
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and cycloaddition reactions of strained alkynes derived from 2,2′-dihydroxy-1,1′-biaryls†
Anish Mistrya,
Richard C. Knighton a,
Sam Forshawa,
Zakaria Dualeha,
Jeremy S. Parkerb and
Martin Wills*a
a,
Sam Forshawa,
Zakaria Dualeha,
Jeremy S. Parkerb and
Martin Wills*a
aDepartment of Chemistry, The University of Warwick, Coventry, CV4 7AL, UK. E-mail: m.wills@warwick.ac.uk
bEarly Chemical Development, Pharmaceutical Sciences, IMED Biotech Unit, AstraZeneca, Macclesfield, SK10 2NA, UK
First published on 12th November 2018
Abstract
A series of strained alkynes, based on the 2,2′-dihydroxy-1,1′-biaryl structure, were prepared in a short sequence from readily-available starting materials. These compounds can be readily converted into further derivatives including examples containing fluorescent groups with potential for use as labelling reagents. The alkynes are able to react in cycloadditions with a range of azides without the requirement for a copper catalyst, in clean reactions with no observable side reactions.
Introduction
The use of highly reactive strained alkynes, typically within eight-membered rings,1 in cycloaddition reactions with azides is now a well-established reaction with numerous applications in materials chemistry and in bioconjugation applications.2 Such reagents are ideal for these applications because the cycloaddition reactions take place spontaneously and without the need for a catalyst to be added – in contrast to the reactions of unstrained terminal alkynes with azides in which case a copper-based catalyst is generally required.3Widely adopted cyclooctyne reagents such as 1–3 and their derivatives (Fig. 1),4–6 are highly reactive, and can be used at the low concentrations which are often required in bioconjugation applications, particularly for in vivo reactions.7 In applications where the concentration of reagents is more typical of synthetic reactions e.g. 0.01–0.5 M, and on larger scales, less reactive larger-ring molecules, which can be prepared through a short synthetic sequence, have also proven to be synthetically valuable reagents.8 Earlier and less reactive cyclooctynes remain synthetically important, for example (2-cyclooctyn-1-yloxy)acetic acid (a derivative of ‘OCT’) was the subject of a successful multigram scale up optimisation study reported in 2018.5d Some highly strained derivatives are also prone to addition of thiols.5e
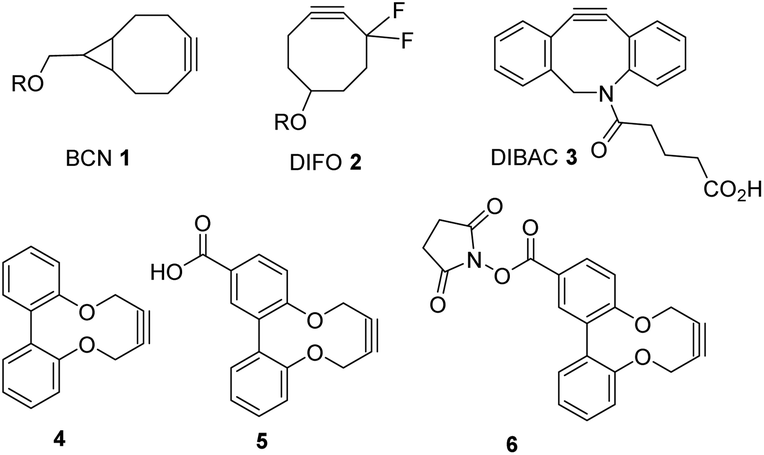 | ||
| Fig. 1 Strained alkynes 1–3, dioxabiaryldecyne 4 and its derivatives 5 and 6. | ||
In a recent paper, we reported the synthesis and applications of a class of strained alkyne based on the 10-membered structure 4, derived from 2,2′-dihydroxy-1,1′-biaryl compounds.9 The unfunctionalised compound, 8,13-dioxatricyclo[12.4.0.02,7]octadeca-1(14),2,4,6,15,17-hexaen-10-yne (dioxabiaryldecyne) 4 and its close derivatives are readily prepared in one step through the reaction of 2,2′-biphenol with but-2-yne-1,4-diyl bis(4-methylbenzene)sulfonate in the presence of potassium carbonate.9 Before our studies, the reactions of alkynes such as 4 with azides had not been reported, and just three papers could be identified which reported the synthesis of the same heterocyclic structure.10 In addition, we demonstrated that reagents such as 4 and its derivatives react with azides, without the need for a Cu catalyst, at rates similar to unfunctionalised cyclooctyne, although lower than the most reactive and recently reported strained alkynes. Significantly, although longer reaction times are required than would be the case for reagents such as 1–3, our alkynes reacted with azides in clean reactions with no visible decomposition when followed by 1H-NMR. We also reported the synthesis of acid 5 and the activated ester 6 derivatives and demonstrated applicability to bioconjugation through its attachment to a number of peptides and one protein in in vitro studies.9 Following our report, another group reported the preparation of some of the same derivatives, as well as N-containing heterocyclic variants, together with a comprehensive molecular modelling study to explain the enhanced reactivity of the reagents.11 This group also demonstrated that the dioxabiaryldecynes do not rapidly undergo reactions with thiols.11
In this paper we report the synthesis of a series of functionalised analogues of the strained alkyne structure 4, in as little as two steps, from readily available and inexpensive starting materials, their subsequent functionalisation and representative applications to a number of cycloaddition reactions with several azides.
Results and discussion
In order to develop an extended synthesis of dioxabiaryldecyne reagents, we employed the reported coupling reactions of iodobenzaldehyde reagents 7 and 8 (iodovanillin) and 4-hydroxy-3′-iodoacetophenone 9 with 2-(hydroxyphenyl)boronic acid 10,12 to give diols 11–13 respectively. This was followed by the cyclisation reactions with ditosylate 14 using our previously-reported procedure (Scheme 1).9 Strained alkynes 15, 16 and 17 were isolated respectively (Scheme 1). 3-Iodo-4-hydroxybenzaldehyde was prepared from 4-hydroxybenzaldehyde through careful iodination using ICl/acetic acid.13 Iodovanillin can be prepared by the same method but is readily commercially available.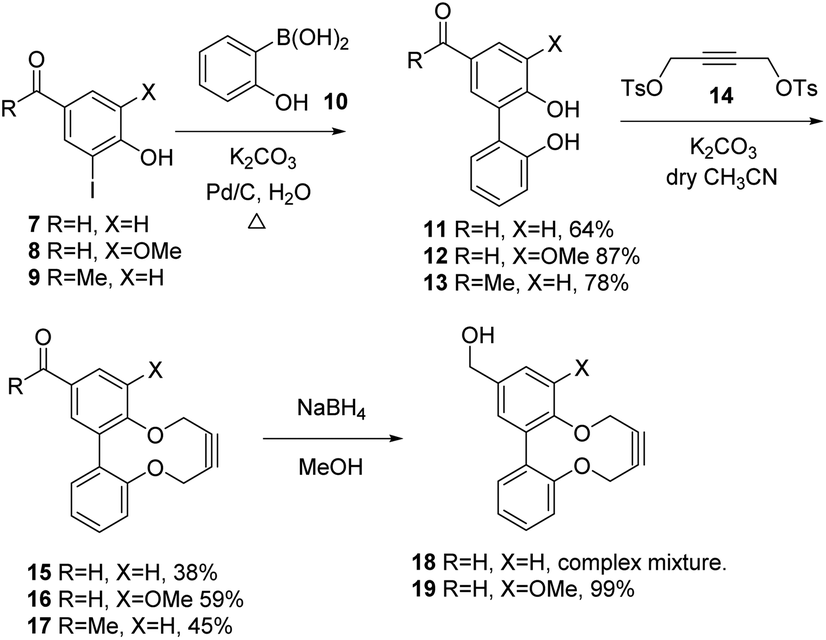 | ||
| Scheme 1 Synthesis of aldehyde-functionalised CBD strained alkynes 15–17 and alcohol 19. | ||
Both the Pd-catalysed coupling and the cyclisation to form aldehydes 15 and 16 worked more efficiently for the product containing a methoxy group adjacent to the strained alkyne, giving a product in unoptimised but acceptable yield in each case. In the case of the transformation of aldehyde 12 to 16, we followed the reaction over time using chiral HPLC, which resolved the two non-interconverting enantiomers of product and allowed the conversion to be monitored over time (see ESI† for HPLC details and graph of conversion over time). The X-ray crystallographic structures of aldehyde 16 (Fig. 2) and ketone 17 (Fig. 3) revealed the strained nature of the alkyne within the constrained ring.
