
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Azodicarboxylate-free esterification with triphenylphosphine mediated by flavin and visible light: method development and stereoselectivity control†
Michal
März
 *a,
Michal
Kohout
a,
Tomáš
Neveselý
a,
Josef
Chudoba
b,
Dorota
Prukała
c,
Stanislaw
Niziński
d,
Marek
Sikorski
c,
Gotard
Burdziński
d and
Radek
Cibulka
*a
*a,
Michal
Kohout
a,
Tomáš
Neveselý
a,
Josef
Chudoba
b,
Dorota
Prukała
c,
Stanislaw
Niziński
d,
Marek
Sikorski
c,
Gotard
Burdziński
d and
Radek
Cibulka
*a
aDepartment of Organic Chemistry, University of Chemistry and Technology, Prague, Technická 5, 166 28 Prague, Czech Republic. E-mail: cibulkar@vscht.cz
bCentral Laboratories, University of Chemistry and Technology, Prague, Technická 5, 166 28 Prague, Czech Republic
cFaculty of Chemistry, Adam Mickiewicz University in Poznan, Umultowska 89b, 61 614 Poznan, Poland
dQuantum Electronics Laboratory, Faculty of Physics, Adam Mickiewicz University in Poznan, Umultowska 85, 61 614 Poznan, Poland
First published on 3rd September 2018
Abstract
Triphenylphosphine (Ph3P) activated by various electrophiles (e.g., alkyl diazocarboxylates) represents an effective mediator of esterification and other nucleophilic substitution reactions. We report herein an aza-reagent-free procedure using flavin catalyst (3-methyl riboflavin tetraacetate), triphenylphosphine, and visible light (448 nm), which allows effective esterification of aromatic and aliphatic carboxylic acids with alcohols. Mechanistic study confirmed that photoinduced electron transfer from triphenylphosphine to excited flavin with the formation of Ph3P˙+ is a crucial step in the catalytic cycle. This allows reactive alkoxyphosphonium species to be generated by reaction of an alcohol with Ph3P˙+ followed by single-electron oxidation. Unexpected stereoselectivity control by the solvent was observed, allowing switching from inversion to retention of configuration during esterification of (S)- or (R)-1-phenylethanol; for example with phenylacetic acid, the ratio shifting from 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90 (retention:inversion) in trifluoromethylbenzene to 99.9:0.1 in acetonitrile. Our method uses nitrobenzene to regenerate the flavin photocatalyst. This new approach to flavin re-oxidation has also been successfully proved in benzyl alcohol oxidation, which is a “standard” process among flavin-mediated photooxidations.
:90 (retention:inversion) in trifluoromethylbenzene to 99.9:0.1 in acetonitrile. Our method uses nitrobenzene to regenerate the flavin photocatalyst. This new approach to flavin re-oxidation has also been successfully proved in benzyl alcohol oxidation, which is a “standard” process among flavin-mediated photooxidations.
Introduction
Nucleophilic substitution of a hydroxy function mediated by triphenylphosphine (Ph3P) is among the most powerful tools in organic synthesis.1 Such substitution is allowed as an alcohol molecule is transformed into an alkoxytriphenylphosphonium intermediate, which reacts smoothly with nucleophiles by virtue of the energetically favourable formation of phosphine oxide with a strong P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond (540 kJ mol−1). Alcohols do not react with triphenylphosphine directly. For the formation of alkoxyphosphonium species, triphenylphosphine must be activated with an electrophile/oxidant, for example with a tetrahalomethane in the Appel reaction2 or with a dialkyl azodicarboxylate in Mitsunobu esterification1c,3 (see Scheme 1A). Under certain conditions, the carboxylic function can be activated analogously, resulting in the formation of acyloxyphosphonium species capable of undergoing acyl substitution reaction.3b,4 Whether the hydroxy or the carboxy group is activated significantly influences the stereoselectivity of the Mitsunobu esterification. Usually, an alkoxyphosphonium species is employed, which results in inversion of configuration of the chiral alcohol. In some cases, the acyloxyphosphonium species prevails, which leads to retention of configuration.5,6
O bond (540 kJ mol−1). Alcohols do not react with triphenylphosphine directly. For the formation of alkoxyphosphonium species, triphenylphosphine must be activated with an electrophile/oxidant, for example with a tetrahalomethane in the Appel reaction2 or with a dialkyl azodicarboxylate in Mitsunobu esterification1c,3 (see Scheme 1A). Under certain conditions, the carboxylic function can be activated analogously, resulting in the formation of acyloxyphosphonium species capable of undergoing acyl substitution reaction.3b,4 Whether the hydroxy or the carboxy group is activated significantly influences the stereoselectivity of the Mitsunobu esterification. Usually, an alkoxyphosphonium species is employed, which results in inversion of configuration of the chiral alcohol. In some cases, the acyloxyphosphonium species prevails, which leads to retention of configuration.5,6
 | ||
| Scheme 1 Activation of triphenylphosphine (Ph3P) for esterification reactions. | ||
One-electron oxidation to Ph3P˙+ could be a novel straightforward way to activate Ph3P for substitution reactions. Such an approach would not require a dialkyl azadicarboxylate (or a tetrahalomethane), thus avoiding safety and stability problems and making the triphenylphosphine-mediated procedure more sustainable.‡2a,7
There are several indications that Ph3P˙+ has the required properties to mediate substitutions of the OH group. Electrochemically generated Ph3P˙+ has been shown to react with alcohols to form alkoxyphosphonium species.8 Ph3P˙+ generated from Ph3P by photoinduced electron transfer to an excited acridinium salt, dicyanoanthracene, pyrylium salts, or Eosin Y has been proved to form triphenylphosphine oxide by reaction with oxygen or water.9 Despite these promising observations, to the best of our knowledge neither electrochemically nor photochemically generated Ph3P˙+ has hitherto been utilized in organic synthesis. The only exception is a “dark” aerobic esterification reaction mediated by an iron phthalocyanine catalyst and 4-methoxypyridine N-oxide (MPO) as co-catalyst, as reported by Taniguchi (Scheme 1B),10 in which the carboxylic function is activated by Ph3P˙+, thus affording an ester with retention of configuration.
Herein, we report the first azodicarboxylate-free esterification procedure based on photocatalytic activation of Ph3P. We use 3-methyl riboflavin tetraacetate (1) as a photoredox catalyst and visible light (Scheme 1c; Fig. 1). It is shown that this esterification allows switching from retention to inversion of configuration of chiral alcohols, depending on the structure and acidity of the acid and alcohol and on the solvent used.
