
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Magnolol dimer-derived fragments as PPARγ-selective probes†
Dominik
Dreier
 a,
Mirta
Resetar
b,
Veronika
Temml
c,
Lukas
Rycek
a,
Nicolas
Kratena
a,
Michael
Schnürch
a,
Daniela
Schuster
de,
Verena M.
Dirsch
b and
Marko D.
Mihovilovic
*a
a,
Mirta
Resetar
b,
Veronika
Temml
c,
Lukas
Rycek
a,
Nicolas
Kratena
a,
Michael
Schnürch
a,
Daniela
Schuster
de,
Verena M.
Dirsch
b and
Marko D.
Mihovilovic
*a
aInstitute of Applied Synthetic Chemistry, TU Wien, Getreidemarkt 9/163, A-1060 Vienna, Austria. E-mail: marko.mihovilovic@tuwien.ac.at
bDepartment of Pharmacognosy, University of Vienna, Althanstraße 14, 1090 Vienna, Austria
cInstitute of Pharmacy/Pharmacognosy and Center for Molecular Biosciences Innsbruck, University of Innsbruck, Innrain 80/82, 6020 Innsbruck, Austria
dInstitute of Pharmacy/Pharmaceutical Chemistry and Center for Molecular Biosciences Innsbruck, University of Innsbruck, Innrain 80/82, 6020 Innsbruck, Austria
eParacelsus Medical University Salzburg, Institute of Pharmacy, Department of Pharmaceutical and Medicinal Chemistry, Strubergasse 21, 5020 Salzburg, Austria
First published on 20th September 2018
Abstract
Partial agonists of the transcription factor PPARγ (peroxisome proliferator-activated receptor γ) have shown potential for the treatment of metabolic and inflammatory conditions and novel activators serve as valuable tool and lead compounds. Based on the natural product magnolol (I) and recent structural information of the ligand–target interaction we have previously developed magnolol dimer (II) which has been shown to have enhanced affinity towards PPARγ and improved selectivity over RXRα (retinoid X receptor α), PPARγ's heterodimerization partner. In this contribution we report the synthesis and evaluation of three fragments of the dimeric lead compound by structural simplifications. Sesqui magnolol A and B (III and IV) were found to exhibit comparable activities to magnolol dimer (II) and selectivity over RXRα persisted. Computational studies suggest a common pharmacophore of the distinctive biphenyl motifs. Truncated magnolol dimer (V) on the other hand does not share this feature and was found to act as an antagonist.
Introduction
PPARs (Peroxisome Proliferator-Activated Receptors) are members of the nuclear receptor family of transcription factors. They require ligand-binding and heterodimerization with the nuclear receptor RXR (Retinoid X Receptor) to activate gene transcription. PPARγ has an established role in adipocyte differentiation and function, lipid and glucose metabolism, and macrophage activation.1,2 PPARγ agonists have been proposed to exhibit beneficial effects on treating some forms of cancer, metabolic and inflammatory conditions.3 The only clinically available PPARγ-targeting drugs, TZDs (thiazolidinediones, e.g. troglitazone, rosiglitazone and pioglitazone) have been used to treat type 2 diabetes due to their insulin sensitizing and anti-hyperlipidemic effects.4 Unfortunately, TZD administration was accompanied by side effects such as weight gain, fluid retention, bone fractures and their usage was later restricted due to hepatotoxicity and heart failure concerns.3,5 The proven beneficial agonistic effects of PPARγ call for development of new ligands, but the challenge remains to develop sufficiently efficacious ligands that would act in a target- and tissue-specific manner. Current efforts are directed towards developing selective PPAR agonists, which would activate desired physiological responses in tissues of interest, exclusively. It has been shown that different partial agonists cause distinct conformational changes that lead to different coactivator recruitment and activation of discrete sets of genes.3 In this work, we report a rational design approach and synthesis towards new synthetic ligands, derived from the structure of the known PPARγ agonist magnolol (I) and magnolol dimer (II), developed previously in our laboratory, employing a modified fragment approach (Fig. 1).6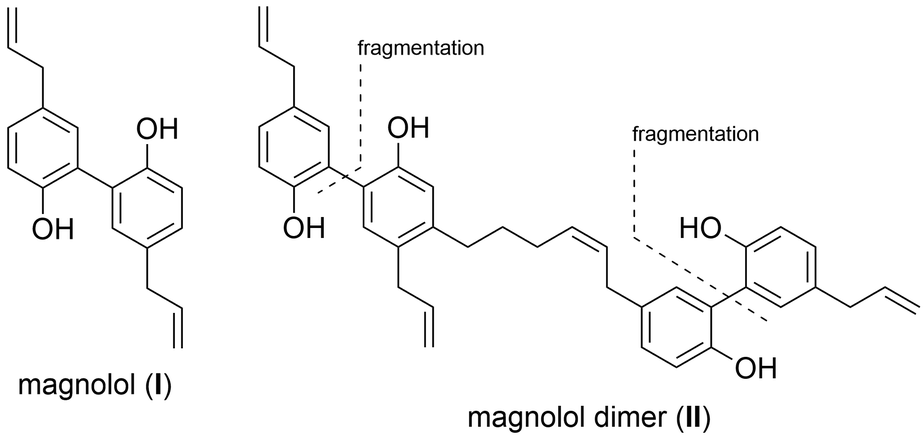 | ||
| Fig. 1 Structure of PPARγ agonists magnolol (I) and magnolol dimer (II). | ||
Magnolol (I) is a lignan found in the bark of the plant Magnolia officinalis which has been used in traditional Chinese medicine as potential anti-inflammatory, anti-cancer, and neuromodulatory agent as well as a lead structure for synthetic derivatives.7,8 We previously demonstrated that magnonol (I) is a partial PPARγ agonist.9 However, magnolol (I) was found to activate RXRα as well.10 Even though RXR agonists themselves can activate PPAR:RXR heterodimers, given that they also activate other permissive heterodimers, and can thus elicit unwanted responses, we aimed to develop PPARγ-specific ligands. In order to do so, we utilized a particular feature of the PPARγ ligand binding pocket – its size, which is substantially larger than in other nuclear receptors.11 Fakhrudin et al. have predicted, which was later confirmed by crystallography, that two magnolol molecules bind into the PPARγ ligand binding site.9,10 Within a trans-disciplinary project aiming at the exploitation of bioactive natural compounds from plants12 as starting point for the design of pharmacological probes,13 we became interested in this particular binding mode of magnolol at the target receptor. Thus, in our previous work, we computationally designed and synthesized magnolol dimer (II) inspired by the dual occupation of the binding site. Magnolol dimer (II) was shown to bind to the PPARγ ligand binding pocket, but not to the one of RXRα.14 Using reporter gene assays, we demonstrated that magnolol dimer (II) can specifically transactivate PPARγ. In the current contribution, we further developed three novel magnolol dimer-based ligands following a reversed fragment based approach. By simplifying the dimeric structure upon “cutting” various rings, we aimed to investigate the influence of the individual aryl moieties on the resulting activity. “Sesqui” magnolol A (III) and sesqui magnolol B (IV) were found to display PPARγ partial agonism while truncated magnolol dimer (V) can act as a PAPRγ antagonist. Here, we report the synthesis, biological evaluation and in silico binding of these compounds, and propose them to be interesting candidates for further evaluation.
