
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorinated triazole-containing sphingosine analogues. Syntheses and in vitro evaluation as SPHK inhibitors†
Margarita
Escudero-Casao
a,
Adrià
Cardona
 a,
Raúl
Beltrán-Debón
b,
Yolanda
Díaz
*a,
M. Isabel
Matheu
*a and
Sergio
Castillón
a
a,
Raúl
Beltrán-Debón
b,
Yolanda
Díaz
*a,
M. Isabel
Matheu
*a and
Sergio
Castillón
a
aDepartament de Química Analítica i Química Orgànica, Universitat Rovira i Virgili, C/Marcel lí Domingo n° 1, 43007, Tarragona, Spain. E-mail: maribel.matheu@urv.cat
bDepartament de Bioquímica i Biotecnologia, Universitat Rovira i Virgili, C/Marcel lí Domingo n° 1, 43007 Tarragona, Spain
First published on 24th September 2018
Abstract
Sphingosine analogues with a rigid triazole moiety in the aliphatic chain and systematic modifications in the polar head and different degrees of fluorination at the terminus of the alkylic chain were synthesized from a common alkynyl aziridine key synthon. This key synthon was obtained by enantioselective organocatalyzed aziridination and it was subsequently ring opened in a regioselective manner in acidic medium. Up to 16 sphingosine analogues were prepared in a straightforward manner. The in vitro activity of the obtained products as SPHK1 and SPHK2 inhibitors was evaluated, displaying comparable activity to that of DMS.
Introduction
Sphingosine kinase (SPHK) is a key regulator of the sphingolipid rheostat (Scheme 1),1 a dynamic balance between ceramide (Cer) and sphingosine 1-phosphate (S1P) that guides the cell toward either an apoptotic or a survival process (Scheme 1).2,3 SPHK catalyses the ATP-dependent phosphorylation of sphingosine (Sph) to generate S1P and exists in two isoforms, SPHK1 and SPHK2.4 The interest in SPHK inhibitors5 as potential drugs stems from the fact that SPHK1 is usually up-regulated in several forms of cancer and that its genetic ablation leads to the sensitization of cancer cells to chemotherapeutic agents and its implication in several diseases.6,7 Similarly, different pieces of evidence support the role of SPHK2 in cancer.8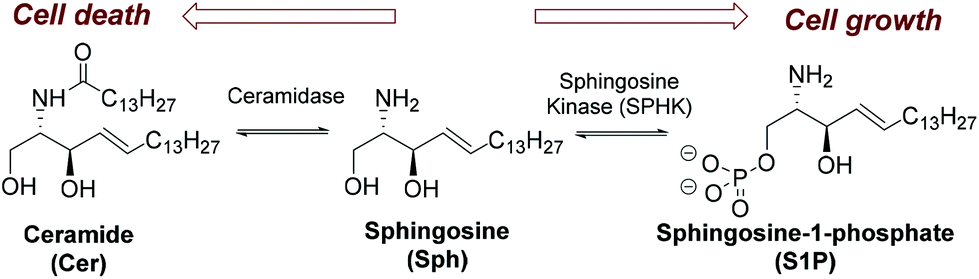 | ||
| Scheme 1 Ceramide/sphingosine-1-phosphate rheostat. | ||