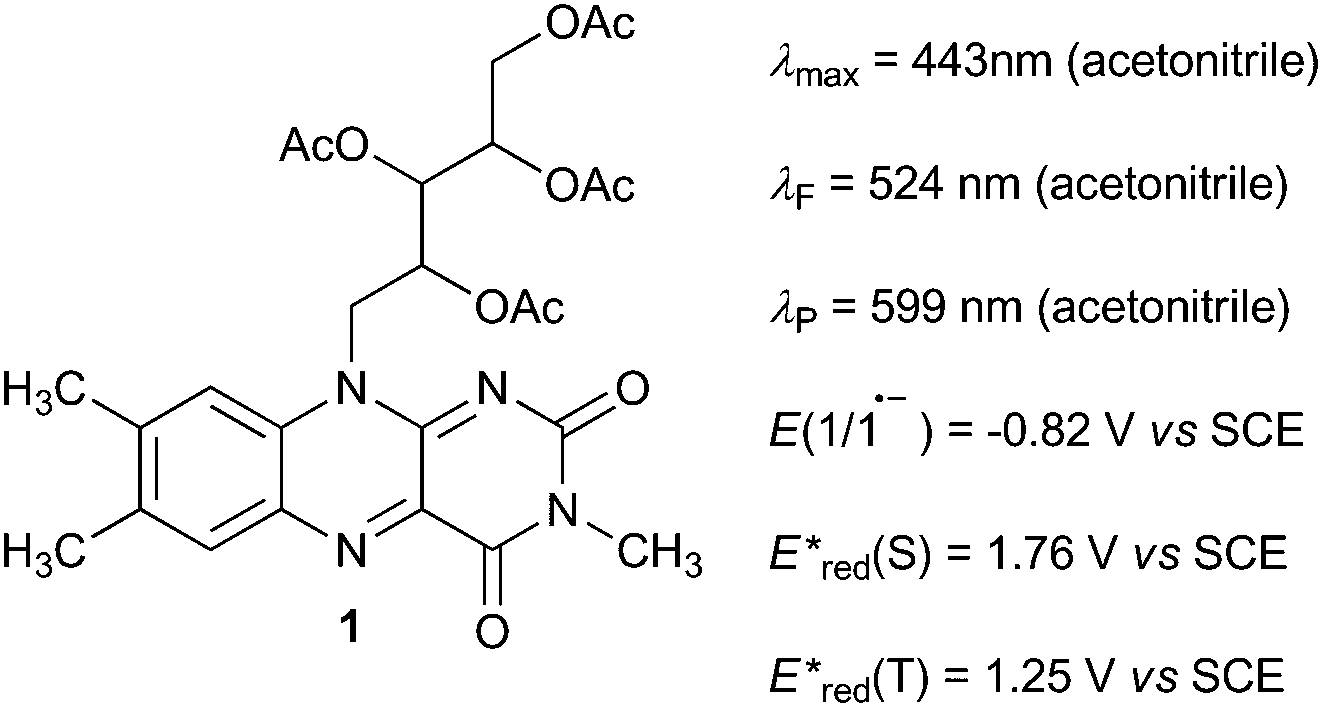 | ||
| Fig. 1 Catalyst 1 and its characteristics. Catalyts 1 was selected since flavins are known versatile catalysts12 and photocatalysts13 in oxidation reactions absorbing in visible region and being of appropriate redox properties. Derivative 1 is known for photostability among flavins.13k For the characteristics measurements, see ESI.† | ||
Results and discussion
Catalytic system design
The design of the photocatalytic system for photoesterification reaction was inspired by our preliminary observations during development of the photocatalytic Mitsunobu reaction.11 In that case, a dialkyl azodicarboxylate was used in a catalytic amount and regenerated by aerial oxidation mediated by flavin photocatalyst 1 and visible light (448 nm) in the presence of molecular sieves 4A (MS 4A).§ Surprisingly, we also observed remarkable ester formation when a mixture of carboxylic acid, alcohol, triphenylphosphine, and a catalytic amount of flavin 1 was irradiated under oxygen in the absence of a dialkyl azodicarboxylate or its precursor, dialkyl hydrazine-1,2-dicarboxylate. This led us to investigate the azodicarboxylate-free system in more detail (Table 1).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Atmosphere (temperature) | Additive | Solvent | Conversion [%] |
| a Conditions: n(alcohol) = 0.15 mmol, n(acid) = 0.18 mmol, n(1) = 0.015 mmol, n(PhNO2) = 0.15 mmol, n(Ph3P) = 0.3 mmol, MS 4 Å (150 mg), 2 mL solvent, 448 nm, 24 h; conversion was determined by 1H NMR. b In the absence of MS. c 4-Chlorobenzyl 3-nitrobenzoate was observed as a sole product. d Beside 4-chlorobenzyl 3-nitrobenzoate (main product) formation of N-(3-carboxyphenyl) 3-nitrobenzamide was also observed. e Beside 4-nitrobenzyl benzoate (main product) formation of N-[4-(hydroxymethyl)phenyl] benzamide was observed. | ||||||
| 1 | NO2 | Cl | O2 (25 °C) | — | CH3CN | 90c |
| 2 | NO2 | Cl | Ar (25 °C) | — | CH3CN | 80d |
| 3 | H | NO2 | Ar (25 °C) | — | CH3CN | 33e |
| 4 | H | Cl | Ar (25 °C) | — | CH3CN | 5 |
| 5 | H | H | Ar (25 °C) | — | CH3CN | 3 |
| 6 | H | H | Ar (25 °C) | PhNO2 | CH3CN | 32 |
| 7 | H | H | Ar (25 °C) | PhNO2 | CH2Cl2 | 32 |
| 8 | H | H | Ar (25 °C) | PhNO2 | Toluene | 38 |
| 9 | H | H | Ar (25 °C) | PhNO2 | THF | 15 |
| 10 | H | H | Ar (25 °C) | PhNO2 | BTF | 47 |
| 11 | H | H | Ar (40 °C) | PhNO2 | BTF | 66 |
| 12b | H | H | Ar (40 °C) | PhNO2 | BTF | 10 |
| 13 | CF3 | H | Ar (40 °C) | PhNO2 | BTF | 84 |
| 14 | CF3 | Cl | Ar (40 °C) | PhNO2 | BTF | 95 |

We started with simple model substrates, 3-nitrobenzoic acid and benzyl alcohol, using 2 equivalents of triphenylphosphine and 10 mol% of 1 (entry 1). An interesting result was that almost the same ester conversion was obtained under argon atmosphere as under oxygen, showing that the flavin catalyst must have been regenerated in another way (cf. entries 1 and 2). The aromatic nitro group seemed to be responsible for this process, which was confirmed by the following results: (i) significantly more than 10% of the ester (expected amount when 10 mol% of flavin is used and it is not regenerated) was formed when the nitro group was part of a benzyl alcohol, benzoic acid or externally added in the form of nitrobenzene (entries 2, 3, and 6); (ii) reduction products of nitro compounds were observed in the reaction mixtures: N-3-carboxyphenyl 3-nitrobenzamide – condensation product of the reduced 3-aminobenzoic acid (entry 2) and N-[4-(hydroxymethyl)phenyl] benzamide – condensation product of the reduced 4-nitrobenzyl alcohol (entry 3); (iii) less than 10 mol% of ester was formed in the absence of an aromatic nitro group (entries 4 and 5). Thus, nitrobenzene was used as a sacrificial oxidant in the next experiments.