Results and discussion
Retrosynthetic analysis
Sesqui magnolol A (III) and sesqui magnolol B (IV) were envisaged by omitting one of the outer phenol rings of magnolol dimer (II). Truncated magnolol dimer (V) results from removing both external phenol domains. These three novel fragments of magnolol dimer (II) should provide valuable insight into the binding behaviour of this compound class.The rationale for the synthesis of the sesqui target compounds III and IV is in line with the reported synthetic route towards magnolol dimer (II) and is depicted in Scheme 1. The phenol groups have to remain protected throughout the synthesis. A very robust protecting group like methyl was thus chosen and global deprotection was planned to be the last step. The two aromatic systems were joined in a Wittig olefination; using a non-stabilized alkyl ylide, e.g.9, to secure the desired (Z)-stereochemistry of the formed double bond. The alkyl chain in 9 might be introduced by lithiation/Grignard formation of aryl bromide 7 and subsequent reaction with desired alkyl electrophile. 2-Bromo-estragole (7) can be obtained by Stille cross-coupling of 2-bromo-1-iodo-4-methoxybenzene (6) with an allylstannane. The iodide 6 is formed by iodination of commercially available 3-bromoanisole (5). The truncated magnolol dimer (V) can likewise be prepared by Wittig reaction of aldehyde 11 with 9 followed by methyl ether deprotection.
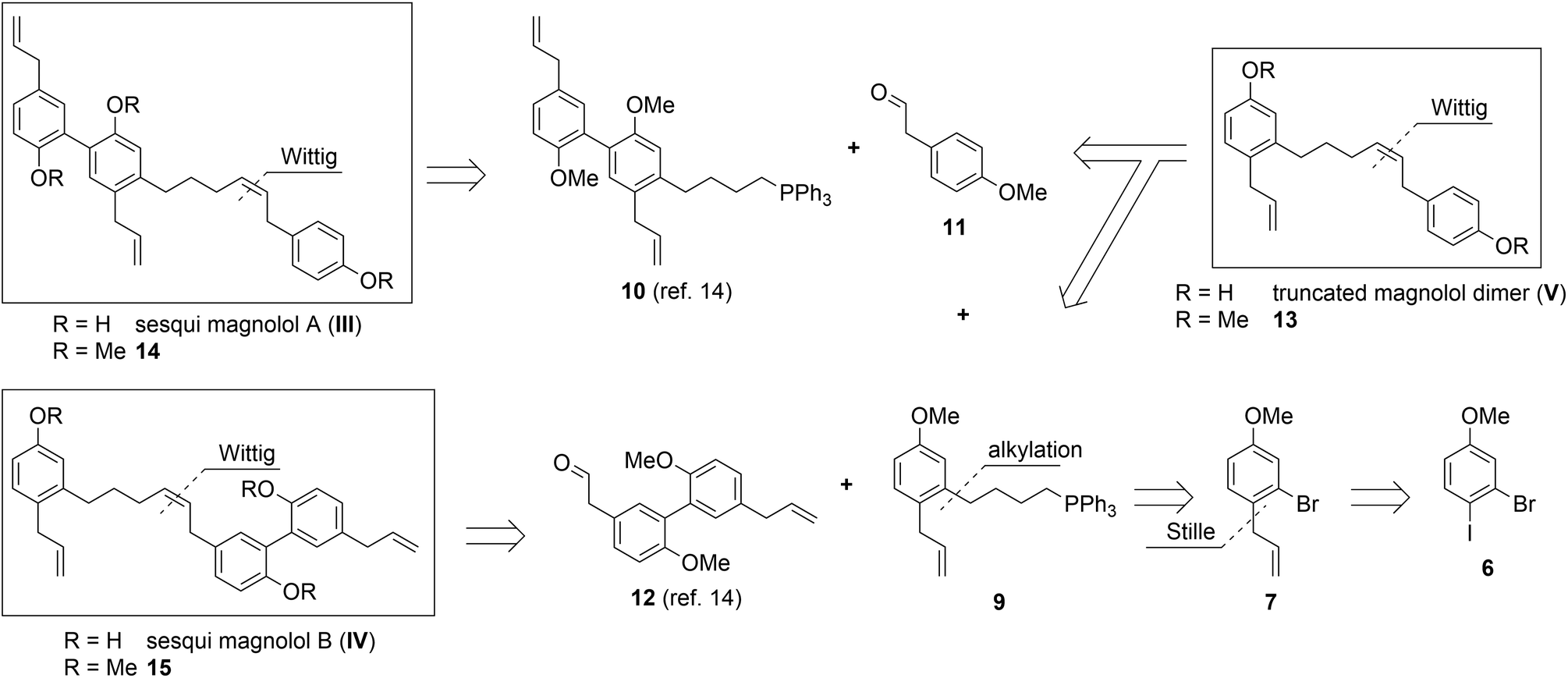 | ||
| Scheme 1 Retrosynthetic analysis of target compounds III, IV and V. | ||
Synthesis
An initial approach towards 9 started from 3-bromo-4-methylaniline 1 (see Scheme 2, 2 and 3 not shown), which was converted to 2-bromo-1-(bromomethyl)-4-methoxybenzene (4) via functional group interconversion, methyl protection, and bromination. Next, dibromide 4 was subjected to Kumada coupling with vinyl magnesium bromide.15,16 Unfortunately, the aryl bromide can undergo facile metal–halogen-exchange under these conditions and formation of the corresponding biaryl dimer and other side-products were observed in considerable quantities. The desired product 7 could be isolated as a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of products in 36% yield.
:1 mixture of products in 36% yield.