There has been extensive focus on the development of effective sphingosine kinase inhibitors (SKIs) in recent years and the progress in this area has been recently reviewed.6,9 Among the described SKIs derived from sphingolipids (Fig. 1), the following SKIs stand out: safingol10 (saturated sphingosine analog with the unnatural threo stereochemistry), N,N-dimethylsphingosine (DMS),11 short-chain sphinganine (C12) and 3-fluorosphingosine analogues,12N-derivatized analogues such as phenethyl isothiocyanate derivatives,13 guanidine analog LCL351,14N-methylated SK1-I,15 the aromatic analogue SG-12,16 enigmol (where the 1-OH of Sph has been shifted to position 5),17 phytosphingosine,18 and FTY720,19 and its vinyl phosphonate derivatives.20
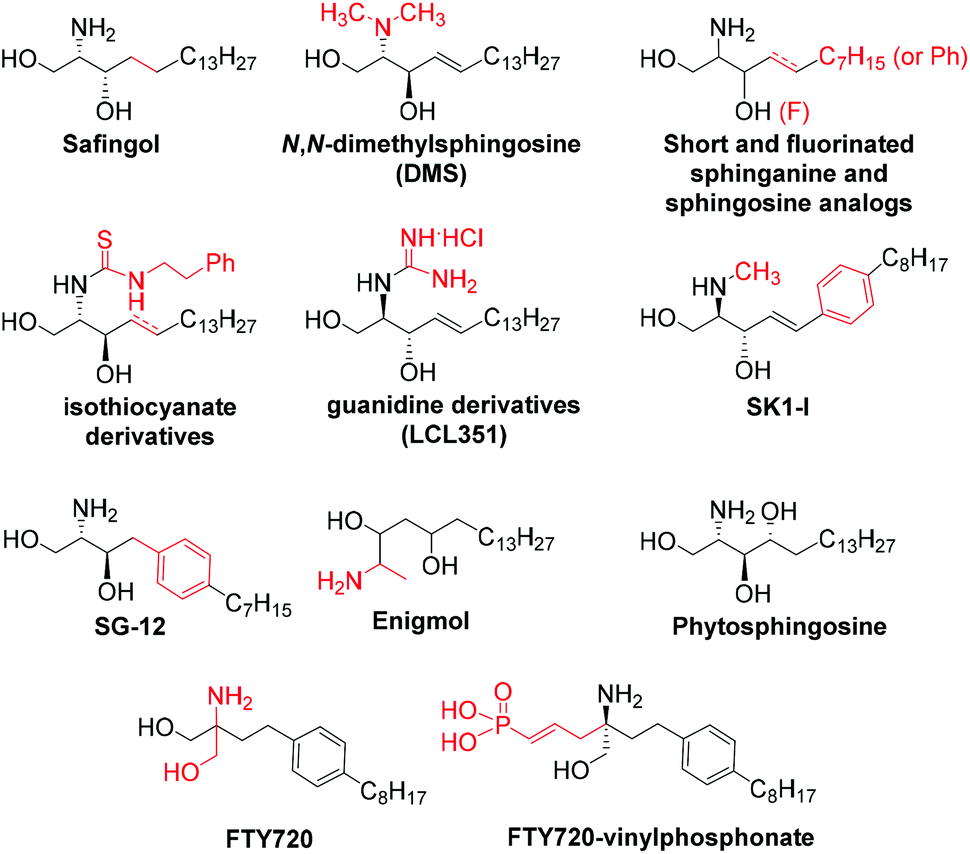 | ||
| Fig. 1 SPHK inhibitors derived from sphingolipids. | ||
Quite recently, the structures of SPHK1 in complexes with ADP, and some inhibitors21 have been disclosed. The hydrophilic head group of the sphingosine is situated at the cleft between two N- and C-domains, leading to hydrogen-bond interactions with Asp and Ser residues. The hydrophobic alkyl chain is accommodated in a J-shaped tunnel, lined by the side chains of mostly nonpolar residues.
Despite the wide array of compounds developed so far and the recent breakthroughs22 in the structural and functional characterization of SPHK, synthesis may provide new architectures that help in gaining insight into the regulation of the complex mechanism that governs SP1 metabolism. In this context, the aim of this work is the syntheses of new lipidic compounds as potential SKIs. The structures proposed for this purpose retain the polar part of the parent compound, sphingosine, maintaining the key aminodiol moiety. The headgroup will be attached to a triazole core, mimicking the trans double bond present in sphingosine23 and allowing additional interactions (Fig. 2). The triazole unit, in turn, will be appended to a modified lipophilic end to enhance hydrophobic interactions with the pocket of SPHK1. Additional SPHK1 affinity might be anticipated to be increased through derivatization of the amino group.
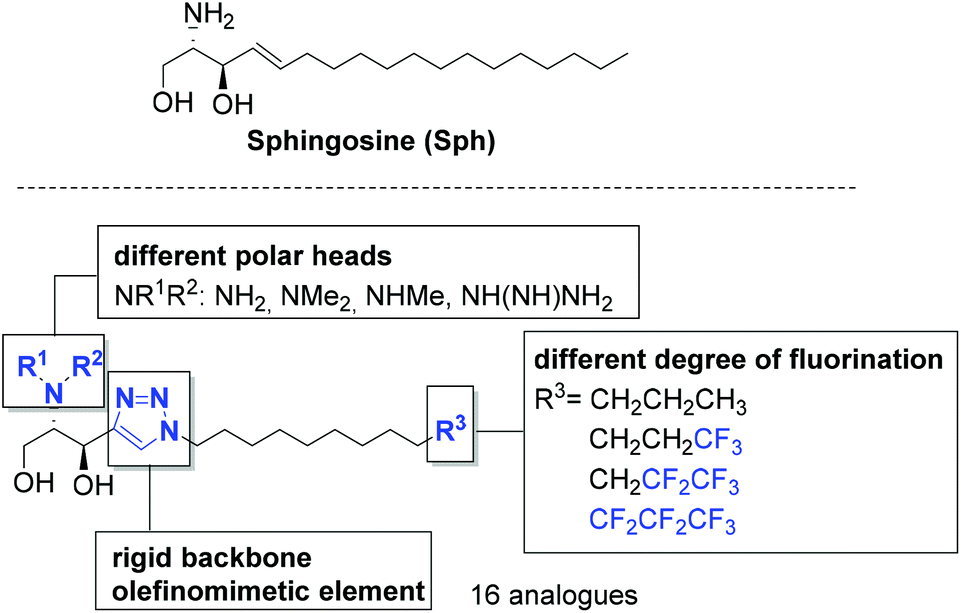 | ||
| Fig. 2 Structural modifications of sphingosine proposed in this work. | ||
Considering our experience in the field of the synthesis of sphingoid bases,24 and aiming to explore new methodologies for obtaining sphingosine derivatives, we envisioned the synthesis of the proposed analogues by a click reaction from a common intermediate, obtained from an alkynyl aziridine (Scheme 2).