 | ||
| Fig. 2 Single crystal X-ray crystallographic structure of 16 (two views; ellipsoids are plotted at the 50% probability level). The bond angles at the sp atoms are 165.2° and 165.3° and the biphenyl torsion angle is 72.2°. | ||
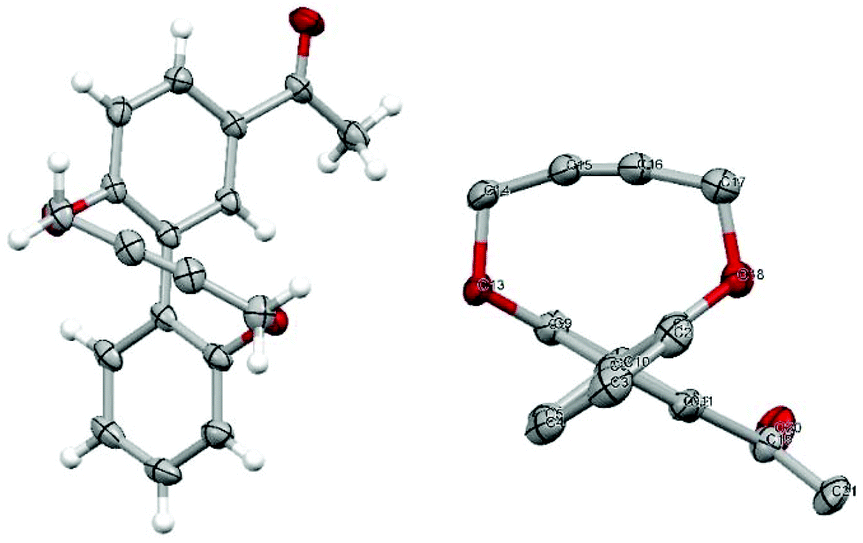 | ||
| Fig. 3 Single crystal X-ray crystallographic structure of 17 (two views: ellipsoids are plotted at the 50% probability level). The bond angles at the sp atoms are 165.1° and 168.6° and the biphenyl torsion angle is 66.4°. | ||
Alkyne 16 could also be reduced to the alcohol 19 using sodium borohydride, which gave a clean product, however attempts to reduce substrate 15, lacking the methoxy group, to 18 gave a complex mixture of products, for reasons that are not clear.14
The strained alkynes prepared in this project are stable solids at rt which can be stored for months without significant decomposition. However a thermal gravimetric analysis (TGA) was carried out in order to examine their stability at higher temperatures. Aldehyde 16 exhibited a drop of ca. 10% mass around 180 °C which may be associated with the loss of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O from the aldehyde, followed by a gradual mass loss of just over 20% as the temperature was raised to 600 °C. The TGA analysis of the previously reported methyl ester of acid 5 was stable to ca. 300 °C then gradually lost ca. 40% of its mass as the temperature was increased to 600 °C (see ESI†).
O from the aldehyde, followed by a gradual mass loss of just over 20% as the temperature was raised to 600 °C. The TGA analysis of the previously reported methyl ester of acid 5 was stable to ca. 300 °C then gradually lost ca. 40% of its mass as the temperature was increased to 600 °C (see ESI†).
Given the improved synthesis of the methoxy-substituted aldehyde 16 over 15, we focussed our studies on the former reagent. Its reaction with a range of functionalised azides was studied (Scheme 2) and in each case the reactions were followed over time, using 1H NMR to monitor the cyclisations of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of reagents in solution; the spectra are in the ESI.† The reaction of 16 with benzylazide 20 was also carried out in MeCN, the product 21 being isolated in 84% yield. In all cases, the cycloadditions proceeded smoothly, with no obvious accompanying decomposition of reagents. Conversions (by NMR) and yields (isolated products) are given in Scheme 2. In all cases the products were formed as inseparable regioisomeric mixtures in ca. 1:1–3:2 ratios. Benzylazide gave a clean product 21 of addition, in analogy with previous reactions.9,11 An azide attached to a red dye, disperse red, 2215a gave a red product 23 from the cycloaddition, which was carried out at 0.128 M, 9 days at rt (95% conversion, 76% isolated yield). An azide containing a PEG-2000 chain, 24, also added cleanly to the strained alkyne 16, and the product in this case (25) was characterised by GPC as well as by NMR, revealing the expected increase to the molar mass of the polymeric product (ESI†). This was gratifying as the reagent concentration (0.025 M) in this example was lower than for other cycloadditions. The cycloaddition of coumarin azide 26 gave a highly fluorescent product 27 (see ESI†) as has been reported previously for this class of reagent.15b,c For improved solubility, deuterated acetonitrile was used as the solvent, and the reaction at 0.11 M proceeded to ca. 80% conversion to 27. Although long reaction times are required relative to the more reactive strained alkynes such as 1–3, the benefits of the catalyst-free conditions and clean cycloadditions make these reagents potentially valuable for the preparation of materials for biological applications.
:1 mixture of reagents in solution; the spectra are in the ESI.† The reaction of 16 with benzylazide 20 was also carried out in MeCN, the product 21 being isolated in 84% yield. In all cases, the cycloadditions proceeded smoothly, with no obvious accompanying decomposition of reagents. Conversions (by NMR) and yields (isolated products) are given in Scheme 2. In all cases the products were formed as inseparable regioisomeric mixtures in ca. 1:1–3:2 ratios. Benzylazide gave a clean product 21 of addition, in analogy with previous reactions.9,11 An azide attached to a red dye, disperse red, 2215a gave a red product 23 from the cycloaddition, which was carried out at 0.128 M, 9 days at rt (95% conversion, 76% isolated yield). An azide containing a PEG-2000 chain, 24, also added cleanly to the strained alkyne 16, and the product in this case (25) was characterised by GPC as well as by NMR, revealing the expected increase to the molar mass of the polymeric product (ESI†). This was gratifying as the reagent concentration (0.025 M) in this example was lower than for other cycloadditions. The cycloaddition of coumarin azide 26 gave a highly fluorescent product 27 (see ESI†) as has been reported previously for this class of reagent.15b,c For improved solubility, deuterated acetonitrile was used as the solvent, and the reaction at 0.11 M proceeded to ca. 80% conversion to 27. Although long reaction times are required relative to the more reactive strained alkynes such as 1–3, the benefits of the catalyst-free conditions and clean cycloadditions make these reagents potentially valuable for the preparation of materials for biological applications.
 | ||
| Scheme 2 Use of aldehyde alkyne 16 in addition reactions with a range of azides. | ||
The addition of benzyl azide to alcohol 19 to give adduct 28 as a 3:2 regioisomeric mixture of products (Fig. 4) proceeded at a similar rate (0.17 M, 5 d at rt, 97% yield, ESI†) indicating that the functional group has minimal influence on the rate of the cycloaddition, probably because of the separation from the alkyne.
 | ||
| Fig. 4 Derivatives of 15–17. | ||
The aldehyde group on 15 and 16 permits their functionalisation with other reagents. The reaction of 15 with benzylhydroxylamine in MeOH overnight at 45 °C gave oxime ether 29 in 66% isolated yield. The formation of oxime ethers represents a valuable method for functionalisation due to their high stability and ease of preparation.16 Also, notably, reductive amination with benzylamine led to the synthesis of amine-containing derivatives 30 and 31. The reaction of methoxy-substituted 30 with benzylazide was found to proceed at a similar rate to aldehyde-containing reagents 16 (ESI†). It was gratifying that these functionalisations could be completed without damaging the strained alkyne group.
The treatment of amine-functionalised polystyrene beads with 16 and sodium cyanoborohydride was followed by reaction of the functionalised beads 32 with disperse red azide 22. After washing, the strong red colour of the dye remained on the beads 33 (Scheme 3). As a control reaction, stirring the solution of red dye-azide 22 with unfunctionalised beads gave only lightly coloured beads after washing, indicating that the cycloaddition had taken place on the dioxabiaryldecyne reagent on the beads (ESI†).
 | ||
| Scheme 3 Functionalisation of beads with disperse red dye. | ||
Other reagents were prepared through reactions of the aldehyde, notably fluorescent groups. The reductive amination of 16 with the amine-functionalised dansyl reagent 34 resulted in formation of 35 (Scheme 4A), which showed strong fluorescent behaviour upon irradiation. A number of BoDIPY derivatives 36–38 were also prepared through the direct reaction of pyrroles with the aldehyde and BF3 in good yield (Scheme 4B).17 Again, the ability to functionalise aldehyde 16 with a variety of reagents, without damaging the strained alkyne, is noteworthy.