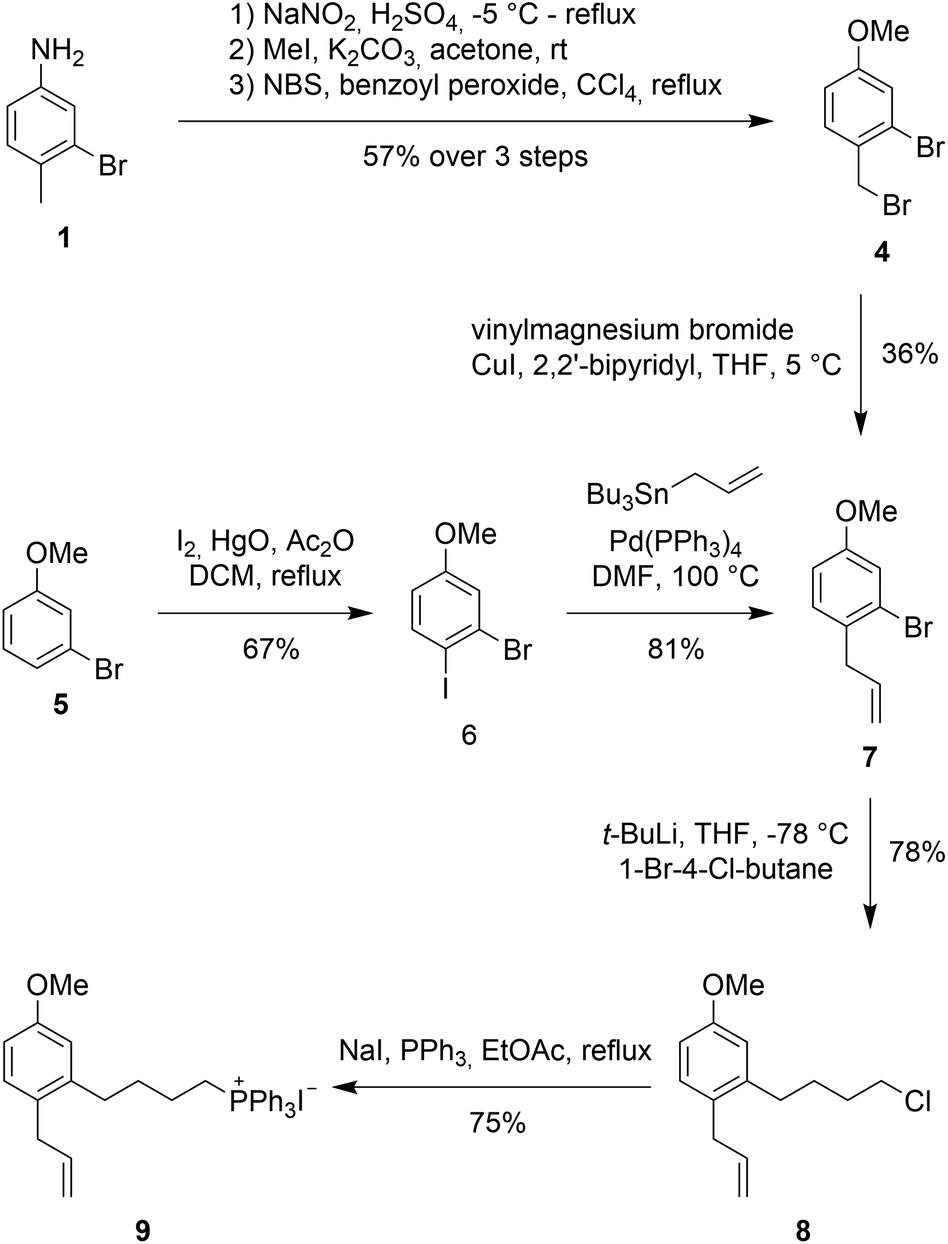 | ||
| Scheme 2 Synthesis of Wittig salt 9. | ||
An improved access to 7 started from 3-bromoanisole (5). Iodination of the aryl system on electronically activated C-2 and C-6 should be suppressed by the steric influences of the methoxy and bromine substituent. Indeed, iodination using in situ formed diiodine monoxide (from HgO and I2)17,18 proceeded to give 2-bromo-1-iodo-4-methoxybenzene (6) in good yield. Less than 10% of the undesired ortho-iodo isomers were formed during this reaction. Employing a Stille coupling with simple tetrakis(triphenylphosphine)palladium in DMF at 100 °C gave allylbenzene 7 after just 30 minutes in 81% yield.
For the introduction of the alkyl side chain we were inspired by a publication by Back et al.19 Lithium halogen exchange of the bromo group in 7 was achieved with 2 equivalents of t-BuLi at −78 °C in THF. Addition of 1-bromo-4-chlorobutane at −78 °C resulted in the desired transformation providing butylchloride 8 as the product in 78% yield.
With alkyl chloride 8 in hand, Wittig salt formation was evaluated. The formation of the phosphonium salt did not proceed directly from the chloride, instead a Finkelstein reaction was required for activation to convert chloride 8 into the corresponding iodide (86% yield). Subsequent substitution with triphenylphosphine proceeded quantitatively. Both transformations could also be performed in a one-pot fashion in EtOAc and delivered the key fragment 9 in 75% yield after recrystallization.
The second building block for the synthesis of sesqui magnolol B (IV) was represented by diaryl aldehyde 12. It was prepared starting from commercially available estragole (16) using a sequence published in earlier work14 (overview see Scheme 3). The first step (oxidative cleavage) gives rise to the second required aldehyde group in 11 (Scheme 1). These 2-phenylacetaldehydes were prone to auto-oxidation as well as degradation to the corresponding benzaldehydes20 and were protected, consequently. Additionally, special conditions were necessary during the coupling of 17 and 18 to prevent double-bond isomerization to the corresponding styrene derivatives.21,22 With all necessary fragments available the synthesis of biological probes could commence. Wittig salt 9 was reacted with aldehydes 11 and 12 giving products 13 and 15, respectively. Diaryl Wittig salt (10) from earlier work14 was reacted with aldehyde 11 furnishing protected sesqui magnolol A (14). Reactions were performed in Et2O using KHMDS as a strong base. Interestingly, upon scale-up this reaction displayed significant olefin isomerization to a number of possible styrene derivatives, which did not occur at a smaller scale. (Z)-Isomers were obtained exclusively in all instances and in good yields (see Scheme 4). Finally, global methoxy deprotection was attained by treatment with BBr3·SMe2 in boiling 1,2-DCE. The di/triphenols III, IV and V were isolated as final products in approximately 60% yield by flash column chromatography.
