scheme 13 Co-Catalysed N-alkylation of arylamines (Kempe). | |
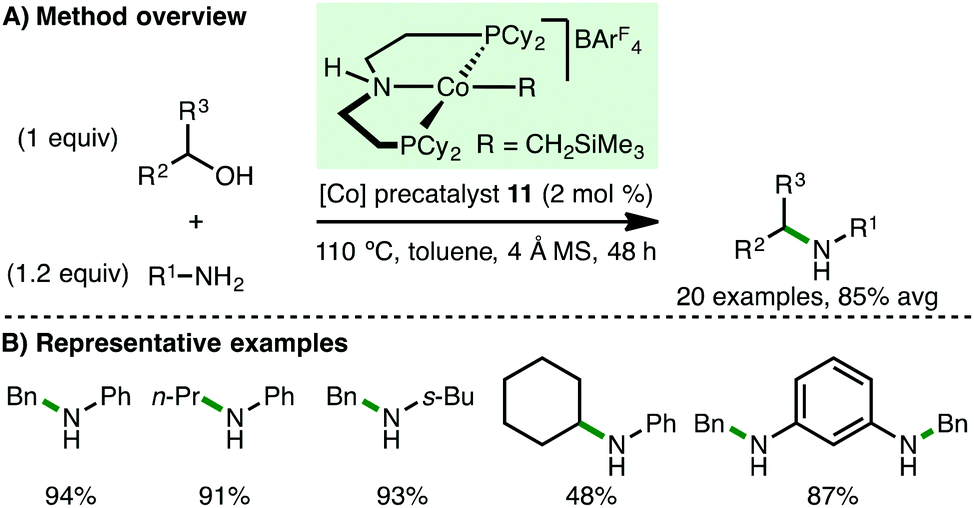
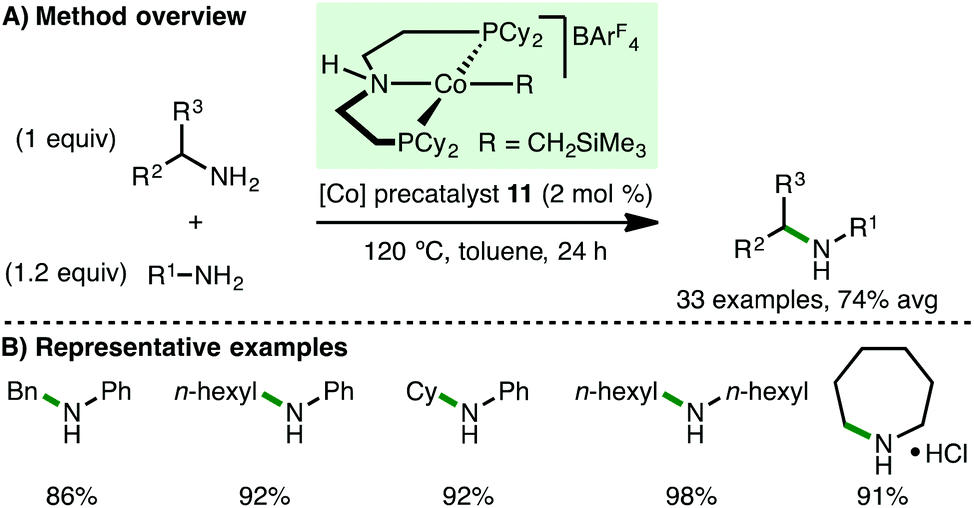
Shortly after the report from Kempe,31 Zhang and co-workers described the use of ionic Co(II) PNP alkyl precatalyst 11 for the N-alkylation of arylamines (Scheme 14).32 In contrast to the catalytic system developed by Kempe, no base is required for efficient N-alkylation, although the addition of 4 Å molecular sieves is necessary. A similar substrate scope was employed, primarily involving electron-rich arylamines coupling with benzylic and aliphatic alcohols. However, it was also possible to mono-N-alkylate aliphatic amines and employ cyclohexanol as the alkylating agent using Zhang's protocol.
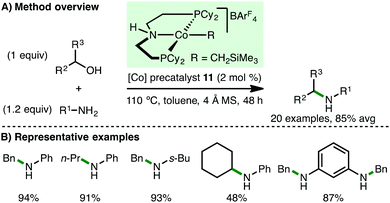 |
| | Scheme 14 Co-Catalysed N-alkylation of amines using alcohols (Zhang). | |
In 2016, Zheng, Zhang and co-workers employed the same ionic Co(II) PNP alkyl precatalyst 11 for the N-alkylation of arylamines using alkylamines as the alkylating agents (Scheme 15).33 This approach, which generates ammonia as by-product, was previously limited to Ru- and Ir-based catalysis, and permitted the N-alkylation of various electron-rich arylamines (including diamines) with benzylamines and both primary and secondary alkylamines, accessing a range of secondary arylamines in good yields. Furthermore, the methodology was applied to the intermolecular homocoupling of primary aliphatic amines and to the synthesis of cyclic secondary amines via intramolecular cyclisation of diamines.
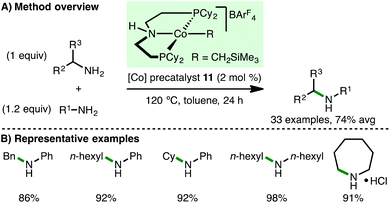 |
| | Scheme 15 Co-Catalysed cross coupling of 1° and 2° amines (Zheng, Zhang). | |
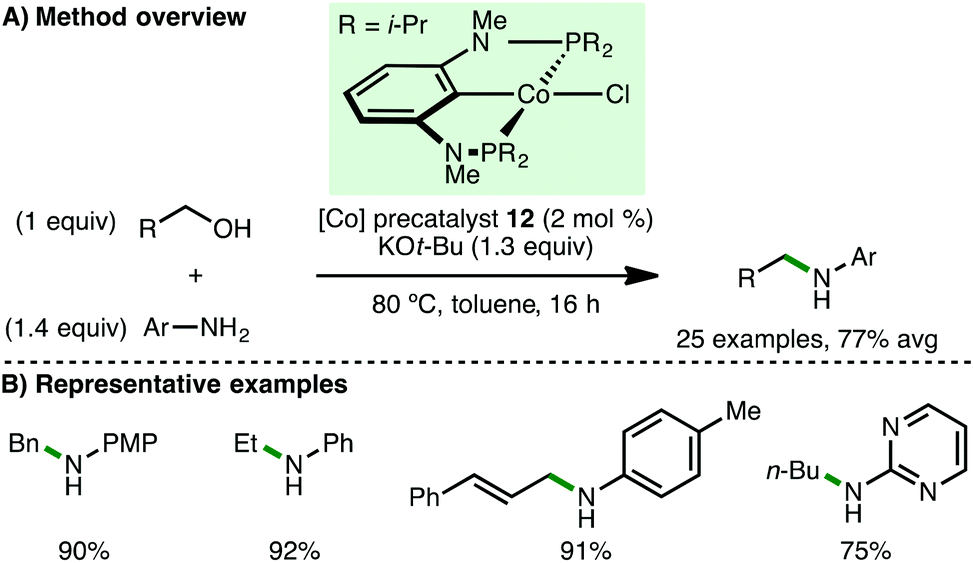
Shortly afterwards, Kirchner and co-workers employed Co(II) complexes that are stabilised by PCP ligands for N-alkylation processes.34 It was found that complex 12 (2 mol%), in the presence of KOt-Bu (1.3 equiv.) at 80 °C, efficiently catalysed the N-alkylation of various arylamines (Scheme 16). Notably, the method was exemplified with a broad range of primary alcohols including the use of ethanol, n-butanol, (R)-citronellol and cinnamyl alcohol as alkylating agents. A related complex bearing the strongly basic CH2SiMe3 coligand obviated the need for KOt-Bu, although 3 Å molecular sieves and increased temperature (130 °C) were required for efficient N-alkylation.