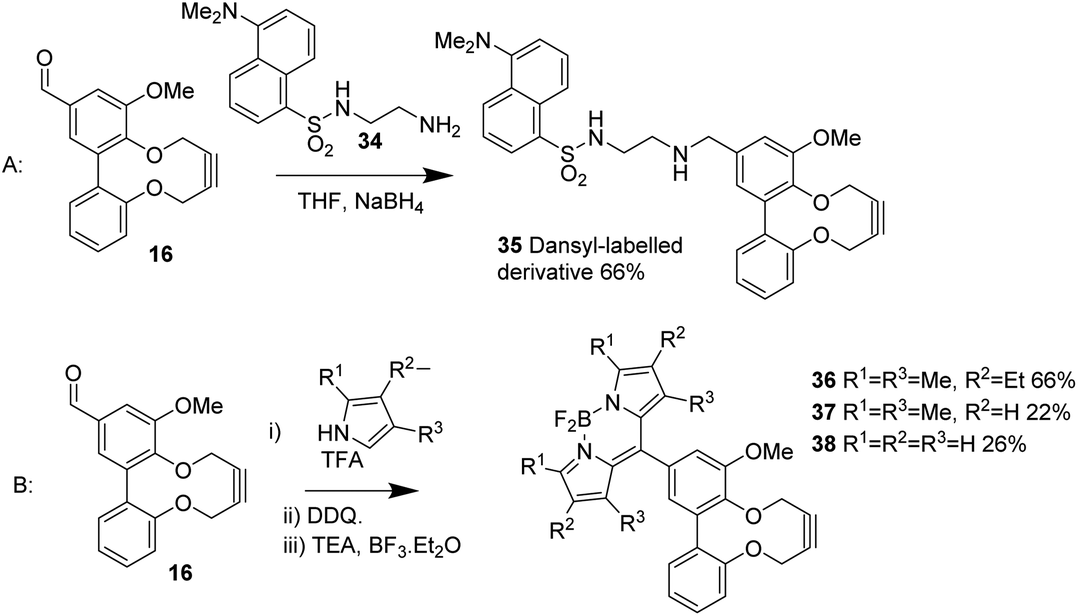 | ||
| Scheme 4 Synthesis of fluorescent derivatives of 16. | ||
The X-ray crystallographic structure of 38 (Fig. 5) revealed the strained nature of the alkyne but also that the BoDIPY component was orientated almost perpendicular to the connected arene ring, presumable with restricted rotation about the connecting C–C bond. This accounts for the observed differences in chemical shifts of the groups attached to the heterocyclic rings of the BoDIPY unit in each of 36–38, which will be in sharply different diastereotopic environments.
 | ||
| Fig. 5 X-ray crystallographic structure of 38 (ellipsoids are plotted at the 50% probability level). The bond angles at the sp atoms are 165.7° and 168.1° and the biphenyl torsion angle is 68.6°. | ||
The fluorescence spectra for compounds 35–38 are given in the ESI.† However the strong and contrasting fluorescence behaviour of the BoDIPY dyes 36–38 is sharply illustrated by their response to UV irradiation. Compound 36 and 37 both show strong fluorescence upon irradiation whereas 38 gives a weaker response (ESI†).
The addition of benzylazide to BoDIPY derivative 36 was tested and worked efficiently to give two regiosiomers 39 and 40 in a 1:1 ratio (Fig. 6). In this case, we were able to separate the isomers by flash chromatography and independently characterise them. We have not unambiguously established which regiosiomer is which, of the two possibilities, however on the basis of the positions of the methylene groups in the 13C-NMR spectra compared to previous examples, we have tentatively assigned them as shown in Fig. 6 (see ESI†).
 | ||
| Fig. 6 Separated cycloaddition products 39 and 40. | ||
Further derivatives were also prepare from the corresponding alcohol 19 using a variety of coupling methods (Fig. 7). These included a biotin-containing reagent 41 which was formed through formation of an ester bond to biotin in one step.
 | ||
| Fig. 7 Functionalised derivatives of alcohol 19 and acid 5 strained alkynes which were prepared in this project. | ||
It was also possible to attach a group through a carbamate i.e.42, using N,N′-disuccinimidyl carbonate (DSC) as a coupling agent to attach alcohol 19 to form the dansyl amine derivative 34.7d,18 Finally, from the alcohol, the direct reaction with an isocyanate could also be employed to create a derivative with a carbamate linkage i.e.43. Formation of derivatives from the acid 5 was also investigated; the in situ formation of isocyanate from acid 5 using diphenylphosphoryl azide and trapping with MeOH gave carbamate derivative 44 through a method that could be used for future functionalisation. Acid 5 was also linked using EDC·HCl to create the disperse-red functionalised 45. These results (Fig. 7) illustrate the range of methods which can be employed to functionalise the strained alkynes.
In conclusion, we have prepared a selection of derivatives, including fluorescently-labelled variants, of a new class of strained alkyne, which benefit from ease of synthesis from readily available and inexpensive starting materials through a short sequence of reactions. We have demonstrated that this class of alkyne undergoes uncatalysed cycloaddition reactions with azides with minimal decomposition or side product formation. Studies of the applications of these reagents are ongoing and further results will be published in due course.
Experimental section
General experimental details, synthesis of intermediates 7,1311,1212,1213,193420 and compounds 29–31 and 41–45 are in the ESI.†Alkyne 15
A series of strained alkynes, based on the 2,2′-dihydroxy-1,1′-biaryl structure, were prepared in a short sequence from readily-available starting materials. This compound is novel.

2′,6-Dihydroxybiphenyl-3-carbaldehyde 11 (3.20 g, 14.9 mmol), potassium carbonate (10.22 g, 73.95 mmol) and but-2-yne-1,2-diyl bis(4-methylbenzenesulfonate) 14 (5.31 g, 13.5 mmol) were added to a clean dry schlenk. The schlenk was then put under nitrogen and purged, thereafter dry acetonitrile (747 mL) was added to the mixture and the reaction left to stir at rt for 10 days. The organics were removed under vacuum, water (500 mL) and DCM (500 mL) were added and the product extracted with DCM (3 × 300 mL). The organic extracts were washed with brine and dried over Na2SO4, filtered and concentrated under vacuum. The crude product was purified by column chromatography (8:2 hexane:ethyl acetate) to afford the product 15 as a white solid (1.50 g, 5.68 mmol, 38%). Mp 143–145 °C; (found (ESI-Q-TOF) [M + Na]+ 287.0675. C17H12O3Na requires 287.0679); νmax 2910, 2863, 1686, 1568, 1495, 1473, 1415, 1345, 1305, 1288, 1188 cm−1; δH (500 MHz, CDCl3) 9.98 (1H, s, CHO), 7.94 (1H, dd, J 8.4, 2.9, ArH), 7.75 (1H, d, J 2.0, ArH), 7.44–7.40 (1H, m, ArH), 7.32 (1H, d, J 8.3, ArH), 7.22–7.19 (3H, m, ArH), 4.63–4.61 (1H, m, CH2), 4.54–4.50 (m, 1H, CH2), 4.41–4.32 (m, 2H, CH2); δC (125 MHz, CDCl3) 191.2, 159.8, 154.4, 136.8, 134.9, 134.6, 132.6, 131.7, 129.7, 129.6, 124.3, 123.6, 122.6, 87.3, 86.0, 63.8, 63.5; m/z (ESI) 287.1 ([M + Na]+).
Alkyne 16
This compound is novel.

2′,6-Dihydroxy-5-methoxybiphenyl-3-carbaldehyde 12 (1.70 g, 6.96 mmol), potassium carbonate (4.76 g, 34.4 mmol) and but-2-yne-1,2-diyl bis(4-methylbenzenesulfonate) 14 (2.47 g, 6.27 mmol) were added to a schlenk. The schlenk was then put under nitrogen and purged, thereafter dry acetonitrile (346 mL) was added to the mixture and the reaction was left to stir at rt for 10–14 days. The organics were removed under vacuum, water (400 mL) and DCM (400 mL) were added and the product extracted with DCM (3 × 200 mL). The organics were collected and washed with brine and dried over Na2SO4, filtered and concentrated under vacuum. The crude product was purified by column chromatography (8:2 hexane:ethyl acetate) to afford 16 as a white solid (1.2 g, 4.08 mmol, 59%). Mp 171–173 °C; (found (ESI-Q-TOF) [M + Na]+ 317.0787. C18H14O4Na requires 317.0784); νmax 2951, 2923, 2852, 1687, 1601, 1580, 1491, 1459, 1426, 1384, 1334, 1290, 1240, 1180, 1163, 1127 cm−1; δH (500 MHz, CDCl3) 9.92 (1H, s, CHO), 7.49 (1H, s, ArH), 7.44–7.40 (1H, m, ArH), 7.33 (1H, s, ArH), 7.22–7.19 (3H, m, ArH), 4.69–4.66 (1H, m, CH2), 4.56–4.60 (2H, m, CH2), 4.37–4.34 (1H, m, CH2) 3.98 (3H, s, OCH3); δC (125 MHz, CDCl3) 191.5, 154.4, 154.2, 148.1, 137.6, 134.7, 132.7, 131.7, 129.7, 129.0, 124.4, 122.6, 108.6, 86.9, 86.7, 63.6, 60.5, 55.9; m/z (ESI) 317.1 ([M + Na]+). This reaction was also monitored by HPLC over 14 days, using 15:85 IPA:Hexane, 1 ml min−1, IB column. Full details are in the ESI.† The X-ray crystallographic structure of this compound was obtained and is described in the ESI.†
Alkyne alcohol 19
This compound is novel.