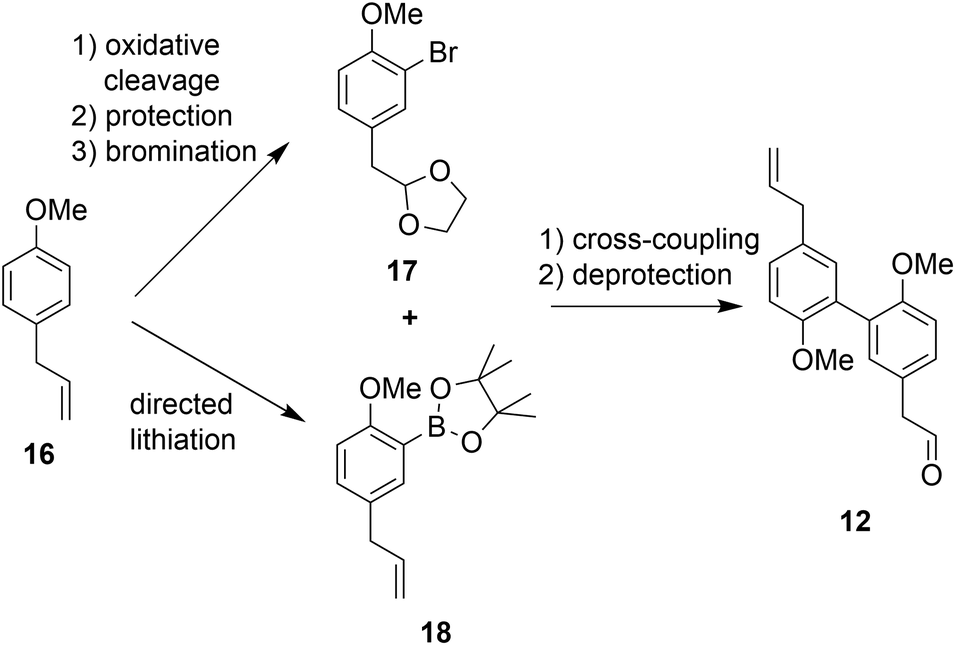 | ||
| Scheme 3 Reported synthesis of building block 12. | ||
 | ||
| Scheme 4 Wittig reaction and deprotection. | ||
Biological evaluation
In order to assess the activation potential of the newly synthesized compounds III, IV and V in a cellular assay, we used a one-hybrid luciferase reporter system. In this model, the PPARγ ligand binding domain (PPARγ-LBD) is combined with the DNA-binding domain of the yeast transcription factor Gal4, which then binds to its response element in the promoter of a luciferase reporter gene. Thus, the ligand-induced transcription of the reporter gene with Gal4 hybrids depends only on ligand binding to the PPARγ-LBD, and not on PPAR:RXR heterodimer formation and permissive activation through RXR.Sesqui magnolol A (III) and sesqui magnolol B (IV) were able to activate the PPARγ-Gal4-dependent transcription of the reporter gene in a dose-dependent manner, similar to magnolol (I, see Fig. 2). Truncated magnolol dimer (V) was unable to induce transactivation. In comparison to magnolol (I: EC50 = 3.24 μM, 95% CI = 1.96–5.33), the two sesqui compounds III and IV were found to be more potent binders (III: EC50 = 1.07 μM, 95% CI = 0.89–1.29, IV: EC50 = 0.77 μM, 95% CI = 0.51–1.17). In comparison to the full PPARγ agonist pioglitazone (not shown on graph), all compounds showed significantly lower Emax values (I: 4.24 fold, 95% CI = 3.35–5.13; III: 3.00 fold, 95%CI = 2.79–3.22, IV: 4.47 fold, 95% CI = 3.82–5.13; pioglitazone: 39.38 fold, 95% CI = 37.31–41.46), suggesting they act as partial agonists which has been a particular goal of this project as partial agonist were proposed to lead to fewer side effects.23
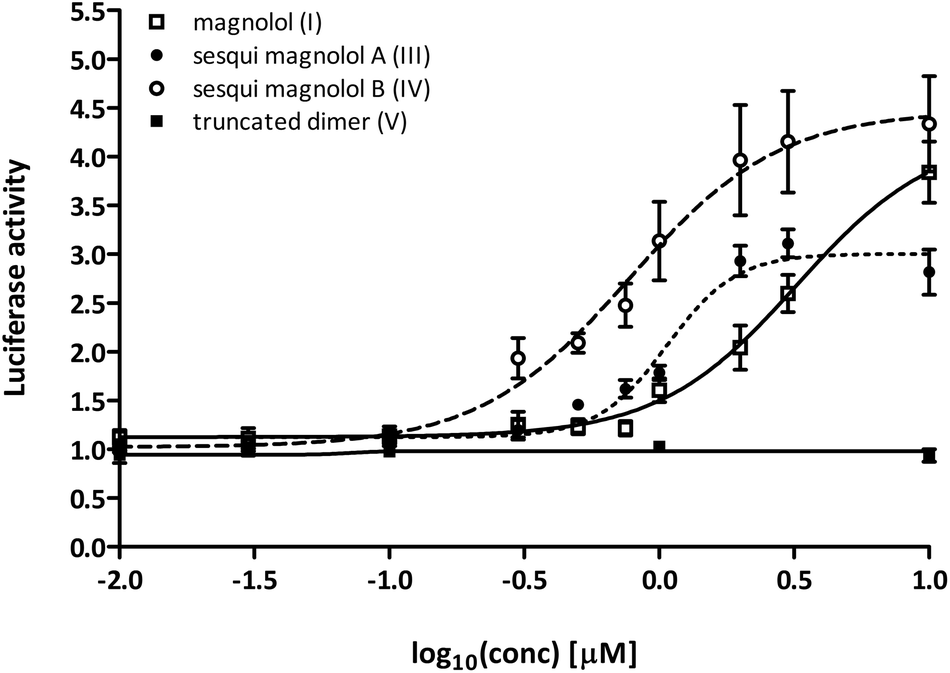 | ||
| Fig. 2 Magnolol (I) and magnolol dimer-based derivatives – sesqui magnolol A (III) and B (IV), but not truncated magnolol dimer (V), induced the PPARγ-Gal4 transactivation of the Luciferase gene expression in a dose-dependent manner. Data points represent mean ± SEM. Concentrations were log-transformed and EC50 and Emax values were obtained using the non-linear fitting of the dose–response data using the variable slope. | ||
We next tested whether the dimer-based derivatives could activate RXRα employing the RXRα-Gal4 hybrid approach. Indeed, none of the dimer-based compounds were able to induce the RXRα-Gal4 transactivation of the Luciferase reporter gene, in contrast to magnolol (I, Fig. 3).