Optimizing the solvent and temperature (see entries 7–11 and the ESI† for details), we found trifluoromethylbenzene (BTF) and elevated temperature (40 °C) to be the best reaction conditions, giving almost quantitative conversion when 3-trifluoromethylbenzoic acid was used as a redox-inactive alternative to electron-poor 3-nitrobenzoic acid (cf. entries 1 vs. 13 and 14). Modification of nitrobenzene (stoichiometric oxidant) by substitution had only a small effect on the ester conversion (see the ESI†). Blank experiments (performed with all substrates in Table 1) showed that esterification did not proceed in the absence of either 1, Ph3P, or light. In the absence of molecular sieves (MS), ester 2 was formed with only 10% conversion (entry 12), probably because of the negative effect of water formed during flavin re-oxidation with nitrobenzene.
Substrate scope and stereoselectivity
Having established the optimized conditions, the scope of the reaction with respect to the structures of the alcohol and acid was investigated through preparative experiments (Table 2).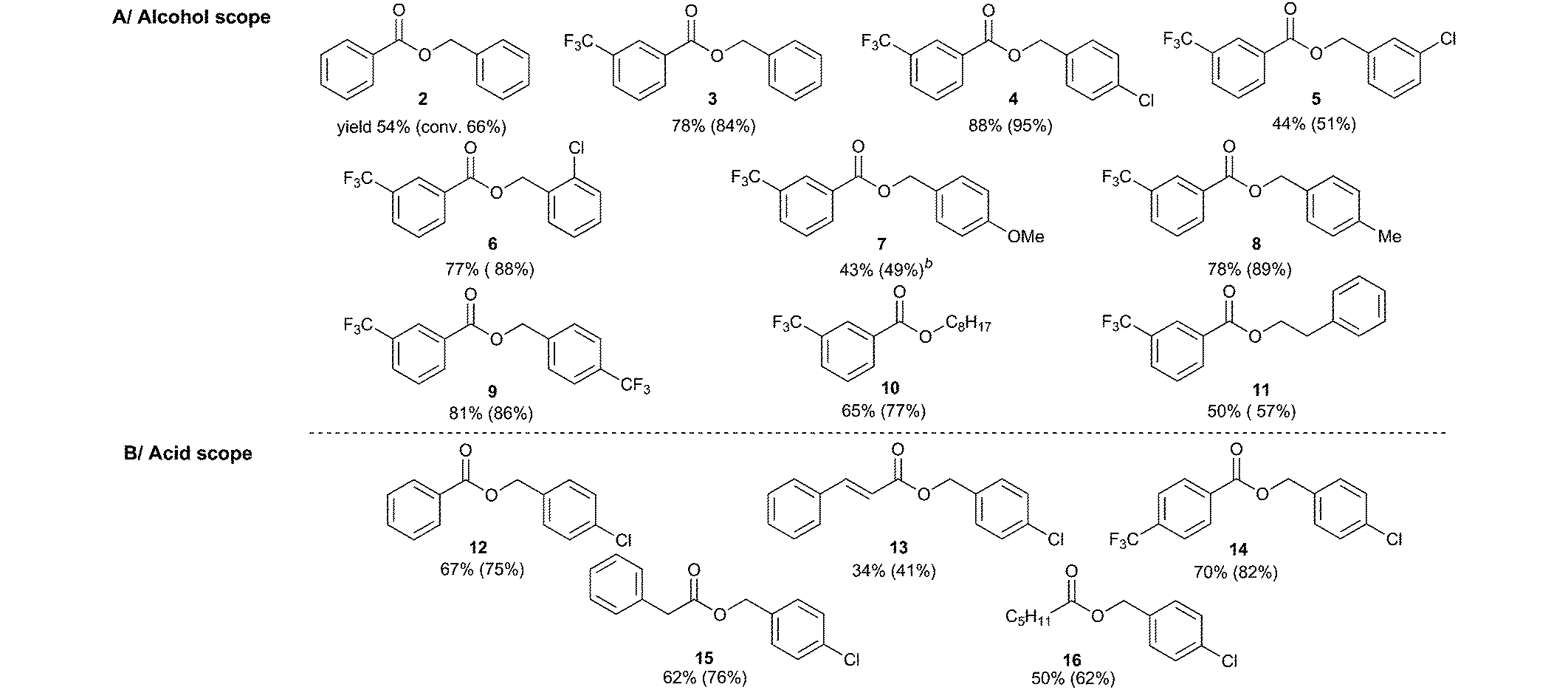
We found that benzyl alcohols react with 3-trifluoromethylbenzoic acid to provide esters 3–9 in moderate to good yields regardless of the nature and position of the substituent. Aliphatic alcohols react smoothly under our conditions (see entries for esters 10 and 11). Besides aromatic acids (see entries for esters 2, 12, and 13), aliphatic acids were also proved to react with 4-chlorobenzyl alcohol (see entries for esters 14–16). The formation of ester 16 is especially noteworthy as it was achieved with significantly higher conversion (62%) compared to the previously described11 aerial photocatalytic Mitsunobu reaction (16%).