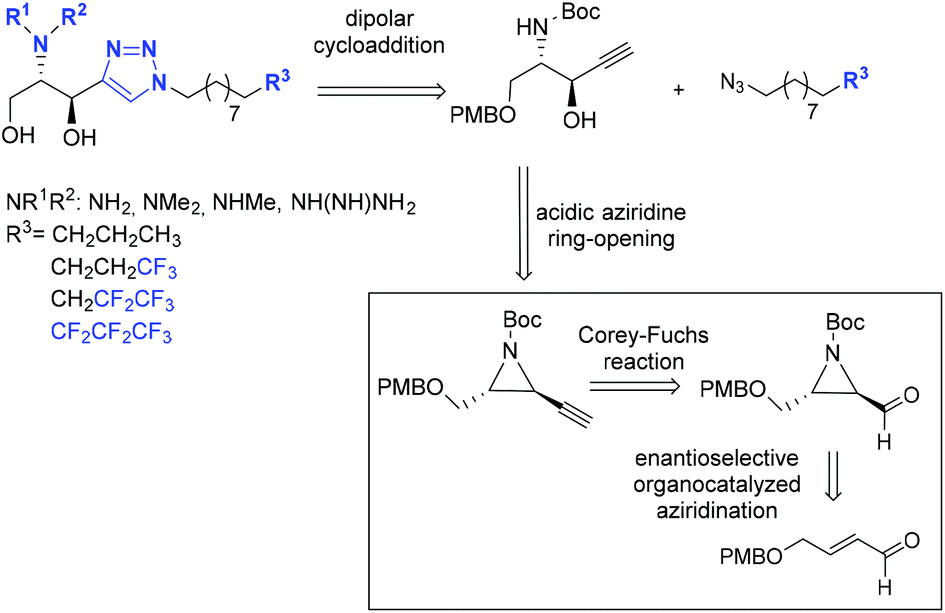 | ||
| Scheme 2 Retrosynthetic proposal: organocatalyzed aziridination and the Corey–Fuchs reaction as the key steps. | ||
Alkynyl aziridines are useful intermediates in many synthetic processes and they have been synthesized through a variety of protocols.25 For some of them the asymmetric version has been reported. Thus, enantioenriched alkynylaziridines have been prepared from chiral alkynylamino alcohols by an intramolecular substitution reaction,26 by addition of guanidium ylides to aldehydes,27 by catalyzed nitrene addition to alkynyl olefins,28 and by catalyzed addition of carbenes to imines.29 Yudin reported that alkynyl aziridines could be obtained from alkynyl aldehydes by one-carbon homologation.30
Here we report that enantioenriched alkynylaziridines can be accessed via a proline organocatalyzed aziridination reaction of α,β-unsaturated aldehydes followed by a homologation reaction (Scheme 2). Regioselective ring opening of this common intermediate followed by triazole click chemistry with the corresponding fatty alkyl azides furnishes a library of sphingosine analogues bearing a triazole unit containing oligofluorinated alkyl chains. This library may be extended by selected derivatizations at the amino moiety.
Results and discussion
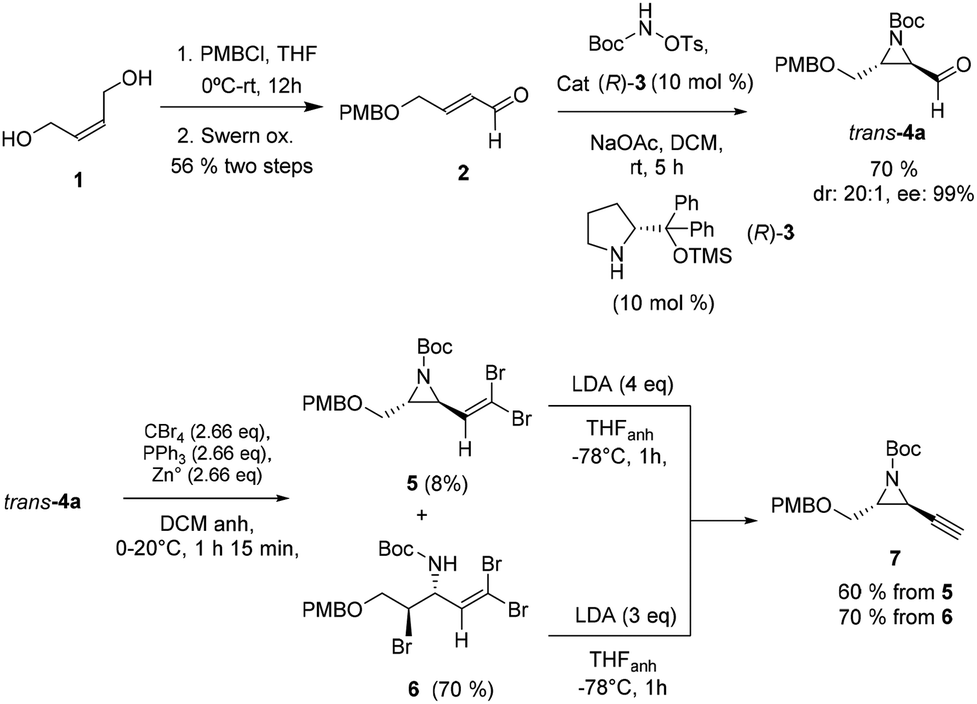
Starting α,β-unsaturated aldehyde substrate 231 for the organocatalyzed aziridination reaction was first synthesized according to the literature via monoprotection of diol 1 as the p-methoxybenzyl ether32 and subsequent Swern oxidation in a 56% global yield (Scheme 3). The enantioselective organocatalytic aziridination33 of the α,β-unsaturated aldehyde 2 with O-tosyl-N-tert-butoxycarbonyl-hydroxylamine catalyzed by TMS-protected diphenylprolinol (R)-334 provided the desired β-formyl aziridine 4a in a 70% yield with 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr and 99 ee%. The enantiomeric excess was established via the formation of the corresponding vinyl derivative via the Wittig reaction (see the ESI†).