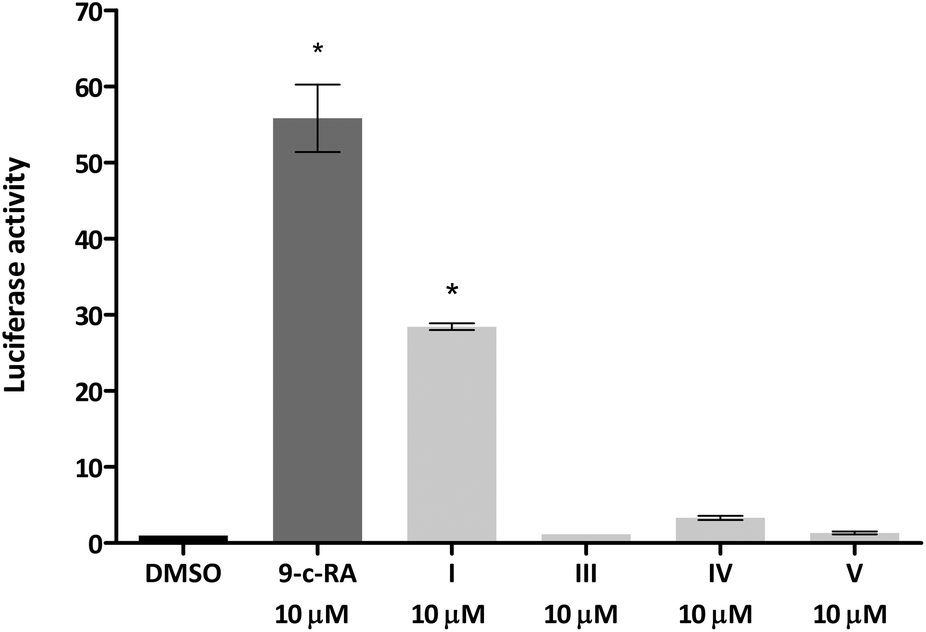 | ||
| Fig. 3 Sesqui magnolol A (III), sesqui magnolol B (IV) and truncated magnolol dimer (V) do not activate the RXRα-Gal4 receptor, in contrast to magnolol (I) and a positive control 9-cis-retinoic acid. Bars show mean ± SEM. *p < 0.01 in comparison to DMSO, one-way ANOVA with Dunett's post hoc test. | ||
Taking the obtained data into account, we hypothesized that sesqui magnolol A (III) and sesqui magnolol B (IV) bind similarly to PPARγ as they display comparable activation. Although they share different molecular regions with the parent compound magnolol dimer (II), one intact biphenyl fragment in III and IV led to a matching behavior. In consequence, we were intrigued whether the two molecules might share a common pharmacophore where the recognition of the biphenyl fragment is the major contribution to the resulting activity. Concerning the inactivity of truncated magnolol dimer (V), we speculated that the molecule's structure is unable to undergo a sufficient number of interactions with the protein due to an amplified simplification of the initial architectures I and II, respectively.
Computational assessment
To test our hypothesis we decided to carry out appropriate in silico studies. The synthesized compounds sesqui magnolol A (III), sesqui magnolol B (IV) and truncated magnolol dimer (V) were computationally docked into the binding site of PPARγ10 to analyze their fit and binding site interactions.The two magnolol molecules in the original crystal structure are displayed in cyan (Fig. 4–6) and referred to as magnolol A (left molecule) and magnolol B (right molecule). Magnolol A of the original crystal structure forms hydrogen bond interactions with Ser342 and a co-crystallized water molecule H2O41, while magnolol B binds to Ser289, Met364 and H2O35. Docking of sesqui magnolol B (IV) exhibited that the intact magnolol fragment aligned to magnolol A and formed the same interactions. The other half of the molecule filled the space occupied by magnolol B in the crystal structure of PPARγ (GoldPLP score: 110.79, Fig. 4). Sesqui magnolol A (III) is indeed predicted to bind in a similar pose with the intact magnolol fragment aligning with magnolol A too and forming similar interactions. (GoldPLP score: 108.21, Fig. 5) The most significant factor of the binding seems to be, whether the structure fills the available space in the binding site, especially where magnolol A binds. Both III and IV form some of the polar interactions of magnolol A and B, while V aligns with the two individual molecules only partly, leaving a large part of the pocket empty, albeit with considerable predicted interactions. This leads to many different proposed docking poses within the binding site and a lower score of 90.80. The program not being able to predict a stable energetic minimum for ligand binding points towards a lower affinity for the target, which corresponds with the score and the experimental findings.