Special attention was paid to the stereoselectivity of the new esterification with chiral secondary alcohols, (R)- and (S)-1-phenylethan-1-ol and (R)- and (S)-ethyl lactate (Table 3, method A, first column). Interestingly, the stereoselective course varied significantly depending on the acidity and steric hindrance of the acid, shifting from dominant retention for ester 21 (90:10, entry 5) to highly selective inversion for 17 (0.1:99.1, entry 1). It was found that higher acidity of the carboxylic acid favoured production of the ester with retention of configuration (cf. entries 2–4), whereas sterically demanding acids favoured inversion of configuration (entry 1). These results can be explained in terms of two mechanistic pathways involving either alkoxyphosphonium or acyloxyphosphonium intermediates, which have been proved to be in equilibrium by several studies.3b,4,5c,6 Most probably, for more acidic acids, acyloxyphosphonium prevails, thus favouring acyl substitution, which results in the product with retention of configuration (see entries 2–4). Formation of the acyloxyphosphonium species and thus higher selectivity for retention of configuration seems to be supported when the nucleophilicity of the hydroxy function is decreased by the electron-withdrawing ethoxycarbonyl group (entry 5). With less acidic and/or sterically hindered acids, the alkyloxyphosphonium species is preferred, which results in inversion of configuration with very good to excellent stereoselectivities (entries 1 and 2). The esterification of non-chiral benzyl alcohol and benzoic acid was inspected by an experiment with 18O-labelled acid, which showed the ester to be formed by acyl vs. alcohol activation in a ratio varying from 25:75 in BTF to 66:34 in acetonitrile (see the ESI†).
| Entry | Product | Method Aa (from this work) | Method Bb (according to ref. 11) | Method Cc (non-catalytic M.R.) | |||
|---|---|---|---|---|---|---|---|
| Yieldd [%] | ere | Yieldd [%] | ere | Yieldd [%] | ere | ||
|
a For conditions, see Table 2.
b
n(alcohol) = 0.15 mmol, n(acid) = 0.18 mmol, n(1) = 0.015 mmol, n(DIADH2) = 0.015 mmol, n(Ph3P) = 0.3 mmol, MS 4 Å (150 mg), 2 mL CH3CN, 50 °C, 448 nm, 72 h.
c Condition of non-catalytic reaction: n(alcohol) = 0.15 mmol, n(acid) = 0.18 mmol, n(DIAD) = 0.2 mmol, n(Ph3P) = 0.3 mmol, 2 ml CH3CN, 25 °C, 24 h.
d Preparative yields.
e Retention:inversion from HPLC, average value from experiments with (S)- and (R)- alcohol, see ESI.
|
|||||||
| 1 |

|
33 | 0.1:99.9 |
0 | — | 38 | 10:90 |
| 2 |

|
19 | 10:90 |
23 | 99.9:0.1 |
1 | 10:90 |
| 3 |

|
52 | 40:60 |
28 | 99.9:0.1 |
65 | 13:87 |
| 4 |

|
42 | 68:32 |
50 | 99:1 |
63 | 9:91 |
| 5 |
|
24 | 90:10 |
19 | 99.1:0.1 |
43 | 0.1:99.9 |
The steric effect of the alcohol and/or acid structure on the stereoselectivity of Mitsunobu esterification has previously been observed in a few studies.1a,5c,6 Nevertheless, the results obtained by this new azocarboxylate-free photoesterification (Table 3, method A) are surprising compared to those from flavin-based photocatalytic esterification under Mitsunobu conditions with a catalytic amount of dialkyl azodicarboxylate (method B) and those from “dark” stoichiometric Mitsunobu reaction (method C), which clearly showed either retention or inversion of configuration, respectively, with all investigated substrates. The question then arose as to the origin of this difference. Although solvent has never been observed as the dominant factor in determining the stereochemistry of Mitsunobu esterification, we firstly examined whether substitution of acetonitrile (used in method B) by BTF (used in method A) could play a role in defining the stereochemistry. Results with trifluorobenzoic acid and (S)-1-phenylethan-1-ol generating ester 19 (Table 4, first column) confirmed this premise, showing the stereochemistry of the new esterification (method A) to significantly vary with the solvent. In polar solvents such as acetonitrile or acetone, the reaction predominantly proceeded with retention of configuration, whereas in non-polar solvents, inversion of configuration prevailed. An even more significant effect of the solvent was observed with phenylacetic acid in forming ester 18, for which the stereoselectivity shifted from 99.9:0.1 in acetonitrile to 10:90 in BTF. On the contrary, the stereoselectivity of the “dark” Mitsunobu reaction leading to ester 19 was found to be unaffected by solvent (er 13:87 and 7:93 in acetonitrile and BTF, respectively) and thus our photocatalytic process remains unique in this respect.


Mechanistic investigation
The key step of the photoesterification is oxidation of Ph3P to Ph3P˙+ by single-electron transfer to the excited flavin 1 (Fl*; see Scheme 2 for the proposed mechanism). Flavin 1 in the singlet excited state (1Fl*) is strong enough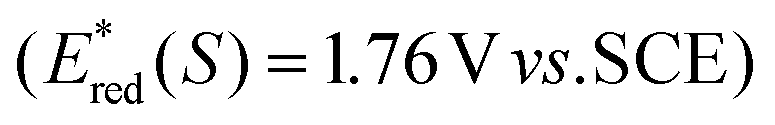 to induce the oxidation of Ph3P (Eox = 1.0 V vs. SCE, ref. 9c). This process was confirmed by the observation of efficient quenching of the steady-state fluorescence emission of 1 upon addition of Ph3P. The value of the quenching rate constant (kq = 6 × 109M−1 s−1) was obtained both from fluorescence intensity as well as lifetime measurements in the presence of various amounts of Ph3P (for the Stern–Volmer plots, see ESI†).
to induce the oxidation of Ph3P (Eox = 1.0 V vs. SCE, ref. 9c). This process was confirmed by the observation of efficient quenching of the steady-state fluorescence emission of 1 upon addition of Ph3P. The value of the quenching rate constant (kq = 6 × 109M−1 s−1) was obtained both from fluorescence intensity as well as lifetime measurements in the presence of various amounts of Ph3P (for the Stern–Volmer plots, see ESI†).
 | ||
| Scheme 2 Proposed mechanism of flavin- and Ph3P-mediated photocatalytic esterification. In the scheme, flavin 1 is labeled as Fl as the mechanism is expected as general for flavin derivatives. | ||
The driving force for photoinduced electron transfer from Ph3P to the flavin triplet state (3Fl*) estimated from corresponding redox potentials  shows that it is also an exergonic process and, indeed, laser-flash photolysis experiments gave direct evidence that flavin in its triplet excited state participates in electron transfer. Although the excited triplet state of flavin exists in dry acetonitrile for tens of microseconds, with an estimated lifetime of 68 μs (Fig. 2A), this is significantly shortened by the addition of Ph3P (Fig. 2B). The quenching rate of the triplet excited state of flavin by Ph3P was estimated to be 4.1 × 109M−1 s−1 from a Stern–Volmer plot (Fig. 2C). The reaction pathway of photoinduced ET from Ph3P to 3Fl* can be expected to predominate under conditions of esterification because of the much longer intrinsic lifetime of 3Fl* (68 μs) compared to that of 1Fl*(5.7 ns). For instance, at the Ph3P concentration of 10−2M, over 99% of 3Fl* undergoes quenching, while only approximately 20% in the case of 1Fl*. Electron transfer from Ph3P to flavin 1 in an excited state leads to flavin radical anion Fl˙−, formation of which was detected by transient spectra (see ESI†). Unfortunately, Ph3P˙+ was not clearly observed in our transient spectroscopy experiments, most probably because of its very low molar absorption coefficient (see ref. 9c) and overlap with the signals of flavin anion species (see ESI†).