:1 dr and 99 ee%. The enantiomeric excess was established via the formation of the corresponding vinyl derivative via the Wittig reaction (see the ESI†).
 | ||
| Scheme 3 Synthesis of ethynyl aziridine 7 from diol 1. | ||
With aldehyde 4a in hand, we set up one-carbon homologation to render the alkyne moiety. When aldehyde 4a was treated under Corey–Fuchs conditions35 with CBr4, triphenylphosphine and zinc dust in dichloromethane the corresponding dibromoalkene 5 was obtained only in an 8% yield (Scheme 3), the tribrominated alkene 6 being the main product (70% yield). Product 6 might result from the bromide ring opening of aziridine 4a.36
Gratifyingly, both products, 5 and 6, when subjected to LDA treatment led to the corresponding ethynylaziridine 7via metal-halogen exchange and ulterior α-elimination,37 and from 6 through an additional ring closing process in the basic media.
Ring opening of aziridine 7 with acetic acid at room temperature followed by basic hydrolysis of the acetate group in 8 furnished the N-protected amino alcohol 9 in excellent yield (Scheme 4). The observed regioselectivity can be rationalized considering the selective nucleophilic attack at the propargylic position (see ref. 25 for a general review dealing with the synthesis and opening of alkynyl (and vinyl) aziridines). This effect has also been described in the ring-opening reactions of vinyloxirane and activated vinylaziridine derivatives.38
 | ||
| Scheme 4 Synthesis of intermediate 9 by aziridine ring opening. | ||
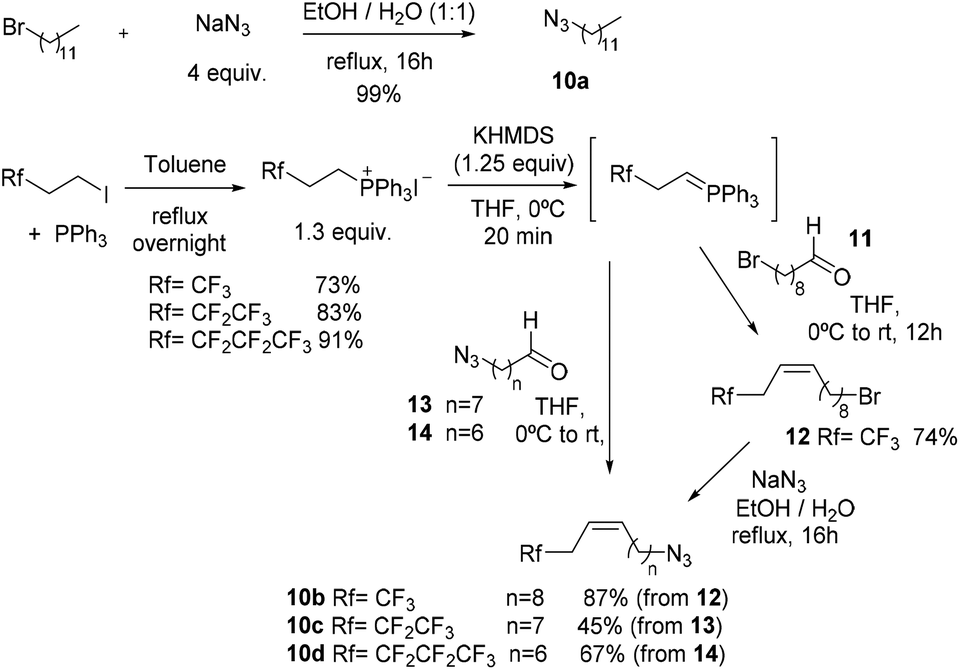
In order to perform click triazole syntheses, azides 10a–d were synthesized. 1-Azidododecane 10a was obtained quantitatively from 1-bromododecane by treatment with sodium azide in a 1:1 mixture of refluxing EtOH:H2O. Azide 10b was synthesized from 9-bromononanal 11via the Wittig reaction with the corresponding trifluorinated phosphorus ylide and ulterior bromide displacement with sodium azide (Scheme 5). For practical reasons,39 azides 10c and 10d were synthesized by the Wittig reaction with the corresponding azido aldehydes 13 and 14. All aldehydes were obtained from the respective alcohols by Swern oxidation.