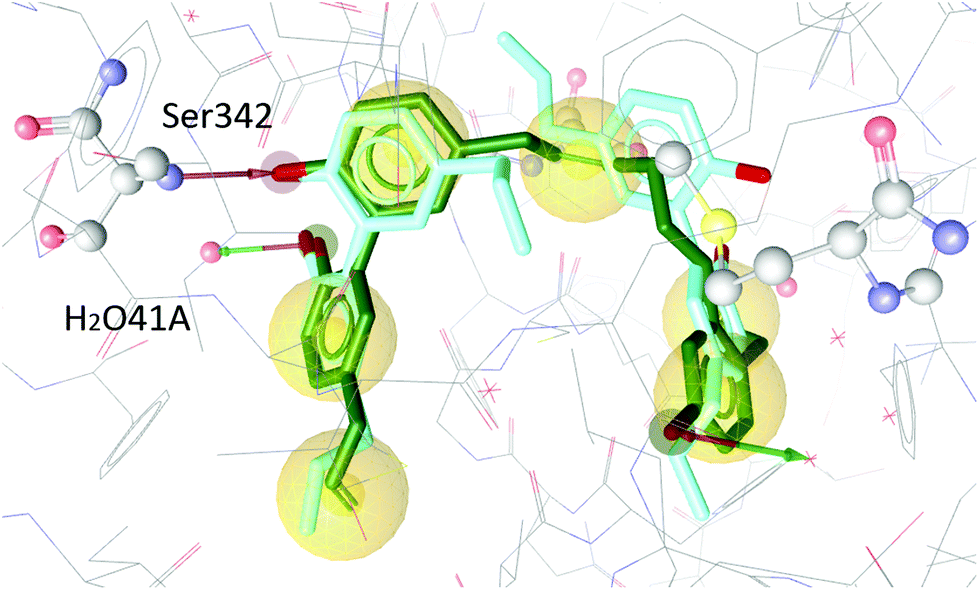 | ||
| Fig. 4 Two magnolol molecules (cyan, A left, B right) co-crystallized in the binding site of PPARγ and the best-ranked docking pose of IV (green) and their interactions with the binding site. Green and red arrows signify hydrogen bonds, yellow spheres mark hydrophobic contacts. Interactions with Ser342 and H2O41 are predicted. | ||
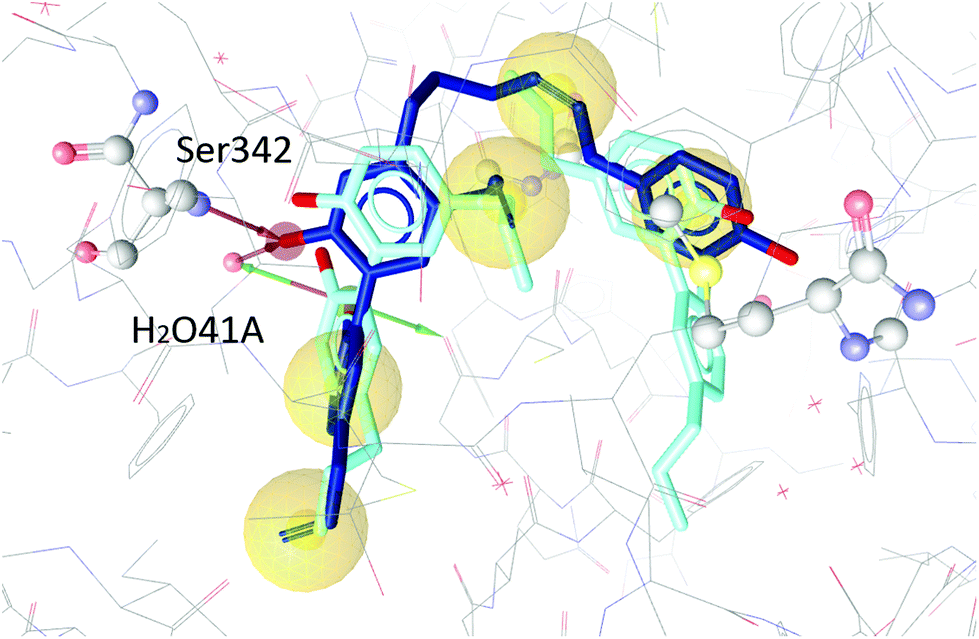 | ||
| Fig. 5 Two magnolol molecules (cyan, A left, B right) co-crystallized in the binding site of PPARγ and the best-ranked docking pose of III (blue) and their interactions with the binding site. Interactions with Ser342 and H2O41 are predicted. | ||
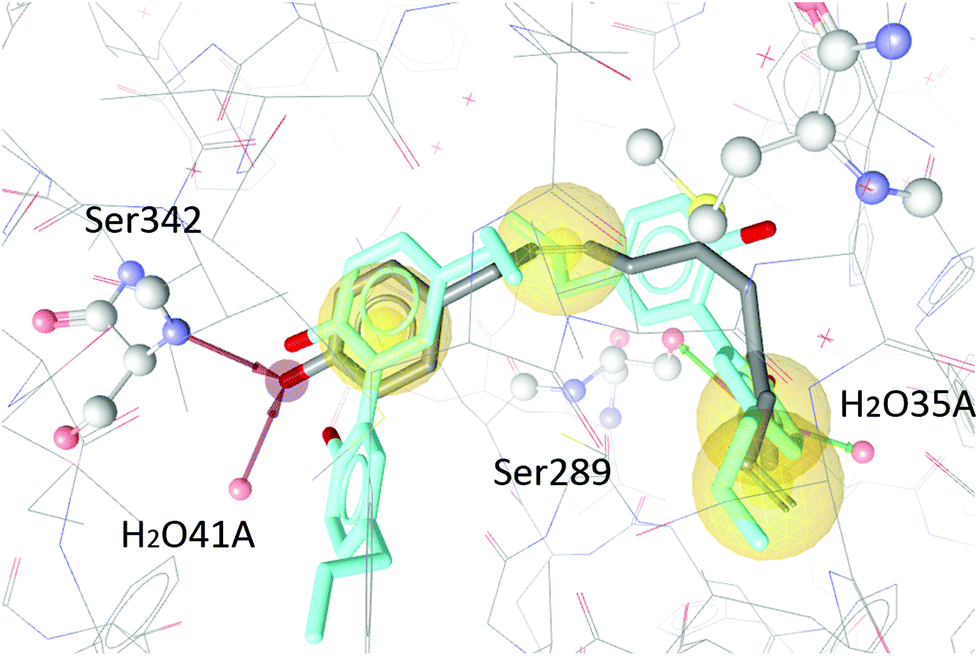 | ||
| Fig. 6 Two magnolol molecules (cyan, A left, B right) co-crystallized in the binding site of PPARγ and the best-ranked docking pose of V (grey) and their interactions with the binding site. Interactions with Ser342, Ser289, H2O41 and H2O35 are predicted. | ||
Given that truncated magnolol dimer (V) did not show activation of the PPARγ-Gal4 receptor, but docking studies indicated that V can fit into the PPARγ ligand binding pocket, this suggested that V could interfere with the PPARγ agonist binding and thus act as an antagonist. We tested this possibility by employing the same approach by co-treating the cells with constant dose of PPARγ full agonist pioglitazone (10 μM) and V as indicated in Fig. 7. Indeed, V showed a dose-dependent inhibition of the pioglitazone induced PPARγ-Gal4 activation.
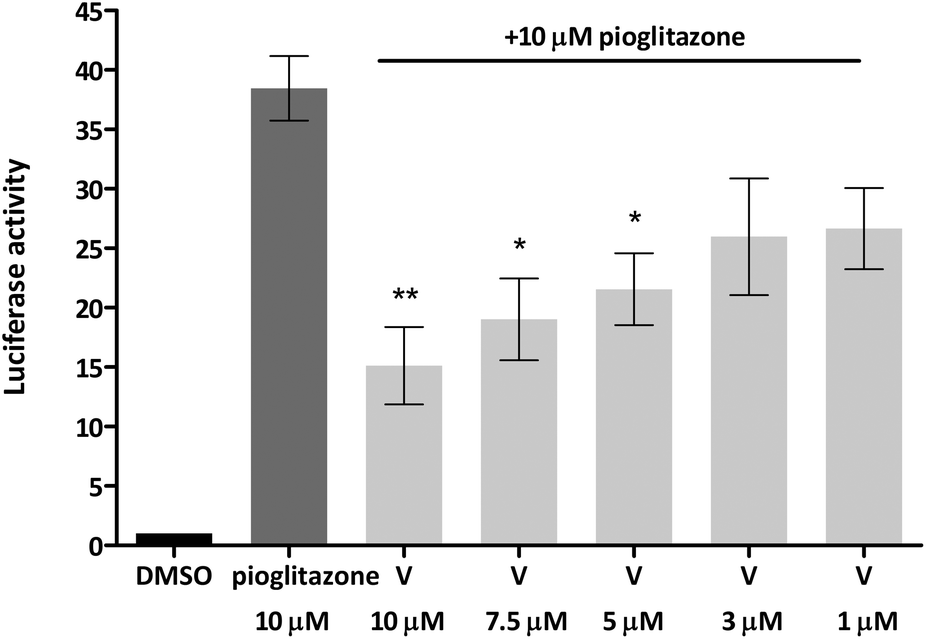 | ||
| Fig. 7 Truncated magnolol dimer (V) dose-dependently inhibits the pioglitazone-induced PPARγ-Gal4 mediated Luciferase gene expression. Bars show mean ± SEM. **p < 0.01, *p < 0.05 in comparison to pioglitazone, one-way ANOVA with Tukey's post hoc test. | ||
In order to understand the antagonistic binding behaviour of V, the compound was additionally docked into an antagonist structure of PPARγ, cocrystallized with betulinic acid.24 In this antagonist structure, V was docked in more conclusive poses than in the agonist structure, moving deeper into the binding pocket and forming different interactions. Compound V occupied a similar space in the pocket as the antagonist betulinic acid and forms interactions with Leu340 and Tyr473 and several water molecules (see Fig. 8).