shows that it is also an exergonic process and, indeed, laser-flash photolysis experiments gave direct evidence that flavin in its triplet excited state participates in electron transfer. Although the excited triplet state of flavin exists in dry acetonitrile for tens of microseconds, with an estimated lifetime of 68 μs (Fig. 2A), this is significantly shortened by the addition of Ph3P (Fig. 2B). The quenching rate of the triplet excited state of flavin by Ph3P was estimated to be 4.1 × 109M−1 s−1 from a Stern–Volmer plot (Fig. 2C). The reaction pathway of photoinduced ET from Ph3P to 3Fl* can be expected to predominate under conditions of esterification because of the much longer intrinsic lifetime of 3Fl* (68 μs) compared to that of 1Fl*(5.7 ns). For instance, at the Ph3P concentration of 10−2M, over 99% of 3Fl* undergoes quenching, while only approximately 20% in the case of 1Fl*. Electron transfer from Ph3P to flavin 1 in an excited state leads to flavin radical anion Fl˙−, formation of which was detected by transient spectra (see ESI†). Unfortunately, Ph3P˙+ was not clearly observed in our transient spectroscopy experiments, most probably because of its very low molar absorption coefficient (see ref. 9c) and overlap with the signals of flavin anion species (see ESI†).
 | ||
| Fig. 2 Transient absorption spectra of 1 (c = 5.8 × 10−5 mol L−1) in the absence (A) and in the presence (B) of Ph3P (c = 7 × 10−4 mol L−1) in various times after laser pulse excitation (λ = 355 nm, 1 mJ) and reciprocal of the triplet excited state lifetime in function of Ph3P concentration (C). Dry acetonitrile has been used as a solvent. | ||
Additionally, we observed Ph3P to react with water under argon atmosphere or with oxygen under air in dry solvent to form triphenylphosphine oxide in acetonitrile or BTF in the presence of 10 mol% of flavin 1 when the solution was irradiated (see Scheme 3). Under argon atmosphere in the absence of water (in dry solvent) or in the dark, oxidation reaction was not observed. These results provide further evidence that excited flavin is able to generate Ph3P˙+, which can then react with an oxygen source or a nucleophile. It should be noted that the formation of triphenylphosphine oxide from Ph3P has been proved several times to occur through Ph3P˙+, which is believed to react smoothly with water or oxygen.9
 | ||
| Scheme 3 Flavin-mediated photocatalytic oxidations of triphenylphosphine in the presence of oxygen or water. | ||
It is reasonable to assume that flavin radical anion Fl˙− (pKa of conjugated acid is 8.4, ref. 14) will be immediately protonated by a carboxylic acid to form radical FlH˙ (Scheme 2). Subsequently, Ph3P˙+ can react with an alcohol (analogously to the abovementioned reaction with water) or carboxylate to form a phosphoranyl radical Ph3P˙-X, which is likely to be rapidly oxidized to corresponding alkyloxy- or acyloxyphosphonium species. The redox potential of the Ph3P+-X/Ph3P˙-X redox couple can be estimated based on reduction half-wave potentials of related compounds, tetraphenylphosphonium (E° = −2.16 V vs. SCE) or methyltriphenylphosphonium (E° = −2.22 V vs. SCE).15 Several agents in the reaction mixture are sufficiently oxidizing to induce this oxidation, e.g. nitrobenzene (E(PhNO2/PhNO2˙−) = −1.22 V vs. SCE, ref. 16) or flavin 1, even in its ground state (E(1/1˙−) = −0.82 V vs. SCE).
In a separate experiment, we confirmed that an alkoxyphosphonium species is formed from Ph3P in the presence of an alcohol and flavin, as evidenced by the 31P NMR spectrum of the mixture following irradiation with visible light. Only a low concentration of alkoxyphosphonium species was observed under conditions of photoesterification, showing that this reactive species reacts smoothly with a nucleophile. In an excess of alcohol (experiment performed in methanol), a significant amount of alkoxyphosphonium salt was detected together with methyltriphenylphosphonium, the product of alkylation of Ph3P with methoxytriphenylphosphonium (see Fig. 3).
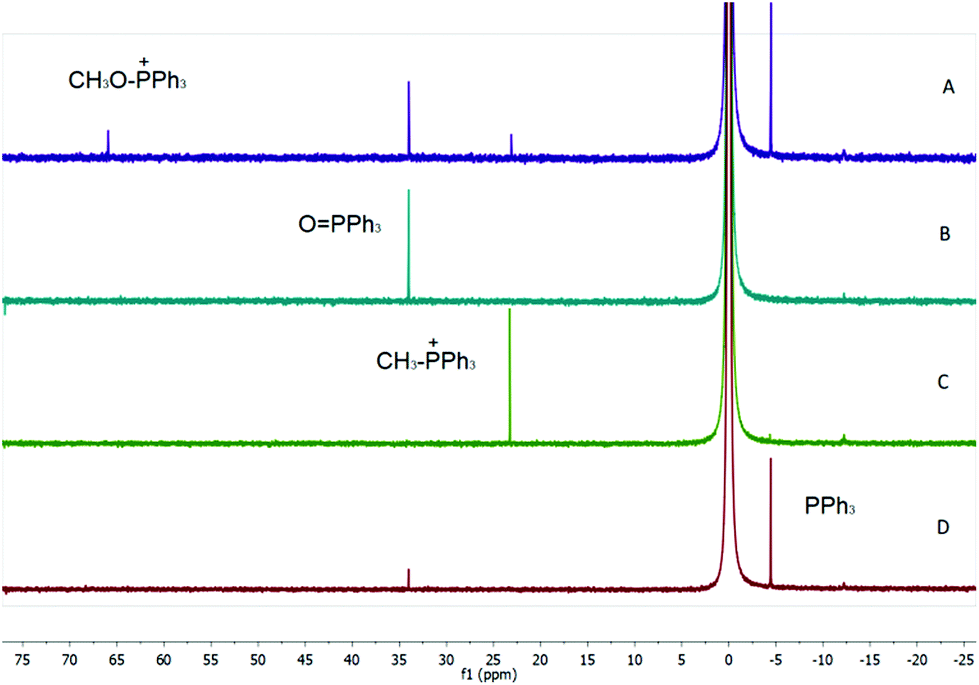 | ||
| Fig. 3
31P NMR spectrum of Ph3P irradiated in the presence of 1 in CH3OH (A), spectrum of Ph3PO (B, standard); spectrum of Ph3P in the presence of CH3I (C) and spectrum of Ph3P (D, standard containing traces of Ph3PO). | ||
A final question remained unanswered about functioning of nitrobenzene in the photocatalytic esterification. In studying the reaction of 3-trifluoromethylbenzoic acid with benzyl alcohol in the absence of nitrobenzene using various amounts of flavins (0–20 mol% with respect to the substrate), we observed ester formation in slightly lower amounts (possibly as a result of flavin bleaching) than might have been expected by taking into account the amount of flavin that was not regenerated; e.g. 5% or 7% ester employing 10% and 20% of 1, respectively (see the ESI†). Thus, nitrobenzene seems to be crucial for flavin re-oxidation, allowing 84% of the ester to be formed under the same conditions. On the other hand, the fact that some ester is also formed in the absence of nitrobenzene shows that its side-role, if indeed there is one, is rather minor; e.g. nitrobenzene could participate in the oxidation of Ph3P˙-X to Ph3P+-X.