 | ||
| Scheme 5 Synthesis of azides 10a–d. | ||
A copper catalysed cycloaddition reaction of azides 10a–d with the alkyne 9 previously prepared afforded triazoles 15a–d in yields ranging from 66% to 86% (Scheme 6). With these protected triazole-based sphingosine analogues in hand, the corresponding modifications in the amine functionality were carried out. Thus, the preparation of the non-fluorinated N-monomethyl amine 18a was accomplished by reduction of the carbamate group and ulterior p-methoxybenzyl ether deprotection. Inverting the sequence of events, as was done for the fluorinated analogues (see below), leads in this case to worse yields. For obtaining fluorinated N-methyl derivatives 18b–d an additional double bond reduction step was required. This transformation was performed using 15b–d with Pd/C, H2, with concomitant PMB hydrogenolysis, and the Boc group was subsequently reduced (Scheme 7). Boc deprotection of compounds 17b–d led to free amino analogues 19b–d. Non-fluorinated aminodiol 19a, in turn, can be directly obtained from the non-olefinated compound 15a by simultaneous Boc and PMB deprotection with TFA.
 | ||
| Scheme 6 Azide–alkyne cycloaddition. | ||
 | ||
| Scheme 7 Last steps and N-derivatization for the syntheses of target triazole analogues. | ||
N,N-Dimethyl derivatives 20a–d were synthesized from 19a–d by reductive amination with p-formaldehyde (Scheme 7). The synthesis of N-guanidine sphingosines was accomplished by reaction of amines 19a–d with N,N′-di-Boc-1H-pyrazole-1-carboxiamidine to afford N-Boc derivatives 21a–d, followed by deprotection with a 1:1 mixture of TFA/CH2Cl2.
Although purification of guanidine derivatives was performed under basic conditions, and despite that an ulterior treatment with aqueous NaOH had been carried out, compounds 22b–d were obtained as the trifluoroacetate salts.
The half maximal inhibitory concentration (IC50) of each compound was determined using an in vitro time-resolved fluorescence energy transfer (TR-FRET) analysis (Table 1 and Fig. 1 and 2 of the ESI†). This experiment was performed independently for each kinase and dimethyl sphingosine was used as a reference.
|
|
|||||
|---|---|---|---|---|---|
| Inhibitor | SPHK1 | SPHK2 | Inhibitor | SPHK1 | SPHK2 |
| Values are expressed as concentrations (μM), and no value (“—”) means low or no inhibitory activity detected. | |||||
| 18a | 61.5 | 22.9 | 20a | 110.2 | 22.2 |
| 18b | 48.9 | 41.9 | 20b | — | 56.8 |
| 18c | 30.0 | 39.5 | 20c | — | 70.8 |
| 18d | 51.6 | 10.7 | 20d | — | 15.6 |
| 19a | 97.1 | 25.6 | 22a | 41.9 | 7.9 |
| 19b | — | 40.5 | 22b | — | — |
| 19c | — | — | 22c | 51.1 | 19.1 |
| 19d | — | 28.5 | 22d | 60.0 | 18.1 |
| DMS | 27.3 | 3.8 | |||

None of the compounds showed a higher inhibitory activity than the reference DMS, but two of them showed an IC50 value close to DMS. Thus, the non-fluorinated guanidine derivative 22a showed an IC50 = 7.9 μM for SPHK2 and the pentafluoro N-monomethyl compound 18c showed an IC50 = 30.0 μM for SPHK1. However, these activities were not selective and these compounds showed also inhibitory activity against the other kinase.
More interestingly, some compounds actually showed modest but selective inhibitory activity for SPHK2 (free amino trifluoromethyl and heptafluoro compounds 19b and 19d as well as N,N-dimethylamino fluorinated derivatives 20b, 20c and 20d). As a general trend, the family of compounds constituted of heptafluoroderivatives (18d, 19d, 20d and 22d) showed a good and selective inhibitory activity against SPHK2, the N,N-dimethyl derivative 22d being the best compound in this series in terms of activity/selectivity.