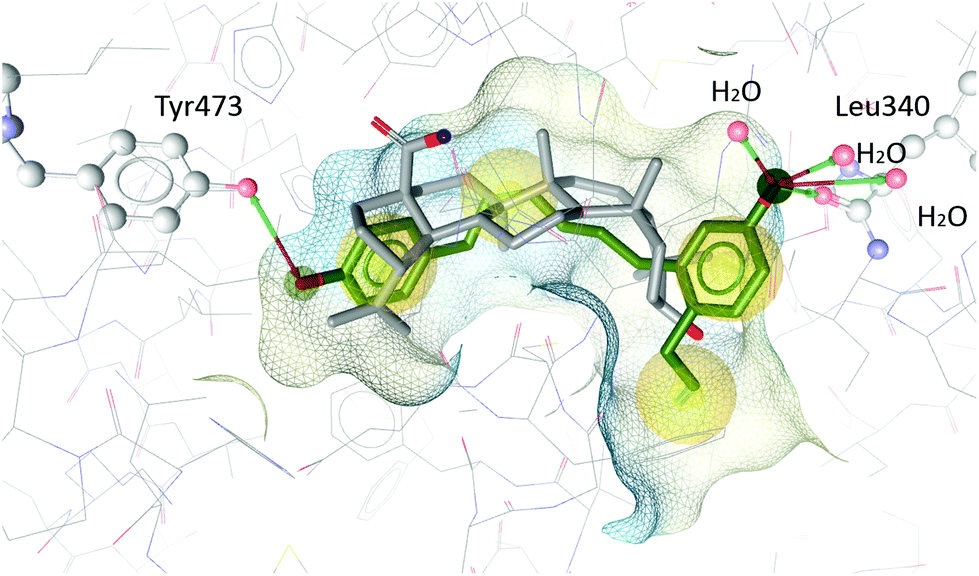 | ||
| Fig. 8 Betulinic acid (grey) co-crystallized in the binding site of PPARγ and the best-ranked docking pose of V (green). Interactions with Tyr473, Leu340 and three water molecules are predicted. | ||
Conclusions
In this report we have designed three novel fragments of the previously disclosed magnolol dimer (II) structure as molecular probes for PPARγ. The new ligands have been successfully synthesized in a straight-forward fashion. In a cell-based reporter gene assay the sesqui magnolol analogs III and IV displayed very similar activity among them and compared to magnolol dimer (II)14 as well acting as partial agonists. The hypothesized common pharmacophore of III and IV was further substantiated by computational docking studies. This suggests that a single biphenyl motif in combination with another aryl domain is sufficient for receptor activation and interactions with Ser342 and H2O41 emerged as crucial features for activity from in silico methods. In contrast to the natural product magnolol (I), the synthetic ligands did not activate RXRα and, hence, serve as a tool for selective PPARγ activation. In truncated magnolol dimer (V) the biphenyl domain is not intact anymore which results in a complete loss of activation. Due to reasonable interaction predicted by docking studies, we experimentally found V to act as a PPARγ antagonist instead. These novel tool compounds, designed by a reversed fragment-based strategy, serve as promising selective PPARγ activators and in combination with the gained insight into ligand–receptor interactions should guide future ligand design.Experimental
General methods
Chemicals were purchased from commercial suppliers and were used without further purification unless otherwise noted. For thin layer chromatography (TLC) aluminium backed silica gel 60 F254 (from Merck) was used. Flash column chromatography was performed with a Büchi Sepacore MPLC system using silica gel 60 M (particle size 40–63 μm) or conventional glass columns. Preparative TLC was performed on glass backed silica gel GF uniplates (1000 microns) from Analtech. Melting points were measured on a Büchi B-545 melting point apparatus and are uncorrected. 1H NMR and 13C NMR spectra were recorded from CDCl3 solutions on a Bruker AC 200 (200 MHz), a Bruker Avance UltraShield 400 (400 MHz) or a Bruker Avance III HD 600 (600 MHz) spectrometer. Chemical shifts are reported in ppm relative to the nominal residual solvent signals of CDCl3: 1H NMR: 7.26 ppm, 13C NMR: 77.16 ppm. HRMS data was measured with a Shimadzu HPLC-IT-TOF mass spectrometer (ESI).HEK293T cells were purchased from ATCC (Manassas, USA), Dulbeccós Modified Eagle Medium (DMEM; 4.5 g L−1 glucose) from Lonza (Basel; Switzerland) and Fetal Bovine Serum (FBS) from ThermoFisher Scientific (Waltham, USA). Plasmids pCMX-Gal4-hPPARγ, pCMX-Gal4-hRXRα, tk(MH1000)-4xLuc and tk-PPREx3-Luc were a gift from Dr Ronald Evans (Salk Institute, La Jolla, USA). Plasmids pSG5-hPPARγ1 and pSG5-hRXRα were a gift from Prof. Walter Wahli and Prof. Beatrice Desvergne (Center for Integrative Genomics, University of Lausanne, Switzerland). pEGFP-N1 was purchased from Takara Bio USA (Mountain View, USA). Magnolol was isolated as described in the literature.9 Pioglitazone was purchased from Molekula (Munich, Germany), 9-cis-retinoic acid from Cayman Chemical (Ann Arbor, USA). All compounds are stored as solutions dissolved in DMSO.
Biological evaluation methods
Luciferase reporter assays were performed in the HEK293 cell line. Cells were cultivated under standard conditions (37 °C, 5% CO2, passage every 3 days) in DMEM Complete containing 10% FBS, 2 mM glutamine, 100 U mL−1 benzylpenicillin and 100 μg mL−1 streptomycin. For transfections, 6 × 106 cells were seeded in 15 cm Petri dishes and incubated overnight. Cells were then transfected using the calcium phosphate method with the following plasmids: for PPARγ-Gal4 and RXRα-Gal4 experiments: 6 μg of pCMX-Gal4-hPPARγ or pCMX-Gal4-hRXRα, respectively and 6 μg tk(MH1000)-4xLuc reporter plasmid were used. Additionally, all cells were co-transfected with 3 μg of pEGFP-N1 to control for transfection efficiency. After 6 hours, cells were washed with PBS and medium was replaced with 5% charcoal-stripped serum DMEM (supplemented with glutamine and antibiotics as DMEM Complete). For compound treatments, 3 × 105 cells per well were seeded in a 96-well plate. Cells were then treated with 0.1% DMSO as vehicle control or with appropriate dilutions of compounds and incubated for 18 hours. Cells were lysed using the Promega Luciferase Cell Culture Lysis 5X Reagent (Mannheim, Germany). Luminescence and fluorescence values were measured on a Tecan Spark instrument (Männedorf, Switzerland). Luminescence values were normalized to EGFP fluorescence and expressed as fold changes relative to the DMSO control. All experiments were performed minimum three times, each with four technical replicates. Data was analyzed using the GraphPad Prism 4 Software (La Jolla).Docking
The ligands were constructed using ChemDraw 15 Professional (PerkinElmer Inc.) and a Pipeline Pilot 8 (Dassault Systems Inc.) protocol translating .cdx into .sd files. The docking simulations were carried out in Gold 5.2 (CCDC, http://www.ccdc.cam.ac.uk, 2015; Cambridge Crystallographic Data Centre, UK; http://www.ccdc.cam.ac.uk/solutions/csd-discovery/components/gold/).27,28 Docking poses and protein–ligand interactions were visualized in LigandScout 4.1 (Inte:Ligand, http://www.inteligand.com/, 2017).For docking into the PPARγ receptor, the X-ray crystal structure of PPARγ 3R5N, a structure of human PPARγ co-crystalized with a magnolol dimer, was employed.10 ChemScore kinase was selected as a configuration template and CHEMPLP was selected as a scoring function.