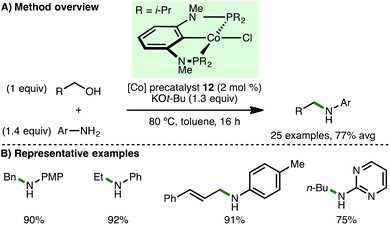 |
| | Scheme 16 Co-Catalysed N-alkylation of arylamines (Kirchner). | |
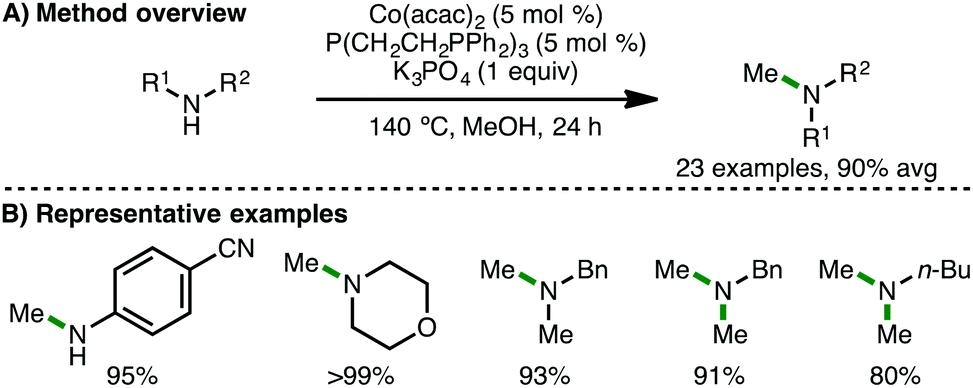
In Kempe's original report on Co-catalyzed N-alkylation of arylamines, it was stated that negligible conversion (3%) was observed when the cobalt precursor, CoCl2, was employed as catalyst.31 However, in 2017, Balaraman and co-workers described a catalytic system for N-alkylation, composed of commercially available CoCl2·6H2O (5 mol%) and PPh3 (10 mol%) as ligand.35 In the presence of KOt-Bu (1.1 equiv.), a variety of electron-rich arylamines couple with benzylic and aliphatic alcohols, giving the corresponding secondary amines in high isolated yields. Soon after, Liu and co-workers employed a closely related catalytic system for the cobalt-catalysed methylation of amines (Scheme 17).36 Using Co(acac)2 (5 mol%) and tetradentate phosphine ligand P(CH2CH2PPh2)3 (5 mol%) in the presence of K3PO4 (1 equiv.) as base, a variety of arylamines and secondary amines undergo efficient mono-N-methylation. Interestingly, electron-poor 4-aminobenzonitrile underwent smooth N-methylation without any nitrile reduction observed. Furthermore, it was shown that butylamine and a range of benzylamines undergo dimethylation under these reaction conditions. The authors performed a number of control experiments to gain mechanistic insight, which provided evidence for formaldehyde (obtained via methanol dehydrogenation) as a plausible reaction intermediate, and that K3PO4 is important for its formation. Furthermore, a cobalt hydride species was proposed based on deuterium-labeling experiments using CD3OD.
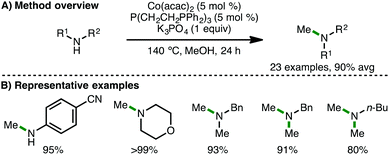 |
| | Scheme 17 Co-Catalysed N-methylation of primary and secondary amines (Liu). | |
3.2
C-Alkylation processes
In 2016, Kempe and co-workers disclosed a cobalt-catalysed C-alkylation of unactivated amides and esters via the borrowing hydrogen approach (Scheme 18).37 These represent challenging classes of nucleophile for borrowing hydrogen processes due to their relatively low CH-acidic nature, compared to the more commonly studied ketones, and that esters can undergo side reactions. Employing Co(II) PNP pincer precatalysts 10 and 13 (2.5–5 mol%) and KOt-Bu or NaOt-Bu (1.2–1.5 equiv.) as base, t-butyl acetate and a variety of acetamides undergo C-alkylation with benzylic and aliphatic alcohols, giving the products in high yields. No α-branched amides or esters were synthesised. The authors also reported the derivatisation of the amide products to ketones and aldehydes via treatment with alkyl/aryl lithium reagents and diisobutylaluminium hydride, respectively.
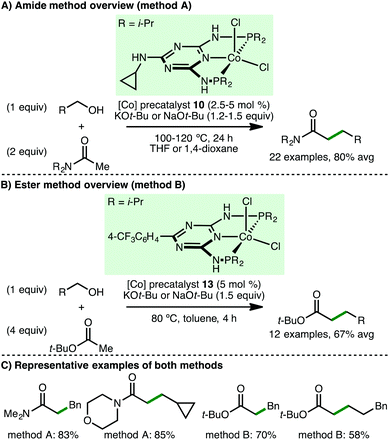 |
| | Scheme 18 Co-Catalysed α-alkylation of unactivated amides and esters (Kempe). | |
Subsequently, Zhang and co-workers employed ionic Co(II) PNP alkyl precatalyst 11 for the α-alkylation of ketones (Scheme 19).38 Using only 2 mol% of precatalyst 11 and 5 mol% of KOt-Bu as base, a variety of aryl ketones undergo alkylation with both benzylic and aliphatic alcohols, giving the products in high yields. One example employing an aliphatic ketone, 2-heptanone, as the nucleophile was described, but the alkylated product was only obtained in 35% yield.