The presence of polyfluorinated alkyl fragments in a compound is known to affect the lipophilicity and conformational rigidity of the parent molecule along with other effects.40 In our case, the terminal heptafluoropropyl unit can confer a lower conformational mobility to this family of sphingolipid analogues, which might make a proper fitting of the alkylic chain into the J-shaped tunnel of SPHK1 difficult. This fact could explain the observed selective inhibitory activity against SPHK2 if the cavity of this kinase was straighter than that of SPHK 1. However, the crystal structure of SPHK2 has not been elucidated yet, so this hypothesis cannot be confirmed. Nonetheless, these results postulate heptafluoro-derivatization as a starting point for future SPHK2 selective inhibitors.
Conclusions
In summary, we have reported an efficient method for obtaining enantioenriched alkynylaziridines by a proline organocatalyzed aziridination reaction of α,β-unsaturated aldehydes followed by a homologation reaction. This approach has allowed easy access to a library of 16 sphingosine analogues bearing a common triazole motif, including systematic modifications at the amino moiety and a different degree of fluorination at the end of the alkyl chain. The products obtained showed comparable in vitro activity to that of DMS as SPHK1 and SPHK2 inhibitors, although the family constituted of a heptafluoro tail exhibits selective inhibitory activity towards SPHK2, postulating this derivatization as a starting point for future SPHK2 selective inhibitors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Spanish Government (CTQ2014-58664-R and CTQ2017-89750-R) for financial support. A. C. thanks Universitat Rovira i Virgili for a research fellowship (PIPF-DL003535).Notes and references
- T. A. Taha, Y. A. Hannun and L. M. Obeid, J. Biochem. Mol. Biol., 2006, 39, 113 CAS.
- M. M. Young, M. Kester and H.-G. Wang, J. Lipid Res., 2013, 54, 5 CrossRef CAS PubMed; M. Maceyka, K. B. Harikumar, S. Milstien and S. Spiegel, Trends Cell Biol., 2012, 22, 50 CrossRef PubMed.
- S. Ponnusamy, M. Meyers-Needham, C. E. Senkal, S. A. Saddoughi, D. Sentelle, S. P. Selvam, A. Salas and B. Ogretmen, Future Oncol., 2010, 6, 1603 CrossRef CAS PubMed; W. Jiang and B. Ogretmen, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2014, 1841, 783 CrossRef PubMed.
- N. C. Hait, C. A. Oskeritzian, S. W. Paugh, S. Milstien and S. Spiegel, Biochim. Biophys. Acta, 2006, 1758, 2016 CrossRef CAS PubMed; S. M. Pitson, Trends Biochem. Sci., 2011, 36, 97 CrossRef PubMed.
- T. A. Taha, Y. A. Hannun and L. M. Obeid, J. Biochem. Mol. Biol., 2006, 39, 113 CAS.
- W. L. Santos and K. R. Lynch, ACS Chem. Biol., 2015, 10, 225 CrossRef CAS PubMed.
- D. Shida, K. Takabe, D. Kapitonov, S. Milstien and S. Spiegel, Curr. Drug Targets, 2008, 9, 662 CrossRef CAS PubMed; C. R. Gault and L. M. Obeid, Crit. Rev. Biochem. Mol. Biol., 2011, 46, 342 CrossRef PubMed; K. Takabe, S. W. Paugh, S. Milstien and S. Spiegel, Pharmacol. Rev., 2008, 60, 181 CrossRef PubMed.
- P. Gao and C. D. Smith, Mol. Cancer Res., 2011, 9, 1509 CrossRef CAS PubMed; C. T. Wallington-Beddoe, J. A. Powell, D. Tong, S. M. Pitson, K. F. Bradstock and L. J. Bendall, Cancer Res., 2014, 74, 2803 CrossRef PubMed.
- Z. Wang, X. Min, S.-H. Xiao, S. Johnstone, W. Romanow, D. Meininger, H. Xu, J. Liu, J. Dai, S. An, S. Thibault and N. Walker, Structure, 2013, 21, 798 CrossRef CAS PubMed.
- M. Hamada, T. Sumi, S. Iwai, M. Nakazawa and Y. Yura, Apoptosis, 2006, 11, 47 CrossRef CAS PubMed; A. Friedman, J. C. Allegood, E. Wang, A. H. Merrill Jr. and G. K. Schwartz, Autophagy, 2009, 5, 184 CrossRef; L. U. Ling, K. B. Tan, H. Lin and G. N. Chiu, Cell Death Dis., 2011, 2, e129 CrossRef PubMed.
- K. A. Orr Gandy and L. M. Obeid, Biochim. Biophys. Acta, 2013, 1, 157 CrossRef CAS PubMed; E. A. Sweeney, C. Sakakura, T. Shirahama, A. Masamune, H. Ohta, S. Hakomori and Y. Igarashi, Int. J. Cancer, 1996, 66, 358 CrossRef PubMed; T. Shirahama, E. A. Sweeney, C. Sakakura, A. K. Singhal, K. Nishiyama, S. Akiyama, S. Hakomori and Y. Igarashi, Clin. Cancer Res., 1997, 3, 257 Search PubMed.