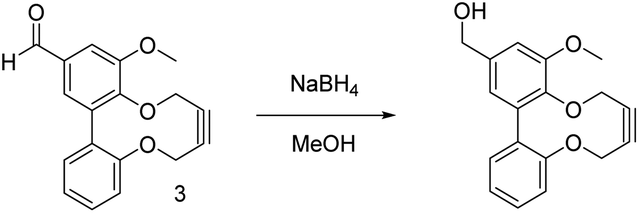
NaBH4 (15 mg, 0.41 mmol, 1.0 eq.) was added carefully at 0 °C to a stirring solution of 16 (0.12 g, 0.41 mmol, 1.0 eq.) in methanol (10 mL) under a nitrogen atmosphere and the reaction was left for 1 hour to react at rt. The methanol was removed under vacuum and the residue was redissolved in ethyl acetate (15 mL). The organic extracts were washed with sat. NH4Cl (15 mL) and then brine (15 mL). The organics collected, dried over Na2SO4, filtered and concentrated under vacuum to afford a white solid. This was purified by column chromatography (1:1 hexane:ethyl acetate) to afford the product 19 as a white solid (0.12 g, 0.40 mmol, 99%). Mp 165–168 °C; (found (ESI-Q-TOF) [M + Na]+ 319.0940. C18H16O4Na requires 319.0941); νmax 3522, 2927, 2866, 2835, 1587, 1494, 1446, 1421, 1338, 1264, 1246, 1192, 1165, 1133, 1122 cm−1; δH (400 MHz, CDCl3) 7.39–7.34 (1H, m, ArH), 7.19–7.17 (2H, m, ArH), 7.14 (1H, d, J 7.9, ArH), 7.01 (1H, d, J 1.7, ArH), 6.74 (1H, d, J 1.8, ArH), 4.65 (2H, s, CH2OH), 4.63–4.57 (1H, m, CH2), 4.53–4.47 (1H, m, CH2), 4.41–4.36 (1H, m, CH2), 4.34–4.28 (1H, m, CH2), 3.91 (3H, s, OCH3); δC (100 MHz, CDCl3) 154.2, 153.3, 141.7, 137.0, 136.8, 135.8, 131.9, 129.1, 124.2, 122.4, 122.2, 109.9, 87.5, 85.9, 65.0, 63.5, 60.2, 55.7; m/z (ESI) 319.1 ([M + Na]+).
Ketoalkyne 17
This compound is novel.
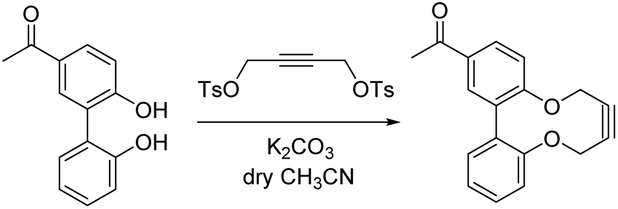
In a round bottom flask under nitrogen atmosphere 13 (550 mg, 2.41 mmol, 1.2 equiv.) and but-2-yne-1,4-diyl-bis(4-methylbenzenesulfonate) (792 mg, 2.01 mmol) were dissolved in anhydrous acetonitrile (111 mL). K2CO3 (1.39 g, 10.0 mmol) was added and the mixture was stirred at RT for 14 days. The volatiles were removed in vacuum and H2O (50 mL) was added. The product was extracted with DCM (3 × 50 mL). The combined organic layers were washed with brine (30 mL), dried over MgSO4, filtered and solvent removed via rotary evaporation. The product was purified by column chromatography on silica (hexane/EtOAc = 4:1) to give 17 (252 mg, 0.906 mmol, 45%) as a crystalline white solid. Crystals suitable for X-ray spectroscopy were grown by vapour diffusion of hexane into a CHCl3 solution of the compound. Mp 137–139 °C. (Found (ESI-Q-TOF) [M + Na]+ 301.0834. C18H14O3Na requires 301.0835); νmax 2158, 1679, 1499, 1357, 1307, 1251, 1188, 1106, 963 cm−1; δH (500 MHz, CDCl3); 8.00 (1H, d, J 8.4, ArH), 7.82 (1H s, ArH), 7.42–7.39 (1H, m, ArH), 7.25–7.18 (4H, m, ArH), 4.60–4.50 (2H, m, CH2), 4.39–4.32 (2H, m, CH2), 2.57 (3H, s, CH3); δC (125 MHz, CDCl3) 197.3 (CO), 158.7 (C), 154.2 (C), 136.1 (C), 135.0 (C), 133.5 (C), 132.9 (CH), 131.8 (CH), 129.1 (CH), 129.0 (CH), 124.8 (CH), 123.2 (CH), 123.0 (CH), 87.5 (C), 86.5 (C), 64.0 (CH2). 63.7 (CH2), 26.8 (CH3); m/z (ESI) 278.08 ([M]+), 301.08 ([M + Na]+).
Cycloadduct 21
This compound is novel.

Alkyne 16 (30 mg, 0.102 mmol) and benzyl azide 20 (13.8 mg, 13 μL, 0.102 mmol) were stirred in MeCN (0.6 mL) for 6 days at rt (ca. 0.17 M), monitoring each day by TLC. At the end of this time the solvent was removed under vacuum and the product purified by flash chromatography on silica gel (hexane:EtOAc, 7:3) to yield the product 21 as a white solid (40 mg, 0.94 mmol, 84%). TLC (hexane:EtOAc, 7:3), silica, Rf 0.15; M.p. 181–184 °C; (found (ESI+): [M + Na]+ 450.1426. C25H21N3NaO4 requires 450.1424); νmax 1684, 1575, 1493, 1155, 722 cm−1; δH (500 MHz, CHCl3, two regioisomers 3:2); 9.94 (0.6H, s, CHO, major regiosomer). 9.91 (0.4H, s, CHO, minor regiosomer), 7.52 (1H, s, ArH), 7.47–7.43 (2H, m, ArH), 7.35–7.30 (3H, m, ArH), 7.20–7.05 (2.6H, m, ArH, 3 × major and 2 × minor regiosiomer), 7.01 (0.6H, t, J 7.0, ArH, major regiosomer), 6.96 (0.4H, t, J 7.5, ArH, minor regiosomer), 6.90 (0.4H, t, J 7.5, ArH, minor regiosomer), 6.09 (0.4H, d, J 8.0, CH, minor regiosomer), 5.87 (0.4H, d, J 11.0, CH, minor regiosomer), 5.80–5.72 (1.2H, m, CH, 2 × major regiosomer), 5.57 (0.6H, d, J 13.0, CH, minor regiosomer), 5.52–5.45 (1.4H, m, CH, 1 × major and 2 × minor regioisomer), 5.20 (0.6H, d, J 15.0, CH, major regiosomer), 5.04 (0.4H, d, J 14.0, CH, minor regiosomer), 4.96 (0.4H, d, J 11.0, CH, minor regiosomer), 4.83 (0.6H, d, J 13.0, CH, major regiosomer), 4.02 (3H, s, OCH3), 3.95 (3H, s, OCH3); δC (125 MHz, CDCl3) 191.0 (CH), 191.0 (CH), 153.8 (C), 153.6 (C), 152.7 (C), 152.6 (C), 151.7 (C), 145.0 (C), 144.2 (C), 134.9 (C), 134.1 (C), 133.6 (C), 133.1 (C), 133.0 (C), 132.6 (C), 132.4 (C), 131.4 (CH), 130.7 (CH), 129.5, (CH), 129.3 (CH), 129.0 (CH), 128.5 (C), 128.0 (C), 127.7 (CH), 127.1 (CH), 126.7 (CH), 122.3 (CH), 121.9 (CH), 113.2 (CH), 111.2 (CH), 110.6 (CH), 109.9 (CH), 67.5 (CH2), 62.6 (CH2), 61.0 (CH2), 57.7 (CH2), 56.2 (CH3), 56.1 (CH3), 53.0 (CH2), 52.0 (CH2). m/z (ES-API+) 450.0 ([M + Na]+). The reaction was also followed over time by 1H NMR and full details are in the ESI.† Alkyne 16 (15 mg, 51.0 μmol) and benzyl azide 20 (6.4 mg, 51.0 μmol) were added together in deuterated chloroform (0.4 mL) (0.128 M in both reagents) and the reaction was followed at rt by 1H NMR.
Disperse red cycloadduct 23
This compound is novel.