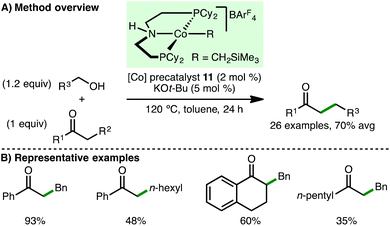 |
| | Scheme 19 Co-Catalysed α-alkylation of ketones (Zhang). | |
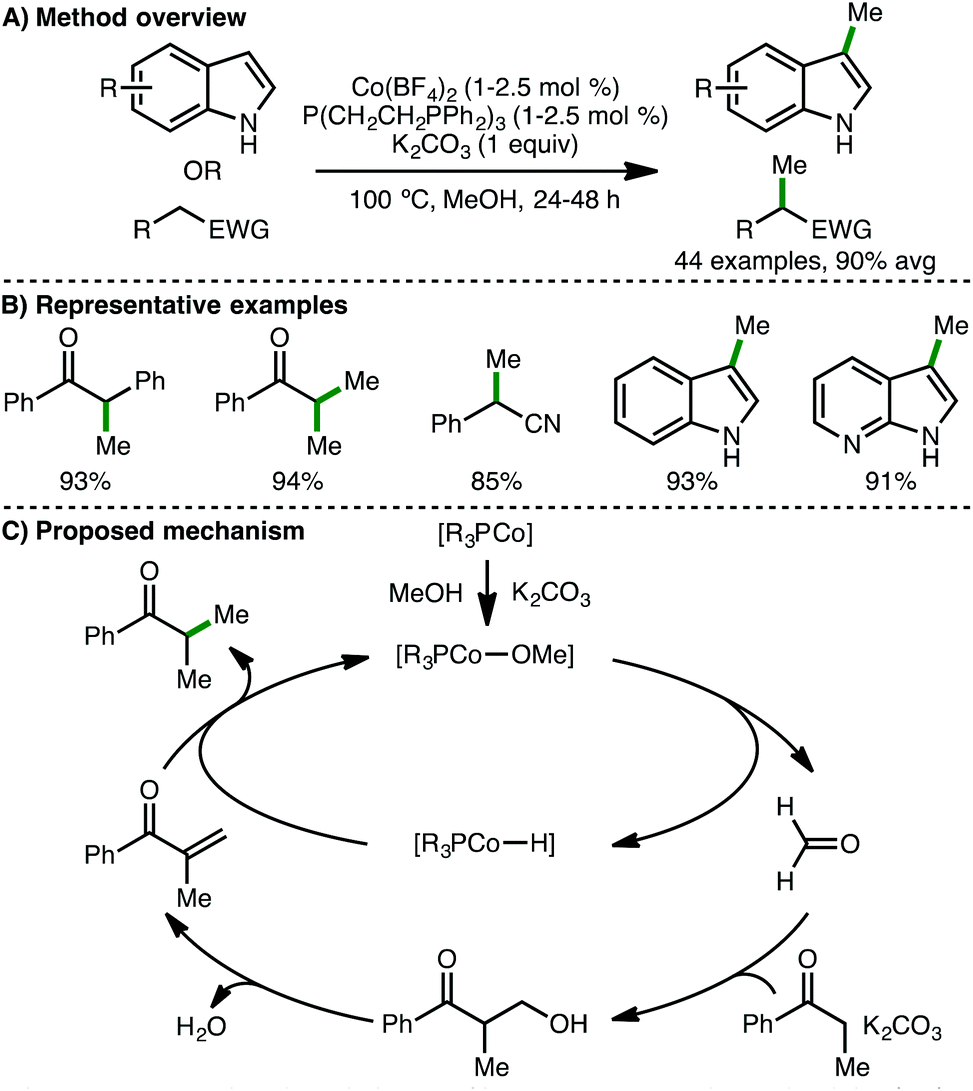
Soon after the report from Zhang,38 Liu and co-workers described a cobalt-based catalytic system for C-methylation using methanol as a sustainable C1 building block (Scheme 20).39 A catalyst system composed of commercially available Co(BF4)2·6H2O (1–2.5 mol%) and tetradentate phosphine ligand P(CH2CH2PPh2)3 (1–2.5 mol%) in the presence of K2CO3 (1 equiv.) as base, enabled mono- and dimethylation of aryl ketones, accessing the products in excellent isolated yields. Furthermore, it was shown that aryl acetonitriles and indoles also undergo monomethylation under the same reaction conditions. Supported by various mechanistic experiments, the authors proposed a catalytic cycle involving initial base-assisted coordination of the alcohol to form a Co-methoxy complex, which undergoes β-hydride elimination to form a Co-hydride species and formaldehyde. A subsequent base-assisted aldol condensation between the aryl ketone and formaldehyde generates an enone, which is reduced by the Co-H intermediate. Protodemetalation in the presence of methanol gives the methylated product with regeneration of the Co-alkoxy species.
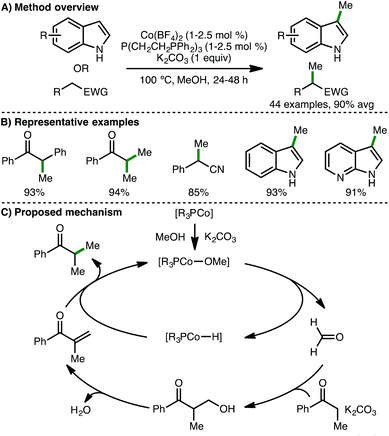 |
| | Scheme 20 Co-Catalysed methylation of ketones, acetonitriles and indoles (Liu). | |
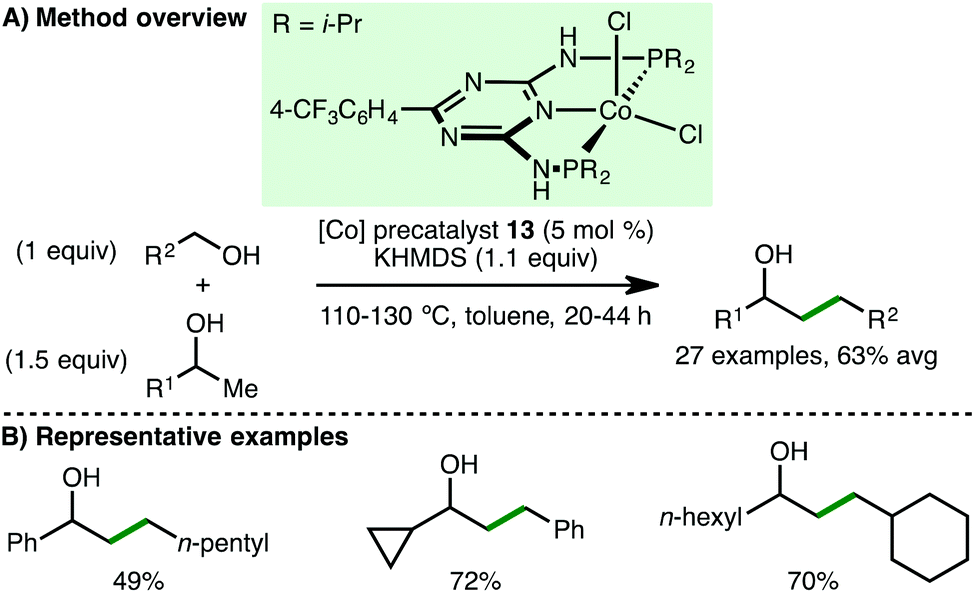
In 2017, Kempe and co-workers disclosed the cobalt-catalysed coupling of secondary alcohols with primary alcohols via the borrowing hydrogen approach (Scheme 21).40 Using Co(II) PNP pincer precatalyst 13 (5 mol%) and KHMDS (1.1 equiv.) as base, a variety of benzylic and aliphatic secondary and primary alcohols are tolerated, accessing higher alcohols in good isolated yields. A particular highlight of this methodology was the heterocoupling of different aliphatic alcohols. Mechanistic investigations performed by the authors support a borrowing hydrogen mechanism over a potential Meerwein–Ponndorf–Verley–Oppenauer-type redox mechanism.