In looking for products of nitrobenzene reduction during esterification under common conditions, we invariably detected only a small amount of the amide of the corresponding acid (1–5% with respect to the amount expected) and unreacted nitrobenzene (up to 15%) as the only compounds containing nitrogen. Interestingly, a significant amount (48%) of amide was found in the reaction mixture when an excess (2 equiv.) of carboxylic acid was added. Most probably, aniline formed as a final product of nitrobenzene reduction was trapped by acyloxyphosphonium to form an amide. If there is insufficient amount of acid, aniline will probably polymerize under the reaction conditions (e.g., on the surface of molecular sieves).17
A more substantial amount of amide was also formed when an aniline was present in an excess; for example competitive reaction of benzoic acid with benzyl alcohol and aniline (alcohol–aniline ratio 1:1) afforded mixture of corresponding ester and amide in the ratio around 1:1 though conversion of alcohol to ester was only 22% (Scheme 4A); most importantly, reaction of trifluorobenzoic acid with 4-trifluoromethylaniline (instead of an alcohol) under our conditions in the presence of nitrobenzene as oxidant provided 71% of trifluoromethylanilide and only a small amount (25%) of amide with aniline being slowly released by nitrobenzene reduction during the catalytic process (Scheme 4B). This result suggests that our photocatalytic procedure may eventually be used for amide synthesis.
 | ||
| Scheme 4 Competitive reaction of benzoic acid with benzyl alcohol and aniline (A) and controlled amide formation by flavin-Ph3P-mediated photocatalytic process (B). | ||
Not only aniline, but also other intermediates of nitrobenzene reduction can undergo polymerization reactions. Moreover, nitrobenzene and nitrosobenzene are reported to be deoxygenated with Ph3P with the formation of Ph3PO.18 This undesired process also occurs under our conditions, as evidenced by a blank experiment conducted in the absence of alcohol and carboxylic acid (see the ESI†). Deoxygenation of nitrobenzene and nitrosobenzene explains why an excess of nitrobenzene is necessary in our photocatalytic system and also the fact that 48% of aniline (see above) was captured in the form of amide in esterification with an excess of carboxylic acid which is remarkably more than expected amount 33% (relative to amount ester formed). Theoretically, only one-third of an equivalent of nitrobenzene could be enough, considering that all reduction intermediates, nitrosobenzene (E(PhNO/PhNO˙−) = −1.07 V vs. SCE, ref. 16), azoxybenzene (E(PhNN(O)Ph/PhNN(O)Ph˙−) = −1.44 V vs. SCE, ref. 16), and azobenzene (E(PhNNPh/PhNNPh˙) = −1.42 V vs. SCE, ref. 16), could participate in flavin re-oxidation, based on their reduction potentials. Indeed, the high conversion to ester 3 corroborated flavin catalyst recycling by these agents (Scheme 5). It should be noted that by using 1 equivalent of nitrobenzene relative to alcohol (triple excess relative to theoretical amount) we always observed nitrobenzene (up to 15%) remaining in the reaction mixture after photoesterification. However any attempt to reduce nitrobenzene loading led to lower ester yields (see the ESI†).
 | ||
| Scheme 5 Photocatalytic esterification using various oxidizing agents. | ||
The use of a nitro compound as a sacrificial oxidizing agent in flavin catalysis is still unknown in artificial systems.19 To gain insight into this process, we investigated it in flavin-mediated 4-methoxybenzyl alcohol oxidation, which is a “standard” oxidation process studied by many authors.20 Whereas in the absence of a sacrificial oxidant, a small amount of aldehyde was formed in an irradiated mixture containing a catalytic amount of flavin 1 (Table 5, entry 1), nitrobenzene substantially increased the amount of aldehyde formed (entries 2 and 3). With an excess of nitrobenzene, quantitative chemoselective formation of aldehyde (no overoxidation) was observed (entry 4). In this case, approximately 20% of benzaldehyde was transformed to an imine by aniline formed from nitrobenzene. Importantly, similar results were obtained when nitrosobenzene, azoxide, and azobenzene (entries 5–7) were used instead, thus confirming their possible participation in the flavin regeneration process.
|
|
||
|---|---|---|
| Entry | Oxidant | Conv. to aldehyde (or imine) [%] |
|
a Conditions: n(alcohol) = 0.15 mmol, n(1) = 0.015 mmol, 2 ml BTF, 40 °C, 24 h; conversion was determined by 1H NMR.
b Aldehyde and imine was present in the reaction mixture in the ratio 8:2.