Aldehyde 16 (15 mg, 51.0 μmol) and azide 22 (19 mg, 51.0 μmol) were combined in deuterated chloroform (0.4 mL) and the reaction (ca. 0.128 M in both reagents) was left at rt. The progression of the reaction was monitored daily. The progression of the reaction was monitored daily by 1H NMR (ESI†). Upon completion, the reaction was worked up and the product purified by column chromatography (DCM→85:15 DCM:EtOAc) to give 23 as a red solid (26 mg, 0.039 mmol, 76%). TLC DCM, silica, Rf 0.05; M.p. 155–158 °C; (found (ESI+): [M + Na]+ 690.1843. C34H30ClN7NaO6 requires 690.1838); νmax 1688, 1597, 1513, 1333, 1121, 745 cm−1; δH (300 MHz, CHCl3) (two regioisomers 1:1); 9.95 (0.5H, s, CHO). 9.90 (0.5H, s, CHO), 8.50 (1H, s, ArH), 8.20–8.15 (1H, m, ArH), 8.05–8.00 (1H, m, ArH), 7.95–7.90 (1H, m, ArH), 7.80–7.75 (1H, m, ArH), 7.45 (1H, d, J 13.0, ArH), 7.30–7.24 (3H, m, ArH), 7.15–7.05 (1H, m, ArH), 6.90–6.85 (1H, m, ArH), 6.80 (1H, d, J 12.0, ArH), 6.75 (1H, d, J 12.0 ArH), 4.85 (1H, d, J 14.0, CH), 5.50–4.50 (5H, m, OCH and NCH), 4.10–3.80 (5H, m, NCH + OMe), 3.35–3.00 (1H, m, NCH), 2.60–2.40 (1H, m, NCH), 1.15 (3H, t, J 6.5, CH3), 0.88 (3H, t, J 6.5, CH3); δC (125 MHz, CDCl3) 190.9, 190.8, 154.1, 153.8, 153.0, 152.8, 152.6, 152.5, 151.7, 151.3, 150.7, 150.5, 149.5, 147.3, 144.8, 144.7, 144.5, 134.3, 133.5, 133.0, 132.8, 132.8, 132.3, 131.5, 130.8, 129.1, 129.1, 129.0, 128.0, 127.0, 126.6, 126.0, 124.7, 122.6, 118.1, 116.0, 118.1, 113.1, 112.1, 111.4, 110.7, 109.9, 62.1, 60.8, 60.1, 58.6, 56.4, 50.4, 50.3, 46.0, 45.4, 11.8; m/z (ES-API+) 690.2 ([M + Na]+).
PEG-2000 azide cycloadduct 23
This compound is novel.

PEG2000-azide 24 (25 mg, 12.5 μmol) and alkyne 16 (3.0 mg, 12.5 μmol) were added together in deuterated chloroform (0.5 mL) and the reaction left at r.t. (ca. 0.025 M in both reagents). The progression of the reaction was monitored daily. The final product was also analysed by GPC, ca. 14 days for 100% conversion. The stacked spectra and the graph of conversion/time, as well as GPC data, are in the ESI.† Characteristic peaks of product were observed as follows: δH (300 MHz, CDCl3) (ca. 1:1 ratio) 9.82 + 9.80 (1H, s × 2, CHO), 7.80–6.80 (6H, m, ArHs), 5.80–4.50 (6H, m, 2 × OCH2, NCH2 of addition product), 3.98 (3H, s, OCH3), 3.90 (3H, s, PEG OCH3), 3.60–3.30 (ca. 190H, m, PEG OCH2 groups). Due to its heterogeneous nature, only the 1H NMR and GPC data was recorded for this complex, and the product was not purified further.
Fluorescent coumarin dye cycloadduct 27
This compound is novel.

Compound 16 (13 mg, 44.2 μmol) and coumarin azide 26 (9.0 mg, 44.2 μmol) were added together in deuterated acetonitrile (0.4 mL) and the reaction left at r.t. (ca. 0.11 M in both reagents). The progression of the reaction was monitored daily. Stacked NMR spectra and the conversion/time graph are in the ESI.† At the end of this time (80% conversion after 12 d) the solvent was removed and the product 27 was purified by column chromatography using a gradient of EtOAc in hexane (14 mg, 28 μmol, 64%,). TLC hexane:EtOAc 3:7, silica, Rf 0.60; M.p. 222–228 °C; (found (ESI+): [M + Na]+ 520.1119. C27H19N3NaO5 requires 520.1115); νmax 1725, 1688, 1605, 1574, 1223, 1133, 966, 684 cm−1; δH (500 MHz, CHCl3) (two regioisomers ca. 3:2); 9.89 (1H, s, CHO), 8.30–8.25 (0.4H, brs, NH or OH; by HSQC, minor regioisomer), 8.12 (0.4H, s, CH, minor regioisomer), 8.10–8.05 (0.6H, brs, NH or OH; by HSQC, major regioisomer), 7.95 (0.6H, s, CH, major regioisomer), 7.50–7.45 (1H, m, ArH), 7.42 (0.6H, s, ArH, major regioisomer), 7.35–7.30 (1H, m, ArH), 7.25–7.15 (2H, m, ArH), 6.95–6.85 (2H, m, ArH), 6.82–6.80 (1.8H, m, ArH, 1 × major, 3 × minor regioisomer), 6.65 (0.6H, d, J 10.0, ArH, major regioisomer), 5.88 (0.4H, d, J 13.0, OCH, minor regioisomer), 5.75 (0.6H, d, J 13.0, OCH, major regioisomer), 5.52 (0.4H, d, J 12.0, OCH, minor regioisomer), 5.35–5.25 (1H, m, OCH), 5.25 (0.6H, d, J 13.0, OCH, major regioisomer), 5.05–4.95 (1H, m, 0.4 OCH, minor regioisomer + 0.6 OCH, major regioisomer), 3.98 (1.2H, s, OCH3, minor regioisomer); 3.88 (1.8H, s, OCH3, major regioisomer), 1.65 (1H, brs, OH); δC (125 MHz, CDCl3), 191.3 (CH), 191.2 (CH), 162.6 (C), 162.3 (C), 157.7 (C), 157.5 (C), 155.9 (C), 153.8 (C), 152.7 (C), 152.5 (C), 151.4 (C), 147.5 (C), 142.5 (CH), 141.1 (CH), 136.0 (C). 134.9 (C), 132.7 (C), 132.5 (C), 131.6 (CH), 130.9 (CH), 130.8 (CH), 129.4 (C), 128.9 (C), 128.5 (CH), 128.1 (CH), 127.2 (C), 126.7 (C), 122.5 (CH), 122.1 (CH), 118.9 (C), 118.5 (C), 115.2 (CH), 114.9 (CH), 113.0 (CH), 111.1 (C), 111.0 (C), 110.9 (CH). 110.7 (CH), 110.1 (CH), 103.6 (CH), 103.5 (CH), 67.1 (CH2), 63.2 (CH2), 60.7 (CH2), 58.4 (CH2), 56.2 (CH3), 56.1 (CH3); m/z (ES-API+) 520.0 ([M + Na]+).
Alcohol 19/benzyl azide Cycloadduct 28
This compound is novel.

Alcohol 19 (30 mg, 0.096 mmol) and benzyl azide 20 (13 mg, 12 μL, 0.096 mmol) were stirred in MeCN (0.6 mL) for 5 days, monitoring each day by TLC (0.16 M in each reagent). At the end of this time the solvent was removed under vacuum and the product purified by flash chromatography on silica gel (hexane:EtOAc, 7:3) to yield the product 28 as a white solid (40 mg, 0.093 mmol, 97%). TLC hexane:EtOAc, 7:3, silica, Rf 0.1; M.p. 126–129 °C; (found (ESI+): [M + Na]+ 452.1579. C25H23N3NaO4 requires 452.1581); νmax 1589, 1492, 1444, 1423, 1335, 1273, 1161, 1138, 1108, 1004, 845, 798, 721 cm−1; δH (500 MHz, CHCl3) (two regioisomers 3:2); 7.45–7.40 (1H, m, ArH), 7.40–7.70 (10H, m, ArH), 5.95 (0.4H, d, J 8.0, CH, minor regioisomer), 5.70–5.55 (2H, m, CH), 5.45–5.25 (2H, m, CH), 5.05 (0.6H, d, J 14.0, CH, major regioisomer), 4.90 (0.4H, d, J 16.0, CH, minor regioisomer). 4.70 (0.4H, d, J 12.0, CH, minor regioisomer). 4.62 (0.6H, d, J 13.0, CH, major regioisomer), 4.58 (1.2H, brs, OCH2Ar, 2 × major regioisomer), 4.54 (0.8H, brs, OCH2Ar, 2 × minor regioisomer), 3.82 (1.4H, s, OCH3, minor regioisomer), 3.80 (1.6H, s, OCH3, major regioisomer), 2.20 (1H, brs, OH). δC (125 MHz, CDCl3) 152.7 (C), 152.4 (C), 151.0 (C), 150.9 (C), 145.5 (C), 144.9 (C), 143.8 (C), 143.7 (C), 136.5 (C), 135.7 (C), 134.0 (C), 133.1 (C), 131.7 (C), 130.9 (C), 130.5 (C), 129.8 (CH), 128.5 (CH), 128.1 (C), 127.9 (C), 127.8 (CH), 127.7, (CH), 127.4 (CH), 127.3 (CH), 126.8 (CH), 126.2 (CH), 124.8 (CH), 123.5 (CH), 121.1 (CH), 112.0 (CH), 110.1 (CH), 110.0 (CH), 109.4 (CH), 66.2 (CH2), 64.0 (CH2), 63.9 (CH2), 61.4 (CH2), 59.8 (CH2), 56.5 (CH2), 55.0 (CH3), 54.8 (CH3), 51.9 (CH2), 50.9 (CH2).
m/z (ES-API+) 452.0 ([M + Na]+). The reaction was followed over time in a separate run using alcohol 19 (15 mg, 0.050 mmol) and benzyl azide (6.5 mg, 0.050 mmol) in CDCl3 (0.4 mL) ([19] ca. 0.13 M). The conversion/time graph for a separate reaction is in the ESI.†
01Dansyl amine alkyne 35
This compound is novel.