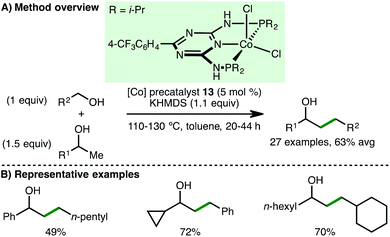 |
| | Scheme 21 Co-Catalysed cross coupling of 2° and 1° alcohols (Kempe). | |
4. Manganese-catalysed borrowing hydrogen
4.1
N-Alkylation processes
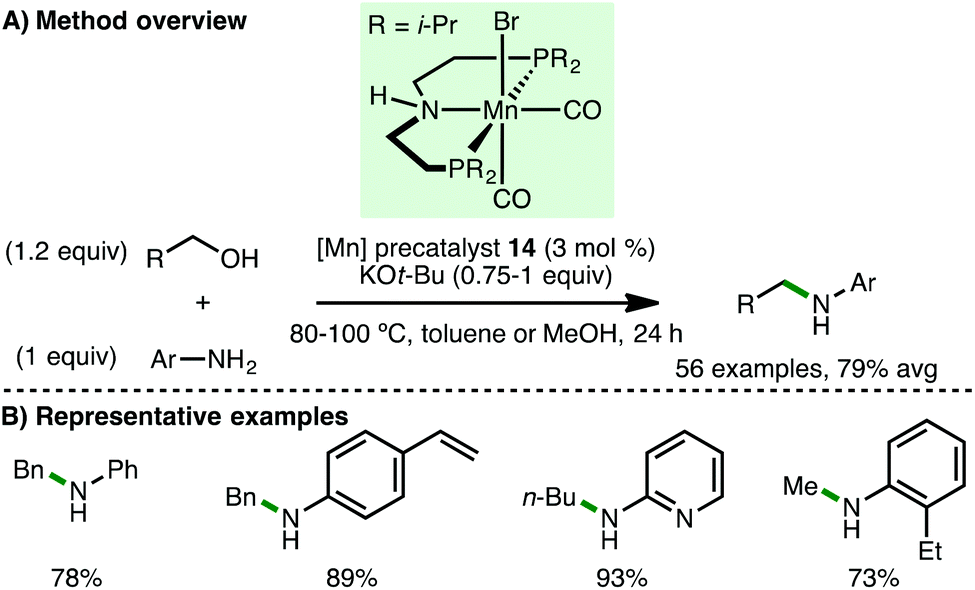
In 2016, Beller and co-workers reported the first homogeneous borrowing hydrogen manganese-catalysed N-alkylation of amines using alcohols.41 Using Mn(I) PNP pincer precatalyst 14 (3 mol%) with KOt-Bu (0.75 equiv.) as base, a variety of arylamines undergo N-alkylation with benzylic and aliphatic alcohols at 80 °C in toluene (Scheme 22). Amines containing alkenes were alkylated smoothly with high chemoselectivity. Furthermore, by increasing the reaction temperature to 100 °C and using methanol as solvent, the authors disclosed the first non-noble metal-catalysed N-methylation of amines using methanol. In 2017, the groups or Beller42 and Sortais43 concurrently reported second-generation pyridine-based manganese PNP-pincer complexes, which allow the N-methylation of arylamines to be performed using reduced loadings of catalyst and/or base. Sortais also applied his system to the N-methylation of sulfonamides, which required 1.2 equivalents of KOt-Bu and 60 h reaction time.
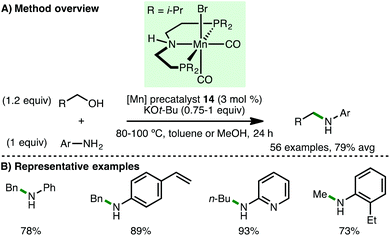 |
| | Scheme 22 Mn-Catalysed N-alkylation of arylamines (Beller). | |
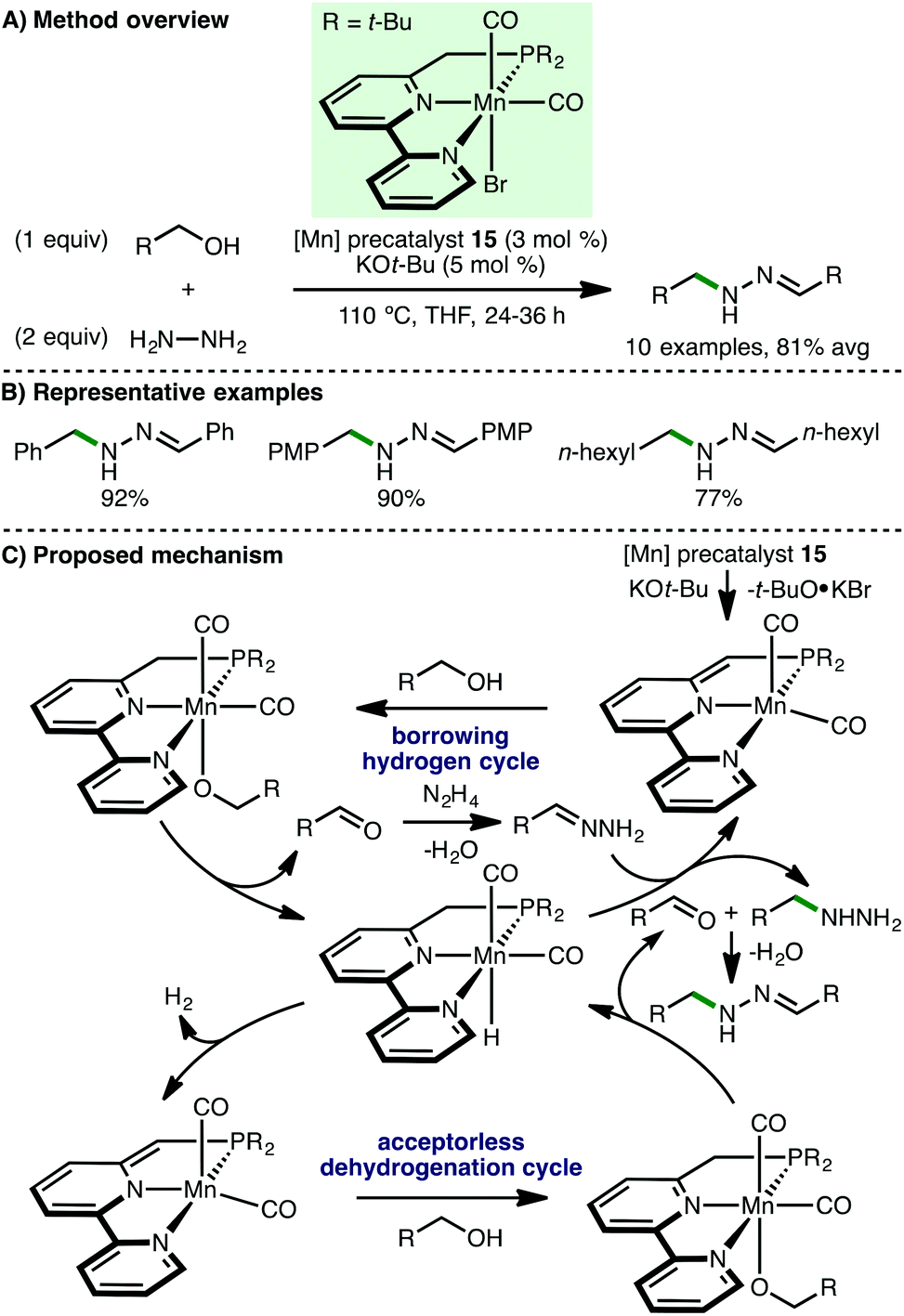
Recently, Milstein and co-workers reported the Mn-catalyzed synthesis of N-substituted hydrazones from hydrazine and alcohol starting materials.44 Using a novel Mn(I) PNN pincer precatalyst 15 (3 mol%) and KOt-Bu (5 mol%), hydrazine was converted to a variety of N-substituted hydrazones in excellent isolated yields via reaction with both benzylic and aliphatic alcohols (Scheme 23). Supported by several mechanistic experiments, the authors proposed a catalytic cycle that involves initial deprotonation of 15 by KOt-Bu to form the active catalyst, which abstracts hydrogen from the alcohol by metal–ligand cooperation. Hydrazine undergoes N-alkylation with the formed aldehyde, completing a borrowing hydrogen cycle, followed by condensation with an additional aldehyde molecule (formed via an acceptorless alcohol dehydrogenation cycle) to form the observed N-substituted hydrazone.