- S. De Jonghe, I. Van Overmeire, S. Poulton, C. Hendrix, R. Busson, S. Van Calenbergh, D. De Keukeleire, S. Spiegel and P. Herdewijn, Bioorg. Med. Chem. Lett., 1999, 9, 3175 CrossRef CAS PubMed.
- C. R. Johnson, J. Chun, R. Bittman and W. D. Jarvis, J. Pharmacol. Exp. Ther., 2004, 309, 452 CrossRef CAS PubMed.
- Z. M. Szulc, A. Bielawska, L. M. Obeid, Y. A. Hannun, J. Norris and L. Xiang, WO2010078247, 2010 CrossRef CAS PubMed; A. K. Sharma, Expert Opin. Ther. Pat., 2011, 21, 807 CrossRef CAS PubMed.
- S. W. Paugh, B. S. Paugh, M. Rahmani, D. Kapitonov, J. A. Almenara, T. Kordula, S. Milstien, J. K. Adams, R. E. Zipkin, S. Grant and S. A. Spiegel, Blood, 2008, 112, 1382 CrossRef CAS PubMed.
- J. W. Kim, Y. W. Kim, Y. Inagaki, Y. A. Hwang, S. Mitsutake, Y. W. Ryu, W. K. Lee, H. J. Ha, C. S. Park and Y. Igarashi, Bioorg. Med. Chem., 2005, 13, 3475 CrossRef CAS PubMed.
- H. Symolon, A. Bushnev, Q. Peng, H. Ramaraju, S. G. Mays, J. C. Allegood, S. T. Pruett, M. C. Sullards, D. L. Dillehay, D. C. Liotta and A. H. Merrill Jr., Mol. Cancer Ther., 2011, 10, 648 CrossRef CAS PubMed.
- H. Liu, M. Sugiura, V. E. Nava, L. C. Edsall, K. Kono, S. Poulton, S. Milstien, T. Kohama and S. Spiegel, J. Biol. Chem., 2000, 275, 19513 CrossRef CAS PubMed.
- T. Ubai, H. Azuma, Y. Kotake, T. Inamoto, K. Takahara, Y. Ito, S. Kiyama, T. Sakamoto, S. Horie, S. Muto, S. Takahara, Y. Otsuki and Y. Katsuoka, Anticancer Res., 2007, 27, 75 Search PubMed; T. Zheng, X. Meng, J. Wang, X. Chen, D. Yin, Y. Liang, X. Song, S. Pan, H. Jiang and L. Liu, J. Cell. Biochem., 2010, 111, 218 CrossRef CAS PubMed; C. White, H. Alshaker, C. Cooper, M. Winkler and D. Pchejetski, Oncotarget, 2016, 7, 23106 Search PubMed.
- K. G. Lim, F. Tonelli, E. Berdyshev, I. Gorshkova, T. Leclercq, S. M. Pitson, R. Bittman, S. Pyne and N. J. Pyne, J. Biochem. Cell Biol., 2012, 44, 1457 CrossRef CAS PubMed.
- J. Wang, S. Knapp, N. J. Pyne, S. Pyne and J. M. Elkins, ACS Med. Chem. Lett., 2014, 5, 1329 CrossRef CAS PubMed; A. Delgado, J. Casas, A. Llebaria, J. L. Abad and G. Fabriàs, Biochim. Biophys. Acta, 2006, 1758, 1957 CrossRef PubMed; P. Gangoiti, L. Camacho, L. Arana, A. Ouro, M. H. Granado, L. Brizuela, J. Casas, G. Fabriás, J. L. Abad, A. Delgado and A. Gómez-Muñoz, Prog. Lipid Res., 2010, 49, 316 CrossRef PubMed; S. Pyne, R. Bittman and N. J. Pyne, Cancer Res., 2011, 71, 6576 CrossRef PubMed; D. Plano, S. Amin and A. K. Sharma, J. Med. Chem., 2014, 57, 5509 CrossRef PubMed; D. Canals and Y. A. Hannun, Handb. Exp. Pharmacol., 2013, 215, 211 Search PubMed; N. N. Patwardhan, E. A. Morris, Y. Kharel, M. R. Raje, M. Gao, J. L. Tomsig, K. R. Lynch and W. L. Santos, J. Med. Chem., 2015, 58, 1879 CrossRef PubMed.
- D. Adams, S. Pyne and N. J. Pyne, Trends Biochem. Sci., 2016, 41, 395 CrossRef CAS PubMed.
- To the best of our knowledge, there is only one example of triazole-containing ceramides, the heterocycle being a bioisostere of the amide group in these sphingolipids: S. Kim, M. Cho, T. Lee, S. Lee, H.-Y. Min and S. K. Lee, Bioorg. Med. Chem. Lett., 2007, 17, 4584 CrossRef CAS PubMed.