The protonation state of His323 was set to NE2 and the water molecule no. 35 was set to “toggle and spin”. This means that the docking algorithm can choose to turn, keep or delete this water molecule depending on which setting gives the best docking results. For the docking, both magnolol ligands were removed from the binding site and used to define the binding site location. Docking settings were validated by re-docking of magnolol into the receptor. Magnolol was docked either at the location of one or the other co-crystallized magnolol binding sites with an average RMSD of 0.823 Å.
For docking into the antagonist PPARγ structure, the X-ray crystal structure of PPARγ 5LSG, a structure of human PPARγ co-crystalized with betulinic acid, was employed.24
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Austrian Science Fund (FWF): national research network Drugs from Nature Targeting Inflammation (S 107) and doctural program Molecular Drug Targets (W 1232) to D. Dreier. Theresa Steinacher provided the docking workflow she prepared for her diploma thesis.Notes and references
- M. Lehrke and M. A. Lazar, Cell, 2005, 123, 993 CrossRef PubMed.
- E. D. Rosen, P. Sarraf, A. E. Troy, G. Bradwin, K. Moore, D. S. Milstone, B. M. Spiegelman and R. M. Mortensen, Mol. Cell, 1999, 4, 611 CrossRef PubMed.
- M. B. Wright, M. Bortolini, M. Tadayyon and M. Bopst, Mol. Endocrinol., 2014, 28, 1756 CrossRef PubMed.
- B. Gross, M. Pawlak, P. Lefebvre and B. Staels, Nat. Rev. Endocrinol., 2017, 13, 36 CrossRef PubMed.
- P. Home, Diabetes Care, 2011, 34, 215 CrossRef PubMed.
- D. Fattori, Drug Discovery Today, 2004, 9, 229–238 CrossRef PubMed.
- Y.-J. Lee, Y. M. Lee, C.-K. Lee, J. K. Jung, S. B. Han and J. T. Hong, Pharmacol. Ther., 2011, 130, 157 CrossRef PubMed.
- L. Rycek, R. Puthenkalam, M. Schnuerch, M. Ernst and M. D. Mihovilovic, Bioorg. Med. Chem. Lett., 2015, 25, 400–403 CrossRef PubMed.
- N. Fakhrudin, A. Ladurner, A. G. Atanasov, E. H. Heiss, L. Baumgartner, P. Markt, D. Schuster, E. P. Ellmerer, G. Wolber, J. M. Rollinger, H. Stuppner and V. M. Dirsch, Mol. Pharmacol., 2010, 77, 559 CrossRef PubMed.
- H. Zhang, X. Xu, L. Chen, J. Chen, L. Hu, H. Jiang and X. Shen, PLoS One, 2011, 6, e28253 CrossRef PubMed.
- J. Uppenberg, C. Svensson, M. Jaki, G. Bertilsson, L. Jendeberg and A. Berkenstam, J. Biol. Chem., 1998, 273, 31108 CrossRef PubMed.
- A. G. Atanasov, B. Waltenberger, E.-M. Pferschy-Wenzig, T. Linder, C. Wawrosch, P. Uhrin, V. Temml, L. Wang, S. Schwaiger, E. H. Heiss, J. M. Rollinger, D. Schuster, J. M. Breuss, V. Bochkov, M. D. Mihovilovic, B. Kopp, R. Bauer, V. M. Dirsch and H. Stuppner, Biotechnol. Adv., 2015, 33, 1582–1614 CrossRef PubMed.
- B. Waltenberger, A. G. Atanasov, E. H. Heiss, D. Bernhard, J. M. Rollinger, J. M. Breuss, D. Schuster, R. Bauer, B. Kopp, C. Franz, V. Bochkov, M. D. Mihovilovic, V. M. Dirsch and H. Stuppner, Monatsh. Chem., 2016, 147, 479–491 CrossRef PubMed.
- D. Dreier, L. Rycek, M. Schnurch, M. D. Mihovilovic, S. Latkolik, A. Dymakova, A. G. Atanasov, A. Ladurner, E. H. Heiss, V. M. Dirsch, A. Dymakova, A. G. Atanasov, H. Stuppner and D. Schuster, Sci. Rep., 2017, 7, 13002 CrossRef PubMed.
- J. Knight and P. J. Parsons, J. Chem. Soc., Perkin Trans. 1, 1989, 979 RSC.
- H. Hazimeh, J.-M. Mattalia, C. Marchi-Delapierre, F. Kanoufi, C. Combellas and M. Chanon, Eur. J. Org. Chem., 2009, 2775 CrossRef.
- K. Orito, T. Hatakeyama, M. Takeo and H. Suginome, Synthesis, 1995, 1273 CrossRef.
- C. P. Forbes, A. Goosen and H. A. H. Laue, J. Chem. Soc., Perkin Trans. 1, 1974, 2350 RSC.
- T. G. Back and J. E. Wulff, Chem. Commun., 2002, 1710 RSC.
- J. Duchek, T. G. Piercy, J. Gilmet and T. Hudlicky, Can. J. Chem., 2011, 89, 709 CrossRef.
- R. M. Denton, J. T. Scragg, A. M. Galofre, X. Gui and W. Lewis, Tetrahedron, 2010, 66, 8029 CrossRef.
- X. Jing, W. Gu, X. Ren, P. Bie and X. Pan, J. Chin. Chem. Soc., 2001, 48, 59 CrossRef.
- S. M. Rangwala and M. A. Lazar, Sci. STKE, 2002, 2002, pe9 Search PubMed.
- G. Brusotti, R. Montanari, D. Capelli, G. Cattaneo, A. Laghezza, P. Tortorella, F. Loiodice, F. Peiretti, B. Bonardo, A. Paiardini, E. Calleri and G. Pochetti, Sci. Rep., 2017, 7, 5777 CrossRef PubMed.
- H. J. Lucas and N. F. Scudder, J. Am. Chem. Soc., 1928, 50, 244 CrossRef.
- J. F. Bower, P. Szeto and T. Gallagher, Org. Biomol. Chem., 2007, 5, 143 RSC.
- G. Jones, P. Willett and R. C. Glen, J. Mol. Biol., 1995, 245, 43 CrossRef PubMed.
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ob01745j |
| This journal is © The Royal Society of Chemistry 2018 |