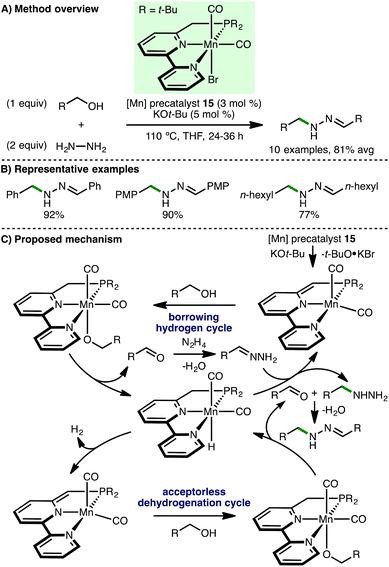 |
| | Scheme 23 Mn-Catalysed formation of N-substituted hydrazones (Milstein). | |
4.2
C-Alkylation processes
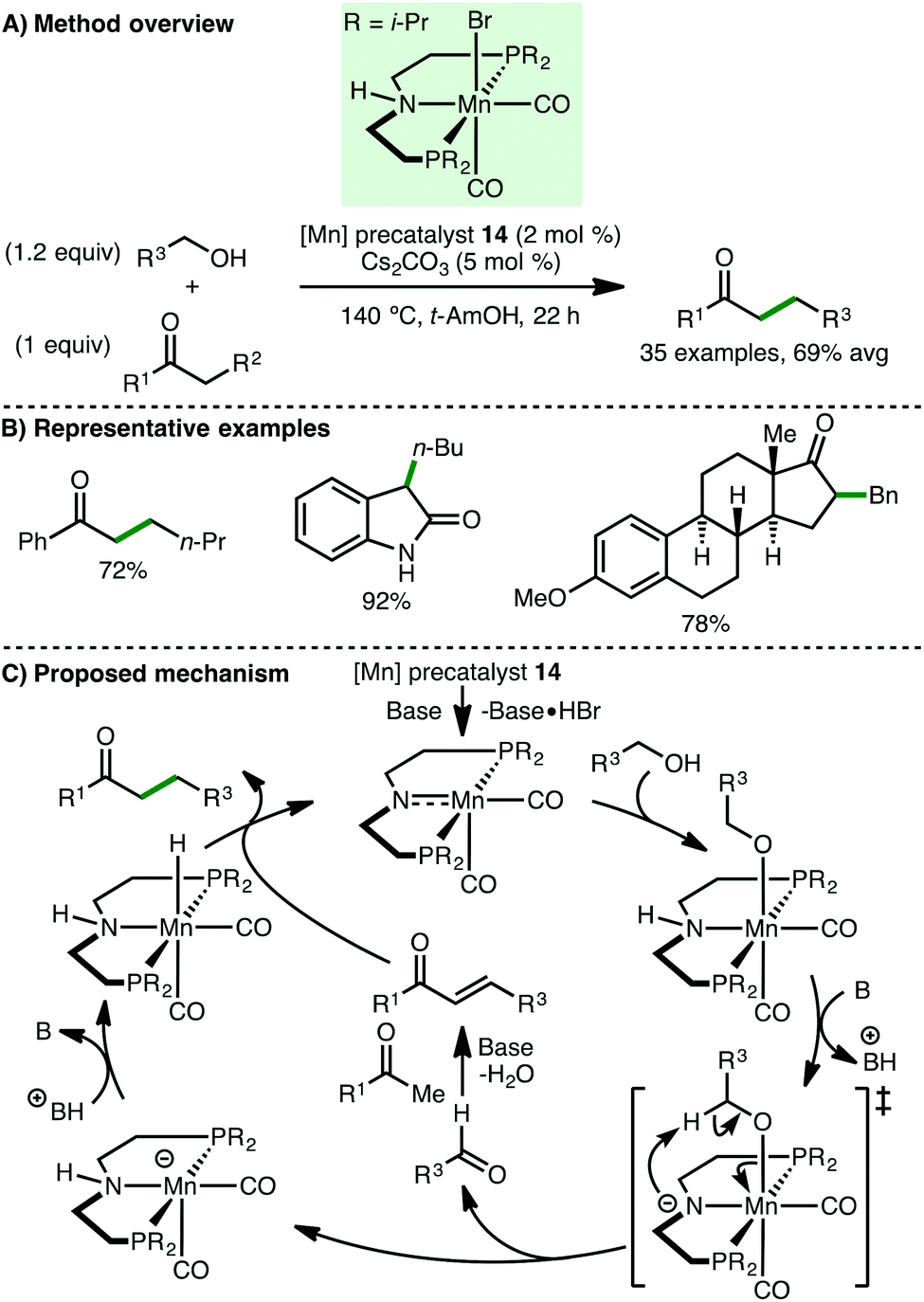
In 2016, Beller and co-workers reported the manganese-catalyzed C-alkylation of ketones with primary alcohols (Scheme 24).45 Using Mn(I) PNP pincer precatalyst 14 (2 mol%) with Cs2CO3 (5 mol%) as base, a variety of aryl ketones undergo alkylation with both benzylic and aliphatic alcohols, giving the products in high yields. One example employing an aliphatic ketone as the nucleophile was described but the alkylated product was only obtained in 35% yield. Interestingly, this method was extended to the C(3)-alkylation of oxindoles and to the late-stage functionalisation of hormones, namely estrone and testosterone. Based on a selection of control experiments, the authors proposed a mechanism that initiates with activation of precatalyst 14 with base in a dehydrobromination reaction, followed by alcohol coordination to form an alkoxo-type complex. In the basic medium, an amidate is formed, which undergoes abstraction of the β-hydrogen atom, forming an aldehyde that is converted to an enone via base-promoted aldol condensation. Protonation of the resulting Mn-complex forms a manganese hydride species, which reduces the enone to give the final product with regeneration of the catalytically active species.
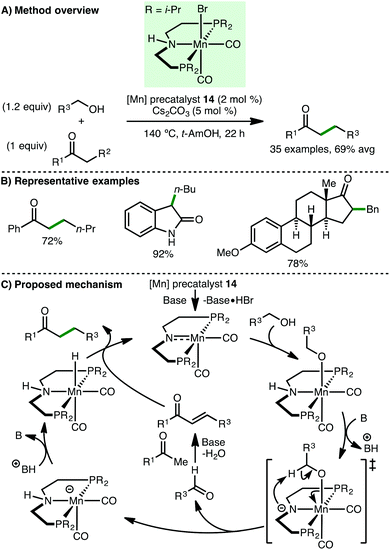 |
| | Scheme 24 Mn-Catalysed C-alkylation of ketones and oxindoles (Beller). | |
In 2017, Liu and co-workers described the upgrading of ethanol to 1-butanol via a manganese-catalyzed Guerbet reaction (Scheme 25).46 Employing a remarkably low catalyst loading of Mn(I) PNP pincer precatalyst 14 (0.0001 mol%, 8 ppm), 1-butanol was formed in 11.2% conversion with 92% C4 selectivity after 168 h reaction time at 160 °C. The TON (114120) and TOF (3078 h−1) are the highest reported to date for ethanol upgrading via the Guerbet reaction.