- J. Guasch, I. Giménez-Nueno, I. Funes-Ardoiz, M. Bernús, M. I. Matheu, F. Maseras, S. Castillón and Y. Díaz, Chem. – Eur. J., 2018, 24, 4635 CrossRef CAS PubMed; J. Llaveria, A. Beltrán, W. M. C. Sameera, A. Locati, M. M. Díaz-Requejo, M. I. Matheu, S. Castillón, F. Maseras and P. J. Pérez, J. Am. Chem. Soc., 2014, 136, 5342 CrossRef PubMed; J. Guasch, Y. Díaz, M. I. Matheu and S. Castillón, Chem. Commun., 2014, 55, 7344 RSC; J. A. Morales-Serna, J. Llaveria, Y. Díaz, M. I. Matheu and S. Castillón, Curr. Org. Chem., 2010, 14, 2483 CrossRef; J. Llaveria, A. Beltrán, M. M. Díaz-Requejo, M. I. Matheu, S. Castillón and P. J. Pérez, Angew. Chem., Int. Ed., 2010, 14, 2483 Search PubMed; J. A. Morales-Serna, Y. Díaz, M. I. Matheu and S. Castillón, Synthesis, 2009, 710 Search PubMed; J. Llaveria, Y. Díaz, M. I. Matheu and S. Castillón, Org. Lett., 2009, 11, 205 CrossRef PubMed; J. A. Morales-Serna, J. Llaveria, Y. Díaz, M. I. Matheu and S. Castillón, Org. Biomol. Chem., 2008, 6, 4502 RSC.
- H. Ohno, Chem. Rev., 2014, 114, 7784 CrossRef CAS PubMed.
- P. Li, C. D. Evans and M. M. Jouillié, Org. Lett., 2005, 7, 5325 CrossRef CAS PubMed.
- W. Disadee and T. Ishikawa, J. Org. Chem., 2005, 70, 9399 CrossRef CAS PubMed.
- H. Kawabata, K. Omura, T. Uchida and T. Katsuki, Chem. – Asian J., 2007, 2, 248 CrossRef CAS PubMed.
- Y. Guan, M. P. López-Alberca, Z. Lu, Y. Zhang, A. A. Desai, A. P. Patwardhan, Y. Dai, M. J. Vetticatt and W. D. Wulff, Chem. – Eur. J., 2014, 20, 13894 CrossRef CAS PubMed.
- Z. He and A. K. Yudin, Angew. Chem., Int. Ed., 2010, 49, 1607 CrossRef CAS PubMed.
- B. Thirupathi and D. K. Mohapatra, Org. Biomol. Chem., 2016, 14, 6212 RSC.
- B. M. Trost, J. D. Chisholm, S. T. Wrobleski and M. Jung, J. Am. Chem. Soc., 2002, 124, 12420 CrossRef CAS PubMed.
- L. Deiana, P. Dziedzic, G.-L. Zhao, J. Vesely, I. Ibrahem, R. Rios, J. Sun and A. Córdova, Chem. – Eur. J., 2011, 17, 7904 CrossRef CAS PubMed; J. Vesely, I. Ibrahem, G.-L. Zhao, R. Rios and A. Córdova, Angew. Chem., Int. Ed., 2007, 46, 778 CrossRef PubMed.
- K. L. Jensen, G. Dickmeiss, H. Jiang, L. Albrecht and K. A. Jørgensen, Acc. Chem. Res., 2011, 45, 248 CrossRef PubMed.
- E. J. Corey and P. L. Fuchs, Tetrahedron Lett., 1972, 13, 3769 CrossRef.
- For regioselective ring opening of activated 2-acyl aziridines see: C. Bonini, Tetrahedron Lett., 1996, 37, 6893 CrossRef CAS; G. Righi, P. Bovicelli, M. Barontini and I. Tirotta, Green Chem., 2012, 14, 495 RSC; G. Righi and S. Catullo, Synth. Commun., 2004, 34, 85 CrossRef.
- B. Sahu, R. Muruganantham and I. N. N. Namboothiri, Eur. J. Org. Chem., 2007, 2477 CrossRef CAS.
- B. Olofsson and P. Somfai, J. Org. Chem., 2002, 67, 8574 CrossRef CAS PubMed; B. Olofsson, U. Khamrai and P. Somfai, Org. Lett., 2000, 2, 408 CrossRef; G. Righi, S. Ciambrone, E. Esuperanzi, F. Montini and R. Pelagalli, J. Heterocycl. Chem., 2010, 47, 564 Search PubMed.
- In some Wittig attempts for obtaining (Z)-12-bromo-1,1,1-trifluorododec-3-ene the formation of a by-product arising from a dehydrobromination process was observed. Although obtaining this by-product was minimized in this case by lowering the excess of the base to 1.25 equiv., the alternative process (Wittig reaction by using the corresponding azidoaldehyde) was the method of choice for obtaining highly fluorinated azides.
- E. Prchalová, O. Štěpánek, S. Smrček and M. Kotora, Future Med. Chem., 2014, 6, 1201 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR spectra and additional graphics. See DOI: 10.1039/c8ob01867g |
| This journal is © The Royal Society of Chemistry 2018 |