|
||
| 1 | — | 20 |
| 2 | PhNO2 (1 equiv.) | 66 |
| 3 | PhNO2 (2 equiv.) | 80 |
| 4 | PhNO2 (5 equiv.) | quant.b |
| 5 | PhNO (5 equiv.) | quant. |
| 6 | PhN(O)NPh (5 equiv.) |
quant. |
| 7 | PhNNPh (5 equiv.) |
67 |

Finally, to gain direct evidence concerning flavin re-oxidation with nitrobenzene, we generated the reduced flavin 1 by catalytic hydrogenation (see the ESI†). After filtering off palladium and addition of 1 equivalent of nitrobenzene (all operations performed in a glove-box), we did not observe the formation of oxidized flavin by 1H NMR. On the other hand, after adding 50 equivalents (corresponding to the amount of nitrobenzene used in flavin-catalyzed benzyl alcohol oxidations), we observed a fast colour change and green fluorescence typical for the oxidized form of flavins. 1H NMR analysis showed that besides nitrobenzene and a product of its reduction, only oxidized flavin 1 was present in the mixture and not its reduced form (see the ESI†). Inspecting the redox potentials of participating species, nitrobenzene (and its reduction products) can oxidize fully reduced flavin to radical species, but it is not strong enough to oxidize the flavin radical to the fully oxidized form (Scheme 6). This can be explained in terms of the re-oxidation of fully reduced flavin (FlH−) to radical FlH˙, which then disproportionates to the reduced FlH− and oxidized flavin Fl (cf.Scheme 2). Similar disproportionation was proved to occur in flavin-mediated benzyl alcohol photooxidations.20b
 | ||
| Scheme 6 Schematic description of redox behaviour of flavin 1 (Fl) and nitrobenzene. | ||
Conclusions
We have proved that the triphenylphosphine radical cation Ph3P˙+, generated by photoinduced electron transfer to flavin 1, can be utilized for the activation of alcohols or acids to promote ester formation by nucleophilic substitution reaction (both SN or SNAc). It should be noted that photooxidation to phosphine oxides has hitherto been the sole reported photoredox procedure with triphenylphosphine.9 Thus, our finding could provide impetus for the design of other methods utilizing photoactivated phosphine in various organic transformations. It could be expected that not only flavins, but also other dyes, might be suitable for this purpose. In the present case, application of this new approach has led to the development of an effective esterification method based on flavin photocatalyst, triphenylphosphine, and visible light, which has proved applicable to a broad range of substrates, including less reactive aliphatic acids and alcohols. Regarding stereoselectivity, we observed an unusual switch from preferential inversion to retention of configuration in the esterification of chiral alcohols which was caused merely by changing the solvent. Previously, such a change has been mainly associated with a change of alcohol or acid structure.1a,5c,6 It should be noted that also such a preferential inversion of configuration in aza-reagent-free esterification is unique.10 The presented method uses nitrobenzene as a novel sacrificial oxidant to regenerate the flavin catalyst. It could also be helpful in other flavin-based oxidative procedures, especially with substrates sensitive to oxygen, which has hitherto been the sole stoichiometric oxidant used in flavin photocatalysis. This would seem to be of great value, in view of the recent growing interest in flavin photocatalysis.13,21Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the Czech Science Foundation (Grant No. 16-09436S), and Ministry of Education, Youth and Sports of the Czech Republic (Specific university research no 21-SVV/2018). M. S. would like to thank support by the research grant 2017/27/B/ST4/02494 from the National Science Centre, Poland (NCN). Authors thank Pavlína Kyjaková for chiral-GC experiments.Notes and references
- (a) K. C. K. Swamy, N. N. B. Kumar, E. Balaraman and K. V. P. P. Kumar, Chem. Rev., 2009, 109, 2551–2651 CrossRef PubMed; (b) L. F. Pedrosa, Synlett, 2008, 1581–1582 CrossRef; (c) J. An, R. M. Denton, T. H. Lambert and E. D. Nacsa, Org. Biomol. Chem., 2014, 12, 2993–3003 RSC.
- (a) H. A. Van Kalkeren, F. L. Van Delft and F. P. J. T. Rutjes, Pure Appl. Chem., 2013, 85, 817–828 Search PubMed; (b) R. Appel, Angew. Chem., Int. Ed. Engl., 1975, 14, 801–811 CrossRef.
- (a) S. Fletcher, Org. Chem. Front., 2015, 2, 739–752 RSC; (b) A. J. Reynolds and M. Kassiou, Curr. Org. Chem., 2009, 13, 1610–1632 CrossRef; (c) T. Y. S. But and P. H. Toy, Chem. – Asian J., 2007, 2, 1340–1355 CrossRef PubMed; (d) M. Oyo and Y. Masaaki, Bull. Chem. Soc. Jpn., 1967, 40, 2380–2382 CrossRef.
- S. Schenk, J. Weston and E. Anders, J. Am. Chem. Soc., 2005, 127, 12566–12576 CrossRef PubMed.
- (a) G. Wang, J.-R. Ella-Menye, M. St. Martin, H. Yang and K. Williams, Org. Lett., 2008, 10, 4203–4206 CrossRef PubMed; (b) A. B. Hughes and M. M. Sleebs, J. Org. Chem., 2005, 70, 3079–3088 CrossRef PubMed; (c) J. McNulty, A. Capretta, V. Laritchev, J. Dyck and A. J. Robertson, Angew. Chem., Int. Ed., 2003, 42, 4051–4054 CrossRef PubMed; (d) A. B. Smith, I. G. Safonov and R. M. Corbett, J. Am. Chem. Soc., 2002, 124, 11102–11113 CrossRef PubMed; (e) C. Ahn and P. DeShong, J. Org. Chem., 2002, 67, 1754–1759 CrossRef PubMed.
- (a) J. Dyck, S. Zavorine, A. J. Robertson, A. Capretta, V. Larichev, J. Britten and J. McNulty, J. Organomet. Chem., 2005, 690, 2548–2552 CrossRef; (b) J. McNulty, A. Capretta, V. Laritchev, J. Dyck and A. J. Robertson, J. Org. Chem., 2003, 68, 1597–1600 CrossRef PubMed.
- (a) D. Hirose, M. Gazvoda, J. Košmrlj and T. Taniguchi, Chem. Sci., 2016, 7, 5148–5159 RSC; (b) D. Hirose, T. Taniguchi and H. Ishibashi, Angew. Chem., Int. Ed., 2013, 52, 4613–4617 CrossRef PubMed; (c) T. Y. S. But and P. H. Toy, J. Am. Chem. Soc., 2006, 128, 9636–9637 CrossRef PubMed.
- (a) H. Maeda, T. Koide, T. Maki and H. Ohmori, Chem. Pharm. Bull., 1995, 43, 1076–1080 CrossRef; (b) H. Ohmori, H. Maeda, M. Kikuoka, T. Maki and M. Masui, Tetrahedron, 1991, 47, 767–776 CrossRef; (c) H. Ohmori, S. Nakai, M. Sekiguchi and M. Masui, Chem. Pharm. Bull., 1980, 28, 910–915 CrossRef.