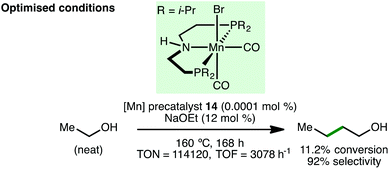 |
| | Scheme 25 Mn-Catalysed upgrading of ethanol to 1-butanol (Liu). | |
5. Nickel-catalysed borrowing hydrogen
5.1
N-Alkylation processes
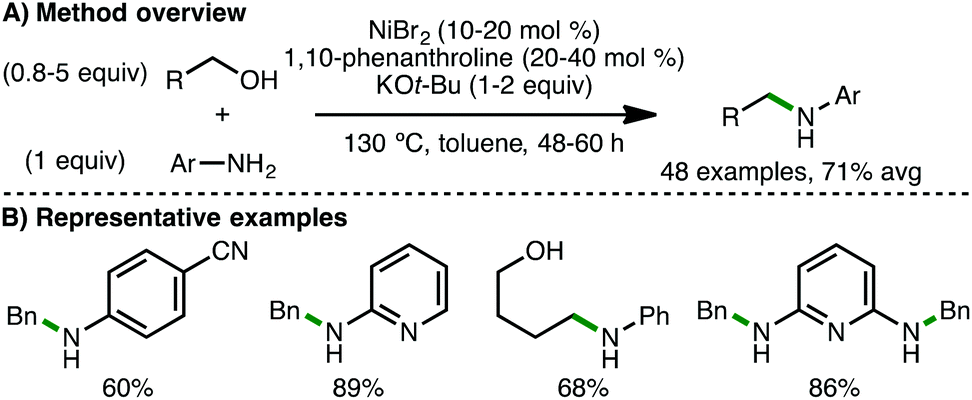
In 2017, the research teams of Banerjee,47 and Tang and Zhou48 independently reported the first examples of homogeneous Ni-catalysed borrowing hydrogen processes for the N-alkylation of amines. Banerjee and co-workers employed a catalytic system involving NiBr2 (10–20 mol%), 1,10-phenanthroline (20–40 mol%) and KOt-Bu (1–2 equiv.).47 At 130 °C for 48–60 h in toluene, a variety of arylamines (including electron-poor anilines) undergo N-alkylation with (hetero)aromatic and aliphatic primary alcohols, including diols and amino alcohols (Scheme 26). Double alkylation of diamines was also possible using this method. Drawbacks of this protocol include the relatively high quantities of Ni(II) salt, ligand, base and alcohol required in addition to extended reaction times. In situ NMR studies revealed the simultaneous presence of starting alcohol and amine, intermediate aldehyde, and N-alkylated amine product, providing evidence for a borrowing hydrogen mechanism. The same group later employed the same catalytic system, except using K3PO4 (2 equiv.) as base, for the N-alkylation of benzamides with both benzylic and aliphatic alcohols.49
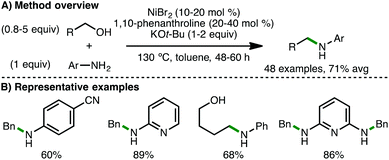 |
| | Scheme 26 Ni-Catalysed N-alkylation of arylamines (Banerjee). | |
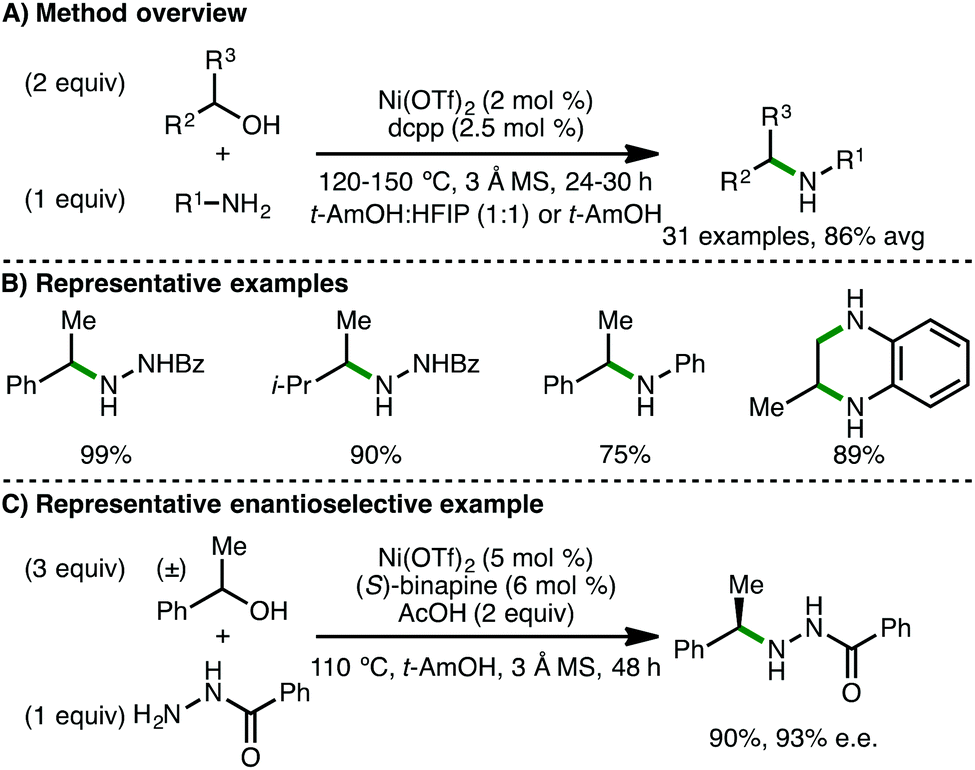
In contrast, Tang, Zhou and co-workers reported a base-free catalytic system composed of Ni(OTf)2 (2 mol%) and 1,3-bis(dicyclohexylphosphine)propane (dcpp) (2.5 mol%) as ligand.48 At 120–150 °C for 24 h in t-AmOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HFIP (1:1) with 3 Å molecular sieves, a variety of hydrazides undergo selective mono-N-alkylation at the terminal N-atom with racemic secondary alcohols (Scheme 27). Electron-rich arylamines also undergo N-alkylation with secondary alcohols and diols using t-AmOH as solvent. Impressively, by employing (S)-binapine as ligand, the authors reported a rare example of a highly enantioselective borrowing hydrogen process, accessing medicinally important benzylamines in high enantiomeric excess, despite the high reaction temperature (110 °C). A selection of deuterium-labeling reactions were performed, supporting a borrowing hydrogen mechanism over direct nucleophilic substitution or a radical pathway.
:HFIP (1:1) with 3 Å molecular sieves, a variety of hydrazides undergo selective mono-N-alkylation at the terminal N-atom with racemic secondary alcohols (Scheme 27). Electron-rich arylamines also undergo N-alkylation with secondary alcohols and diols using t-AmOH as solvent. Impressively, by employing (S)-binapine as ligand, the authors reported a rare example of a highly enantioselective borrowing hydrogen process, accessing medicinally important benzylamines in high enantiomeric excess, despite the high reaction temperature (110 °C). A selection of deuterium-labeling reactions were performed, supporting a borrowing hydrogen mechanism over direct nucleophilic substitution or a radical pathway.