N-(2-Aminoethyl)-5-(dimethylamino)naphthalene-1-sulfonamide 34 (100 mg, 0.34 mmol) was added to aldehyde strained alkyne 16 (100.3 mg, 0.34 mmol) in THF (1 mL) and heated to reflux for two hours. The reaction was cooled to ambient temperature, NaBH4 (25.7 mg, 0.68 mmol) was added and stirred for 18 hours. The reaction was diluted with EtOAc (5 mL), washed with sat. aq. NaHCO3 (3 × 5 mL), and dried over MgSO4. Purification by column chromatography (silica; EtOAc/Hex; 20:80 → 50:50) afforded 35 as a waxy green solid (128 mg, 0.22 mmol, 66%). Mp 67–73 °C. (Found (ESI) [M + H]+, 572.2212. C32H34N3O5S requires 572.2214). νmax 2923, 2784, 1587, 1489, 1234, 1102, 1003 and 757 cm−1; δH (500 MHz, CDCl3) 8.52 (1H, d, J 8.5, ArH), 8.31–8.23 (2H, m, ArH), 7.54–7.48 (1H, m, ArH), 7.45–7.37 (2H, m, ArH), 7.22–7.15 (3H, m, ArH), 7.08 (1H, d, J 7.5, ArH), 6.79 (1H, d, J 2.0, ArH), 6.53 (1H, d, J 1.9, ArH), 4.62–4.49 (2H, m, OCH2), 4.43–4.27 (2H, m, OCH2), 3.90 (3H, s, OCH3), 3.45 (2H, d, J 5.3, NCH2), 2.95 (2H, t, J 5.6, NCH2), 2.85 (6H, s, N(CH3)2). δC (126 MHz, CDCl3) 154.5, 153.3, 152.1, 141.5, 136.9, 136.1, 135.9, 134.7, 132.1, 130.5, 130.0, 129.9, 129.7, 129.3, 128.6, 124.4, 123.3, 123.3, 122.7, 118.8, 115.3, 110.9, 87.7, 86.2, 63.7, 60.4, 55.9, 53.0, 47.3, 45.5, 42.6. m/z (ESI) 570 ([M + H]+, 100%) and 606 ([M + Na]+, 10%); UV-Vis (MeCN) λmax (ε/M−1 cm−1): 338 (10500), 234 ((36200) nm; fluorescence (MeCN; λex = 367 nm); λem 510 nm.
2,4-Dimethyl-3-ethyl BoDIPY strained alkyne 36
This compound is novel.

To a solution of alkyne 16 (100 mg, 0.340 mmol, 1.0 eq.) in DCM (22 mL) was added 3-ethyl-2,4-dimethylpyrrole (87.3 mg, 0.1 mL, 0.71 mmol, 2.1 eq.), at which point the solution became red, TFA (3.9 mg, 2 μL, 34 μmol, 0.1 eq.) was added, resulting in the formation of a green solution which was stirred for 3 h, during which time it returned to a red colour. The solution was then washed with a saturated solution of NaHCO3 (20 mL), turning the organic layer yellow, and brine (20 mL). The organic layer was then dried over MgSO4, which was subsequently removed by filtration, and the solvent was evaporated. The residue was then dissolved in toluene (12.3 mL) and a suspension of DDQ (84 mg, 0.37 mmol, 1.1 eq.) in toluene (6 mL) was added, resulting in the solution turning purple. The mixture was then stirred for 1 h before TEA (141 mg, 0.20 mL, 1.4 mmol, 4.1 eq.) was added along with BF3·Et2O (290 mg, 0.25 ml, 2.04 mmol, 6.0 eq.) and the mixture was refluxed at 75 °C for 45 min. The mixture was then cooled before being filtered through a silica plug eluted with DCM. The resultant solution was then concentrated under vacuum to afford the crude product. The product was purified by column chromatography using an eluent of 2:8 EtOAc:pet. ether to afford the pure product 36 as a red/green metallic solid (128 mg, 0.225 mmol, 66%). (Found (ESI) [M + Na]+ 591.2609 C34H35BF2N2NaO3 requires 591.2601; vmax 2962, 2928, 2868, 1536, 1454, 1315, 1182, 972 and 960 cm−1; δH (500 MHz, CDCl3) 7.38 (1H, t, J 7.5, ArH), 7.17–7.22 (1H, m, ArH), 7.14 (1H, d, J 7.9, ArH), 6.87 (1H, d, J 1.5, ArH), 6.78 (1H, d, J 1.7, ArH), 4.62–4.71 (1H, m, CHH), 4.46 (2H, d, J 13.9, 2 × CHH), 4.31–4.39 (1H, m, CHH), 3.89 (3H, s, OCH3), 2.54 (3H, s, CH3), 2.51 (3H, s, CH3), 2.35 (2H, q, J 7.5, CH2CH3), 2.29 (2H, q, J 7.6, CH2CH3), 1.64 (3H, s, CH3), 1.45 (3H, s, CH3), 1.03 (3H, t, J 7.6, CH2CH3), 0.97 (3H, t, J 7.5, CH2CH3); δC (125 MHz, CDCl3) 154.4, 154.1, 153.8, 153.7, 142.7, 139.5, 138.7, 138.2, 138.0, 135.3, 132.7, 131.7, 131.6, 130.8, 130.7, 129.4, 124.3, 124.1, 122.6, 111.2, 87.2, 86.1, 63.3, 60.5, 56.1, 17.2, 17.1, 14.7, 14.6, 12.5, 11.8 and 11.4; δF (376 MHz, CDCl3) −146.53–−145.15 (brm); m/z (ESI) 591 ([M + Na]+). Fluorescence (MeCN; λex = 524 nm); λem = 534 nm; UV-Vis (MeCN) λmax (ε/M−1 cm−1): 520 (14700) nm.
2,4-Dimethyl BoDIPY Strained alkyne 37
This compound is novel.

To a solution of aldehyde 16 (100 mg, 0.340 mmol, 1.0 eq.) in DCM (22 mL), 2,4-dimethylpyrrole (67 mg, 0.71 mmol, 2.1 eq.), TFA (3.8 mg, 34 μmol, 0.1 eq.) was added and the solution was stirred for 3 h. The solution was then washed with a saturated solution of NaHCO3 (20 mL) and brine (20 mL). The organic layer was then dried over MgSO4, which was subsequently removed by filtration, and the solvent evaporated. The residue was then dissolved in toluene (12.3 mL) and a suspension of DDQ (84 mg, 0.37 mmol, 1.1 eq.) in toluene (6 mL) was added. The mixture was then stirred for 1 h before TEA (141 mg, 1.4 mmol, 4.1 eq.) was added along with BF3·Et2O (290 mg, 2.04 mmol, 6.0 eq.) and the mixture was refluxed at 80 °C for 45 min. The mixture was then cooled to rt, before being filtered through a silica plug eluted with DCM. The resulting solution was then concentrated under vacuum to afford the crude product. The crude product was purified by flash column chromatography to afford the pure product 37 as an orange/green metallic solid (38 mg, 0.074 mmol, 22%). Rf = 0.44 (Hexane–EtOAc 3:1); (found (ESI)) [M + Na]+ 535.1983. C30H27BF2N2NaO3 requires 535.1980; vmax 2955, 2922, 2853, 1536, 1504, 1456, 1264 and 964 cm−1; δH (500 MHz, CDCl3) 7.35–7.41 (1H, m, ArH), 7.23–7.25 (1H, m, ArH), 7.17–7.22 (1H, m, ArH), 7.11–7.16 (1H, m, ArH), 6.86 (1H, s, ArH), 6.77 (1H, s, ArH), 6.03 (1H, s, MeCCHCMe), 5.97 (1H, s, MeCCHCMe), 4.66 (1H, d, J 15.4, CHH), 4.40–4.48 (2H, m, 2 × CHH), 4.33 (1H, d, J 15.4, CHH), 3.88 (3H, s, OCH3), 2.56 (3H, s, CH3), 2.53 (3H, s, CH3); δC (125 MHz, CDCl3) 155.5, 155.4, 154.4, 154.2, 143.5, 142.9, 141.0, 138.2, 135.2, 131.5, 131.3, 130.7, 129.4, 124.3, 123.8, 122.6, 121.2, 121.1, 110.8, 87.2, 86.1, 63.2, 60.5, 56.1, 14.5 and 14.1; m/z (ESI) 535 ([M + Na]+); fluorescence (MeCN; λex = 504 nm); λem = 510 nm; UV-Vis (MeCN) λmax (ε/M−1 cm−1): 498 (18407) nm.