- (a) S. Yasui and M. Tsujimoto, J. Phys. Org. Chem., 2013, 26, 1090–1097 CrossRef; (b) O. Kei, N. Takashi and F. Shunichi, Bull. Chem. Soc. Jpn., 2006, 79, 1489–1500 CrossRef; (c) M. Nakamura, M. Miki and T. Majima, J. Chem. Soc., Perkin Trans. 2, 2000, 1447–1452 RSC; (d) S. Yasui, K. Shioji, A. Ohno and M. Yoshihara, J. Org. Chem., 1995, 60, 2099–2105 CrossRef; (e) Y. B. Zhang, C. Ye, S. J. Li, A. S. Ding, G. X. Gu and H. Guo, RSC Adv., 2017, 7, 13240–13243 RSC; (f) S. M. Bonesi, S. Protti and A. Albini, J. Org. Chem., 2016, 81, 11678–11685 CrossRef PubMed; (g) S. Yasui, S. Tojo and T. Majima, Org. Biomol. Chem., 2006, 4, 2969–2973 RSC.
- T. Taniguchi, D. Hirose and H. Ishibashi, ACS Catal., 2011, 1, 1469–1474 CrossRef.
- M. März, J. Chudoba, M. Kohout and R. Cibulka, Org. Biomol. Chem., 2017, 15, 1970–1975 RSC.
- (a) Y. Arakawa, K. Yamanomoto, H. Kita, K. Minagawa, M. Tanaka, N. Haraguchi, S. Itsuno and Y. Imada, Chem. Sci., 2017, 8, 5468–5475 RSC; (b) H. Iida, Y. Imada and S. I. Murahashi, Org. Biomol. Chem., 2015, 13, 7599–7613 RSC; (c) R. Cibulka, Eur. J. Org. Chem., 2015, 915–932 CrossRef; (d) T. Ishikawa, M. Kimura, T. Kumoi and H. Iida, ACS Catal., 2017, 7, 4986–4989 CrossRef.
- (a) G. Tang, Z. Gong, W. Han and X. Sun, Tetrahedron Lett., 2018, 59, 658–662 CrossRef; (b) V. Mojr, G. Pitrová, K. Straková, D. Prukała, S. Brazevic, E. Svobodová, I. Hoskovcová, G. Burdziński, T. Slanina, M. Sikorski and R. Cibulka, ChemCatChem, 2018, 10, 849–858 CrossRef; (c) B. Mühldorf and R. Wolf, ChemCatChem, 2017, 9, 920–923 CrossRef; (d) J. B. Metternich, D. G. Artiukhin, M. C. Holland, M. von Bremen-Kühne, J. Neugebauer and R. Gilmour, J. Org. Chem., 2017, 82, 9955–9977 CrossRef PubMed; (e) R. Martinez-Haya, M. A. Miranda and M. L. Marin, Eur. J. Org. Chem., 2017, 2164–2169 CrossRef; (f) M. Jirásek, K. Straková, T. Neveselý, E. Svobodová, Z. Rottnerová and R. Cibulka, Eur. J. Org. Chem., 2017, 2139–2146 CrossRef; (g) T. Neveselý, E. Svobodová, J. Chudoba, M. Sikorski and R. Cibulka, Adv. Synth. Catal., 2016, 358, 1654–1663 CrossRef; (h) B. Mühldorf and R. Wolf, Angew. Chem., Int. Ed., 2016, 55, 427–430 CrossRef PubMed; (i) J. B. Metternich and R. Gilmour, J. Am. Chem. Soc., 2016, 138, 1040–1045 CrossRef PubMed; (j) T. Morack, J. B. Metternich and R. Gilmour, Org. Lett., 2018, 20, 1316–1319 CrossRef PubMed; (k) M. Insińska-Rak, E. Sikorska, J. L. Bourdelande, I. V. Khmelinskii, W. Prukała, K. Dobek, J. Karolczak, I. F. Machado, L. F. V. Ferreira, E. Dulewicz, A. Komasa, D. R. Worrall, M. Kubicki and M. Sikorski, J. Photochem. Photobiol., A, 2007, 186, 14–23 CrossRef.
- A. Ehrenberg, F. Müller and P. Hemmerich, Eur. J. Biochem., 1967, 2, 286–293 CrossRef PubMed.
- L. Horner, F. Röttger and H. Fuchs, Chem. Ber., 1963, 96, 3141–3147 CrossRef.
- H. Youqin and L. Jean, Electroanalysis, 2016, 28, 2716–2727 CrossRef.
- F. Márquez, R. Roque-Malherbe, J. Ducongé and W. del Valle, Surf. Interface Anal., 2004, 36, 1060–1063 CrossRef.
- (a) V. S. Khursan, V. A. Shamukaev, E. M. Chainikova, S. L. Khursan and R. L. Safiullin, Russ. Chem. Bull., 2013, 62, 2477–2486 CrossRef; (b) J. I. G. Cadogan, Q. Rev., Chem. Soc., 1968, 22, 222–251 RSC.
- Flavin-based nitro group reductions are known from biological systems, see: K. Durchschein, M. Hall and K. Faber, Green Chem., 2013, 15, 1764–1772 RSC.
- (a) B. Mühldorf and R. Wolf, Chem. Commun., 2015, 51, 8425–8428 RSC; (b) C. Feldmeier, H. Bartling, K. Magerl and R. M. Gschwind, Angew. Chem., Int. Ed., 2015, 54, 1347–1351 CrossRef PubMed; (c) U. Megerle, M. Wenninger, R.-J. Kutta, R. Lechner, B. König, B. Dick and E. Riedle, Phys. Chem. Chem. Phys., 2011, 13, 8869–8880 RSC; (d) S. Fukuzumi, K. Yasui, T. Suenobu, K. Ohkubo, M. Fujitsuka and O. Ito, J. Phys. Chem. A, 2001, 105, 10501–10510 CrossRef.
- I. K. Sideri, E. Voutyritsa and C. G. Kokotos, Org. Biomol. Chem., 2018, 16, 4596–4614 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental, NMR and HPLC data, electrochemical and spectral characteristics of flavin 1, additional laser-flash photolysis and kinetic experiments. See DOI: 10.1039/c8ob01822g |
| ‡ It should be noted that Mitsunobu reactions catalytic in the dialkyl azadicarboxylate represents an alternative way how to reduce its amount.7 |
| § Molecular sieves were needed to decompose hydrogen peroxide (a possible substrate for Mitsunobu reaction) formed during aerial flavin re-oxidation and, consequently, to remove water generated by H2O2 decomposition. |
| This journal is © The Royal Society of Chemistry 2018 |