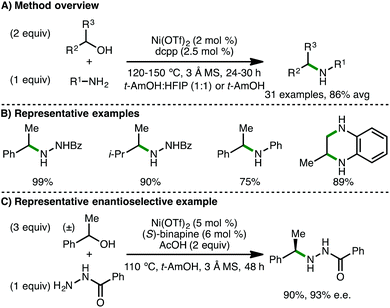 |
| | Scheme 27 Ni-Catalysed N-alkylation of using secondary alcohols (Tang, Zhou). | |
6. Copper-catalysed borrowing hydrogen
6.1
N-Alkylation processes
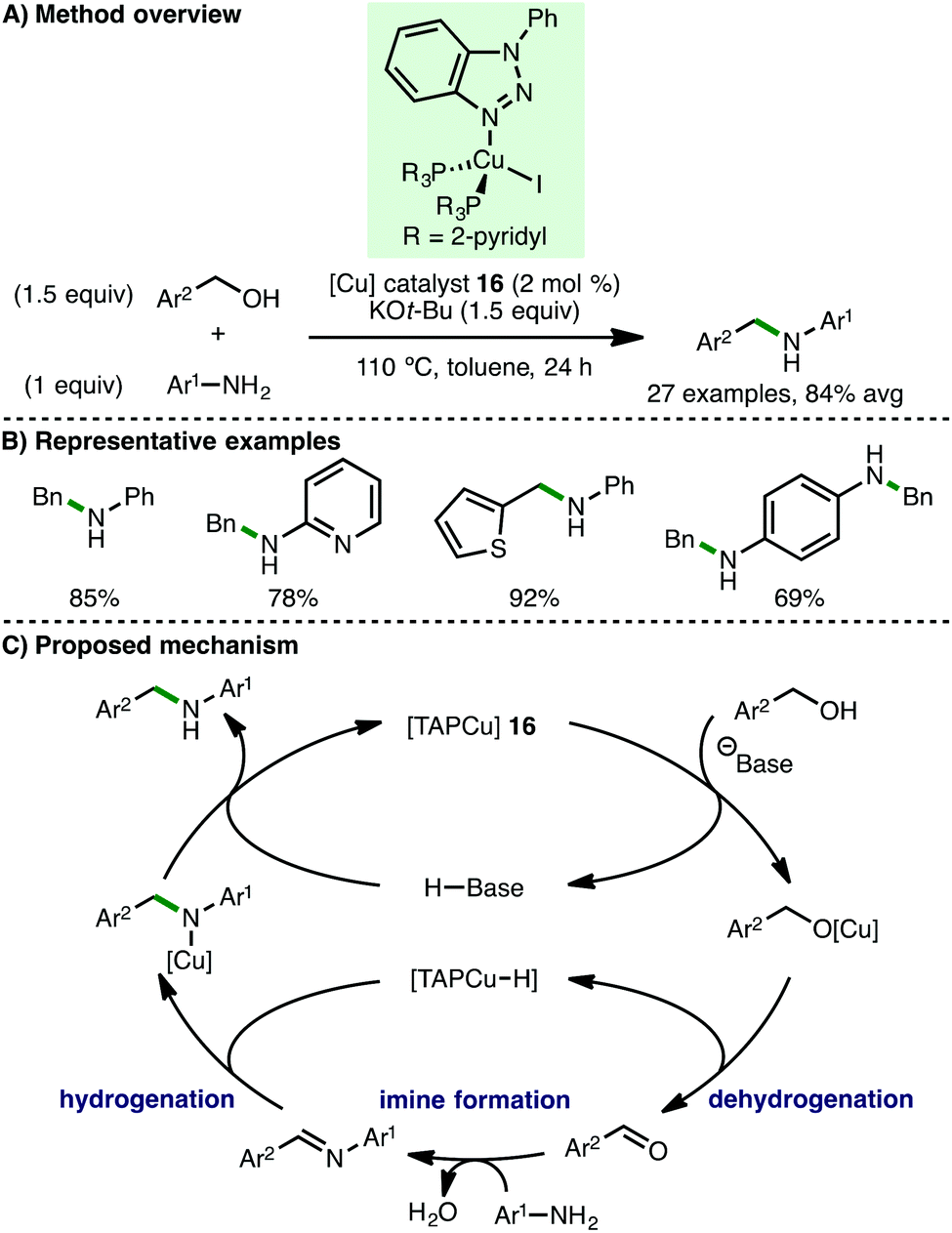
In 2017, Wang and co-workers described the use of a triazole-phopshine-copper [TAPCu] complex 16 (2 mol%) for the N-alkylation of electron-rich arylamines using benzylic alcohols as the alkylating agent and KOt-Bu (1 equiv.) as base (Scheme 28).50 A deuterium labelling experiment involving d7-benzyl alcohol implicated a copper hydride species as a catalytic intermediate, which was tentatively supported by solid state infrared spectra (absorptions at 912 cm−1). Based on their mechanistic studies, the authors proposed a catalytic cycle involving initial base-assisted coordination of the alcohol to 16. Subsequent β-hydride elimination of the resulting Cu-alkoxy species forms a copper hydride intermediate and the aldehyde, which undergoes condensation with the arylamine to form an aldimine. Subsequent reduction by the Cu–H intermediate followed by protodecupration gives the secondary amine product with regeneration of the catalyst.
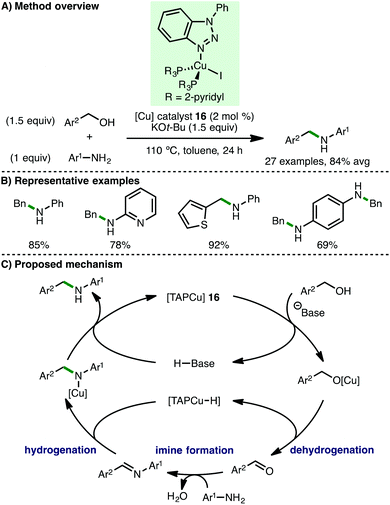 |
| | Scheme 28 Cu-Catalysed N-alkylation of arylamines (Wang). | |
7. Outlook and conclusion
This review highlights that the area of homogeneous borrowing hydrogen catalysis using earth-abundant first row transition metals for C–C and C–N bond forming processes has received considerable attention in recent years (2013-present). Furthermore, the drive to replicate the reactivity of precious metal catalysts using catalysts based on Mn, Fe, Co, Ni and Cu, is likely to increase in the future. Despite the range of innovative strategies that have been developed in this area, challenges and opportunities remain. The rational design of novel well-defined complexes based on earth-abundant transition metals with improved activity is required in order to lower catalyst loadings and be competitive with established precious metal catalysts. It is likely that careful mechanistic studies, including computational analysis, will play an important role in this rational design process. Furthermore, there remain limited examples of highly enantioselective borrowing hydrogen processes. With increased catalyst efficiency and functional group tolerance, it may be possible to lower the reaction temperature, base loading, and reaction time, allowing enantioselective processes to be developed more readily. Due to the diverse nature of the reaction processes disclosed using earth-abundant metal catalysts, it is clear that many new applications remain to be discovered in the coming years.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge the School of Chemistry, Cardiff University, for generous support, the EPSRC Doctoral Training Grant for a part-funded Ph.D. studentship (B.G.R-B., EP/M507842/1) and the EPSRC-funded Bath/Bristol/Cardiff Catalysis Centre for Doctoral Training (K.P., EP/L016443/1).
Notes and references
- A. Corma, J. Navas and M. J. Sabater, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS and references therein.
- A. J. A. Watson and J. M. J. Williams, Science, 2010, 329, 635–636 CrossRef CAS and references therein.
- C. Gunanathan and D. Milstein, Science, 2013, 341, 1229712 CrossRef PubMed and references therein.
- L. L. Schafer, P. Mountford and W. E. Piers, Dalton Trans., 2015, 44, 12027–12028 RSC.