BoDIPY strained alkyne 38
This compound is novel.

To a solution of aldehyde 16 (100 mg, 0.340 mmol, 1.0 eq.) in DCM (22 mL) pyrrole (47 mg, 0.71 mmol, 2.1 eq.), TFA (3.8 mg, 34 μmol, 0.1 eq.) was added and the solution was stirred for 3 h. The solution was then washed with a saturated solution of NaHCO3 (20 mL) and brine (20 mL). The organic layer was then dried over MgSO4, which was subsequently removed by filtration, and the solvent evaporated. The residue was then dissolved in toluene (12.3 mL) and a suspension of DDQ (84 mg, 0.37 mmol, 1.1 eq.) in toluene (6 mL) was added. The mixture was then stirred for 1 h before TEA (141 mg, 1.4 mmol, 4.1 eq.) was added along with BF3·Et2O (290 mg, 2.04 mmol, 6.0 eq.) and the mixture was refluxed at 80 °C for 45 min. The mixture was then cooled to rt, before being filtered through a silica plug eluted with DCM. The resulting solution was then concentrated under vacuum to afford the crude product. The crude product was purified by flash column chromatography to afford the pure product 38 as an orange/green metallic solid (42 mg, 0.089 mmol, 26%). Rf = 0.55 (DCM); (found (ESI)) [M + Na]+ 479.1351 C26H19BF2N2NaO3 requires 479.1354; vmax 1545, 1410, 1385, 1261, 1114, 1078 and 956 cm−1; δH (500 MHz, CDCl3) 7.94 (2H, brs, 2 × NCHCHCH), 7.40–7.45 (1H, m, ArH), 7.27–7.31 (2H, m, ArH), 7.23–7.26 (1H, m, ArH), 7.20–7.22 (1H, m, ArH) 7.18–7.20 (2H, m, ArH + NCHCHCH), 7.08–7.10 (1H, m, NCHCHCH), 6.59 (2H, brs, 2 × NCHCHCH), 4.71 (1H, d, J 15.4, CHH), 4.62 (1H, d, J 15.4, CHH), 4.50 (1H, d, J 15.4, CHH), 4.41 (1H, d, J 15.4, CHH), 3.97 (3H, s, OCH3); δC (125 MHz, CDCl3) 154.5, 153.2, 146.8, 145.1, 143.9, 137.3, 134.9, 131.7, 129.8, 126.7, 127.0, 124.4, 122.6, 118.6, 113.6, 87.2, 86.7, 63.7, 60.6, 56.1; m/z (ESI) 479 ([M + Na]+; fluorescence (MeCN; λex = 508 nm); λem = 519 nm; UV-Vis (MeCN) λmax (ε/M−1 cm−1): 497 (58563) nm.
(Me, Et, Me) BoDIPY clicked product 39 and 40
These compounds are novel. We have not confirmed which regioisomer is which, however tentative assignments have been made; full details are in the ESI.†

To a solution of (Me, Et, Me) BoDIPY alkyne (28.4 mg, 0.05 mmol, 1 eq.) in CDCl3 (0.5 mL), benzylazide (0.56 mg, 5.2 μL, 0.05 mmol, 1 eq.) was added. The reaction was followed by 1H NMR. The solvent was then removed under vacuum to give the crude product. This was then purified by flash column chromatography (eluent EtOAc–Hexane gradiant 1:4–1:1), to give the pure products 39 and 40 in a 1:1 ratio as two isolable regioisomers A (first to elute) and B (second to elute). Regioisomer A: (13 mg, 0.0185 mmol, 37%). Rf = 0.4 (EtOAc–Hexane 1:1); (found (ESI)) [M + Na]+ 724.3238. C41H42BF2N5NaO3 requires 724.3248; νmax 2960, 2923, 2853, 1539, 1472, 1314, 1181, 973 and 751 cm−1; δH (500 MHz, CDCl3) 7.29–7.33 (3H, m, ArH), 7.21–7.25 (1H, m, ArH), 7.16 (1H, d, J 8.4, ArH), 7.09–7.14 (2H, m, ArH), 7.04 (1H, d, J 7.0, ArH), 6.95, (1H, t, J 7.4, ArH), 6.87 (1H, s, ArH), 6.82 (1H, s, ArH), 5.75 (1H, d, J 15.6, NCHH), 5.49 (1H, d, J 14.3, OCHH), 5.51 (1H, d, J 12.8, OCHH), 5.43 (1H, d, J 15.6, NCHH), 5.19 (1H, d, J 14.3, OCHH), 4.77 (1H, d, J 12.8, OCHH), 3.87 (3H, s, OCH3), 2.54 (3H, s, CH3), 2.52 (3H, s, CH3), 2.25–2.38 (4H, m, 2 × CH2CH3),1.52 (3H, s, CH3), 1.45 (3H, s, CH3) 1.03 (3H, t, J 7.4, CH2CH3), 0.98 (3H, t, J 7.4, CH3); δC (125 MHz, CDCl3) 154.0, 152.5, 146.9, 145.2, 145.2, 139.4, 135.1, 134.3, 132.9, 132.5, 131.7, 130.6, 129.1, 129.0, 128.7, 128.4, 127.0, 122.4, 121.7, 113.4, 111.5, 63.0, 61.4, 56.2, 52.0, 23.6, 17.1, 12.5, 11.9 and 11.6; m/z (ESI) 724 ([M + Na]+); fluorescence (MeCN; λex = 528 nm); λem = 535 nm; UV-Vis (MeCN) λmax (ε/M−1 cm−1): 521 (14466) nm. Regioisomer B: (13 mg, 0.0185 mmol, 37%). Rf = 0.3 (EtOAc–Hexane 1:1); (Found (ESI)) [M + Na]+ 724.3245 C41H42BF2N5NaO3 requires 724.3241; νmax 2960, 2923, 2853, 1539, 1472, 1314, 1181, 973 and 751 cm−1; δH (500 MHz, CDCl3) 7.42–7.46 (3H, m, ArH), 7.27–7.33 (3H, m, ArH), 7.16 (1H, d, J 8.4, ArH), 7.01–7.06 (1H, m, ArH), 6.90–6.93 (2H, m, ArH), 6.86, (1H, d, J 2.0, ArH), 6.73 (1H, d, J 2.0, ArH), 6.82 (1H, s, ArH), 5.80 (1H, d, J 11.6, OCHH), 5.73 (1H, d, J 14.3, NCHH), 5.53 (1H, d, J 14.3, NCHH), 5.38 (1H, d, J 14.5, OCHH), 5.02 (1H, d, J 14.5, OCHH), 4.95 (1H, d, J 11.6, OCHH), 3.91 (3H, s, OCH3), 2.49–2.55 (6H, m, 2 × CH3), 2.24–2.37 (4H, m, 2 × CH2CH3), 1.53 (3H, s, CH3), 1.39 (3H, s, CH2CH3), 0.94–1.06 (6H, m, 2 × CH3); δc (125 MHz, CDCl3) 153.9, 153.8, 147.3, 144.6, 139.7, 134.3, 133.4, 133.1, 131.4, 130.8, 129.5, 129.3, 129.0, 128.9, 128.8, 127.6, 127.0, 122.3, 122.1, 113.5, 112.1, 67.6, 58.1, 56.3, 52.9, 17.1, 12.5, 12.0, 11.6; m/z (ESI) 724 ([M + Na]+); fluorescence (MeCN; λex = 528 nm); λem = 535 nm; UV-Vis (MeCN) λmax (ε/M−1 cm−1): 521 (14466) nm.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank EPSRC for funding of RCK through a project grant (Ref. EP/M006670/1) and EPSRC/AstraZeneca for funding SF through an NPIF PhD studentship, EPSRC are thanked for supporting AM through the Warwick Impact Acceleration Account (IAA EP/K503848/1) and Warwick University for support of project student ZD. We are grateful for the Polymer Characterisation RTP and Dan Lester for providing access to the GPC and TGA instruments described in the General Experimental. Crystallographic data were collected using an instrument (described in the General Experimental) purchased through support from Advantage West Midlands (AWM) and the European Regional Development Fund (ERDF).References
- Strained alkynes. (a) J. D. Thomas, H. Cui, P. J. North, T. Hofer, C. Rader and T. R. Burke Jr., Bioconjugate Chem., 2012, 23, 2007–2013 CrossRef CAS PubMed; (b) J. Dommerholt, F. P. J. T. Rutjes and F. L. van Delft, Top. Curr. Chem. (Z), 2016, 374, 16 CrossRef PubMed; (c) J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC; (d) M. F. Debets, S. S. van Berkel, J. Dommerholt, A. J. Dirks, F. P. J. T. Rutjes and F. L. van Delft, Acc. Chem. Res., 2011, 44, 805–815 CrossRef CAS PubMed; (e) M. Boudiemeline, C. D. McNitt, T. A. Singleton, V. V. Popik and A. P. Kostikov, Org. Biomol. Chem., 2018, 10, 363–366 RSC; (f) I. Dovgan, S. Erb, S. Hessmann, S. Ursuegui, C. Michel, C. Muller, G. Chaubet, S. Cianférani and A. Wagner, Org. Biomol. Chem., 2018, 10, 1305–1311 RSC; (g) J. C. Brendel, G. Gody and S. Perrier, Polym. Chem., 2016, 7, 5536–5543 RSC.