- For a highlight on the earliest Fe- and Co-catalysed borrowing hydrogen processes, see: A. Quintard and J. Rodriguez, ChemSusChem, 2016, 9, 28–30 CrossRef CAS PubMed and references therein.
- K.-I. Shimizu, Catal. Sci. Technol., 2015, 5, 1412–1427 RSC and references therein.
- G. A. Filonenko, R. van Putter, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2018, 47, 1459–1483 RSC and references therein.
- F. Kallmeier and R. Kempe, Angew. Chem., Int. Ed., 2018, 57, 46–60 CrossRef CAS PubMed and references therein.
- Q. Yang, Q. Wang and Z. Yu, Chem. Soc. Rev., 2015, 44, 2305–2329 RSC and references therein.
- M. Bala, P. Kumar Verma, U. Sharma, N. Kumar and B. Singh, Green Chem., 2013, 15, 1687–1693 RSC.
- T. Yan, B. L. Feringa and K. Barta, Nat. Commun., 2014, 5, 5602 CrossRef CAS.
- T. Yan, B. L. Feringa and K. Barta, ACS Catal., 2016, 6, 381–388 CrossRef CAS.
- T. Yan, B. L. Feringa and K. Barta, Sci. Adv., 2017, 3, eaao6494 CrossRef.
- A. J. Rawlings, L. J. Diorazio and M. Wills, Org. Lett., 2015, 17, 1086–1089 CrossRef CAS.
- T. J. Brown, M. Cumbes, L. J. Diorazio, G. J. Clarkson and M. Wills, J. Org. Chem., 2017, 82, 10489–10503 CrossRef CAS.
- H.-J. Pan, T. Wei Ng and Y. Zhao, Chem. Commun., 2015, 51, 11907–11910 RSC.
- B. Emayavaramban, M. Roy and B. Sundararaju, Chem. – Eur. J., 2016, 22, 3952–3955 CrossRef CAS.
- B. Emayavaramban, M. Sen and B. Sundararaju, Org. Lett., 2017, 19, 6–9 CrossRef CAS PubMed.
- M. Vayer, S. P. Morcillo, J. Dupont, V. Gandon and C. Bour, Angew. Chem., Int. Ed., 2018, 57, 3228–3232 CrossRef CAS PubMed.
- K. Polidano, B. D. W. Allen, J. M. J. Williams and L. C. Morrill, ACS Catal., 2018, 8, 6440–6445 CrossRef CAS.
- M. Mastalir, M. Glatz, N. Gorgas, B. Stöger, E. Pittenauer, G. Allmaier, L. F. Veiros and K. Kirchner, Chem. – Eur. J., 2016, 22, 12316–12320 CrossRef CAS.
- M. Mastalir, B. Stöger, E. Pittenauer, M. Puchberger, G. Allmaier and K. Kirchner, Adv. Synth. Catal., 2016, 358, 3824–3831 CrossRef CAS.
- A. Quintard, T. Constantieux and J. Rodriguez, Angew. Chem., Int. Ed., 2013, 52, 12883–12887 CrossRef CAS.
- A. Quintard, M. Roudier and J. Rodriguez, Synthesis, 2018, 50, 785–792 CrossRef CAS.
- M. Roudier, T. Constantieux, A. Quintard and J. Rodriguez, Org. Lett., 2014, 16, 2802–2805 CrossRef CAS.
- M. Roudier, T. Constantieux, A. Quintard and J. Rodriguez, ACS Catal., 2016, 6, 5236–5244 CrossRef CAS.
- S. Elangovan, J.-B. Sortais, M. Beller and C. Darcel, Angew. Chem., Int. Ed., 2015, 54, 14483–14486 CrossRef CAS.
- C. Seck, M. Diagne Mbaye, S. Coufourier, A. Lator, J.-F. Lohier, A. Poater, T. R. Ward, S. Gaillard and J.-L. Renaud, ChemCatChem, 2017, 9, 4410–4416 CrossRef CAS.
- G. Di Gregorio, M. Mari, F. Bartocchini and G. Piersanti, J. Org. Chem., 2017, 82, 8769–8775 CrossRef CAS.
- M. Mastalir, E. Pittenauer, G. Allmaier and K. Kirchner, J. Am. Chem. Soc., 2017, 139, 8812–8815 CrossRef CAS.
- S. Rösler, M. Ertl, T. Irrgand and R. Kempe, Angew. Chem., Int. Ed., 2015, 54, 15046–15050 CrossRef PubMed.
- G. Zhang, Z. Yin and S. Zheng, Org. Lett., 2016, 18, 300–303 CrossRef CAS.
- Z. Yin, H. Zeng, J. Wu, S. Zheng and G. Zhang, ACS Catal., 2016, 6, 6546–6550 CrossRef CAS.
- M. Mastalir, G. Tomsu, E. Pittenauer, G. Allmaier and K. Kirchner, Org. Lett., 2016, 18, 3462–3465 CrossRef CAS.
- S. P. Midya, A. Mondal, A. Begum and E. Balaraman, Synthesis, 2017, 49, 3957–3961 CrossRef CAS.
- Z. Liu, Z. Yang, X. Yu, H. Zhang, B. Yu, Y. Zhao and Z. Liu, Adv. Synth. Catal., 2017, 359, 4278–4283 CrossRef CAS.
- N. Deibl and R. Kempe, J. Am. Chem. Soc., 2016, 138, 10786–10789 CrossRef CAS.
- G. Zhang, J. Wu, H. Zeng, S. Zhang, Z. Yin and S. Zheng, Org. Lett., 2017, 19, 1080–1083 CrossRef CAS.
- Z. Liu, Z. Yang, X. Yu, H. Zhang, B. Yu, Y. Zhao and Z. Liu, Org. Lett., 2017, 19, 5228–5231 CrossRef CAS.
- F. Freitag, T. Irrang and R. Kempe, Chem. – Eur. J., 2017, 23, 12110–12113 CrossRef CAS PubMed.
- S. Elangovan, J. Neumann, J.-B. Sortais, K. Junge, C. Darcel and M. Beller, Nat. Commun., 2016, 7, 12641 CrossRef.
- J. Neumann, S. Elangovan, A. Spannenberg, K. Junge and M. Beller, Chem. – Eur. J., 2017, 23, 5410–5413 CrossRef CAS.
- A. Bruneau-Voisine, D. Wang, V. Dorcet, T. Roisnel, C. Darcel and J.-B. Sortais, J. Catal., 2017, 347, 57–62 CrossRef CAS.
- U. Kumar Das, Y. Ben-David, Y. Diskin-Posner and D. Milstein, Angew. Chem., Int. Ed., 2018, 57, 2179–2182 CrossRef.
- M. Peña-López, P. Piehl, S. Elangovan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 14967–14971 CrossRef.
- S. Fu, Z. Shao, Y. Wang and Q. Liu, J. Am. Chem. Soc., 2017, 139, 11941–11948 CrossRef CAS.
- M. Vellakkaran, K. Singh and D. Banerjee, ACS Catal., 2017, 7, 8152–8158 CrossRef CAS.
- P. Yang, C. Zhang, Y. Ma, C. Zhang, A. Li, B. Tang and J. Steve Zhou, Angew. Chem., Int. Ed., 2017, 56, 14702–14706 CrossRef CAS.
- J. Das and D. Banerjee, J. Org. Chem., 2018, 83, 3378–3384 CrossRef CAS.
- Z. Xu, D.-S. Wang, X. Yu, Y. Yang and D. Wang, Adv. Synth. Catal., 2017, 359, 3332–3340 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*