- Bioorthogonal reactions. (a) E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed; (b) P. Thirumurugan, D. Matosiuk and K. Jowiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef CAS PubMed; (c) J. M. Palomo, Org. Biomol. Chem., 2012, 10, 9309–9318 RSC . Bioorthogonal: Y. Gong and L. Pan, Tetrahedron Lett., 2015, 56, 2123–2332 Search PubMed; (d) C. S. McKay and M. G. Finn, Chem. Biol., 2014, 21, 1075–1101 CrossRef CAS PubMed.
- Cu-Catalysed. (a) E. Haldon, M. C. Nicasio and P. J. Perez, Org. Biomol. Chem., 2015, 13, 9528–9550 RSC; (b) H. Li, R. Aneja and I. Chaiken, Molecules, 2013, 18, 9797–9817 CrossRef CAS PubMed; (c) D. Y. Jackson, Org. Process Res. Dev., 2016, 20, 852–866 CrossRef CAS.
- BCN. (a) J. Dommerholt, S. Schmidt, R. Temming, L. J. A. Hedriks, F. P. J. T. Rutjes, J. C. M. van Hest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422–9425 CrossRef CAS PubMed; (b) G. Stefanetti, Q.-Y. Hu, A. Usera, Z. Robinson, M. Allan, A. Singh, H. Imase, J. Cobb, H. Zhai, D. Quinn, M. Lei, A. Saul, R. Adamo, C. A. MacLennan and F. Micoli, Angew. Chem., Int. Ed., 2015, 54, 13198–13203 CrossRef CAS PubMed.
- (a) Oct: N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed; (b) DIFO: J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Nat. Acad. Sci. U. S. A., 2007, 104, 16793–16797 CrossRef CAS PubMed; (c) DIBO: X. Ning, J. Guo, M. Wolfert and G.-J. Boons, Angew. Chem., Int. Ed., 2008, 47, 2253–2255 CrossRef CAS PubMed; (d) OCT derivative: M. Burk, S. Rothstein and P. Dube, Org., Process Res. Dev., 2010, 22, 108–110 CrossRef; (e) B. D. Fairbanks, E. A. Sims, K. S. Anseth and C. N. Bowman, Macromolecules, 2010, 43, 4113–4119 CrossRef CAS.
- (a) DIBAC: M. F. Debets, S. S. van Berkel, S. Schoffelen, F. P. J. T. Rutjes, J. C. M. van Hest and F. L. van Delft, Chem. Commun., 2010, 46, 97–99 RSC; (b) BARAC: J. C. Jewitt, E. M. Sletten and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 3688–3690 CrossRef PubMed; (c) BARAC C. G. Gordon, J. L. Mackey, J. C. Jewett, E. M. Sletten, K. N. Houk and C. R. Bertozzi, J. Am. Chem. Soc., 2012, 134, 9199–9208 CrossRef CAS PubMed.
- In vivo applications. (a) D. R. Spiciarich, R. Nolley, S. L. Maund, S. C. Purcell, J. Herschel, A. T. Iavarone, D. M. Peehl and C. R. Bertozzi, Angew. Chem., Int. Ed., 2017, 56, 8992–8997 CrossRef CAS PubMed; (b) J. Park, B. Andrade, Y. Seo, M.-J. Kim, S. C. Zimmerman and H. Kong, Chem. Rev., 2018, 118, 1664–1690 CrossRef CAS PubMed; (c) X. Zhang and Y. Zhang, Molecules, 2013, 18, 7145 CrossRef CAS PubMed; (d) K. Lang, L. Davis, S. Wallace, M. Mahesh, D. J. Cox, M. L. Blackman, J. M. Fox and J. W. Chin, J. Am. Chem. Soc., 2014, 134, 10317–10320 CrossRef PubMed.
- (a) K. Kaneda, R. Naruse and S. Yamamoto, Org. Lett., 2017, 19, 1096–1099 CrossRef CAS PubMed; (b) J. Tummatorn, P. Batsomboon, R. J. Clark, I. V. Alabugin and G. B. Dudley, J. Org. Chem., 2012, 77, 2093–2097 CrossRef CAS PubMed; (c) B. Gold, P. Batsomboon, G. B. Dudley and I. V. Alabugin, J. Org. Chem., 2014, 79, 6221–6232 CrossRef CAS PubMed; (d) R. R. Ramsubhag and G. B. Dudley, Org. Biomol. Chem., 2016, 14, 5028–5031 RSC; (e) R. Ni, N. Mitsuda, T. Kashiwagi, K. Igawa and K. Tomooka, Angew. Chem., Int. Ed., 2014, 54, 1190–1194 CrossRef PubMed.
- A. Del Grosso, L.-D. Galanopoulos, C. K. C. Chiu, G. J. Clarkson, P. B. O'Connor and M. Wills, Org. Biomol. Chem., 2017, 15, 4517–4521 RSC.
- (a) U. Koch-Pomeranz, H.-J. Hansen and H. Schmid, Helv. Chim. Acta, 1971, 56, 2981–3004 CrossRef; (b) G. Proni, G. P. Spada, P. Lustenberger, R. Welti and F. Diederich, J. Org. Chem., 2000, 65, 5522–5527 CrossRef CAS PubMed; (c) P. Lustenberge, E. Martinborough, T. M. Denti and F. Diederich, J. Chem. Soc., Perkin Trans. 2, 1998, 747–760 RSC.
- T. Harris, G. dos Passos Gomes, S. Ayad, R. J. Clark, V. V. Lobodin, M. Tuscan, K. Hanson and I. V. Alabugin, Chem., 2017, 3, 629–640 CAS.
- B. Schmidt, M. Riemer and M. Karras, J. Org. Chem., 2013, 78, 8680–8688 CrossRef CAS PubMed.
- R. S. Harapanhalli, L. W. McLaughlin, R. W. Howell, D. V. Rao, S. J. Adelstein and A. I. Kassis, J. Med. Chem., 1996, 39, 4804–4809 CrossRef CAS PubMed.
- Attempted reduction of the ketone with NaBH4 and with an asymmetric transfer hydrogenation (ATH) catalyst was attempted but these reactions were not clean, with evidence of alkyne reduction to alkene. There is precedent for strained alkyne reduction by alcohols; A. Krebs and H. Colberg, Chem. Ber., 1980, 113, 2007–2014 CrossRef CAS.
- (a) M. Aigner, M. Hartl, K. Fauster, J. Steger and K. Bister, ChemBioChem, 2011, 12, 47–51 CrossRef CAS PubMed; (b) K. Sivakumar, F. Xie, B. M. Cash, S. Long, H. N. Barnhill and Q. Wang, Org. Lett., 2004, 6, 4603–4606 CrossRef CAS PubMed; (c) M. T. Gokmen, J. Brassinne, R. A. Prasath and F. E. Du Prez, Chem. Commun., 2011, 47, 4652–4654 RSC.
- (a) S. Sabir, G. Kumar and J. L. Jai, Org. Biomol. Chem., 2018, 10, 3314–3327 RSC; (b) E. M. Brustad, E. A. Lemke, P. G. Schultz and A. A. Deniz, J. Am. Chem. Soc., 2008, 130, 17664–17665 CrossRef CAS PubMed; (c) P. Wu, W. Shui, B. L. Carlson, N. Hu, D. Rabuka, J. Lee and C. R. Bertozzi, Proc. Nat. Acad. Sci. U. S. A., 2009, 106, 3000–3005 CrossRef CAS PubMed.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- A. F. Trindade, R. F. M. Frade, E. M. S. Maçôas, C. Graça, C. A. B. Rodriques, J. M. G. Martinho and C. A. M. Afonso, Org. Biomol. Chem., 2014, 12, 2182–2190 RSC.
- J. P. Bachelet, P. Demerseman and R. Royer, J. Heterocycl. Chem., 1977, 14, 1409–1411 CrossRef CAS.
- R. C. Knighton, M. R. Sambrook, J. C. Vincent, S. A. Smith, C. J. Serpell, J. Cookson, M. S. Vickers and P. D. Beer, Chem. Commun., 2013, 49, 2293–2295 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental details, synthesis of intermediates 7,13 11,12 12,12 13,19 34,20 and compounds 29–31 and 41–45, 1H and 13C NMR spectra, graphs of conversion/time, fluorescence spectra, functionalisation of amino-loaded beads and X-ray crystallographic data. CCDC 1852221, 1852222 and 1852224. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ob01768a. The research data supporting this publication can be accessed at http://wrap.warwick.ac.uk/. |
| This journal is © The Royal Society of Chemistry 2018 |